

Abstract
RnBeads is a software tool for large-scale analysis and interpretation of DNA methylation data, providing a user-friendly analysis workflow that yields detailed hypertext reports (http://rnbeads.mpi-inf.mpg.de). Supported assays include whole genome bisulfite sequencing, reduced representation bisulfite sequencing, Infinium microarrays, and any other protocol that produces high-resolution DNA methylation data. Important applications of RnBeads include the analysis of epigenome-wide association studies and epigenetic biomarker discovery in cancer cohorts.
Keywords: DNA methylation analysis, computational epigenetics, whole genome bisulfite sequencing, reduced representation bisulfite sequencing, epigenotyping microarrays, Illumina Infinium HumanMethylation450 assay, bioinformatics software, epigenome-wide association studies, medical epigenomics
DNA methylation is an important epigenetic mark and widely studied in the context of biological processes and diseases. Several assays are now available for mapping DNA methylation genome-wide, at high resolution and in a large number of samples. Whole genome bisulfite sequencing (WGBS) provides comprehensive genome-wide coverage of the approximately 28 million CpGs in the human genome - at the cost of resequencing the whole genome1. Reduced representation bisulfite sequencing (RRBS) focuses the sequencing on a defined subset of DNA fragments that contain at least one CpG each, thereby covering 2-3 million individual CpGs in the human genome2. The Infinium HumanMethylation450 (“450k”) assay uses an adapted genotyping microarray to measure DNA methylation at approximately 0.5 million CpGs3. In addition, enrichment-based assays such as MeDIP-seq4 and restriction enzyme-based assays such as MRE-seq5 can be combined with bioinformatic algorithms to infer high-resolution DNA methylation data for a large proportion of genomic CpGs6. The technical accuracy and reproducibility of these assays is generally high5, 7, but bioinformatic analysis of the resulting datasets remains a complex task with many pitfalls8.
We developed the RnBeads software with the goal of establishing a user-friendly workflow for the analysis and interpretation of large-scale DNA methylation data. RnBeads builds upon extensive prior research on bioinformatic and statistical methods for DNA methylation analysis. We have reviewed the features of 22 related software tools (Supplementary Table 1), and based on our assessment of existing algorithms and software we defined the following key elements of RnBeads: (i) Support for all genome-scale and genome-wide DNA methylation assays that provide single-basepair resolution; (ii) extensive functionality for high-level DNA methylation analysis, including data visualization, quality control, exploratory analysis, handling of batch effects, correction for tissue heterogeneity, and differential DNA methylation analysis; (iii) generation of interactive reports that allow users to select results and adjust parameters without having to rerun the analysis; (iv) implementation of a standardized pipeline mode that is essentially self-configuring, with the additional option to adapt the workflow using custom parameter settings and/or custom scripts; (v) flexibility to run RnBeads on a personal computer, on high-performance computing infrastructure, via a web-based service and in a cloud computing environment, depending on the scale of the analysis; (vi) sufficient performance to process – on a suitable scientific computing cluster – the largest DNA methylation datasets that are currently available (10s of WGBS profiles, 100s of RRBS profiles, or 1000s of Infinium 450k profiles); (vii) reproducibility and easy results sharing through automatic documentation of parameters and analysis methods in the RnBeads report.
To be able to support all protocols for large-scale DNA methylation mapping, RnBeads builds upon existing software tools that can convert raw data into high-resolution DNA methylation profiles. Sequencing data should be preprocessed prior to running RnBeads using software tools such as Bismark9, BSMAP10 and/or Bis-SNP11 (for WGBS/RRBS), MEDIPS12, MEDUSA13 or BayMeth14 (for MeDIP-seq), or methylCRF6 (for MRE-seq). Raw IDAT files from Infinium 450k experiments can be imported directly into RnBeads, in which case the preprocessing and normalization are performed by RnBeads using low-level functionality imported from other R/Bioconductor packages (methylumi, minfi and wateRmelon, cf. Supplementary Table 1). Performing Infinium data normalization in directly RnBeads has practical advantages (e.g., it allows RnBeads to use Infinium quality control probes for pinpointing problematic samples), but it is also possible to load already normalized Infinium data into RnBeads, for example when importing data from the Gene Expression Omnibus resource or from the Illumina GenomeStudio software. In addition to data import, the core workflow of RnBeads comprises quality control, preprocessing and filtering, generation of genome browser tracks and data tables, optional inference of confounding covariates (e.g., different cell type compositions), exploratory analysis and differential DNA methylation analysis (Figure 1 and Online Methods).
Figure 1. RnBeads workflow for analyzing large-scale DNA methylation data.

The RnBeads workflow consists of seven modules and is essentially self-configuring based on a sample annotation table provided by the user. Each module generates part of the RnBeads hypertext report, which includes method descriptions, diagrams, and links to data tables. Furthermore, all data and annotations are stored in RnBSet objects to facilitate custom analysis workflows in R, and they are exported for visualization using genome browsers and follow-up analyses using other software tools.
IDAT, signal intensity data; GS, Illumina GenomeStudio data; TAB, TAB-delimited data; GEO, Gene Expression Omnibus data.
RnBeads is straightforward to run, and the standard pipeline requires an R installation (http://r-project.org) but no prior R programming experience. An RnBeads analysis can be launched using a single command in R: rnb.run.analysis(), which takes a user-provided sample annotation table as input and extracts relevant information needed to automatically configure the analysis. For example, annotation columns containing many unique labels are interpreted as sample identifiers, while columns with several different labels are regarded as sample groups that are to be compared against each other. It is also possible to run some or all steps of the RnBeads workflow interactively and to write R scripts that operate directly on the RnBSet object containing all DNA methylation data and sample annotations of a given analysis. The main result of the RnBeads pipeline is an interactive hypertext report with publication-quality figures (box plots, bar charts, heatmaps, dendrograms, histograms, density plots, quantile-quantile plots, scatter plots, deviation plots, volcano plots, word clouds, etc.) and tables (DNA methylation profiles, ranked lists of differentially methylated regions, attribute enrichment scores, etc.) covering a broad spectrum of topics and analyses. These reports can be viewed from a local directory or over the Internet, and they facilitate data integration with web-based tools such as the UCSC Genome Browser15, Ensembl16, Galaxy17, the WashU Epigenome Browser18, and EpiExplorer19.
To illustrate the practical use of RnBeads, we applied the software to two datasets for which the underlying biology is relatively well understood. The resulting RnBeads reports are available online (http://rnbeads.mpi-inf.mpg.de/examples.php), and it is straightforward to rerun these analyses using the rnb.run.example() function in RnBeads. The first example is based on Infinium 450k profiles for 124 glioblastoma patients generated by The Cancer Genome Atlas (TCGA) project20. We show how RnBeads can identify and characterize samples with glioblastoma CpG island methylator phenotype, an epigenetically defined subtype of brain tumors (Supplementary Figure 1 and Supplementary Note).
The second example focuses on an RRBS dataset describing the DNA methylation dynamics of blood and skin stem cell differentiation in mice21. This dataset comprises 13 blood and 6 skin cell populations with biological replicates and DNA methylation data for slightly more than two million CpGs in each sample. The global distribution of DNA methylation is characteristically bimodal, and discrete peaks at 33%, 50% and 67% DNA methylation disappear after filtering out CpGs with low sequencing coverage (Figure 2a). Exploratory analysis confirms that the difference between blood and skin cell types dominates the analysis (Figure 2b), and DNA methylation levels are generally higher in blood cells than in skin cells when taking regional averages over all annotated genes (Figure 2c). Hierarchical clustering perfectly discriminates between blood and skin cell types (Figure 2d), confirming that DNA methylation patterns tend to be determined more strongly by cellular lineage than by other properties such as cell proliferation or differentiation status.
Figure 2. Analysis of DNA methylation during adult stem cell differentiation.
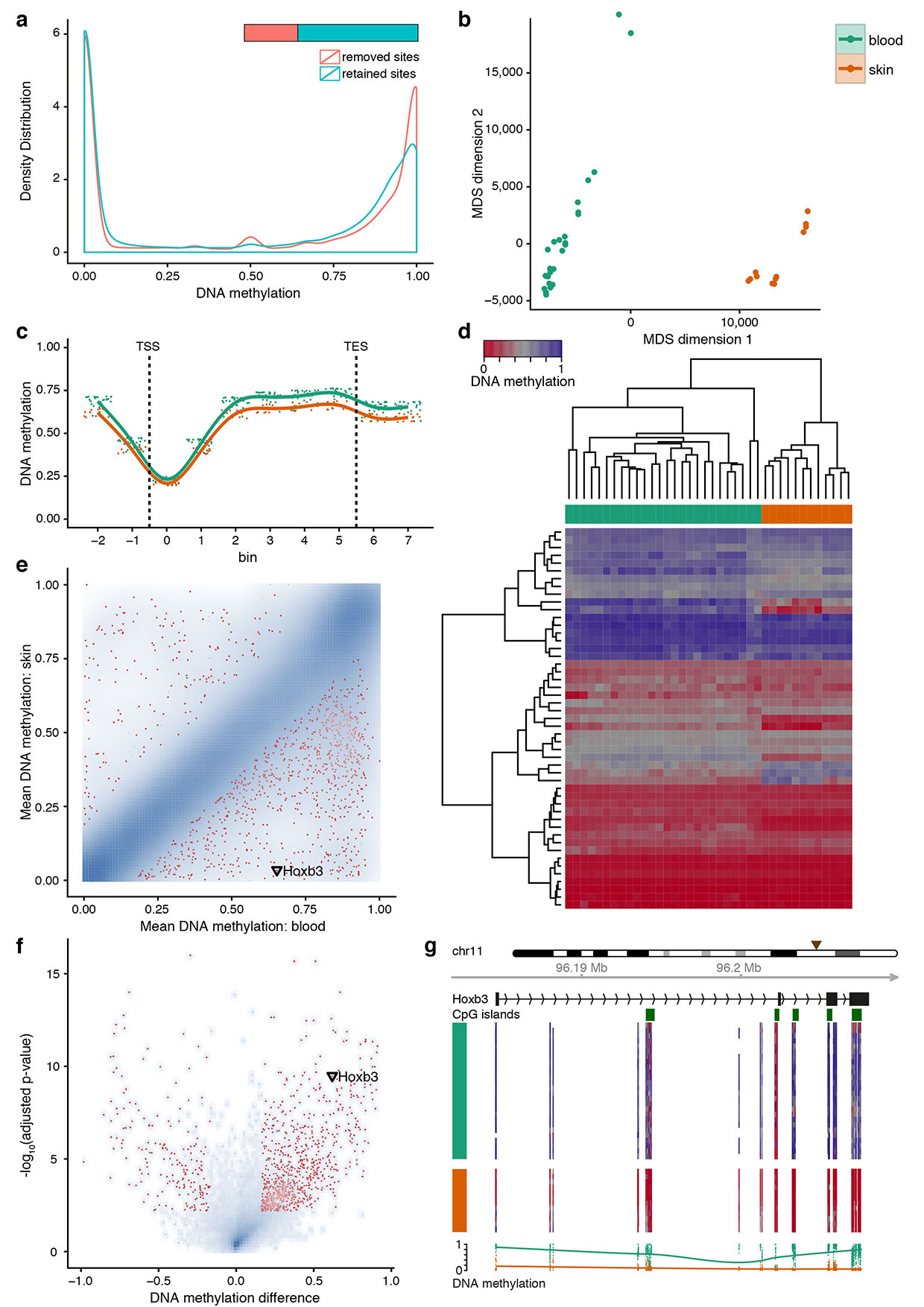
RnBeads was used to re-analyze an RRBS dataset comprising 19 cell types of the blood and skin lineages21. All diagrams shown were calculated by RnBeads but have been reformatted according to journal standards.
(a) Global distribution of DNA methylation levels among retained and removed CpGs after the preprocessing step.
(b) Relative similarity and differences of DNA methylation profiles between cell types. Two maximally informative dimensions were calculated using multi-dimensional scaling (MDS) based on the matrix of average methylation levels in 5kb tiling regions. Samples are color-coded according to cell type.
(c) Composite plot of DNA methylation levels in blood (green) and skin (orange) cell types averaged across all genes. Each gene was covered by six equally sized bins and by two flanking regions of the same size. Smoothing was done using cubic splines.
(d) Heatmap with hierarchical clustering of DNA methylation levels among lineage marker genes that are specifically expressed in the blood lineage. Clustering used average linkage and Manhattan distance.
(e) Scatterplot of groupwise mean DNA methylation levels across genes, with the 1,000 highest-ranking differentially methylated genes highlighted in red. Point density is shown as blue shading.
(f) Volcano plot illustrating effect size and statistical significance across genes, with the 1,000 highest-ranking differentially methylated genes highlighted in red. Point density is shown as blue shading.
(g) DNA methylation profile of the Hoxb3 gene locus on Chromosome 11 (brown triangle). Heatmaps show DNA methylation levels of single CpGs according to the color scheme in panel d. Smoothing of DNA methylation levels (bottom) was done using cubic splines.
RnBeads also identifies differentially methylated regions (DMRs) that are statistically significant and exhibit pronounced DNA methylation differences between the two lineages. This analysis is performed for single CpGs and also for sets of pre-defined genomic regions such as CpG islands, genes, promoters and genome-wide tiling regions. Such region-of-interest based DMR analyses provide an effective way of increasing the statistical power to detect differential DNA methylation, and it also increases the interpretability of identified DMRs8. RnBeads’ priority-ranked list of DMRs between blood and skin cell types comprises many genes with established roles in blood and skin biology, such as members of the homeobox and keratin gene families. Scatterplots provide a convenient way of visualizing the overall frequency of DMRs for a region type of interest (Figure 2e shows data for gene loci as regions of interest), and volcano plots illustrate the relationship between effect size and significance of the DMRs (Figure 2f). In Figure 2, the Hoxb3 gene is highlighted as an example of blood-specific DNA methylation, and we can use the rnb.plot.locus.profile(…) function of RnBeads to produce a genomic view of this locus – thus providing an example of custom R scripting on top of the RnBSet object calculated by the standard pipeline (Figure 2g).
In addition to these two relatively small examples, we assessed the performance of RnBeads when applied to large-scale datasets from the ENCODE, TCGA, IHEC, Roadmap Epigenomics, and BLUEPRINT consortia. All analyses could be completed within reasonable time on a standard scientific computing cluster (Supplementary Table 2), and the resulting Methylome Resource (Supplementary Figure 2 and http://rnbeads.mpi-inf.mpg.de/methylomes.php) provides comprehensive analysis reports for some of the largest publicly available DNA methylation datasets. On this website we also provide preconfigured RnBeads analyses for these large-scale epigenome collections, which can be run along as reference maps when analyzing custom DNA methylation datasets. Such reference-based analyses are particularly valuable for researchers who have generated a specialized DNA methylation dataset and who want to assess the data quality and/or biological relevance in context with a broad range of reference methylomes. The concept of preconfigured and re-runnable analyses of reference epigenome data also provides a means of making data from large-scale epigenome mapping projects more useful for smaller-scale and mechanism-centered studies, thereby contributing to reproducibility, data sharing, and the broader relevance of large-scale epigenome mapping projects22.
Online Methods
RnBeads software overview
RnBeads is written in the R programming language (http://www.r-project.org). It is available under the GPLv3 open source license and has been submitted for inclusion in Bioconductor23. RnBeads follows a modular design that supports automated pipeline workflows as well as flexible interactive analyses. The default RnBeads workflow is executed by running the rnb.run.analysis(…) command, either in an interactive R session or via R’s support for scripted analyses. Optionally, an XML configuration file can be provided in order to execute RnBeads analyses with selected parameter sets. RnBeads analyses can also be run on the Internet using either the RnBeads web service (http://rnbeads.mpi-inf.mpg.de/webservice.php, which is restricted to small datasets, or using the Galaxy integration of RnBeads available from Galaxy tool shed (http://toolshed.g2.bx.psu.edu). On a sufficiently powerful computing infrastructure, RnBeads can process very large cohorts (Supplementary Table 2). To exploit the parallelization options of RnBeads and to avoid out-of-memory problems, users who want to run large analyses should carefully review the corresponding sections in the RnBeads documentation.
When used with default options on small to medium-scale datasets, RnBeads is essentially self-configuring: It parses a user-provided sample annotation table, configures the analysis accordingly and then executes the RnBeads modules as shown in Figure 1. RnBeads workflows can also be fine-tuned using global configuration parameters, which are specified using rnb.options(…). During execution of an RnBeads analysis, each step is tracked by extensive logging functionality. Upon successful completion, the modules write their results into an interactive report comprising method descriptions, publication-quality diagrams and links to data tables. The reports generated by RnBeads use client-side scripting and the dynamic features of XHTML to enable interactive data exploration of pre-calculated results. RnBeads can also save the analysis options and data objects in binary RData objects, which makes it straightforward to rerun an analysis with the same parameters and to comply with the paradigm of reproducible research24. Finally, custom workflows can be designed by running the analysis modules individually or by using R functions that operate directly on RnBSet objects (these objects are instances of an R S4 class and constitute the RnBeads representation of all DNA methylation and metadata within a given dataset). For instance, Figure 2g was created using the function rnb.plot.locus.profile(…) for plotting genome browser like views of individual genomic loci.
The following paragraphs describe the methodology and functionality behind RnBeads and its modules in more detail. Further information on RnBeads is also available in the package vignette (http://rnbeads.mpi-inf.mpg.de/data/RnBeads.pdf), from the example reports (http://rnbeads.mpi-inf.mpg.de/examples.php), and from the other materials on the RnBeads website (http://rnbeads.mpi-inf.mpg.de). RnBeads was also compared with 22 related software tools for DNA methylation analysis in terms of supported assays, data and analysis types, visualizations and other functionalities (Supplementary Table 1). This comparison includes the following software tools: BEAT25, BiSeq26, Bisulfighter27, BSmooth28, ChAMP29, COHCAP30, CpGassoc31, DMEAS32, EpiDiff (QDMR)33, Illumina GenomeStudio34, FastDMA35, Hum-Meth27QCReport36, IMA37, LumiWCluster38, MethLAB39, methyAnalysis (http://www.bioconductor.org/packages/release/bioc/html/methyAnalysis.html)40, methylkit41, MethylSig42, methylumi (http://bioconductor.org/packages/release/bioc/html/methylumi.html)43, minfi44, shinyMethyl (http://shinymethyl.com/)45 and wateRmelon46. For each software tool, the supported features were determined by manual review of the respective publication, software documentation and supplementary material.
Data import
RnBeads supports a broad range of DNA methylation assays, comprising the Illumina Infinium microarray platform (in both its 450k and 27k version), various types of bisulfite sequencing (including WGBS and RRBS) and other sequencing-based methods that can be used to bioinformatically infer DNA methylation measurements at the level of single CpGs (such as MeDIP, MDB-seq and MRE-seq). RnBeads analyses are configured by providing a user-generated sample annotation table that not only identifies the input data files but also includes columns with analysis-relevant information such as tissue types or disease states. RnBeads accepts a broad range of tab-separated or comma-separated text files, and concrete examples of such sample annotation tables are available from the RnBeads website. The data import module of RnBeads parses the annotation table and uses the contained information to configure the analysis, for example identifying the data files that are to be loaded and inferring which specific comparisons may be of interest to the user.
For Infinium microarrays, it is recommended to start the analysis from signal intensity data (IDAT) files and to let RnBeads perform the normalization and DNA methylation calling. Alternatively, RnBeads can load pre-normalized data from Illumina GenomeStudio report files, import Infinium datasets directly from the Gene Expression Omnibus (GEO) database, or read preprocessed data in one of several tabular formats. When IDAT files are loaded into RnBeads, the R/Bioconductor package methylumi43 is internally used for performing the low-level processing. RnBeads offers several alternative options for signal intensity-based normalization, which is an important step to reduce probe biases that could interfere with the analysis. The RnBeads default for Infinium data normalization is SWAN47, which is implemented in the minfi package44 and which – in our experience – provides a good balance of accuracy, robustness, runtime performance and software stability. In addition, RnBeads supports Illumina’s standard normalization procedure as implemented in methylumi, the BMIQ normalization method48, and all modular normalization algorithms that are available in the wateRmelon package46. RnBeads also supports the background correction techniques implemented in methylumi49, which can optionally be combined with the normalization algorithms.
For sequencing-based methods, data preparation requires steps that are highly protocol-dependent, including sequence alignment and DNA methylation calling for single CpGs8. These steps need to be completed prior to loading the data into RnBeads; and the RnBeads analysis starts with importing BED files or data tables that provide the number of methylated and unmethylated observations for each covered CpG. For example, bisulfite sequencing data can be preprocessed with the Bismark software9, whose export format for DNA methylation values is directly supported by RnBeads without the need for file conversion. Furthermore, the combination of BSMAP10, 50 and Bis-SNP11 is well-suited for preprocessing reduced representation bisulfite sequencing data, and the output format of Bis-SNP is also a supported input format for RnBeads. Enrichment-based and restriction-enzyme based assays require specialized algorithms for inferring DNA methylation levels at single-basepair resolution. Software tools such as MEDIPS51, MEDUSA13 and methylCRF6 give rise to DNA methylation tables that can be imported into RnBeads as BED files or in one of several other data file formats.
After the DNA methylation data have been loaded from any of the supported input formats, RnBeads combines the data of all samples into a single RnBSet object that constitutes the basis for all further analysis steps. The RnBeads data matrices become very large when performing genome-wide analyses in large numbers of samples (e.g., up to 100 GB for some of the benchmarking analyses shown in Supplementary Table 2). RnBeads thus provides the option to maintain these matrices on hard disk rather than in main memory using the ff package (http://cran.r-project.org/web/packages/ff/index.html)52, which is essential for performing large analyses on computers with limited memory. The RnBSet object also links the DNA methylation data to genome annotations such as CpG islands, genes and promoters, genome-wide tiling regions and user-defined genomic region sets. RnBeads currently supports the human, mouse and rat genomes with auxiliary data packages named RnBeads.hg19, RnBeads.mm9, RnBeads.mm10 and RnBeads.rn5. The FAQ section on the RnBeads website describes how users can prepare additional genome assemblies for DNA methylation analysis with RnBeads. The RnBSet object primarily stores DNA methylation levels as beta values, which are used by most modules; nevertheless, RnBeads also calculates M-values53 and uses them for the limma analysis as part of the differential DNA methylation module.
Quality control
RnBeads helps the user identify certain technical and biological biases that are common in large-scale DNA methylation datasets, which includes technical assay failures, sample mix-ups, and batch effects (the latter are addressed by the Exploratory Analysis module and described in the corresponding section below). Quality issues are highlighted in the RnBeads reports, but it is ultimately left to the user to handle them appropriately, e.g. by excluding samples with low technical data quality, by resolving sample mix-ups using genotyping data, or by statistically correcting for batch effects. When RnBeads reports significant quality issues it is typically advisable to consult with an experienced statistician, in order to assess whether or not these issues may be symptoms of more severe problems with the study design or the assay that was used.
The detection of technical failures is assay-specific and differs between sequencing-based and microarray-based analyses. For Infinium data, RnBeads plots the microarray’s quality control probes to monitor technical parameters such as bisulfite conversion efficiency and unspecific probe hybridization. For sequencing-based datasets, the quality assessment is largely focused on sequencing coverage, given that bisulfite conversion and clonal read rates are typically dealt with already during alignment and DNA methylation calling.
RnBeads also addresses the relatively common problem of sample mix-ups54, for example using the genotyping probes that are present on the Infinium microarray to confirm sample identity. As illustrated in Supplementary Figure 1a, clustered heatmaps based on genotype measurements provide a straightforward graphical approach for identifying sample duplications and mix-ups, genetically related individuals, and other types of genetic similarity. RnBeads also calculates inter-sample distances based on these genetic data, which enables users to quantitatively compare sample pairs with respect to their genetic similarity. In addition, RnBeads uses DNA methylation data to predict which samples were derived from male and female donors, based on their X-inactivation status and the presence or absence of measurements on the Y-chromosome. This classifier makes it easy to detect discrepancies between gender information from the sample annotation table and the biological sex of the analyzed samples, which are often indicative of sample mix-ups.
Preprocessing
To minimize the risk of measurement biases affecting the analysis, RnBeads implements a framework for rule-based filtering of samples, CpG sites and DNA methylation measurements. Filtering is performed in two steps, in order to provide flexibility and to avoid biasing the normalization procedure of Infinium analyses with problematic samples. First, RnBeads removes low-quality data that could bias an analysis, discarding samples and CpGs that contain a substantial fraction of measurements with low technical quality (e.g., bad detection p-value for Infinium data or low sequencing coverage in the case of bisulfite sequencing data) as well as CpGs and measurements that may be unreliable for other reasons. For example, RnBeads can remove Infinium probes overlapping SNPs that stand a high chance of influencing DNA methylation measurements; and the default pipeline implements a previously published heuristic for identifying such probes55. Users who wish to apply different criteria can also switch off the default filtering in RnBeads and instead provide a custom list of probes or CpGs that should always be excluded. In a second step, RnBeads discards those samples and CpGs that should be included in the normalization but not in the analysis. Examples are CpGs with too many missing values or with zero variability in their methylation values. Furthermore, users can configure additional filtering rules and define a custom blacklist of CpGs that should always be excluded and/or a whitelist of CpGs that should always be retained. The default filtering criteria of RnBeads were chosen relatively conservatively with the goal of reducing the risk of spurious or misleading results. For datasets with significant quality issues, it can be worthwhile to change the filtering criteria in order to remove problematic probes and samples more aggressively, whereas low-coverage bisulfite sequencing data may require more lenient filtering criteria. All filtering is tracked in the RnBeads report, and before-after plots visualize any changes in the global distribution of DNA methylation levels that may arise from the filtering.
Tracks and Tables
Before proceeding with detailed data analysis, RnBeads exports the preprocessed and quality-controlled data in several formats, thus facilitating data visualization with genome browsers and complementary analyses with other software tools. On the one hand, RnBeads provides track hubs that that can be loaded into various genome browsers, thus providing a common reference point for exploring the bigBed and bigWig data tracks that RnBeads generates. On the other hand, the software aggregates the preprocessed data in CSV and BED files that can be loaded and analyzed with custom scripts and with web-based tools such as Galaxy17, the Genomic HyperBrowser56, EpiGRAPH57, and EpiExplorer19. Furthermore, sample-wise statistics including the number of assayed CpGs and genomic regions, the number of assayed CpGs per region type, and the average read coverage (for sequencing data) are summarized in a dedicated table.
Exploratory Analysis
Global changes in DNA methylation can often be identified by visual inspection of the normalized and quality-controlled DNA methylation data, prior to in-depth analysis of differential DNA methylation. To facilitate this type of exploratory analysis, RnBeads visualizes sample-specific DNA methylation profiles at the single-CpG level and for genomic regions of interest. The global distribution of DNA methylation levels is summarized by density plots, which help identify samples and sample groups that deviate from the characteristic bimodal shape with its clear-cut distinction between highly methylated loci and essentially unmethylated loci (e.g., due to global gain or loss of DNA methylation). RnBeads also provides two types of visualization for DNA methylation variation within and across sample groups, which facilitates the detection of hypervariable samples (e.g., due to technical issues or biological effects such as high tissue heterogeneity). The aforementioned DNA methylation profiles are computed not only based on single CpG measurement values, but also based on methylation levels in predefined regions such as gene promoters or CpG islands. Furthermore, if the user includes biological or technical replicates in the analysis and identifies them as such (as described in the package vignette), RnBeads calculates pairwise correlations and visualizes them as scatterplots, thereby providing a global assessment of the reproducibility between the experiments.
Hierarchically clustered heatmaps provide a global assessment of sample subtypes in the dataset. This analysis is quantitatively supported by various distance metrics, by the calculation of silhouette statistics to identify the best fitting number of clusters, and by systematic association testing between the obtained clusters and the user-provided sample annotations. Dimension reduction using principal component analysis and multidimensional scaling is also available within RnBeads. In combination with interactive sample coloring, this functionality provides a powerful way of visualizing associations between sample annotations and global trends in DNA methylation data. Finally, RnBeads generates composite plots of DNA methylation levels around genes and other genomic regions; and these plots can for example help detect global changes in DNA methylation that affect gene promoters differently compared to intragenic or intergenic regions.
The analysis of global trends and associations is also helpful for detecting batch effects, which can arise from technical confounders such as date and duration of sample processing, the person running the assay, and the sample origin. Batch effects are not uncommon in large-scale DNA methylation datasets, in particular among those generated with microarrays or with enrichment sequencing protocols such as MeDIP and MBD-seq. To systematically detect batch effects, RnBeads runs tests for significant association between user-provided sample annotations (we recommend to include at least the sample collection date, the processing date, and the sample origin) and the directions of largest variance identified in a principal component analysis of the DNA methylation dataset. Statistical testing is also performed to identify significant associations among the sample annotations (e.g., in order to identify problematic confounding between collection date and sample type or disease status) and with quality control indicators such as bisulfite conversion rates and non-specific binding (for Infinium data). In these comparisons, RnBeads automatically selects the appropriate statistical test (Fisher’s exact test, Wilcoxon rank sum test, Kruskal-Wallis one-way analysis of variance, or Pearson correlation coupled with a permutation test) based on the type of annotation data. All results are visualized in the RnBeads report, thus providing a systematic assessment of associations between trends in the DNA methylation data and sample annotations.
Differential DNA methylation
DNA methylation differences can be analyzed not only at the level of individual CpGs, but also by combining measurements across larger genomic regions, which increases statistical power and can result in more interpretable sets of differentially methylated regions8, 58. In each comparison defined by the sample annotation table, RnBeads initially computes p-values for all covered CpGs. By default, this analysis is performed with hierarchical linear models as implemented in the limma package59 and using M-values53, which exhibit a distribution that is more consistent with limma’s statistical model assumptions than the beta values that RnBeads uses in most parts of its analysis. Alternatively, by configuring the differential.site.test.method option, p-values can also be calculated using two-sided t-tests or the RefFreeEWAS method60, which is described in more detail in the section on covariate inference below. In addition to the default unpaired analysis, RnBeads also supports paired-samples analysis, which can substantially increase statistical power when analyzing matched pairs such as tumor versus normal or disease-discordant twins. The CpG-level p-values are corrected for multiple testing using the false discovery rate (FDR) method. Furthermore, to obtain aggregate p-values at the level of predefined genomic regions, the uncorrected, CpG-specific p-values within a given region are combined using an extension of Fisher’s method61. This procedure results in a single aggregate p-value for each region, and the aggregate p-values are subjected to multiple testing correction using the FDR method.
In order to address the problem that minimal but consistent differences tend to receive low p-values that do not reflect biological significance, RnBeads ranks the differentially methylated regions according to the combination of statistical significance and effect size. The effect size is estimated in two ways, namely as the absolute difference in DNA methylation and as the relative ratio of mean DNA methylation levels between sample groups. These two measurements differ in their relevance for regions with low versus high DNA methylation levels and thus complement each other. In regions of the genome that exhibit DNA methylation values near 0%, the DNA methylation ratio between sample groups tends to overestimate the effect size, and the absolute DNA methylation difference is a more appropriate measure. The opposite is true for high DNA methylation values near 100%, where the relative ratio is the more stringent and appropriate measure of effect size.
In summary, RnBeads combines statistical testing with a priority ranking scheme that is based on the absolute and relative effect size of the differences between sample groups; and it assigns a combined rank score for differential DNA methylation to each analyzed CpG site and genomic region. This combined rank is defined as the maximum (i.e. worst) of three individual rankings: (i) by absolute difference in mean DNA methylation levels, (ii) by the relative difference in mean DNA methylation levels, which is calculated as the absolute value of the logarithm of the quotient of mean DNA methylation levels, and (iii) by the CpG-based or region-based p-value calculated as described above. The priority-ranked lists can be used directly for downstream analysis, such as manual inspection of the top-ranking regions in a genome browser, or for web-based analysis using tools such as Galaxy and EpiExplorer. In addition to the ranking of differential DNA methylation, RnBeads visualizes the observed differences using scatterplots and volcano plots, and it performs enrichment analysis for Gene Ontology (GO) terms associated with strongly differentially methylated regions.
Covariate inference
Even well-designed studies performed with accurate DNA methylation assays can include confounders and potential sources of batch effects. For example, the samples in an epigenome-wide association study may be collected using different preprocessing steps in different countries or from genetically distinct populations. Furthermore, many large cohort studies are currently being conducted on whole blood, which is characterized by significant cellular heterogeneity. RnBeads implements a number of methods for data correction that can be used to help control such biases.
Batch effects arise from variation in the sample origin or sample handling62, and their effect on the measurements can obscure biologically relevant differences. As long as the batch effects are not too strongly confounded with the biological comparisons of interest, RnBeads together with specialized statistical tools can correct for the resulting biases. To that end, known sources of batch effects (e.g., sample processing date, the microarray slide or the sequencing machine, the origin of clinical samples or the person performing the sample preparation) should be documented by dedicated columns in the sample annotation table, and these columns can then be specified as known confounders when performing the limma-based analysis of differential DNA methylation. RnBeads also integrates the surrogate variable analysis method as implemented in the sva package63 as an optional step of the standard workflow, which can detect batch effects of unknown origin and annotate them in such a way that they can be controlled for as covariates during limma analysis. Furthermore, other methods for batch effect detection such as ComBat64, ISVA65 and RUV-266 can be applied to RnBSet objects as part of custom RnBeads workflows. Any such adjustments should be carefully monitored to avoid introducing additional biases, and it is typically advisable to consult with an experienced statistician when strong batch effects are detected in a dataset.
DNA methylation differences between heterogeneous samples (such as blood, tumor tissue, and most other types of tissue biopsies) can arise not only from cell-intrinsic differences in DNA methylation but also from differences in the cell composition between cases and controls. It is often important to distinguish between these two causes of DNA methylation differences, particularly because they give rise to different biological interpretations67. RnBeads supports three alternative methods for handling cell type heterogeneity in the context of analyzing differential DNA methylation. First, for certain sample types such as whole blood it is possible to purify reference populations of the most prevalent cell types in the heterogeneous sample and to use their DNA methylation as reference for quantifying differences in cell composition between samples68. The estimated cell composition percentages can then be included as covariates in the limma-based analysis of differential DNA methylation. This method is most commonly used for epigenome wide association studies performed on patient cohorts for which only whole blood samples are available69. Suitable reference maps have been generated for the Infinium 450k assay70. Any such reference maps must be generated with the same assay and processed in the same RnBeads analysis to minimize bias. It is also important to assess whether there are any strong batch effects between the reference samples and the samples that are to be analyzed, which can be a major issue when using published reference datasets rather than reestablishing the reference populations in-house. Second, the RefFreeEWAS method has recently been proposed for inferring global trends indicative of cell type heterogeneity directly from the data60. RnBeads supports this method as an alternative to limma and t-tests in the differential DNA methylation module. Third, the FaST-LMMEWASher software provides an alternative approach to reference-free analysis of tissue heterogeneity71, and RnBeads can export preprocessed DNA methylation data in a format that can be directly loaded into FaST-LMM-EWASher. However, users should be aware that especially the reference-free methods are still relatively new and susceptible to various biases in the data, such that the results of these analyses – and in fact of any analysis that attempts to correct for tissue heterogeneity – should be carefully checked for statistical as well as biological plausibility.
Implementation details and package design
RnBeads and its companion data packages currently comprise a code base of approximately 32,000 lines of R code, and they export over 200 functions, classes and methods. To structure all functionality in a flexible and easily understandable way, RnBeads utilizes elements of object-oriented programming available in R, and all DNA methylation data are organized in an R S4 class hierarchy. Each analysis module is implemented as an independent unit operating on an RnBSet object, and the modules write their results into a hypertext report that employs XHTML and JavaScript to enable self-contained interactivity. The RnBeads reports are organized by figures, which are collections of related plots spanning relevant parts of the parameter space. This setup allows users to dynamically explore each figure without the need to rerun the analysis. The ggplot2 package72 is used to generate publication-grade plots, which are incorporated in the reports as bitmaps for quick visualization and as vector graphics for high-resolution printing and for custom postprocessing using vector graphics software. Heatmaps are visualized using the heatmap.2 functionality of the gplots package73. Genome browser like views are created using the Gviz package74.
Scalability and performance
RnBeads has been designed to be scalable to large sample sizes and efficient in its use of computational resources. Parallel computation is implemented using the foreach and doParallel packages; and large R objects can be maintained directly on hard disk using the ff package, which leads to a massive reduction of the memory required for large analyses. Small RnBeads analyses can be completed on a standard personal computer, while large analyses should be run a scientific computing cluster or on adequately powered cloud computing infrastructure. For users who prefer a web-based workflow, a web server supporting analyses with up to 24 samples is available on the RnBeads website. Furthermore, it is relatively straightforward to run RnBeads in an academic or commercial cloud computing environment using an instance of Galaxy CloudMan75, as described in the FAQ section on the RnBeads website. RnBeads has been tested successfully on Infinium datasets comprising thousands of samples, on reduced presentation bisulfite sequencing datasets with hundreds of samples and on whole genome bisulfite sequencing datasets with dozens of deeply sequenced methylomes. Nevertheless, analyses of this scale require careful planning and configuration to avoid out-of-memory problems or excessive runtime. Supplementary Table 2 lists runtime measurements of RnBeads for several large datasets, and the RnBeads documentation provides additional instructions on how to set up large analyses.
Methylome resource
The Methylome Resource on the RnBeads website (http://rnbeads.mpi-inf.mpg.de/methylomes.php) was established by applying RnBeads to the largest public datasets that are currently available for whole genome bisulfite sequencing, for reduced representation bisulfite sequencing, and for the Infinium 450k assay. This resource provides a reference of large-scale DNA methylation analyses that can be used in various ways. For example, researchers can browse through the reports online, explore biological hypotheses, and investigate relevant aspects of the data visually or through custom data analysis with R or other software tools. Furthermore, researchers can download the data and configuration files of the Methylome Resource, add their own DNA methylation data, and then run RnBeads in order to analyze their data in the context of high-quality methylome datasets that span a broad set of tissue types.
For whole genome bisulfite sequencing, the Methylome Resource covers DNA methylation profiles of 41 samples with coverage of 28,158,385 CpGs76. These methylomes are compiled from several sources, including the activities of the Roadmap Epigenomics Project and the International Human Epigenome Consortium77, and they span a broad range of human cell types. For reduced representation bisulfite sequencing, we obtained DNA methylation profiles for 216 samples with coverage of 2,295,083 CpGs from the ENCODE project78, which comprises cell lines and primary samples of various normal and cancerous tissue types79. Finally, for the Illumina Infinium 450k assay, we downloaded raw intensity files for 4,034 primary tumor and normal control samples with microarray coverage of 482,421 CpGs, which have been collected by the TGCA consortium20. All data were processed according to a standardized RnBeads workflow, and these analyses could be completed in a few days on a standard scientific computing cluster (Supplementary Table 2).
Availability and website
Additional materials, including the RnBeads download, the package vignette, the source code, an RnBeads web service, commands and configurations for cloud-based RnBeads analysis, example analysis reports, the methylome resource, documentation and FAQs are available on the RnBeads website (http://rnbeads.mpi-inf.mpg.de).
Supplementary Material
Acknowledgements
We would like to thank D. Brocks, H. Hernandez-Vargas, A. Houseman, E. Schneider, A. Schönegger and all users of RnBeads for their extensive testing and feedback. Furthermore, we would like to thank G. Friedrich, J. Büch and the Information Services and Technology team at the Max Planck Institute for technical support. This work is funded in part by the European Union’s Seventh Framework Programme (FP7/2007-2013) grant agreement No. 282510 (BLUEPRINT) and grant agreement No. 267038 (NOTOX) as well as by the German Science Ministry grant No. 01KU1216A (DEEP).
References
- 1.Lister R, et al. Nature. 2009;462:315–322. doi: 10.1038/nature08514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gu H, et al. Nat. Methods. 2010;7:133–136. doi: 10.1038/nmeth.1414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bibikova M, et al. Genomics. 2011;98:288–295. doi: 10.1016/j.ygeno.2011.07.007. [DOI] [PubMed] [Google Scholar]
- 4.Down TA, et al. Nat. Biotechnol. 2008;26:779–785. doi: 10.1038/nbt1414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Harris RA, et al. Nat. Biotechnol. 2010;28:1097–1105. doi: 10.1038/nbt.1682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Stevens M, et al. Genome Res. 2013;23:1541–1553. doi: 10.1101/gr.152231.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bock C, et al. Nat. Biotechnol. 2010;28:1106–1114. doi: 10.1038/nbt.1681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bock C. Nat. Rev. Genet. 2012;13:705–719. doi: 10.1038/nrg3273. [DOI] [PubMed] [Google Scholar]
- 9.Krueger F, Andrews SR. Bioinformatics. 2011;27:1571–1572. doi: 10.1093/bioinformatics/btr167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Xi Y, et al. Bioinformatics. 2012;28:430–432. doi: 10.1093/bioinformatics/btr668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Liu Y, Siegmund KD, Laird PW, Berman BP. Genome Biol. 2012;13:R61. doi: 10.1186/gb-2012-13-7-r61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lienhard M, Grimm C, Morkel M, Herwig R, Chavez L. Bioinformatics. 2014;30:284–286. doi: 10.1093/bioinformatics/btt650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wilson G, et al. GigaScience. 2012;1:3. doi: 10.1186/2047-217X-1-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Riebler A, et al. Genome Biol. 2014;15:R35. doi: 10.1186/gb-2014-15-2-r35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Meyer LR, et al. Nucleic Acids Res. 2013;41:D64–69. doi: 10.1093/nar/gks1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Flicek P, et al. Nucleic Acids Res. 2013;41:D48–55. doi: 10.1093/nar/gks1236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Giardine B, et al. Genome Res. 2005;15:1451–1455. doi: 10.1101/gr.4086505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhou X, et al. Nat. Methods. 2011;8:989–990. doi: 10.1038/nmeth.1772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Halachev K, Bast H, Albrecht F, Lengauer T, Bock C. Genome Biol. 2012;13:R96. doi: 10.1186/gb-2012-13-10-r96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Weisenberger DJ. J. Clin. Invest. 2014;124:17–23. doi: 10.1172/JCI69740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bock C, et al. Mol. Cell. 2012;47:633–647. doi: 10.1016/j.molcel.2012.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bock C. Genome Med. 2014;6:41. doi: 10.1186/gm557. [DOI] [PMC free article] [PubMed] [Google Scholar]
Methods-only References
- 23.Gentleman RC, et al. Genome Biol. 2004;5:R80. doi: 10.1186/gb-2004-5-10-r80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gentleman R, Temple Lang D. Bioconductor Project Working Paper 2. 2004.
- 25.Akman K, Haaf T, Gravina S, Vijg J, Tresch A. Bioinformatics. 2014;30:1933–1934. doi: 10.1093/bioinformatics/btu142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hebestreit K, Dugas M, Klein HU. Bioinformatics. 2013;29:1647–1653. doi: 10.1093/bioinformatics/btt263. [DOI] [PubMed] [Google Scholar]
- 27.Saito Y, Tsuji J, Mituyama T. Nucleic Acids Res. 2014;42:e45. doi: 10.1093/nar/gkt1373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hansen KD, Langmead B, Irizarry RA. Genome Biol. 2012;13:R83. doi: 10.1186/gb-2012-13-10-r83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Morris TJ, et al. Bioinformatics. 2014;30:428–430. doi: 10.1093/bioinformatics/btt684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Warden CD, et al. Nucleic Acids Res. 2013;41:e117. doi: 10.1093/nar/gkt242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Barfield RT, Kilaru V, Smith AK, Conneely KN. Bioinformatics. 2012;28:1280–1281. doi: 10.1093/bioinformatics/bts124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.He J, Sun X, Shao X, Liang L, Xie H. Bioinformatics. 2013;29:2044–2045. doi: 10.1093/bioinformatics/btt332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhang Y, Su J, Yu D, Wu Q, Yan H. Conf Proc IEEE Eng Med Biol Soc. 2013;2013:655–658. doi: 10.1109/EMBC.2013.6609585. [DOI] [PubMed] [Google Scholar]
- 34.Illumina, Inc. GenomeStudio software data sheet. 2014 http://www.illumina.com/software/genomestudio_software.ilmn.
- 35.Wu D, Gu J, Zhang MQ. PLoS One. 2013;8:e74275. doi: 10.1371/journal.pone.0074275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mancuso FM, Montfort M, Carreras A, Alibes A, Roma G. BMC Res Notes. 2011;4:546. doi: 10.1186/1756-0500-4-546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang D, et al. Bioinformatics. 2012;28:729–730. doi: 10.1093/bioinformatics/bts013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kuan PF, Wang S, Zhou X, Chu H. Bioinformatics. 2010;26:2849–2855. doi: 10.1093/bioinformatics/btq553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kilaru V, Barfield RT, Schroeder JW, Smith AK, Conneely KN. Epigenetics. 2012;7:225–229. doi: 10.4161/epi.7.3.19284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Du P, Bourgon R. methyAnalysis: DNA methylation data analysis and visualization. Reference manual. 2014 http://bioconductor.org/packages/release/bioc/html/methyAnalysis.html.
- 41.Akalin A, et al. Genome Biol. 2012;13:R87. doi: 10.1186/gb-2012-13-10-r87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Park Y, Figueroa ME, Rozek LS, Sartor MA. Bioinformatics. 2014 doi: 10.1093/bioinformatics/btu339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Davis S, Du P, Bilke S, Triche TJ, Jr., Bootwalla M. methylumi: Handle Illumina methylation data. Reference manual. 2014 http://bioconductor.org/packages/release/bioc/html/methylumi.html.
- 44.Aryee MJ, et al. Bioinformatics. 2014.
- 45.Fortin J-P, Hansen KD. shinyMethyl: Interactive visualization for Illumina’s 450k methylation arrays. Reference manual. 2014. http://www.bioconductor.org/packages/devel/bioc/html/shinyMethyl.html. [DOI] [PMC free article] [PubMed]
- 46.Pidsley R, et al. BMC Genomics. 2013;14:293. doi: 10.1186/1471-2164-14-293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Makismovic J, Gordon L, Oshlack A. Genome Biol. 2012;13:R44. doi: 10.1186/gb-2012-13-6-r44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Teschendorff AE, et al. Bioinformatics. 2013;29:189–196. doi: 10.1093/bioinformatics/bts680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Triche TJ, Jr., Weisenberger DJ, Van Den Berg D, Laird PW, Siegmund KD. Nucleic Acids Res. 2013;41:e90. doi: 10.1093/nar/gkt090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Xi Y, Li W. BMC Bioinformatics. 2009;10:232. doi: 10.1186/1471-2105-10-232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chavez L, et al. Genome Res. 2010;20:1441–1450. doi: 10.1101/gr.110114.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Adler D, Gläser C, Nenadic O, Oehlschlägel J, Zucchini W. ff: Memory-efficient storage of large data on disk and fast access functions. Reference manual. 2014. http://cran.r-project.org/web/packages/ff/index.html.
- 53.Du P, et al. BMC Bioinformatics. 2010;11:587. doi: 10.1186/1471-2105-11-587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Westra HJ, et al. Bioinformatics. 2011;27:2104–2111. doi: 10.1093/bioinformatics/btr323. [DOI] [PubMed] [Google Scholar]
- 55.Nordlund J, et al. Genome Biol. 2013;14:r105. doi: 10.1186/gb-2013-14-9-r105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sandve GK, et al. Nucleic Acids Res. 2013;41:W133–141. doi: 10.1093/nar/gkt342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bock C, Halachev K, Büch J, Lengauer T. Genome Biol. 2009;10:R14. doi: 10.1186/gb-2009-10-2-r14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bock C, Walter J, Paulsen M, Lengauer T. Nucleic Acids Res. 2008;36:e55. doi: 10.1093/nar/gkn122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Smyth GK. Stat. Appl. Genet. Mol. Biol. 2004;3 doi: 10.2202/1544-6115.1027. Article 3. [DOI] [PubMed] [Google Scholar]
- 60.Houseman EA, Molitor J, Marsit CJ. Bioinformatics. 2014. [DOI] [PMC free article] [PubMed]
- 61.Makambi KH. Journal of Applied Statistics. 2003;30:225–234. [Google Scholar]
- 62.Leek JT, et al. Nat. Rev. Genet. 2010;11:733–739. doi: 10.1038/nrg2825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Leek JT, Johnson WE, Parker HS, Jaffe AE, Storey JD. Bioinformatics. 2012;28:882–883. doi: 10.1093/bioinformatics/bts034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Johnson WE, Li C, Rabinovic A. Biostatistics. 2007;8:118–127. doi: 10.1093/biostatistics/kxj037. [DOI] [PubMed] [Google Scholar]
- 65.Teschendorff AE, Zhuang J, Widschwendter M. Bioinformatics. 2011;27:1496–1505. doi: 10.1093/bioinformatics/btr171. [DOI] [PubMed] [Google Scholar]
- 66.Gagnon-Bartsch JA, Speed TP. Biostatistics. 2012;13:539–552. doi: 10.1093/biostatistics/kxr034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Jaffe AE, Irizarry RA. Genome Biol. 2014;15:R31. doi: 10.1186/gb-2014-15-2-r31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Houseman EA, et al. BMC Bioinformatics. 2012;13:86. doi: 10.1186/1471-2105-13-86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Michels KB, et al. Nat. Methods. 2013;10:949–955. doi: 10.1038/nmeth.2632. [DOI] [PubMed] [Google Scholar]
- 70.Reinius LE, et al. PLoS One. 2012;7:e41361. doi: 10.1371/journal.pone.0041361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Zou J, Lippert C, Heckerman D, Aryee M, Listgarten J. Nat. Methods. 2014;11:309–311. doi: 10.1038/nmeth.2815. [DOI] [PubMed] [Google Scholar]
- 72.Wickham H. ggplot2: elegant graphics for data analysis. Springer Publishing Company, Incorporated; 2009. [Google Scholar]
- 73.Warnes GR, Bolker B, Bonebakker L, Gentleman R, Huber W, Liaw A, Lumley T, Maechler M, Magnusson A, Moeller S, Schwartz M, Venables B. Various R programming tools for plotting data. Reference manual. 2014. http://cran.r-project.org/web/packages/gplots/gplots.pdf.
- 74.Hahne F, et al. Gviz: Plotting data and annotation information along genomic coordinates. Reference manual. 2014. http://bioconductor.org/packages/release/bioc/html/Gviz.html.
- 75.Afgan E, et al. BMC Bioinformatics. 2010;11(Suppl 12):S4. doi: 10.1186/1471-2105-11-S12-S4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Ziller MJ, et al. Nature. 2013;500:477–481. doi: 10.1038/nature12433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Satterlee JS, Schübeler D, Ng HH. Nat. Biotechnol. 2010;28:1039–1044. doi: 10.1038/nbt1010-1039. [DOI] [PubMed] [Google Scholar]
- 78.ENCODE Project Consortium Science. 2004;306:636–640. doi: 10.1126/science.1105136. [DOI] [PubMed] [Google Scholar]
- 79.Varley KE, et al. Genome Res. 2013;23:555–567. doi: 10.1101/gr.147942.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.