

Abstract
Lipoyl synthase (LS) catalyzes the final step in lipoyl cofactor biosynthesis: the insertion of two sulfur atoms at C6 and C8 of an (N6-octanoyl)-lysyl residue on a lipoyl carrier protein (LCP). LS is a member of the radical SAM superfamily, enzymes that use a [4Fe–4S] cluster to effect the reductive cleavage of S-adenosyl-l-methionine (SAM) to l-methionine and a 5′-deoxyadenosyl 5′-radical (5′-dA•). In the LS reaction, two equivalents of 5′-dA• are generated sequentially to abstract hydrogen atoms from C6 and C8 of the appended octanoyl group, initiating sulfur insertion at these positions. The second [4Fe–4S] cluster on LS, termed the auxiliary cluster, is proposed to be the source of the inserted sulfur atoms. Herein, we provide evidence for the formation of a covalent cross-link between LS and an LCP or synthetic peptide substrate in reactions in which insertion of the second sulfur atom is slowed significantly by deuterium substitution at C8 or by inclusion of limiting concentrations of SAM. The observation that the proteins elute simultaneously by anion-exchange chromatography but are separated by aerobic SDS-PAGE is consistent with their linkage through the auxiliary cluster that is sacrificed during turnover. Generation of the cross-linked species with a small, unlabeled (N6-octanoyl)-lysyl-containing peptide substrate allowed demonstration of both its chemical and kinetic competence, providing strong evidence that it is an intermediate in the LS reaction. Mössbauer spectroscopy of the cross-linked intermediate reveals that one of the [4Fe–4S] clusters, presumably the auxiliary cluster, is partially disassembled to a 3Fe-cluster with spectroscopic properties similar to those of reduced [3Fe–4S]0 clusters.
Lipoyl synthase (LS) catalyzes the final step in the de novo pathway for the biosynthesis of the lipoyl cofactor (N6-(6R-6,8-dimercapto-octanoyl)-lysyl), which consists of the sequential insertion of two sulfur atoms into C–H bonds at C6 and C8 of an octanoyl chain appended to the ε-amino group of a specific lysyl residue of lipoyl carrier proteins (LCPs)1−6 (Figure 1). Five LCPs are known: the E2 subunits of the pyruvate, α-ketoglutarate, branched chain oxo-acid, and acetoin dehydrogenase complexes as well as the H protein of the glycine cleavage system.7−12 In these complexes, the lipoyl cofactor functions as an electron acceptor in oxidative decarboxylation or other oxidative C–C fragmentation reactions while covalently transporting the resulting intermediates to different subunits that catalyze their modification with coenzyme A or attachment to tetrahydrofolate.13−16 The lipoyl cofactor is virtually ubiquitous among organisms that undergo, or are capable of undergoing, aerobic respiration. In addition to its most notable role as an essential cofactor involved in energy metabolism or amino acid degradation, lipoic acid (LA) is gaining prominence as a broad-spectrum antioxidant and regulator of key cellular signaling pathways, as in its regulation of transcription factor NF-κB.1,17,18 Because of its antioxidant properties, it has been reported to be efficacious in the treatment of a number of disorders that are related to oxidative stress, such as diabetic neuropathy19−21 and Alzheimer’s disease.22
Figure 1.
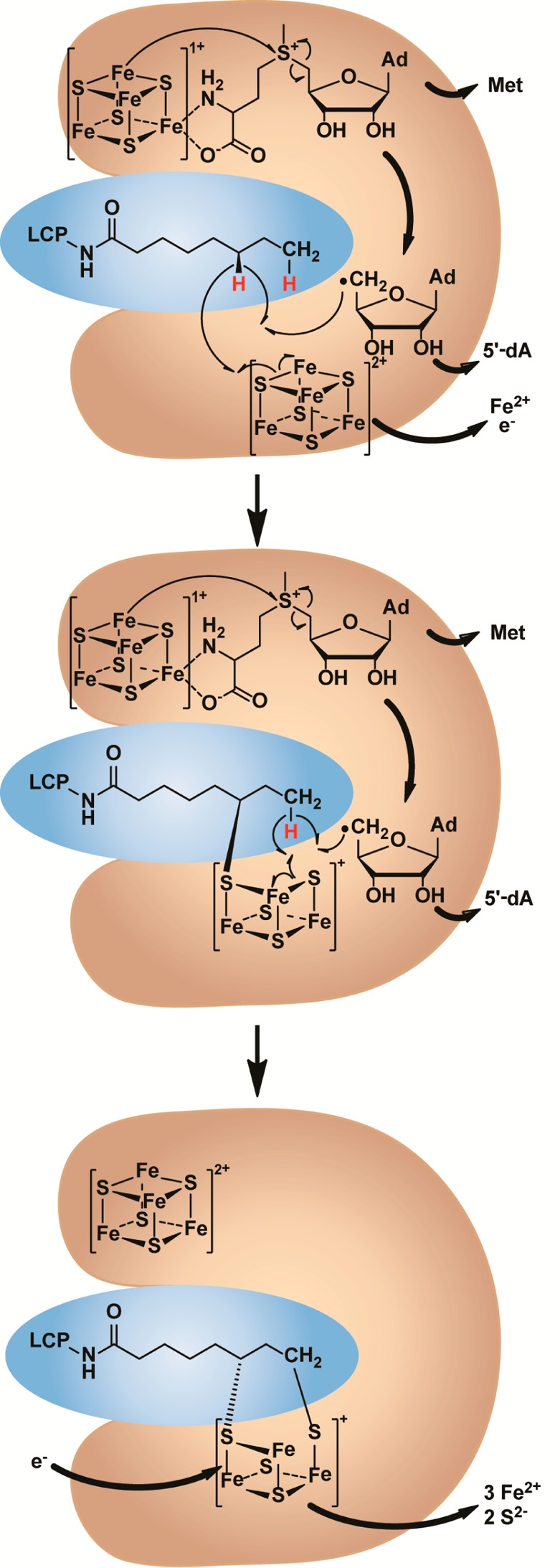
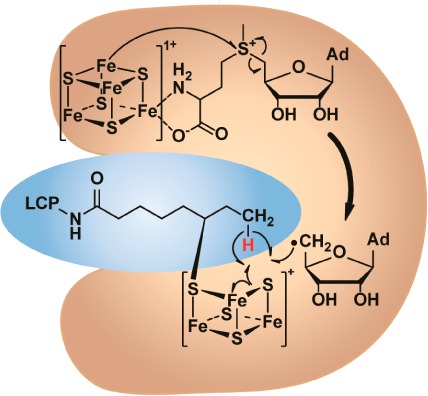
Proposed reaction mechanism of LS. LS and its substrate are shaded in orange and blue, respectively. In each structure, the RS cluster is shown in the upper part of LS, and the auxiliary cluster is shown in the lower part. The assignment of cluster charges is based on the Mössbauer spectroscopic results.
Genetic and biochemical studies conducted primarily in Escherichia coli (Ec) provide evidence for two distinct pathways for lipoyl cofactor biosynthesis, which are generally conserved among organisms that utilize this cofactor.2,23,24 An exogenous pathway allows organisms to salvage LA or acquire it from nutrient sources. In Ec, the bifunctional enzyme, lipoate protein ligase A (LplA), catalyzes the ATP-dependent activation of the free acid to the adenylated species as well as the subsequent attachment of the lipoyl group to target lysyl residues of LCPs.25,26 In mammals and some bacteria, two separate enzymes are utilized: one for activating lipoic acid to lipoyl-AMP (lipoate activating enzyme) and one for transferring the activated lipoyl group to target LCPs (lipoyl transferase).27,28 A second pathway, labeled the endogenous pathway, intersects with type II fatty acid biosynthesis.6,29 The first comitted step in this pathway, catalyzed by octanoyltransferase (LipB), is the transfer of an n-octanoyl chain from octanoyl-acyl carrier protein to an LCP.30−33 The second enzyme, lipoyl synthase (LS), then catalyzes sulfur insertion at C6 and C8 to afford the intact cofactor.3−6 Recent studies describing lipoyl cofactor biosynthesis in Bacillus subtilis and Saccharomyces cerevisiae reveal important perturbations of the pathways described in Ec, showing the centrality of the H protein of the glycine cleavage system in lipoyl cofactor formation on all LCPs in those organisms.34−36 Although genes encoding proteins in both pathways are present in mammals, mice that are homozygous null for the lias gene (lipoyl synthase) die in utero, suggesting that they cannot use maternally supplied LA. On the other hand, mice that are heterozygous null for the lias gene (i.e., they contain one functional copy) survive, but they have increased markers for oxidative stress.37
LS is a member of the radical-SAM (RS) superfamily, enzymes that use an electron donated from a [4Fe–4S]+ cluster to cleave S-adenosyl-l-methionine (SAM) reductively to l-methionine and a 5′-deoxyadenosyl 5′-radical (5′-dA•). 5′-dA• is a potent oxidant used to abstract hydrogen atoms (H•) both from protein and small-molecule substrates, many of which contain inert target C–H bonds.38−41 The [4Fe–4S] cluster is ligated by three Cys residues that reside typically in a CX3CX2C motif, which serves as a signature sequence for RS enzymes.42 LS, like BioB, RimO, MiaB, MtaB, and MTL1, is a member of a subclass of RS enzymes that catalyze sulfur insertion into hydrocarbon backbones. BioB catalyzes sulfur insertion into dethiobiotin to form biotin, whereas RimO, MiaB, MtaB, and MTL1 catalyze insertion of a sulfur atom capped with a methyl group derived from a second molecule of SAM.38,43−45 RimO modifies the β carbon of a conserved aspartyl group (Asp 88 in Ec) in protein S12 of the small subunit of the bacterial ribosome, whereas MiaB and MtaB catalyze methylthiolation of adenosine 37 of certain tRNAs.46−50 A common feature of all RS enzymes that catalyze sulfur insertion into unactivated C–H bonds is a second Fe/S cluster, a [2Fe–2S] cluster in BioB and [4Fe–4S] clusters in all other characterized enzymes, which is proposed to be the source of the inserted sulfur atom. Whether the sulfur atom emanates from one of the bridging μ-sulfido ions of the cluster or an externally ligated sulfide or polysulfide species is the subject of debate and may in fact depend on the individual enzymatic reaction. In the former instance, the cluster would be degraded concomitant with turnover, implying a single turnover reaction, whereas in the latter instance, multiple turnovers can ensue if the ligated sulfur source can be replenished.46,48,51−55
The LS reaction proceeds as two distinct half-reactions (Figure 1).3,5,38 In the first half-reaction, SAM bound to the [4Fe–4S] cluster ligated by Cys residues in the CX3CX2C motif (RS cluster) is reductively cleaved to a 5′-dA•, which is used to abstract an H• from C6 of the octanoyl chain. The resulting carbon-centered radical attacks an activated form of sulfide, believed to be a bridging μ3-sulfido ion of the [4Fe–4S] cluster ligated by Cys residues in the CX4CX5C motif (auxiliary cluster). In the second half-reaction, another equivalent of 5′-dA• is similarly generated, but it abstracts an H• from C8 of the octanoyl chain. The carbon-centered radical at C8 attacks a second form of activated sulfide, presumably from the same [4Fe–4S] cluster.56 Upon addition of two protons, the lipoyl cofactor is formed in its two-electron reduced state. In this mechanism, the auxiliary [4Fe–4S] cluster is degraded during catalysis, and the enzyme is therefore inactivated after a single turnover. This mechanism is consistent with the finding that the protein itself is the source of the inserted sulfur atoms and that both sulfurs derive from the same polypeptide.56 Although a monothiolated species, proposed to be the product of the first half-reaction, has been detected in previous studies, it has not been shown to be catalytically or kinetically competent to form product.3,5 Herein, we provide evidence that the intermediate is in fact competent and is covalently cross-linked to LS. Characterization of the cross-linked species suggests a shared sulfur atom between the monothiolated intermediate and a trinuclear, partially degraded Fe/S cluster.
Materials and Methods
Materials
Kanamycin and ampicillin were from IBI Shelton Scientific (Pecsto, IA) or Gold Biotechnology (St. Louis, MO). N-(2-Hydroxyethyl)piperazine-N′-2-ethanesulfonic acid (HEPES), tris(2-carboxyethyl)phosphine (TCEP), pyridoxal 5′-phosphate (PLP), sodium sulfide, 2-mercaptoethanol (BME), and sodium dithionite were from Sigma Corp. (St. Louis, MO). Ferric chloride was from EMD Biosciences (Gibbstown, NJ), and Coomassie brilliant blue was from ICN Biomedicals (Aurora, OH). Bradford reagent for protein concentration determination as well as the bovine serum albumin standard was from Pierce, Thermofisher Scientific (Rockford, IL). Dithiothreitol (DTT) and isopropyl β-d-1-thiogalactopyranoside were purchased from Gold Biotechnology (St. Louis, MO). 57Fe metal (98%) was from Isoflex USA (San Francisco, CA), and [8,8,8-2H3]octanoic acid (99.7%) was purchased from CDN Isotopes (Pointe-Claire, Canada). Sephadex G-25 resin was obtained from GE Healthcare (Piscataway, NJ), and DE-52 anion-exchange resin was from Whatman (Maidstone, England). Peptide substrates and standards were custom-synthesized by ProImmune Ltd. (Oxford, UK), and their concentrations were estimated by weight using their theoretical molecular masses. All other chemicals were of reagent grade quality or higher.
Hexahistidine-tagged Ec LS was prepared essentially as described previously.3 The final protein was exchanged into buffer A (50 mM HEPES, pH 7.5, 0.3 M KCl, 1 mM DTT, 20% glycerol) using a Sephadex G-25 column, snap-frozen in small aliquots, and stored in liquid N2 until further use. Thermus thermophilus (Tt) LS was expressed and purified as described previously for Ec LS.3,57S-Adenosylhomocysteine nucleosidase (SAHN) and SAM were prepared as described previously.58,59 [8,8,8-2H3]-Octanoyl-H protein (OHP) was synthesized using Ec LplA as described previously.3,32
Methods
UV–vis spectra were recorded on Cary 50 or Cary 300 spectrometers (Varian; Walnut Creek, CA). Low-temperature X-band electron paramagnetic resonance (EPR) spectroscopy was performed on a Bruker Elexsys E-560 spectrometer (Billerica, MA) equipped with a SuperX FT microwave bridge. An ITC503S temperature controller and ESR900 helium cryostat, both from Oxford Instruments (Concord, MA), were used to control and maintain sample temperature. High-performance liquid chromatography (HPLC) with detection by mass spectrometry (LC–MS) was conducted on an Agilent Technologies (Santa Clara, CA) 1200 system coupled to an Agilent Technologies 6410 QQQ mass spectrometer. The system was operated with the associated MassHunter software package, which was also used for data collection and analysis. Fast protein liquid chromatography (FPLC) was conducted on an ÄKTA system (GE Healthcare) housed in a Coy anaerobic chamber (Grass Lake, MI) and fitted with a Mono Q (GE Healthcare) anion-exchange column. Sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) was conducted as described previously60 using a mini vertical electrophoresis unit from Hoefer (Holliston, MA).
Mössbauer spectra were recorded on alternating constant acceleration Mössbauer spectrometers equipped either with a Janis SVT-400 variable-temperature cryostat (weak-field) or a Janis 8TMOSS-OM-12SVT variable-temperature cryostat (strong-field). The external field at the sample is oriented parallel to the γ beam. All isomer shifts are quoted relative to the centroid of the spectrum of α-iron metal at room temperature. Simulation of the Mössbauer spectra was carried out using the WMOSS spectral analysis software (www.wmoss.org, WEB Research, Edina, MN). Simulations of the 3Fe-clusters in external magnetic fields are based on the spin Hamiltonian formalism given by eq 1, in which the first term describes the electron Zeeman effect, the second and third terms represent the zero field splitting of the S = 2 ground state, the fourth term represents the interaction between the electric field gradient and the nuclear quadrupole moment, the fifth term describes the magnetic hyperfine interactions of the electronic spin with the 57Fe nuclei, and the last term represents the nuclear Zeeman interaction. All symbols have their usual meaning.61 Simulations of the two spectroscopically distinct sites (FeIII and Fe22.5+ pair) were carried out with respect to the electronic spin of the ground state, S = 2. Spectra were calculated in the slow relaxation limit.
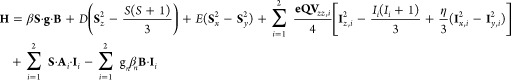 |
1 |
LS Activity Determinations
LS activity assays using OHP as substrate have been described previously.3,32,51,56 When OHP was replaced with the peptide substrate Glu-Ser-Val-(N6-octanoyl)Lys-Ala-Ala-Ser-Asp (peptide 1) or biotin-PEG-Glu-Ser-Val-([8,8,8-2H3]-N6-octanoyl)Lys-Ala-Ala-Ser-Asp (peptide 2) (Figure S1), assays contained the following: 50 mM HEPES, pH 7.5, 300 mM KCl, 10% glycerol, 200 μM LS, 300 μM octanoyl-peptide, 700 μM SAM, 2 mM dithionite, and 500 nM SAHN. After pre-equilibration at the appropriate temperature (37 °C or room temperature), reactions were initiated by addition of dithionite. Aliquots were quenched at designated times in 100 mM sulfuric acid containing 4 mM TCEP and 20 μM external peptide standard (Pro-Met-Ser-Ala-Pro-Ala-Arg-Ser-Met). A standard curve for analysis by LC–MS was prepared in assay buffer and contained 20 μM external peptide standard, 2 mM TCEP, and 3.9–500 μM octanoyl- and lipoyl-peptide. The quantity of mercapto-octanoyl (i.e., monothiolated species) peptide was estimated using the lipoyl-peptide standards. The time-dependent formation and/or decay of octanoyl-, 6-mercapto-, and lipoyl-peptides was fitted individually to eqs 2, 3, or 4, respectively, using IgorPro graphing software (Wavemetrics, Lake Oswego, OR). Equations 2 and 4 describe biphasic behavior, where t is time, A1 and A2, and A4 and A5 correspond to the amplitudes of the two phases, and k1 and k2, and k5 and k6 correspond to the rate constants for the two phases. Equation 3 describes the formation and decay phases of an intermediate (B) in an A → B → C reaction. Here, A3 corresponds to the amount of intermediate formed, whereas k3 and k4 correspond to rate constants for formation and decay of the intermediate.
| 2 |
| 3 |
| 4 |
Analysis of Peptide Products by LC–MS
Substrates and products were separated on an Agilent Technologies Zorbax Rapid Resolution SB-C18 column (2.1 mm × 30 mm, 3.5 μm particle size) equilibrated in 92% solvent A (0.1% formic acid, pH 2.6) and 8% solvent B (acetonitrile). A gradient of 8–19% B was applied from 0 to 1 min and maintained at 19% from 1 to 5 min before returning to 8% solvent B from 5 to 6 min. A flow rate of 0.4 mL/min was maintained throughout the chromatographic procedure. The column was allowed to re-equilibrate for 3 min under initial conditions before subsequent sample injections. The external standard peptide elutes at approximately 1.5 min under these conditions, whereas the lipoyl-, mercapto-octanoyl-, and octanoyl-peptides elute at 3.6, 4.1, and 4.8 min, respectively.
The external standard peptide and octanoyl-, mercapto-octanoyl-, and lipoyl-peptides were detected by MS2 selected-ion monitoring at m/z ratios of 474.4, 932.5, 964.5, and 996.5, respectively (Figure S2A). Detection of analytes was performed using electrospray ionization in positive mode (ESI+) with the following parameters: a nitrogen gas temperature of 340 °C and flow rate of 9.0 L/min; a nebulizer pressure of 40 PSI; and a capillary voltage of 4000 V. The fragmentor voltage was optimized for octanoyl- and lipoyl-peptides at 240 V, which was used for all derivatives. The fragmentor voltage for the external standard peptide was 135 V.
Analysis of Biotin-Tagged Peptide Products by LC–MS
Assay mixtures were separated as described above except that a gradient of 8–35% solvent B was applied from 0 to 2 min and was followed by a gradient of 35–48% solvent B from 2 to 3 min. The gradient was maintained at 48% solvent B for 1 min before returning to 8% solvent B from 4 to 5 min. The column was allowed to re-equilibrate for 2 min under initial conditions before subsequent sample injections. The external standard peptide elutes at approximately 1.4 min under these conditions, whereas the lipoyl-, 6-mercapto-octanoyl-, and octanoyl-peptides elute at 3.5, 3.5, and 3.6 min, respectively.
External standard and octanoyl-, mercapto-octanoyl-, and lipoyl-peptides were detected using an MS2 selected-ion monitoring method at m/z ratios of 474.4, [M + 2H]+2; 1306.6, [M + H]+; 1338.6, [M + H]+; and 1369.6, [M + H]+, respectively (Figure S2B). 8-Mercapto- and lipoyl-peptide standards lacking deuterium labels were monitored at m/z ratios of 1335.5, [M + H]+; and 1367.6, [M + H]+, respectively. Detection of analytes was performed as described above except that the fragmentor voltage was optimized for octanoyl- and lipoyl-peptides at 280 V and for the 8-mercapto-octanoyl-peptide at 300 V.
Assessment of a Covalent Interaction between LS and [8,8,8-2H3]-OHP
Assay mixtures contained 180 μM LS, 50 mM HEPES, pH 7.5, 2 mM dithionite, 1 mM SAM, and 300 μM OHP or [8-2H3]-OHP in a total volume of 1 mL. Reactions were initiated by addition of SAM and incubated for 20 min at 37 °C under anaerobic conditions. After incubation, reactions were immediately injected onto a Mono Q column equilibrated in 50 mM HEPES, pH 7.5, 0.1 M NaCl at a flow rate of 1 mL min–1. A gradient from 0.1 to 1 M NaCl was applied over 60 min, and 50 μL aliquots of each 1 mL fraction were combined with an equal volume of 2× SDS-PAGE gel-loading buffer and boiled for 10 min. After centrifugation, 10 μL aliquots of each sample were analyzed by SDS-PAGE (15% gel) with Coomassie brilliant blue staining.
To generate quantities of cross-linked species suitable for spectroscopic analysis, reaction volumes were increased to 7 mL and contained 50 mM HEPES, pH 7.5, 2 mM dithionite, 400 μM LS, 700 μM SAM, and 200 μM [8,8,8-2H3]-OHP. After incubating at room temperature for 1.5 h, the reaction was diluted 10-fold with buffer (50 mM HEPES, pH 7.5, 300 mM NaCl, 20% glycerol) and loaded onto a DE-52 column (2.5 × 10 cm) pre-equilibrated in cold (<10 °C) 50 mM HEPES, pH 7.5. A 500 mL linear gradient (0.1–1 M NaCl in 50 mM HEPES, pH 7.5) was applied to the column while collecting 5 mL fractions. Each of the fractions was analyzed by SDS-PAGE as described above to determine those that contained the cross-linked species. Appropriate fractions were pooled and concentrated by ultrafiltration and frozen in liquid N2 until further use. Protein, iron, and sulfide concentrations of the cross-linked species were determined as described previously.51 Samples to be analyzed by Mössbauer and EPR spectroscopies were prepared in the anaerobic chamber. Mössbauer samples contained 300 μL of the cross-linked species or unreacted LS. EPR samples of both species were prepared with and without prior incubation (20 min) with 8 mM dithionite before freezing.
Generation of the Cross-Linked Species Using a Peptide Substrate
A reaction containing 50 mM HEPES, pH 7.5, 300 mM KCl, 10% glycerol, 325 μM [8,8,8-2H3]-peptide 2, 275 μM Thermus thermophilus (Tt) LS, and 700 μM SAM was initiated by the addition of 2 mM dithionite (final conc.). After 30 min at 30 °C, the reaction was applied to a 5 mL Streptavidin Mutein column (Roche Diagnostics, Mannheim, Germany) equilibrated in 100 mM KH2PO4, pH 7.2, 150 mM NaCl, and 600 mM (NH4)2SO4. Unreacted LS was removed by washing with 10 column volumes of the same buffer. The cross-linked species was eluted from the column using an identical buffer containing 2 mM d-biotin. Alternatively, a reaction containing 50 mM HEPES, pH 7.5, 300 mM KCl, 10% glycerol, 300 μM [8,8,8-2H3]-peptide 2, 200 μM Ec LS, and 700 μM SAM was initiated with 2 mM dithionite. After 90 min at room temperature (22 °C), a sample was frozen for Mössbauer analysis without any additional chromatography.
Assessment of the Chemical and Kinetic Competence of the Cross-Linked Species
A reaction using peptide 1 as the substrate was performed as described above except that the reaction mixture contained 180 μM SAM and SAHN was omitted. The reaction was allowed to proceed for 30 min and then was applied to a NAP-10 column pre-equilibrated in 50 mM HEPES, pH 7.5, containing 300 mM KCl and 10% glycerol. The protein-containing fraction was collected, and an aliquot (20 μL) was removed and added to an equal volume of 100 mM H2SO4 to quench the reaction and prepare it for analysis by LC–MS. SAHN, SAM, and dithionite were added to the remaining assay mixture to final concentrations of 200 nM, 500 μM, and 2 mM, respectively, to initiate the second half-reaction, and aliquots were quenched at various times and analyzed as described above. Four Mössbauer samples during various stages of the reaction were prepared: (1) prior to initiation with dithionite, (2) before size-exclusion chromatography, (3) after size-exclusion chromatography, and (4) after size-exclusion chromatography and a further 90 min incubation with added SAHN, SAM, and dithionite.
Results
Generation of a Cross-Link between Ec LS and OHP
Previous studies by Cicchillo et al. indicated that use of [octanoyl-2H15]-HP in the LS reaction suppressed formation of LHP, although a species consistent with one inserted sulfur atom was observed by mass spectrometry.3 These findings, in conjunction with the observation that the total amount of 5′-dA formed was ∼50% of that formed when unlabeled OHP was used as a substrate, suggested that one C–D bond, presumably at C6 or C8, was not readily cleaved because of significant isotopic discrimination. Subsequent studies by Douglas et al. using LS from Sulfolobus solfataricus (Ss) showed that deuterium incorporation at C8 of a target (N6-octanoyl)-lysyl residue in a synthetic tripeptide substrate significantly impeded formation of the lipoyl group and that sulfur insertion at C6 precedes insertion at C8.5 This observation is in agreement with earlier findings that showed that [8-3H]-octanoic acid administered to growing Ec is incorporated intact into LA without loss of radioactivity.5,62 Douglas et al. also commented that when the peptide substrate containing an [8,8,8-2H3-octanoyl]-lysyl residue was incubated with Ss LS under turnover conditions, the peptide was poorly liberated from the protein by ultrafiltration unless the protein/peptide complex was denatured with acid, suggesting that it was bound tightly to the protein.5
To understand the nature of the turnover-dependent interaction between LS and its substrate and to provide evidence for a potential intermediate in catalysis, we studied the Ec LS reaction with [8,8,8-2H3]-OHP. Because sulfur insertion takes place at C6 before C8 and because quantitative deuterium substitution at C8 of the octanoyl moiety results essentially in arrest of turnover after the first sulfur insertion, our working model would predict that OHP and LS should become cross-linked through the auxiliary cluster under turnover conditions when using [8,8,8-2H3]-OHP as substrate. Ec HP binds tightly to Ec LS; however, the two proteins can be separated by anion-exchange chromatography. Ec HP (pI = 4.59) adheres strongly to anion-exchange resin under conditions described in Materials and Methods, whereas Ec LS (pI = 8.15) adheres poorly, if at all, under the same conditions. Figure 2A shows an SDS-PAGE analysis of fractions eluting from a Mono Q column under increasing concentrations of NaCl. Ec LS and [8,8,8-2H3]-OHP were applied to the column after their coincubation under turnover conditions in the absence of required low-potential reductant. LS elutes essentially in the void volume of the column (fractions 1–6), whereas [8,8,8-2H3]-OHP begins to elute at approximately 750 mM NaCl. The faint band that migrates slightly higher than [8,8,8-2H3]-OHP corresponds to LHP (or monothiolated LHP), which sometimes contaminates our preparations of apo-HP that are used to prepare OHP. When the proteins are incubated both in the presence of SAM and required reductant (sodium dithionite), a new species appears that displays migratory properties that are intermediate between those of LS and OHP (Figure 2B; fractions 20–25). For reasons that we do not understand, OHP supports no more than 30–40% turnover (i.e., 0.3–0.4 equiv LHP per LS).3 Therefore, the majority of LS and OHP do not react productively and elute in fractions 2–6 and 27–30, respectively. Consistent with a cross-linked species, the intermediate fractions contain both LS and HP (presumably with one sulfur atom inserted). This same cross-linked species is observed under turnover conditions with unlabeled substrate (Figure S3), consistent with sulfur insertion at C8 being rate-limiting.
Figure 2.

SDS-PAGE analysis of fractions from DE-52 column. Lanes are molecular weight markers (M), flow-through during column load (A and B), and samples from eluted 10 mL fractions as the NaCl gradient was increased from 0.1–1 M (1–14). Fractions that were pooled and used in further experiments are boxed.
Analysis of the Cross-Linked Species by Mössbauer Spectroscopy
To generate suitable concentrations of the cross-linked species for analysis by Mössbauer spectroscopy, a large-scale reaction was conducted using [8,8,8-2H3]-OHP as substrate, and the cross-linked species was separated from unreacted proteins by anion-exchange chromatography on DE-52 resin (Figure S4). The 4.2-K/53-mT Mössbauer spectrum of the non-cross-linked fraction of LS (Figure 3A) can be adequately described using a broad quadrupole doublet with parameters characteristic for [4Fe–4S]2+ clusters (isomer shift (δ) of 0.45 mm/s and quadrupole splitting parameter (ΔEQ) of 1.18 mm/s; ∼90% of total Fe, 1.9 clusters per LS monomer; solid line in Figure 3A). These parameters are similar, albeit not identical, to those reported previously for Ec LS.51 In addition to the prominent quadrupole doublet, the spectrum exhibits a weak absorption line at ∼2.5 mm/s, demonstrating that a small amount (∼10%) of N/O-coordinated high-spin FeII is present in the sample.
Figure 3.
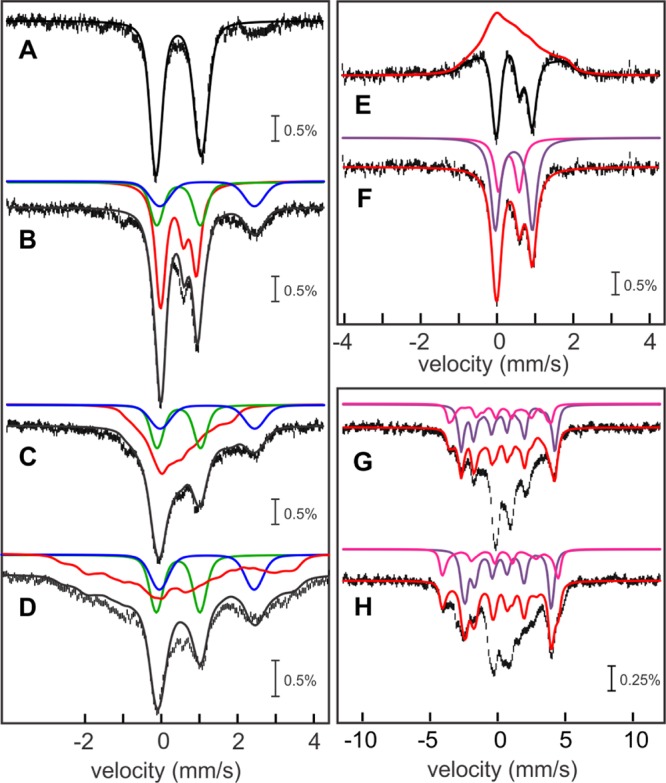
Mössbauer-spectroscopic characterization of the cross-linked species observed during the reaction of Ec LS. 4.2-K/53-mT Mössbauer spectrum of Ec LS (A, vertical bars) and simulation thereof with parameters quoted in the text (A, black line). 4.2 K Mössbauer spectra of the complex obtained upon reacting Ec LS with [8,8,8-2H3]-OHP in the presence of SAM and dithionite recorded in externally applied magnetic fields of 0 mT (B, vertical bars), 53 mT (C, vertical bars), 200 mT (D, vertical bars), 1 T (G, vertical bars), and 4 T (H, vertical bars) oriented parallel to the γ beam. Addition of the features of the 3Fe-clusters in a 53 mT field (E, red line) to the [0–53 mT] difference spectrum (E, vertical bars) yields the zero-field reference spectrum of the 3Fe-clusters (F, vertical bars). The blue and green lines in B–D are simulations of FeII and [4Fe–4S]2+ components, respectively, with parameters quoted in the text. The red lines in B–H are simulations of the 3Fe-clusters with parameters given in Table 1. The individual contributions from the FeIII site and Fe22.5 pair are shown in magenta and purple, respectively, in F–H. The added contribution of the FeII, [4Fe–4S]2+, and 3Fe-clusters are shown as black lines in B–D. The black line in E is the theoretical [0–53 mT] difference spectrum of the 3Fe-clusters.
The spectra of the cross-linked complex after purification by anion-exchange chromatography are distinctly different and reveal the presence of three dominant components: [4Fe–4S]2+ clusters (20% of 8.7 Fe per Ec LS, i.e., 0.4 clusters per LS, Figures 3B–D, green lines), N/O-coordinated FeII (16% of 8.7 Fe, Figures 3B–D, blue lines), and 3Fe-clusters that exhibit spectroscopic properties similar to those of [3Fe–4S]0 clusters63−66 (54% of 8.7 Fe, 1.6 clusters per LS, Figures 3B–H, red lines). (The stoichiometry indicates that both [4Fe–4S] clusters are (at least in part) converted to 3Fe clusters. We speculate that they differ in the number of sulfide ligands because of the proposed role of the auxiliary [4Fe–4S] cluster as sulfur donor. Because the Mössbauer spectra might not be sensitive to the number and nature of the bridging sulfur ligands, the cuboidal [3Fe–4S]0 and the hypothetical [3Fe–3S–1RS]+ clusters, both of which contain one FeIII site and one valence-delocalized Fe22.5+ pair, may exhibit similar Mössbauer spectra. We therefore refer to these cluster forms collectively as 3Fe–clusters). The mismatches between the summations of the theoretical spectra (Figure 3B–D, black lines) of these three components with the experimental data reveal the presence of a minor and poorly defined subspectrum (∼10% intensity). Because this sample does not exhibit EPR features reminiscent of Fe/S clusters with S = 1/2 ground states (i.e., [3Fe–4S]+, [4Fe–4S]+, or [2Fe–2S]+; see Figure S5), we attribute the remaining broad component to unspecifically bound iron.
Comparison of spectra recorded in zero-field and small applied fields (53 and 200 mT) shows that the features associated with the [4Fe–4S]2+ cluster and the N/O-coordinated high-spin FeII are not, or only slightly, broadened by the externally applied field (Figures 3B–D, blue and green lines). By contrast, the 3Fe-clusters exhibit sharp quadrupole doublet features in zero field, which are significantly broadened in weak applied fields, demonstrating that the 3Fe-clusters are associated with a paramagnetic, integer-spin ground state. This behavior is illustrated in the [0–53 mT] difference spectrum (Figure 3E, vertical bars). The zero-field reference spectrum of the 3Fe-clusters (Figure 3F, vertical bars) was generated by adding back 54% of the simulated spectrum of the 3Fe-clusters at 53 mT field (red line in Figure 3E). It can be simulated with two quadrupole doublets with slightly different line widths and assuming a 1:2 ratio with the following parameters: δ = 0.31 mm/s and ΔEQ = 0.54 mm/s (18% of total Fe); and δ = 0.44 mm/s and ΔEQ = 0.97 mm/s (36% of total Fe) (magenta and purple lines in Figure 3F). These parameters are characteristic of high-spin FeIII–S4 sites and valence-delocalized Fe22.5+ pairs, respectively.67 Together with the 1:2 intensity ratio of the two sites, these parameters strongly suggest that the 3Fe-clusters are composed of a valence-delocalized Fe22.5+ unit and a high-spin FeIII-site. Indeed, the spin Hamiltonian parameters of the intermediate are similar to those reported for cuboidal [3Fe–4S]0 clusters,63−66,68 with the exception that the absolute magnitude of the quadrupole splitting parameter of the mixed-valent pair is approximately 0.50 mm/s smaller than the values reported previously (Table 1).
Table 1. Comparison of the Mössbauer Parameters of the 3Fe-Cluster of the LS Monothiolated Intermediate to Those of Cuboidal [3Fe–4S]0 Clusters.
| species | δ (mm/s) | ΔEQ (mm/s) | η | β (deg)a | A (MHz) | ref | |
|---|---|---|---|---|---|---|---|
| Ec LS monothiolated intermediateb | FeIII | 0.31 | –0.55 | –2 | 16 | +13.8, +16.2, +16.2 | this work |
| Fe22.5 | 0.44 | 0.98 | 0.4 | 25 | –19.1, −19.1, −15.7 | ||
| [3Fe–4S]0 cluster of D. gigas ferredoxin IIc | FeIII | 0.32 | –0.52 | –2 | 16 | +13.7, +15.8, +17.3 | (65) |
| Fe22.5 | 0.46 | 1.47 | 0.4 | 20 | –20.5, −20.5, −16.4 | ||
| [3Fe–4S]0 cluster of A. vinelandii ferredoxin (pH 6)c | FeIII | 0.29 | –0.47 | 0.8 | 20 | +15.0, +14.0, +17.3 | (85) |
| Fe22.5 | 0.47 | 1.41 | 1.1 | 24 | –21.0, −25.0, −14.3 | ||
| 0.47 | 1.41 | 0.2 | 24 | –19.0, −22.0, −16.7 | |||
| [3Fe–4S]0 cluster of A. vinelandii ferredoxin (pH 8.5)c | FeIII | 0.29 | –0.47 | 5 | 27 | +16.0, +14.0, +17.2 | (85) |
| Fe22.5 | 0.47 | 1.41 | 1 | 19 | –19.0, −27.0, −16.7 | ||
| 0.47 | 1.41 | 0 | 19 | –20.0, −22.0, −16.7 | |||
| [3Fe–4S]0 cluster of beef heart aconitase | FeIII | 0.30 | 0.49 | nd | nd | nd | (64) |
| Fe22.5 | 0.45 | 1.34 | nd | nd | nd | ||
| [3Fe–4S]0 cluster of human MOCS1A | FeIII | 0.28 | 0.56 | nd | nd | nd | (66) |
| Fe22.5 | 0.47 | 1.44 | nd | nd | nd | ||
Euler angle that rotates the electric field gradient tensor into the frame of the zero-field splitting tensor.
Mössbauer spectra were calculated for S = 2 with gS=2 = (2.0, 2.0, 2.0), DS=2 = −4.0 cm–1, and (E/D)S=2 = 0.23.
Mössbauer spectra were calculated for S = 2 with gS=2 = (2.0, 2.0, 2.0), DS=2 = −2.5 cm–1, and (E/D)S=2 = 0.23.
The features associated with the 3Fe-clusters in spectra collected in externally applied magnetic fields (Figure 3C, D, G, and H) were simulated using the spin Hamiltonian parameters given in Table 1. The parameters of the prototypical cuboidal [3Fe–4S]0 cluster from Desulfovibrio gigas ferredoxin II were used as a starting point for these simulations65 together with the isomer shift and quadrupole splitting values obtained from analysis of the zero-field reference spectrum. Then, selected parameters were adjusted to better match the features of the 3Fe-clusters of the field-dependent spectra. We note that only minor adjustments are required to reproduce the salient features of the 3Fe-clusters. Collectively, the Mössbauer spectra demonstrate the presence of 1.5 equiv of 3Fe-clusters in the cross-linked samples, with spectroscopic properties similar to those of well-studied [3Fe–4S]0 clusters.63−66,69,70
Generation of a Cross-Linked Species with Thermus thermophilus LS and a Synthetic Peptide Substrate
The observation that the sample of the Ec LS/[8,8,8-2H3]-OHP cross-linked species contains 1.5 equiv of 3Fe-clusters rather than the expected 1:1 ratio of a 3Fe-cluster and a [4Fe–4S] cluster suggests that the RS cluster is partially degraded during anion-exchange chromatography, presumably to the [3Fe–4S]0 form by loss of one Fe ion. Therefore, a milder approach was sought to remove the unreacted LS from the cross-linked species, wherein the OHP substrate was replaced with an 8-amino acid synthetic peptide containing a target [8,8,8-2H3]-N6-octanoyl-lysyl residue and an N-terminal biotin tag (peptide 2). The biotin tag was intended for purification of the cross-linked species by affinity chromatography on streptavidin mutein resin; however, the Ec LS/peptide 2 cross-linked complex did not adhere to the column, suggesting that the biotin tag was occluded from binding to the resin. By contrast, when the orthologous LS from Thermus thermophilus (Tt) was incubated with peptide 2 under turnover conditions and the reaction mixture was applied to a streptavidin mutein column, two brown protein fractions were observed: a fast-migrating band, corresponding most likely to unreacted LipA, and a band that was strongly retained on the column. The latter band was subsequently eluted by addition of 2 mM biotin and analyzed by Mössbauer spectroscopy along with a control sample containing as-isolated (AI) Tt LS.
The 4.2-K/53-mT Mössbauer spectrum of AI Tt LS (Figure 4A, vertical bars) can be simulated with a broad quadrupole doublet with parameters (δ = 0.46 mm/s and ΔEQ = 1.13 mm/s, ∼95% of the total Fe; Figure 4A, solid line) typical of [4Fe–4S]2+ clusters. This sample contains 5.3 Fe per Tt LS, resulting in a stoichiometry of 1.3 [4Fe–4S] clusters per Tt LS. The 4.2 K Mössbauer spectra of the sample containing the Tt LS/peptide 2 complex collected in zero field (Figure 4B, vertical bars) and 53 mT (Figure 4C, vertical bars) reveal several new features. First, the spectra are dependent on the presence of the external magnetic field, as is evident from the [0–53 mT] difference spectrum (Figure 4D, vertical bars). This difference spectrum is essentially identical to that observed for the 3Fe-clusters associated with the Ec LS/[8,8,8-2H3]-OHP complex (Figure 4D, solid line, scaled by a factor of 0.78). From the amplitude of the difference spectrum, it is possible to estimate the amount of 3Fe-clusters in the sample containing the Tt LS cross-linked species to be ∼40% of the total Fe. Given that this sample contains 3.6 Fe ions after purification, a stoichiometry of 0.5 3Fe-clusters per protein can be calculated.
Figure 4.
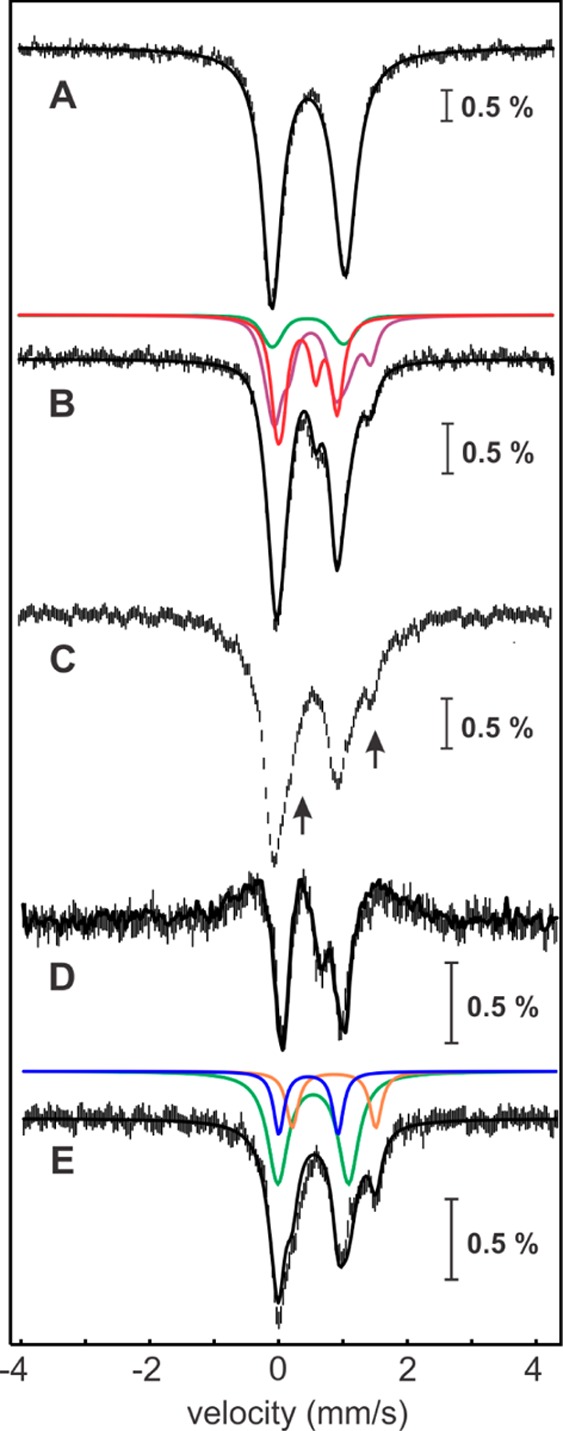
Mössbauer spectroscopic characterization of the monothiolated intermediate in Tt LS. 4.2-K/53-mT Mössbauer spectrum of Tt LS (A, vertical bars) and simulation thereof with parameters quoted in the text (A, solid line). 4.2 K Mössbauer spectra of the Tt LS/peptide 2 complex reacted with SAM and dithionite recorded in externally applied magnetic fields of 0 mT (B, vertical bars) and 53 mT oriented parallel to the γ beam (C, vertical bars). The solid black line in B corresponds to the superposition of the features associated with the 3Fe-cluster (40%, red line), [4Fe–4S]2+ cluster (12%, green line), and site-differentiated [4Fe–4S]2+ cluster (48%, purple line, see below). The [0–53 mT] difference spectrum (C, vertical bars) is similar to that observed for Ec LS (C, solid line is experimental spectrum shown in Figure 3E scaled by 0.75). The reference spectrum of the site-differentiated [4Fe–4S]2+ cluster in Tt LS (E, vertical bars) is obtained by subtracting the contribution of the other components (see text) and can be simulated with three quadrupole doublets representing the valence-delocalized Fe22.5 pair (green line; parameters given in the text) and the two partially localized sites (orange and blue lines; parameters given in Table 2) in a 2:1:1 intensity ratio. The black line in E is the added contribution of the three quadrupole doublets.
Second, the spectra of the Tt LS cross-linked species exhibit two new shoulders (see arrows in Figure 4C). These shoulders are reminiscent of the spectroscopic features observed for SAM-coordinated [4Fe–4S]2+ clusters, in which a pronounced site differentiation and valence localization in one of the two mixed-valent pairs is caused by the coordination of SAM to the unique, non-cysteinyl-ligated Fe site48,71,72 (Table 2). Removal of the contributions of the 3Fe-clusters (40% of spectrum shown in Figure 3E) and of the [4Fe–4S]2+ clusters (36% of the spectrum shown in Figure 4A) from the zero-field spectrum affords a new spectrum with two resolved quadrupole doublets of equal intensity (not shown). This spectrum can be fitted with two quadrupole doublets with the following parameters: δ1 = 0.80 mm/s, ΔEQ1 = 1.30 mm/s; and δ2 = 0.40 mm/s, ΔEQ2 = 0.92 mm/s. Typically, in site-differentiated [4Fe–4S]2+ clusters, the second pair is valence-delocalized, having parameters similar to those of the SAM-free [4Fe–4S]2+ cluster. Thus, the reference spectrum of the site-differentiated [4Fe–4S]2+ cluster is generated by adding 24% of the spectrum of AI Tt LS to that of the two resolved sites (Figure 4E, vertical bars, with individual contributions shown as colored solid lines). This form accounts for 0.4 equiv per LS (48% of 3.6 Fe). The remaining fraction of [4Fe–4S]2+ clusters, which is not associated with the site-differentiated [4Fe–4S]2+ clusters (12%), corresponds to 0.1 equiv of SAM-free [4Fe–4S]2+ clusters.
Table 2. Mössbauer Parameters of the (Partially) Valence-Localized Mixed-Valent Pairs Observed in Site-Differentiated [4Fe–4S]2+ RS Clusters.
Although the approach using the biotinylated peptide substrate for facile isolation of the cross-linked intermediate works well for Tt LS, we note that the cluster content of Tt LS (5.3 Fe corresponding to 1.3 [4Fe–4S]2+ clusters) and of the cross-linked Tt LS/peptide 2 species (3.6 Fe corresponding to 0.5 3Fe-clusters, 0.4 site-differentiated [4Fe–4S] clusters, and 0.1 SAM-free [4Fe–4S]2+ clusters) is significantly substoichiometric, given that approximately half of the expected Fe content is observed. We therefore returned to Ec LS for further studies, focusing on generating the intermediate in sufficiently high yield to obviate the need for purification before spectroscopic analysis.
Generation of the Ec LS/Peptide 2 Intermediate in High Yield
To generate the cross-linked species in high yield, peptide 2 was incubated with Ec LS at room temperature for 90 min, which was sufficient to allow the first sulfur insertion to take place in the absence of detectable insertion of the second sulfur. As shown in Figure 5A, a similar unlabeled synthetic peptide substrate supports almost 1 equiv of lipoyl product (closed, black triangles), which is significantly greater than the lipoyl product generated (0.3–0.4 equiv) when using OHP as a substrate. However, incorporation of deuterium at C8 of the octanoyl group of peptide 2 leads to arrest of the reaction after insertion of the first sulfur atom and therefore accumulation of a monothiolated species. Upon incubation of peptide 2 with Ec LS, a portion of the reaction mixture was analyzed by LC–MS and shown to contain 0.9 equiv of a monothiolated species, while the remainder was analyzed by Mössbauer spectroscopy.
Figure 5.
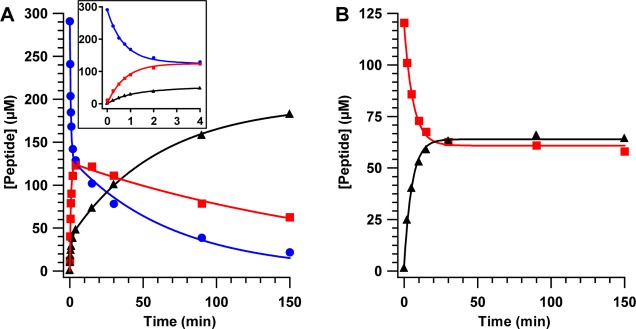
LC–MS analysis of the peptide products of LS. The reaction of 200 μM LS with 300 μM peptide substrate in the presence of SAM and dithionite (A) results in decay of the octanoyl peptide (blue circles), formation and decay of the mercaptooctanoyl species (red squares), and formation of the lipoyl product (black triangles). The lines are fits of the data to the equations described in Materials and Methods with the following kinetic parameters: octanoyl peptide decay: A1 = 157 μM, A2 = 132 μM, k1 = 1.5 min–1, k2 = 0.014 min–1; mercaptooctanoyl peptide formation and decay: A3 = 126 μM, k3 = 1.31 min–1, k4 = 0.005 min–1; and lipoyl peptide formation: A4 = 38 μM, A5 = 157 μM, k5 = 1.1 min–1, k6 = 0.017 min–1. Rapid production of intermediate and product is observed in the first 4 min (inset) followed by a slower phase. The reaction of approximately 120 μM cross-linked intermediate, isolated as described in Materials and Methods, with SAM and dithionite (B) results in decay of the intermediate (red squares) and formation of lipoyl product (black triangles). The lines are fits to a first-order single-exponential equation with the following kinetic parameters: mercaptooctanoyl peptide decay: A = 60.0 μM, k = 0.16 min–1; and lipoyl peptide formation: A = 63.6 μM, k = 0.20 min–1.
The Mössbauer spectrum of the AI Ec LS sample (i.e., before reaction) shows that >95% of the Fe in the sample (7.5 Fe per LS; 1.8 [4Fe–4S]2+ per LS) is in the form of [4Fe–4S]2+ clusters (Figure S6A). The cross-linked species prepared by reacting Ec LS with excess peptide 2 under turnover conditions and without further purification afforded spectra very similar to those of the Tt LS/peptide 2 cross-linked species (Figure S6B,C). The analysis was carried out similarly and reveals that the sample contains 0.68 3Fe-clusters (27% of total Fe), 0.45 SAM-bound [4Fe–4S]2+ clusters (24% of total Fe), and 0.45 [4Fe–4S]2+ clusters per protein, in addition to a significant fraction of N/O-coordinated FeII (25% of total Fe; Figure S6). We note that the parameters of the two unique sites of the SAM-bound [4Fe–4S]2+ cluster are slightly different from those of the Tt LS/peptide 2 intermediate (Table 2). Our finding of 0.9 equiv of monothiolated species by LC–MS is consistent within experimental error with the stoichiometry of 3Fe-clusters (0.68) and [4Fe–4S] clusters (0.9) observed by Mössbauer spectroscopy. Theoretically, we would predict that the stoichiometry of 3Fe-clusters and monothiolated species would be identical if one cluster were sacrificed during turnover and that 0.9 [4Fe–4S] clusters (based on a total of 1.8 [4Fe–4S] clusters in the starting enzyme) and 0.9 3Fe-clusters would remain. The slightly smaller fraction of 3Fe-clusters observed appears to be due to nonproductive degradation of this species to N/O-coordinated FeII.
Catalytic and Kinetic Competence of the Cross-Linked Species
To assess whether the cross-linked species, presumably 6-mercaptooctanoyl-H protein bound to LS via an S–Fe bond to the auxiliary cluster, is catalytically and kinetically competent, LS reactions were conducted with peptide 1, which is not labeled with deuterium at C8. As opposed to the H protein, the smaller size of the peptide substrate allows for facile detection of octanoyl, lipoyl, and 6-mercaptooctanoyl peptides by LC–MS without prior liberation of the modification. A kinetic analysis of the LS reaction using 300 μM peptide 1 and 200 μM LS is shown in Figure 5A. Formation of lipoyl product (black triangles) and decay of the octanoyl substrate (blue circles) follow biphasic behavior, exhibiting the following kinetic parameters, respectively: A4 = 38 μM, k5= 1.1 min–1, A5 = 157 μM, k6 = 0.017 min–1 (lipoyl product); and A1 = 157 μM, k1 = 1.5 min–1, A2 = 132 μM, k2 = 0.014 min–1 (octanoyl substrate). The peptide substrate is converted to the lipoyl product with a rate constant that is similar to that observed in the presence of OHP; however, it supports nearly a full equivalent of lipoyl product (0.9 equiv) after 150 min, significantly higher than that supported by the OHP substrate (Figure 5A). The substrate peptide displays m/z 932.5 [M + H]+, and the addition of two sulfur atoms (in the two-electron reduced state) results in a shift in m/z to 996.5 for the product peptide. A third species (red squares), exhibiting m/z 964.5, forms and decays during the LS reaction with the following kinetic parameters: A3 = 126 μM, k3 = 1.31 min–1, k4 = 0.005 min–1. This mass corresponds to the addition of a single sulfur atom to the octanoyl-peptide, demonstrating the catalytic competence of the mercapto-octanoyl intermediate. As can be observed, the kinetics of the reaction are complex. Both formation of the monothiolated species and loss of the octanoyl substrate take place with similar rate constants and amplitudes for the first phases of their kinetic profiles; however, rate constants for decay of the monothiolated species and formation of the lipoyl product do not match, as would be expected for a scenario in which insertion of the first sulfur atom is much faster than insertion of the second. The observation of a second, much slower, phase of octanoyl substrate decay after nearly 1 equiv is expended in the first phase, suggests that a portion of the enzyme can be reactivated after initial turnover to perform the first step of the reaction during a second turnover, which is consistent with the observation of a substantial concentration of monothiolated intermediate at the completion of the time course. This reactivation may take place as a consequence of some Fe/S clusters being degraded as a result of turnover and the liberated iron and sulfide being used to construct new clusters. Consistent with this premise, this second phase of octanoyl substrate decay occurs with a rate constant that is similar to that of the second, slower, phase for formation of the lipoyl species. Another interesting aspect of the time course is the small burst of lipoyl product (black triangles) corresponding to ∼25% of the total enzyme concentration, which, on this time scale, appears with kinetics similar to those for the monothiolated intermediate. The source of the burst is presently unknown, but it may derive from a fraction of the protein that is optimally suited to react in both steps or perhaps slow protein monomer–dimer interconversion, wherein only one of the two species allows for C8 insertion.
The formation of the lipoyl cofactor from the octanoyl-lysyl substrate requires the expenditure of two molecules of SAM, which are used to generate sequentially the 5′-dA• that abstract the C6 and C8 H• to allow for subsequent sulfur insertion. Therefore, given the slow rate of insertion of the second sulfur atom, use of only 1 equiv of SAM with respect to enzyme concentration in a reaction with excess peptide substrate should result in the formation of a cross-linked species that cannot proceed to product. The cross-linked peptide can then be separated from unreacted peptide substrate using a gel-filtration column, because the unreacted peptide is retained in the column matrix. This strategy obviates the need to use the labeled [8,8,8-2H3]-octanoyl substrate, which essentially induces arrest of the reaction at the intermediate stage, allowing the kinetic competence of the isolated cross-linked species to be evaluated. Upon incubating Ec LS under turnover conditions in the presence of limiting SAM and analyzing the reaction by LC–MS, approximately 0.9 equiv of the cross-linked species (based on quantification of the monothiolated peptide product) was obtained. Control reactions lacking either SAM or dithionite did not contain significant amounts of peptide in the protein fraction (<0.06 equiv each after 10 min of incubation at RT) (Table 3), indicating that the octanoyl peptide does not bind tightly to LS and is removed by gel-filtration chromatography. When excess SAM and reductant were added back to the intermediate complex, clean formation of the lipoyl product was observed, which took place with a rate constant of ∼0.2 min–1 (Figure 5B), which is faster than that of the slowest step in the overall LS reaction (0.04 min–1), indicating that the intermediate formed is both catalytically and kinetically competent to proceed to product. Interestingly, however, although the kinetics of product formation and loss of the intermediate species fit nicely to single-exponential equations, only ∼50% of the intermediate decayed to the product. We speculate that the substoichiometric product yield may be due to an unproductive reaction pathway, which involves the disassembly of the 3Fe-cluster and liberation of Fe and the monothiolated intermediate into solution.
Table 3. Retention of Substrate, Intermediate, and Product by Size-Exclusion Chromatography in LS Reactions.
| sample | equiv intermediate (before column) | equiv intermediate (after column) | equiv substrate (after column) | equiv product (after column) |
|---|---|---|---|---|
| complete reaction | 0.87 | 0.60 | 0.03 | 0.01 |
| no dithionite | 0.01 | 0.03 | 0.04 | 0.00 |
| no SAM | 0.01 | 0.01 | 0.06 | 0.01 |
The Chemically and Kinetically Competent Intermediate Contains a 3Fe-Cluster That Is Disassembled upon Product Formation
The experiments establishing the chemical and kinetic competence of the cross-linked intermediate described in the previous section set the stage to interrogate the configuration of the Fe/S clusters in the intermediate species, as well as their fate upon completion of the reaction, by Mössbauer spectroscopy. The 4.2-K/53-mT Mössbauer spectrum of the anaerobically isolated Ec LS used in this experiment (Figure 6A, vertical bars) reveals the presence of 1.8 [4Fe–4S]2+ clusters (δ = 0.46 mm/s, ΔEQ = 1.16 mm/s, 95% of total Fe, Figure 6A, solid line). The 4.2 K Mössbauer spectra recorded in external fields of 0 and 53 mT (Figure 6B,C, vertical bars) of a sample, in which Ec LS was reacted with peptide 1, dithionite, and 1 equiv of SAM for 15 min, are virtually identical to those of samples prepared by reacting Ec LS with excess peptide 2 under turnover conditions (Figure S6). The [0–53 mT] difference spectrum (Figure 6D, vertical bars) demonstrates the presence of 0.6 3Fe-clusters (27% of total Fe, as determined from the amplitude of the experimental difference spectrum, Figure 3E, solid line). The analysis of the spectra according to the method described above reveals that the sample also contains 0.4 site-differentiated [4Fe–4S]2+ clusters (24% of total Fe) and 0.5 [4Fe–4S]2+ clusters (28% of total Fe). We note that the presence of the site-differentiated [4Fe–4S]2+ cluster is unexpected, because under the reaction conditions there is no excess SAM to bind to the RS cluster. We speculate that binding of the coproduct, methionine, to the RS cluster may lead to similar spectroscopic perturbations. Lastly, ∼12% of total Fe is in the form of N/O-coordinated FeII (δ = 1.25 mm/s and ΔEQ = 3.2 mm/s). The remaining Fe (9%) gives rise to broad features in the Mössbauer spectra. A parallel EPR sample (Figure S8) reveals the presence of a small amount (6% of total Fe) of reduced [4Fe–4S] clusters, which accounts for the majority of the observed broad features. The total yield of 0.6 3Fe-clusters is significant, albeit somewhat less than that of the monothiolated species detected by LC–MS in the analogous reaction (0.9 equiv).
Figure 6.
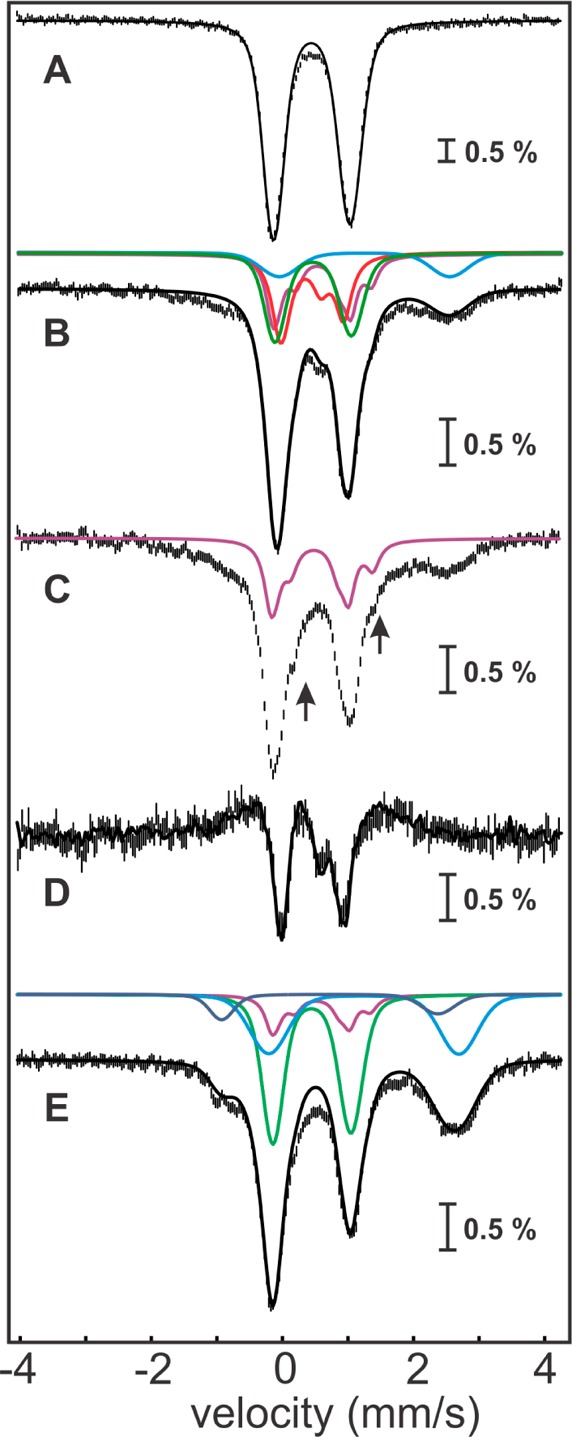
Mössbauer spectroscopic characterization of the reaction of the Ec LS/peptide 1 complex with varying amounts of SAM. 4.2-K/53-mT Mössbauer spectrum of Ec LS (A, vertical bars) and simulation thereof with parameters quoted in the text (A, solid line). 4.2 K Mössbauer spectra of the Ec LS/peptide 1 complex reacted with dithionite and 1 equiv of SAM recorded in externally applied magnetic fields of 0 mT (B, vertical bars) and 53-mT oriented parallel to the γ beam (C, vertical bars). The solid black line in B corresponds to the superposition of the features associated with the 3Fe-clusters (27%, red line), [4Fe–4S]2+ clusters (28%, green line), site-differentiated [4Fe–4S]2+ clusters (24%, purple line, see below), and N/O-coordinated high-spin FeII (12%, light blue line). The arrows in C indicate the shoulders emanating from the spectral features of the site-differentiated [4Fe–4S]2+ cluster (purple line). The [0–53 mT] difference spectrum (D, vertical bars) is similar to that observed for Ec LS (D, solid line is experimental spectrum shown in Figure 3E scaled by 0.53). 4.2 K/53 mT Mössbauer spectrum of the sample of the purified, monothiolated intermediate reacted with dithionite and a second equivalent of SAM (E, vertical bars). The solid black line in E corresponds to the superposition of the features associated with the [4Fe–4S]2+ clusters (41%, green line), site-differentiated [4Fe–4S]2+ clusters (12%, purple line), N/O-coordinated high-spin FeII (28%, light blue line), and S-coordinated high-spin FeII (8%, dark blue line).
The fate of the auxiliary cluster upon completion of the reaction was probed by Mössbauer spectroscopy on an identical sample that was purified from excess reactants. Comparison of spectra recorded before and after purification by gel-filtration chromatography reveals that these two spectra are essentially identical (Figure S7), demonstrating that purification does not lead to decay of the cross-linked intermediate. Addition of a second equivalent of SAM and reductant (dithionite) to the sample of the purified intermediate is associated with significant Mössbauer spectroscopic changes. Spectra collected at 4.2 K in a 53-mT field oriented parallel to the γ beam (Figure 6E, vertical bars) and zero field (Figure S9, vertical bars) are almost identical (see Figure S9 for comparison), thereby demonstrating that the 3Fe-clusters have decayed during the second part of the reaction (we estimate an upper limit of ∼5% of the 3Fe-clusters in this sample). Because the EPR spectrum of an identical sample only reveals that approximately 3% of total Fe in the sample is associated with Fe/S clusters with half-integer ground state (Figure S8), which is below the detection limit in the Mössbauer spectra, we assign the features to quadrupole doublets. The spectrum can be simulated as a superposition of the spectral features of the regular (i.e., non-site-differentiated) [4Fe–4S]2+ cluster (41% of total Fe; 0.6 clusters per LS), site-differentiated [4Fe–4S]2+ clusters (upper limit of 12% of total Fe; 0.17 clusters per LS inferred from spectral subtractions), and two (or more) broad quadrupole doublets with parameters characteristic of high-spin FeII. Although a significant fraction of FeII has parameters typical of N/O-coordinated FeII (∼28% of total Fe, δ ∼1.25 mm/s and ΔEQ ∼2.9 mm/s), we also note a pronounced shoulder at ∼ −1 mm/s, which is at a position expected for a quadrupole doublet associated with an FeII–S4 site (∼8% of total Fe, δ ∼0.73 mm/s and ΔEQ ∼3.3 mm/s).73 The [4Fe–4S]2+ cluster is assigned to the RS cluster, whereas the various FeII complexes are proposed to derive from the Fe sites of the auxiliary cluster, which is (at least partially) disassembled during the reaction.
Discussion
The proposed mechanism of LS involves two reductive cleavages of SAM, each of which is followed by abstraction of an H• from the octanoyl-containing substrate by a 5′-dA• with subsequent sulfur insertion. Notably, the source of the inserted sulfur atom has been suggested to be the auxiliary [4Fe–4S] cluster of the protein, which would result in inactivation of the enzyme after one complete turnover.56 In support of this unusual and provocative role of an Fe/S cluster in enzyme catalysis, studies by Miller et al. and Cicchillo et al. showed that no exogenous source of sulfur is required for the reaction and that both sulfur atoms derive from the same LS polypeptide.4,56 In one key study, in which [octanoyl-d15]-H protein was used as a substrate for the LS reaction, formation of the lipoyl product was almost completely suppressed, whereas analyses by mass spectrometry showed significant formation of a monothiolated species, suggested to be an intermediate in the reaction.3 Subsequent studies, in which substrates specifically labeled with deuterium at C6 or C8 were used, showed that deuterium incorporation only at C8 results in significant suppression of lipoyl product formation and that sulfur insertion at C6 always precedes insertion at C8, even with an unlabeled substrate.5 These observations suggested a possible strategy to test the provocative notion that an enzyme might sacrifice a required cofactor during turnover by contributing a sulfur atom from one of its Fe/S clusters. Indeed, if a suitable substrate containing deuterium only at C8 of its octanoyl chain were used in LS reactions, then it might permit trapping of an intermediate species in a way that would allow for its spectroscopic characterization.
Our initial efforts made use of the size of the OHP substrate to show cross-link formation by SDS-PAGE. Ec LS and Ec HP form a fairly tight complex that will survive molecular sieve chromatography but can be separated by anion-exchange chromatography, wherein Ec LS adheres poorly to the resin, while Ec HP adheres tightly. When the two proteins were incubated under turnover conditions and then applied to the column, a new band exhibiting intermediate migratory properties and containing both proteins was observed. The observation that the proteins in this band migrated according to their native molecular masses rather than as the sum of their molecular masses indicates that the bond that links them, unlike a typical covalent bond, is unstable under conditions of SDS-PAGE. Similar observations were made in studies in which the cross-link was generated with small peptide substrates. In these instances, when LS and the peptide substrate were combined and subjected to gel-filtration chromatography, LS eluted cleanly in the fraction containing large molecules, whereas the peptide substrate eluted cleanly in fractions containing small molecules. However, when LS was incubated with a peptide substrate containing an [8,8,8-d3]-octanoyl moiety or with only 1 equiv of SAM and a peptide substrate containing an unlabeled octanoyl moiety, LS and the peptide substrate eluted together in fractions containing large molecules. The peptide was readily liberated from the protein under acidic conditions, and analysis by mass spectrometry showed it to contain an inserted sulfur atom. These characteristics are consistent with LS and either the protein or peptide substrate being linked by an Fe/S cluster in the isolated intermediate. Moreover, linkage of the two proteins or the protein and peptide via a disulfide bond is unlikely, because the presence of DTT in reaction buffers is expected to reduce any disulfide bonds before the complexes are subjected to anion-exchange or molecular sieve chromatography.
Further insight into the nature of the cross-linked intermediate was obtained from Mössbauer spectroscopy. All samples containing this intermediate exhibit features of a 3Fe-cluster, with an electronic structure similar to that of cuboidal [3Fe–4S]0 clusters, namely, an S = 2 electron spin ground state emanating from antiferromagnetic (AF) coupling between a high-spin FeIII center (S = 5/2) and a valence-delocalized Fe22.5+ unit (S = 9/2). We assign the 3Fe-cluster to the partially disassembled auxiliary [4Fe–4S] cluster. This cluster is proposed to form upon reaction of the putative C6-centered substrate radical (R•) with one of the cluster’s μ3-bridging sulfides, which would yield the C6-monothiolated intermediate (RS–). Formally, this reaction entails recombination of the substrate radical with one electron of the lone pair of one of the μ3-sulfido ligands of the [4Fe–4S]2+ cluster. The second electron of the lone pair is formally donated to one of the Fe22.5+ pairs to yield an Fe2II pair. We formally designate the resulting cluster as [4Fe–3S–1RS]2+ (Scheme 1), in which we consider formally the charges of the inorganic sulfide ligands (2– each), the monothiolated intermediate (1–), and the formal oxidation states of the Fe ions (one Fe2II and one Fe22.5, i.e., 9+) (eq 5a and Scheme 1). Loss of one FeII (eq 5b and Scheme 1) and one electron (eq 5c and Scheme 1) would lead to the experimentally observed trinuclear cluster, which we denote as [3Fe–3S–1RS]1+ and which is best described as consisting of a valence-delocalized Fe22.5+ pair AF coupled to the FeIII site (i.e., a total of eight positive charges), bridged by three sulfides and the monothiolated intermediate.
| 5a |
| 5b |
| 5c |
Scheme 1. Schematic Representation of the Auxiliary Cluster of LS during Turnover.

We note that the reactions shown in eqs 5b and 5c are conceptually similar to the well-known oxidative disassembly of [4Fe–4S]2+ clusters to the cuboidal [3Fe–4S]+ form upon loss of one FeII. Moreover, the electronic structure of the intermediate [3Fe–3S–1RS]+ cluster is consistent with a cuboidal geometry. Thus, this reaction would not require major structural rearrangement of the cluster.
The second distinct spectroscopic feature observed in samples of the monothiolated intermediate are shoulders associated with the quadrupole doublet features typical of a site-differentiated [4Fe–4S]2+ cluster. We tentatively assign this cluster to the RS [4Fe–4S]2+ cluster, of which the unique, non-cysteine-ligated Fe site may be coordinated by SAM or methionine, a coproduct of reductive SAM cleavage. Similar spectroscopic perturbations have been observed in other RS enzymes48,71,72 (Table 2). We also note that the RS [4Fe–4S]2+ cluster can be partially disassembled to the [3Fe–4S]0 form upon purification of the cross-linked intermediate in the presence of excess reductant, because we observe 1.5 equiv of 3Fe-clusters. Loss of Fe from a [4Fe–4S] cluster to yield [3Fe–4S] clusters has been reported for RS enzymes.66,69,74
To assess whether the isolated complex is competent to proceed to product, Ec LS was incubated with an unlabeled peptide substrate and 1 equiv of SAM under turnover conditions, reisolated by molecular sieve chromatography, and then introduced into a second reaction containing excess SAM, dithionite, and SAHN. Samples to be analyzed by Mössbauer spectroscopy were also removed after formation of the intermediate, after isolation of the intermediate by molecular sieve chromatography, and after incubation of the intermediate further under turnover conditions. Analysis of the reaction by LC–MS showed that 0.9 equiv of the intermediate was formed and that ∼50% of the intermediate decayed to the lipoyl product with a rate constant that was faster than the overall rate constant for catalysis. The substoichiometric amount of product may be a consequence of an unproductive reaction pathway involving disassembly of the auxiliary cluster under our nonphysiological reaction conditions and liberation of the cluster’s 3 Fe ions and the monothiolated intermediate into solution. While it has been previously reported that the 6-mercapto-octanoyl species is the stable intermediate generated by LS,5 one possible explanation for the substoichiometric product is that LS generates both the 6-mercapto- and the 8-mercapto-octanoyl species, but only one of these is competent to form product. However, when the intermediate is cleaved from the peptide using lipoamidase from Enterococcus faecalis and subjected to reverse-phase liquid chromatography, it is clear that only 6-MOA is present (Figure S10).
The second step of the LS reaction involves generation of the C8-centered radical (R′•) by a second equivalent of 5′-dA•. The R′• can then attack one of the remaining sulfido groups of the [3Fe–3S–1RS]+ auxiliary cluster of the monothiolated intermediate. Again, this reaction formally entails recombination of the substrate radical with one electron of the lone pair of the attacked sulfido ligand, with the second electron of the lone pair being formally donated to the Fe22.5+ unit of the [3Fe–3S–1RS]+ cluster, giving rise to a [3Fe–2S–1RS/1R′S]+ cluster (eq 6a and Scheme 1), in which the RS/R′S moiety represents the dianionic form of the reduced lipoyl product. We speculate that dissociation of the lipoyl product will destabilize the remainder of the disassembled cluster under our in vitro conditions, resulting in loss of Fe (formally, two FeII and one FeIII ion) and sulfide to solution (eq 6b and Scheme 1). Mössbauer analysis of the sample of the complete reaction suggests that the intermediate’s [3Fe–3S–1RS]+ auxiliary cluster is partially or completely disassembled and reduced to multiple high-spin FeII complexes, including N/O-coordinated FeII (presumably lost from the cluster into solution) and S-coordinated FeII (either Fe remaining in the partially disassembled site of the auxiliary cluster or Fe in solution coordinated by the lipoyl product).
| 6a |
| 6b |
While it has been clear for some time that the source of the inserted sulfur atom in the LS reaction is LS itself, whether the auxiliary iron sulfur cluster is the direct source has not been established. Although the idea of using an Fe/S cluster as an activated source of sulfur that recombines with an organic substrate radical is provocative, significant evidence has been gathered for this role in biotin synthase, which catalyzes similar sulfur insertion steps to form the thiolane ring of the biotin cofactor.54,71,75−78 By contrast, recent studies have provided evidence that in the methylthiotransferases, RimO and MiaB, which attach methythio groups (−SCH3) at sp2- or sp3-hybridized carbon centers and which also contain auxiliary [4Fe–4S] clusters, the sulfur atom of the methylthio group does not derive from one of the μ3-sulfido ions of the cluster but more likely from a sulfide ion that is attached externally to the cluster as a ligand.45
Given that LS is inactivated during catalysis because of cluster destruction, it is likely that other cellular factors serve to rebuild or replace the cluster to allow for multiple turnover. Proteins encoded in the isc or suf operons, responsible for building and inserting Fe/S clusters in Ec and other organisms, are potential candidates for playing such roles. Recently, two genes from the mitochondrial Fe/S cluster assembly system in Saccharomyces cerevisiae (Sc), ISA1 and ISA2, were identified as playing a role in the catalytic cycle of biotin synthase but not in the de novo biosynthesis of the Fe/S clusters of the protein. It was suggested that these proteins might function in regeneration of the sacrificial [2Fe–2S] cluster of biotin synthase.79 In a subsequent study, a third gene, IBA57, that encodes a protein that interacts with Isa1 and Isa2 was identified in Sc.80 Deletion of the IBA57 gene prevented insertion of the Fe/S cluster of aconitase and homoaconitase and also inhibited the in vivo functions of biotin and lipoyl synthases. The same three genes have been identified in humans (ISCA1, ISCA2, and IBA57), and defects are associated with severe disease and premature death.81,82 Silencing of these genes in HeLa cells by RNAi results in misformed mitochondria and decreased activity of a subset of mitochondrial Fe/S proteins, including LS.81 Moreover, homologues of these proteins are also found Ec, suggesting the importance of this pathway in prokaryotes as well.80
A second set of human genes, NFU1 and BOLA3, have been characterized as having similar phenotypes.83,84 Defects in these genes cause decreases in protein lipoylation and a corresponding low activity of lipoyl-dependent multienzyme complexes, again leading to disease and premature death.83,84 NFU1 is postulated to be an alternative Fe/S scaffold, and BOLA3 is hypothesized to be a reductase that aids in the transfer of clusters from the scaffold to the target protein.83 Interestingly, the role of NFU1 appears to be somewhat specific for lipoyl synthase and succinate dehydrogenase given that the activities of other mitochondrial Fe/S proteins were unaffected upon NFU1 knockdown by RNAi. Whether these proteins act in de novo biosynthesis of the Fe/S clusters in LS or cluster regeneration has not been determined.
In summary, we provide strong biochemical and structural evidence for a key proposed intermediate in the biosynthesis of the lipoyl cofactor that involves a covalent bond between the organic substrate and a sulfur atom of a sacrificed [4Fe–4S] cluster. Although the enzyme is inactivated upon turnover because of cluster destruction, ongoing studies of Fe/S cluster biosynthesis in a number of different organisms hint at the possibility of dedicated pathways for regeneration of the auxiliary cluster of LS and other related enzymes, thus demonstrating that the employment of an Fe/S cofactor as a source of sulfur during the reaction of a LS may not be as limiting as initially thought.
Glossary
Abbreviations Used
- 5′-dA
5′-deoxyadenosine
- 5′-dA•
5′-deoxyadenosyl 5′-radical
- AI
as-isolated
- BioB
biotin synthase
- DTT
dithiothreitol
- Ec
Escherichia coli
- EPR
electron paramagnetic resonance
- Fe/S
iron–sulfur
- HEPES
N-(2-hydroxyethyl)piperazine-N′-2-ethanesulfonic acid
- HP
H protein
- HPLC
high-performance liquid chromatography
- IS
internal standard
- LA
lipoic acid
- LCP
lipoyl carrier protein
- LHP
lipoyl-H protein
- LS
lipoyl synthase
- OHP
octanoyl-H protein
- RS
radical SAM
- SAHN
S-adenosylhomocysteine nucleosidase
- SAM
S-adenosyl-l-methionine
- SDS-PAGE
sodium dodecyl sulfate–polyacrylamide gel electrophoresis
- Ss
Sulfolobus solfataricus
- TCA
trichloroacetic acid
- TCEP
tris(2-carboxyethyl)phosphine
- Tt
Thermus thermophilus
- UV–vis
ultraviolet–visible
- WT
wild type
Supporting Information Available
Figures S1 (structure of peptide 2), S2 (extracted ion chromatograms of peptides), S3 (SDS-PAGE of fractions from anion-exchange chromatography of a LipA–OHP mixture), S4 (SDS-PAGE of fractions of anion-exchange chromatography of large-scale EcLipA cross-linked with OHP), S5 (EPR spectra of EcLipA cross-linked with OHP), S6 (Mössbauer spectra of EcLipA cross-linked with peptide 2), S7 (comparison of Mössbauer spectra of EcLipA cross-link generated with 1 equiv of SAM before and after gel-filtration chromatography), S8 (EPR spectra of EcLipA cross-link generated with 1 equiv of SAM), S9 (comparison of Mössbauer spectra in zero-field and 53 mT field of EcLipA cross-link generated with 1 equiv of SAM and reacted with excess SAM), and S10 (LC–MS analysis of 6-MOA and 8-MOA) as well as methods (cleavage of 6-MOA and 8-MOA from peptides and analysis by LC–MS). This material is available free of charge via the Internet at http://pubs.acs.org.
This work was supported by NIH grants GM-63847 and GM-103268 (S.J.B.), the Dreyfus Foundation (Teacher Scholar Award to C.K.), and the Beckman Foundation (Young Investigator Award to C.K.).
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Billgren E. S., Cicchillo R. M., Nesbitt N. M., and Booker S. J. (2010) Lipoic acid biosynthesis and enzymology, in Comprehensive Natural Products II: Chemistry and Biology (Mander L., and Liu H.-W., Eds.) pp 181–212, Elsevier, Amsterdam, The Netherlands. [Google Scholar]
- Cronan J. E. Jr.; Zhao X.; Jiang Y. (2005) Function, attachment and synthesis of lipoic acid in Escherichia coli. Adv. Microb. Physiol. 50, 103–146. [DOI] [PubMed] [Google Scholar]
- Cicchillo R. M.; Iwig D. F.; Jones A. D.; Nesbitt N. M.; Baleanu-Gogonea C.; Souder M. G.; Tu L.; Booker S. J. (2004) Lipoyl synthase requires two equivalents of S-adenosyl-l-methionine to synthesize one equivalent of lipoic acid. Biochemistry 43, 6378–6386. [DOI] [PubMed] [Google Scholar]
- Miller J. R.; Busby R. W.; Jordan S. W.; Cheek J.; Henshaw T. F.; Ashley G. W.; Broderick J. B.; Cronan J. E. Jr.; Marletta M. A. (2000) Escherichia coli LipA is a lipoyl synthase: in vitro biosynthesis of lipoylated pyruvate dehydrogenase complex from octanoyl-acyl carrier protein. Biochemistry 39, 15166–15178. [DOI] [PubMed] [Google Scholar]
- Douglas P.; Kriek M.; Bryant P.; Roach P. L. (2006) Lipoyl synthase inserts sulfur atoms into an octanoyl substrate in a stepwise manner. Angew. Chem., Int. Ed. 45, 5197–5199. [DOI] [PubMed] [Google Scholar]
- Zhao X.; Miller J. R.; Jiang Y.; Marletta M. A.; Cronan J. E. Jr. (2003) Assembly of the covalent linkage between lipoic acid and its cognate enzymes. Chem. Biol. 10, 1293–1302. [DOI] [PubMed] [Google Scholar]
- Robinson J. R.; Klein S. M.; Sagers R. D. (1973) Glycine metabolism. Lipoic acid as the prosthetic group in the electron transfer protein P2 from Peptococcus glycinophilus. J. Biol. Chem. 248, 5319–5323. [PubMed] [Google Scholar]
- Koike M.; Reed L. J. (1960) α-Keto acid dehydrogenation complexes. II. The role of protein-bound lipoic acid and flavin adenine dinucleotide. J. Biol. Chem. 235, 1931–1938. [PubMed] [Google Scholar]
- Reed L. J.; Koike M.; Levitch M. E.; Leach F. R. (1958) Studies on the nature and reactions of protein-bound lipoic acid. J. Biol. Chem. 232, 143–158. [PubMed] [Google Scholar]
- Ambrose-Griffen M. C.; Danson M. J.; Griffen W. G.; Hale G.; Perham R. N. (1980) Kinetic analysis of the role of lipoic acid residues in the pyruvate dehydrogenase multienzyme complex of Escherichia coli. Biochem. J. 187, 393–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angelides K. J.; Hammes G. G. (1978) Mechanism of action of the pyruvate dehydrogenase multienzyme complex from Escherichia coli. Proc. Natl. Acad. Sci. U.S.A. 75, 4877–4880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettit F. H.; Yeaman S. J.; Reed L. J. (1978) Purification and characterization of branched chain α-keto acid dehydrogenase complex of bovine kidney. Proc. Natl. Acad. Sci. U.S.A. 75, 4881–4885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Douce R.; Bourguignon J.; Macherel D.; Neuburger M. (1994) The glycine decarboxylase system in higher plant mitochondria: structure, function and biogenesis. Biochem. Soc. Trans. 22, 184–188. [DOI] [PubMed] [Google Scholar]
- Reed L. J.; Hackert M. L. (1990) Structure–function relationships in dihydrolipoamide acyltransferases. J. Biol. Chem. 265, 8971–8974. [PubMed] [Google Scholar]
- Biewenga G. P., Haenen G. R. M. M. and Bast A. (1997) An overview of lipoate chemistry, in Lipoic Acid in Health and Disease (Fuchs J., Packer L., and Zimmer G., Eds.) pp 1–32, Marcel Dekker, New York. [Google Scholar]
- Krüger N.; Oppermann F. B.; Lorenzl H.; Steinbüchel A. (1994) Biochemical and molecular characterization of the Clostridium magnum acetoin dehydrogenase enzyme system. J. Bacteriol. 176, 3614–3630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiemer A. K., and Diesel B. (2008) Activation of cytoprotective signaling pathways by alpha-lipoic acid, in Lipoic Acid: Energy Production, Antioxidant Activity and Health Effects (Patel M. S., and Packer L., Eds.) CRC Press, Boca Raton, FL. [Google Scholar]
- Han D., Hamilton R. T., Lam P. Y., and Packer L. (2008) Modulation of cellular redox and metabolic status by lipoic acid, in Lipoic Acid: Energy Production, Antioxidant Activity and Health Effects (Patel M. S., and Packer L., Eds.) CRC Press, Boca Raton, FL. [Google Scholar]
- Borcea V.; Nourooz-Zadeh J.; Wolff S. P.; Klevesath M.; Hofmann M.; Urich H.; Wahl P.; Ziegler R.; Trischler H.; Halliwell B.; Nawroth P. P. (1999) α-Lipoic acid decreases oxidative stress even in diabetic patients with poor glycemic control and albuminuria. Free Radical Biol. Med. 26, 1495–1500. [DOI] [PubMed] [Google Scholar]
- Tirosh O.; Sen C. K.; Roy S.; Kobayashi M. S.; Packer L. (1999) Neuroprotective effects of alpha-lipoic acid and its positively charged amide analogue. Free Radical Biol. Med. 26, 1418–1426. [DOI] [PubMed] [Google Scholar]
- Konrad D.; Somwar R.; Sweeney G.; Yaworsky K.; Hayashi M.; Ramlal T.; Klip A. (2001) The antihyperglycemic drug alpha-lipoic acid stimulates glucose uptake via both GLUT4 translocation and GLUT4 activation: potential role of p38 mitogen-activated protein kinase in GLUT4 activation. Diabetes 50, 1464–1471. [DOI] [PubMed] [Google Scholar]
- Hager K.; Marahrens A.; Kenklies M.; Riederer P.; Münch G. (2001) alpha-Lipoic acid as a new treatment option for Alzheimer type dementia. Arch. Gerontol. Geriatr. 32, 275–282. [DOI] [PubMed] [Google Scholar]
- Morris T. W.; Reed K. E.; Cronan J. E. Jr. (1995) Lipoic acid metabolism in Escherichia coli: the lplA and lipB genes define redundant pathways for ligation of lipoyl groups to apoprotein. J. Bacteriol. 177, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reed K. E.; Cronan J. E. Jr. (1993) Lipoic acid metabolism in Escherichia coli: sequencing and functional characterization of the lipA and lipB genes. J. Bacteriol. 175, 1325–1336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green D. E.; Morris T. W.; J G.; Cronan J. E. Jr.; Guest J. R. (1995) Purification and properties of the lipoate protein ligase of Escherichia coli. Biochem. J. 309, 853–862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris T. W.; Reed K. E.; Cronan J. E. Jr. (1994) Identification of the gene encoding lipoate-protein ligase A of Escherichia coli. Molecular cloning and characterization of the lplA gene and gene product. J. Biol. Chem. 269, 16091–16100. [PubMed] [Google Scholar]
- Fujiwara K.; Okamura-Ikeda K.; Motokawa Y. (1994) Purification and characterization of lipoyl-AMP:Nε-lysine lipoyltransferase from bovine liver mitochondria. J. Biol. Chem. 269, 16605–16609. [PubMed] [Google Scholar]
- Fujiwara K.; Takeuchi S.; Okamura-Ikeda K.; Motokawa Y. (2001) Purification, characterization, and cDNA cloning of lipoate-activating enzyme from bovine liver. J. Biol. Chem. 276, 28819–28823. [DOI] [PubMed] [Google Scholar]
- Jordan S. W.; Cronan J. E. Jr. (1997) A new metabolic link. The acyl carrier protein of lipid synthesis donates lipoic acid to the pyruvate dehydrogenase complex in Escherichia coli and mitochondria. J. Biol. Chem. 272, 17903–17906. [DOI] [PubMed] [Google Scholar]
- Jordan S. W.; Cronan J. E. Jr. (2003) The Escherichia coli lipB gene encodes lipoyl (octanoyl)-acyl carrier protein:protein transferase. J. Bacteriol. 185, 1582–1589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma Q.; Eddine A. N.; Geerlof A.; Li X.; Cronan J. E. Jr.; Kaufmann S. H. E.; Wilmanns M. (2006) The Mycobacterium tuberculosis LipB enzyme functions as a cysteine/lysine dyad acyltransferase. Proc. Natl. Acad. Sci. U.S.A. 103, 8662–8667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nesbitt N. M.; Baleanu-Gogonea C.; Cicchillo R. M.; Goodson K.; Iwig D. F.; Broadwater J. A.; Haas J. A.; Fox B. G.; Booker S. J. (2005) Expression, purification, and physical characterization of Escherichia coli lipoyl(octanoyl)transferase. Protein Expression Purif. 39, 269–282. [DOI] [PubMed] [Google Scholar]
- Zhao X.; Miller J. R.; Cronan J. E. Jr. (2005) The reaction of LipB, the octanoyl-[acyl carrier protein]:protein N-octanoyltransferase of lipoic acid synthesis, proceeds through an acyl-enzyme intermediate. Biochemistry 44, 16737–16746. [DOI] [PubMed] [Google Scholar]
- Schonauer M. S.; Kastaniotis A. J.; Kursu V. A.; Hiltunen J. K.; Dieckmann C. L. (2009) Lipoic acid synthesis and attachment in yeast mitochondria. J. Biol. Chem. 284, 23234–23242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin N.; Christensen Q. H.; Mansilla M. C.; Cronan J. E. Jr.; de Mendoza D. (2011) A novel two-gene requirement for the octanoyltransfer reaction of Bacillus subtilis lipoic acid biosynthesis. Mol. Microbiol. 80, 335–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christensen Q. H.; Martin N.; Mansilla M. C.; de Mendoza D.; Cronan J. E. Jr. (2011) A novel amidotransferase required for lipoic acid cofactor assembly in Bacillus subtilis. Mol. Microbiol. 80, 350–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi X.; Maeda N. (2005) Endogenous production of lipoic acid is essential for mouse development. Mol. Cell. Biol. 25, 8387–8392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Booker S. J.; Cicchillo R. M.; Grove T. L. (2007) Self-sacrifice in radical S-adenosylmethionine proteins. Curr. Opin. Chem. Biol. 11, 543–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frey P. A.; Booker S. J. (2001) Radical mechanisms of S-adenosylmethionine-dependent enzymes. Adv. Protein Chem. 58, 1–45. [DOI] [PubMed] [Google Scholar]
- Frey P. A.; Hegeman A. D.; Ruzicka F. J. (2008) The radical SAM superfamily. Crit. Rev. Biochem. Mol. Biol. 43, 63–88. [DOI] [PubMed] [Google Scholar]
- Roach P. L. (2011) Radicals from S-adenosylmethionine and their application to biosynthesis. Curr. Opin. Chem. Biol. 15, 267–275. [DOI] [PubMed] [Google Scholar]
- Sofia H. J.; Chen G.; Hetzler B. G.; Reyes-Spindola J. F.; Miller N. E. (2001) Radical SAM, a novel protein superfamily linking unresolved steps in familiar biosynthetic pathways with radical mechanisms: functional characterization using new analysis and information visualization methods. Nucleic Acids Res. 29, 1097–1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Booker S. J. (2009) Anaerobic functionalization of unactivated C–H bonds. Curr. Opin. Chem. Biol. 13, 58–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atta M.; Mulliez E.; Arragain S.; Forouhar F.; Hunt J. F.; Fontecave M. (2010) S-Adenosylmethionine-dependent radical-based modification of biological macromolecules. Curr. Opin. Struct. Biol. 20, 1–9. [DOI] [PubMed] [Google Scholar]
- Forouhar F.; Arragain S.; Atta M.; Gambarelli S.; Mouesca J.-M.; Hussain M.; Xiao R.; Kieffer-Jaquinod S.; Seetharaman J.; Acton T. B.; Montelione G. T.; Mulliez E.; Hunt J. F.; Fontecave M. (2013) Two Fe–S clusters catalyze sulfur insertion by radical-SAM methylthiotransferases. Nat. Chem. Biol. 9, 333–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arragain S.; García-Serres R.; Blondin G.; Douki T.; Clemancey M.; Latour J.-M.; Forouhar F.; Neely H.; Montelione G. T.; Hunt J. F.; Mulliez E.; Fontecave M.; Atta M. (2010) Post-translational modification of ribosomal proteins: structural and functional characterization of RimO from Thermotoga maritima, a radical S-adenosylmethionine methylthiotransferase. J. Biol. Chem. 285, 5792–5801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anton B. P.; Saleh L.; Benner J. S.; Raleigh E. A.; Kasif S.; Roberts R. J. (2008) RimO, a MiaB-like enzyme, methylthiolates the universally conserved Asp88 residue of ribosomal protein S12 in Escherichia coli. Proc. Natl. Acad. Sci. U.S.A. 105, 1826–1831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee K. H.; Saleh L.; Anton B. P.; Madinger C. L.; Benner J. S.; Iwig D. F.; Roberts R. J.; Krebs C.; Booker S. J. (2009) Characterization of RimO, a new member of the methylthiotransferase subclass of the radical SAM superfamily. Biochemistry 48, 10162–10174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierrel F.; Douki T.; Fontecave M.; Atta M. (2004) MiaB protein is a bifunctional radical-S-adenosylmethionine enzyme involved in thiolation and methylation of tRNA. J. Biol. Chem. 279, 47555–47563. [DOI] [PubMed] [Google Scholar]
- Landgraf B. J.; Arcinas A. J.; Lee K. H.; Booker S. J. (2013) Identification of an intermediate methyl carrier in the radical S-adenosylmethionine methylthiotransferases RimO and MiaB. J. Am. Chem. Soc. 135, 15404–15416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cicchillo R. M.; Lee K. H.; Baleanu-Gogonea C.; Nesbitt N. M.; Krebs C.; Booker S. J. (2004) Escherichia coli lipoyl synthase binds two distinct [4Fe–4S] clusters per polypeptide. Biochemistry 43, 11770–11781. [DOI] [PubMed] [Google Scholar]
- Cosper M. M.; Jameson G. N. L.; Hernández H. L.; Krebs C.; Huynh B. H.; Johnson M. K. (2004) Characterization of the cofactor composition of Escherichia coli biotin synthase. Biochemistry 43, 2007–2021. [DOI] [PubMed] [Google Scholar]
- Hernández H. L.; Pierrel F.; Elleingand E.; García-Serres R.; Huynh B. H.; Johnson M. K.; Fontecave M.; Atta M. (2007) MiaB, a bifunctional radical-S-adenosylmethionine enzyme involved in the thiolation and methylation of tRNA, contains two essential [4Fe–4S] clusters. Biochemistry 46, 5140–5147. [DOI] [PubMed] [Google Scholar]
- Ugulava N. B.; Gibney B. R.; Jarrett J. T. (2001) Biotin synthase contains two distinct iron–sulfur cluster binding sites: chemical and spectroelectrochemical analysis of iron–sulfur cluster interconversions. Biochemistry 40, 8343–8351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ugulava N. B.; Surerus K. K.; Jarrett J. T. (2002) Evidence from Mössbauer spectroscopy for distinct [2Fe–2S]2+ and [4Fe–4S]2+ cluster binding sites in biotin synthase from Escherichia coli. J. Am. Chem. Soc. 124, 9050–9051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cicchillo R. M.; Booker S. J. (2005) Mechanistic investigations of lipoic acid biosynthesis in Escherichia coli: both sulfur atoms in lipoic acid are contributed by the same lipoyl synthase polypeptide. J. Am. Chem. Soc. 127, 2860–2861. [DOI] [PubMed] [Google Scholar]
- Lanz N. D.; Grove T. L.; Gogonea C. B.; Lee K. H.; Krebs C.; Booker S. J. (2012) RlmN and AtsB as models for the overproduction and characterization of radical SAM proteins. Methods Enzymol. 516, 125–152. [DOI] [PubMed] [Google Scholar]
- Iwig D. F.; Booker S. J. (2004) Insight into the polar reactivity of the onium chalcogen analogues of S-adenosyl-l-methionine. Biochemistry 43, 13496–13509. [DOI] [PubMed] [Google Scholar]
- Iwig D. F.; Grippe A. T.; McIntyre T. A.; Booker S. J. (2004) Isotope and elemental effects indicate a rate-limiting methyl transfer as the initial step in the reaction catalyzed by Escherichia coli cyclopropane fatty acid synthase. Biochemistry 43, 13510–13524. [DOI] [PubMed] [Google Scholar]
- Laemmli U. K. (1970) Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227, 680–685. [DOI] [PubMed] [Google Scholar]
- Krebs C.; Price J. C.; Baldwin J.; Saleh L.; Green M. T.; Bollinger J. M. Jr. (2005) Rapid freeze–quench 57Fe Mössbauer spectroscopy: monitoring changes of an iron-containing active site during a biochemical reaction. Inorg. Chem. 44, 742–757. [DOI] [PubMed] [Google Scholar]
- Parry R. J. (1977) Biosynthesis of lipoic acid. 1. Incorporation of specifically tritiated octanoic acid into lipoic acid. J. Am. Chem. Soc. 99, 6464–6466. [Google Scholar]
- Emptage M. H.; Kent T. A.; Huynh B. H.; Rawlings J.; Orme-Johnson W. H.; Münck E. (1980) Nature of the iron–sulfur centers in a ferredoxin from Azotobacter vinelandii – Mössbauer studies and cluster displacement experiments. J. Biol. Chem. 255, 1793–1796. [PubMed] [Google Scholar]
- Kent T. A.; Dreyer J. L.; Kennedy M. C.; Huynh B. H.; Emptage M. H.; Beinert H.; Münck E. (1982) Mössbauer studies of beef heart aconitase: evidence for facile interconversion of iron–sulfur clusters. Proc. Natl. Acad. Sci. U.S.A. 79, 1096–1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papaefthymiou V.; Girerd J. J.; Moura J. J. G.; Moura I.; Münck E. (1987) Mössbauer study of D. gigas ferredoxin II and spin-coupling model for Fe3S4 cluster with valence delocalization. J. Am. Chem. Soc. 109, 4703–4710. [Google Scholar]
- Hänzelmann P.; Hernández H. L.; Menzel C.; García-Serres R.; Huynh B. H.; Johnson M. K.; Mendel R. R.; Schindelin H. (2004) Characterization of MOCS1A, an oxygen-sensitive iron-sulfur protein involved in human molybdenum cofactor biosynthesis. J. Biol. Chem. 279, 34721–34732. [DOI] [PubMed] [Google Scholar]
- Beinert H.; Holm R. H.; Münck E. (1997) Iron–sulfur clusters: nature’s modular, multipurpose structures. Science 277, 653–659. [DOI] [PubMed] [Google Scholar]
- Kent T. A.; Huynh B. H.; Münck E. (1980) Iron–sulfur proteins: spin-coupling model for three-iron clusters. Proc. Natl. Acad. Sci. U.S.A. 77, 6574–6576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krebs C.; Henshaw T. F.; Cheek J.; Huynh B. H.; Broderick J. B. (2000) Conversion of 3Fe–4S to 4Fe–4S clusters in native pyruvate formate-lyase activating enzyme: Mössbauer characterization and implications for mechanism. J. Am. Chem. Soc. 122, 12497–12506. [Google Scholar]
- Huynh B. H.; Moura J. J. G.; Moura I.; Kent T. A.; LeGall J.; Xavier A. V.; Münck E. (1980) Evidence for a three-iron center in a ferredoxin from Desulfovibrio gigas. Mössbauer and EPR studies. J. Biol. Chem. 255, 3242–3244. [PubMed] [Google Scholar]
- Jameson G. N. L.; Cosper M. M.; Hernández H. L.; Johnson M. K.; Huynh B. H. (2004) Role of the (2Fe–2S) cluster in recombinant Escherichia coli biotin synthase. Biochemistry 43, 2022–2031. [DOI] [PubMed] [Google Scholar]
- Yang J.; Naik S. G.; Ortillo D. O.; García-Serres R.; Li M.; Broderick W. E.; Huynh B. H.; Broderick J. B. (2009) The iron–sulfur cluster of pyruvate formate-lyase activating enzyme in whole cells: cluster interconversion and a valence-localized [4Fe–4S]2+ State. Biochemistry 48, 9234–9241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vrajmasu V. V.; Bominaar E. L.; Meyer J.; Münck E. (2002) Mössbauer study of reduced rubredoxin as purified and in whole cells. Structural correlation analysis of spin Hamiltonian parameters. Inorg. Chem. 41, 6358–6371. [DOI] [PubMed] [Google Scholar]
- Krebs C.; Broderick W. E.; Henshaw T. F.; Broderick J. B.; Huynh B. H. (2002) Coordination of adenosylmethionine to a unique iron site of the (4Fe–4S) of pyruvate formate-lyase activating enzyme: a Mössbauer spectroscopic study. J. Am. Chem. Soc. 124, 912–913. [DOI] [PubMed] [Google Scholar]
- Choi-Rhee E.; Cronan J. E. Jr. (2005) Biotin synthase is catalytic in vivo, but catalysis engenders destruction of the protein. Chem. Biol. 12, 461–468. [DOI] [PubMed] [Google Scholar]
- Ugulava N. B.; Sacanell C. J.; Jarrett J. T. (2001) Spectroscopic changes during a single turnover of biotin synthase: destruction of a (2Fe–2S) cluster accompanies sulfur insertion. Biochemistry 40, 8352–8358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson K. J.; Pelletier D. A.; Turner I. M. Sr. (1999) Transfer of sulfur to biotin from biotin synthase (BioB protein). Biochem. Biophys. Res. Commun. 254, 632–635. [DOI] [PubMed] [Google Scholar]
- Tse Sum Bui B.; Mattioli T. A.; Florentin D.; Bolbach G.; Marquet A. (2006) Escherichia coli biotin synthase produces selenobiotin. Further evidence of the involvement of the [2Fe–2S]2+ cluster in the sulfur insertion step. Biochemistry 45, 3824–3834. [DOI] [PubMed] [Google Scholar]
- Mühlenhoff U.; Gerl M. J.; Flauger B.; Pirner H. M.; Balser S.; Richardt N.; Lill R.; Stolz J. (2007) The iron–sulfur proteins Isa1 and Isa2 are required for the function but not for the de novo synthesis of the Fe/S clusters of biotin synthase in Saccharomyces cerevisiae. Eukaryotic Cell 6, 495–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gelling C.; Dawes I. W.; Richhardt N.; Lill R.; Mühlenhoff U. (2008) Mitochondrial Iba57p is required for Fe/S cluster formation on aconitase and activation of radical SAM enzymes. Mol. Cell. Biol. 28, 1851–1861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheftel A. D.; Wilbrecht C.; Stehling O.; Niggemeyer B.; Elsasser H. P.; Mühlenhoff U.; Lill R. (2013) The human mitochondrial ISCA1, ISCA2, and IBA57 proteins are required for [4Fe–4S] protein maturation. Mol. Biol. Cell 23, 1157–1166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ajit Bolar N.; Vanlander A. V.; Wilbrecht C.; Van der Aa N.; Smet J.; De Paepe B.; Vandeweyer G.; Kooy F.; Eyskens F.; De Latter E.; Delanghe G.; Govaert P.; Leroy J. G.; Loeys B.; Lill R.; Van Laer L.; Van Coster R. (2013) Mutation of the iron-sulfur cluster assembly gene IBA57 causes severe myopathy and encephalopathy. Hum. Mol. Genet. 22, 2590–2602. [DOI] [PubMed] [Google Scholar]
- Cameron J. M.; Janer A.; Levandovskiy V.; Mackay N.; Rouault T. A.; Tong W. H.; Ogilvie I.; Shoubridge E. A.; Robinson B. H. (2011) Mutations in iron–sulfur cluster scaffold genes NFU1 and BOLA3 cause a fatal deficiency of multiple respiratory chain and 2-oxoacid dehydrogenase enzymes. Am. J. Hum. Genet. 89, 486–495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navarro-Sastre A.; Tort F.; Stehling O.; Uzarska M. A.; Arranz J. A.; Del Toro M.; Labayru M. T.; Landa J.; Font A.; Garcia-Villoria J.; Merinero B.; Ugarte M.; Gutierrez-Solana L. G.; Campistol J.; Garcia-Cazorla A.; Vaquerizo J.; Riudor E.; Briones P.; Elpeleg O.; Ribes A.; Lill R. (2011) A fatal mitochondrial disease is associated with defective NFU1 function in the maturation of a subset of mitochondrial Fe–S proteins. Am. J. Hum. Genet. 89, 656–667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Z.; Jollie D.; Burgess B. K.; Stephens P. J.; Münck E. (1994) Mössbauer and EPR studies of Azotobacter vinelandii ferredoxin I. Biochemistry 33, 14475–14485. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.