

Abstract
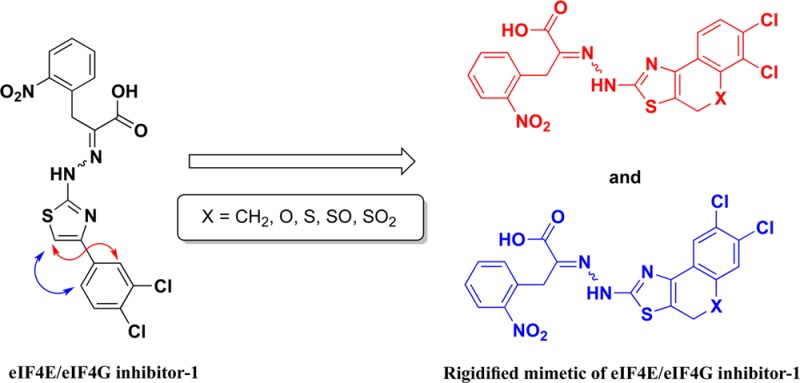
The 4EGI-1 is the prototypic inhibitor of eIF4E/eIF4G interaction, a potent inhibitor of translation initiation in vitro and in vivo and an efficacious anticancer agent in animal models of human cancers. We report on the design, synthesis, and in vitro characterization of a series of rigidified mimetic of this prototypic inhibitor in which the phenyl in the 2-(4-(3,4-dichlorophenyl)thiazol-2-yl) moiety was bridged into a tricyclic system. The bridge consisted one of the following: ethylene, methylene oxide, methylenesulfide, methylenesulfoxide, and methylenesulfone. Numerous analogues in this series were found to be markedly more potent than the parent prototypic inhibitor in the inhibition of eIF4E/eIF4G interaction, thus preventing the eIF4F complex formation, a rate limiting step in the translation initiation cascade in eukaryotes, and in inhibition of human cancer cell proliferation.
Introduction
Translation initiation in eukaryotic cells is a highly regulated process and plays an important role in cell proliferation, differentiation, survival, and maintenance of homeostasis.1 Disruption and/or perturbation of cap-dependent translation is associated with many pathophysiological processes such as Wolcott–Rallison syndrome, fragile X syndrome,2 neurodegenerative disorders such as Alzheimer’s disease, and proliferative disorders such as malignant transformation.3,4 Translation initiation commences with the binding of the eukaryotic initiation factor 4E (eIF4E) to the mRNA 5′-end-cap (m7GpppN, where N is any nucleotide and m is a methyl group) structure, which is present in all mRNAs. Protein–protein interaction between eIF4E and eIF4G, the scaffolding protein, enables the recruitment of eIF4A, a DEAD-box RNA helicase, and formation of the eIF4F complex that unwinds the secondary structure of mRNAs and allows the docking and assembly of the 43S pre-initiation complex.5 The 40S ribosome complex then traverses the 5′ untranslated region (UTR) until it recognizes the initiation codon AUG, followed by the 60S large ribosomal subunit binding to form the 80S initiation complex, which is competent to enter the elongation cycle.6,7
Under normal cellular conditions, eIF4F complex is limited as eIF4E is secluded from eIF4G by binding to hypophosphorylated eIF4E binding proteins (4E-BP). Stimulation of the phosphatidylinositol 3-kinase/AKT/mTOR pathway leads to hierarchical 4E-BP phosphorylation, dislodging hyperphosphorylated 4E-BP from eIF4E and enabling assembly of eIF4F complex. Because both the 4E-BPs and eIF4G share the same binding motif on eIF4E,8−10 the former can function as an endogenous inhibitor of cap-dependent translation initiation. As such, ectopic overexpression of 4E-BP can inhibit cap-dependent protein synthesis, inhibit tumor growth, and revert the malignant phenotype of eIF4E-overexpressing cancer cells.
eIF4F complex assembly is rate limiting for translation initiation and is predominantly dependent on the availability of eIF4E. Although eIF4F complex formation increases the translation of all cap-dependent mRNAs and thereby increases global protein synthesis rate, mRNAs vary widely in their inherent “translatability”, that is, primarily dictated by the length and structure of their 5′-UTRs. mRNAs with long and structurally complex 5′-UTRs (i.e., “weak” mRNAs) are most sensitive to restrictive abundance of eIF4E and therefore to the limited availability of the eIF4F complex. These “weak” mRNAs, which encode proteins that play important roles in cell growth, proliferation, and apoptosis,11,12 are poorly translated when eIF4F complex is scarce, due to inefficient unwinding of “weak” mRNA and subsequently preventing ribosome loading. In contrast, most mRNAs that are characterized by relatively short, unstructured 5′UTRs, the so-called “strong” mRNAs, express housekeeping proteins such as β-actin, continue to be efficiently scanned to achieve robust initiation codon recognition, effective ribosome loading, and efficient translation even when eIF4F complex levels are limiting.13
Dysregulated eIF4E-dependent translational control is implicated in the pathobiology of human disorders including autism,14 fragile X syndrome,15 tuberous sclerosis,16 and some cancers.17 eIF4E function is particularly critical for the expression of a wide array of proteins that contribute to all aspects of malignancy, including growth factors such as c-myc and cyclin D1, angiogenesis factors such as VEGF and FGF-2, and antiapoptotic proteins such as survivin and Bcl-2.13 Inhibition of either eIF4E expression by antisense RNA or the eIF4E/eIF4G interaction by overexpression of 4E-BPs reverses the malignant phenotype in vitro and in vivo.18 Hence, disrupting the formation of eIF4F complex will retard translation initiation in general and in particular translation initiation of “weak” mRNAs that encode a wide array of proteins involved in pathophysiological processes, including pro-oncogenes, growth factors, cell cycle regulators, and transcription factors, will yield powerful molecular probes and may lead to novel drug candidates.19−21
We have previously reported the discovery of (2-(2-(4-(3,4-dichlorophenyl)thiazol-2-yl)hydrazono)-3-(2-nitrophenyl)propanoic acid) (1), the eIF4E/eIF4G inhibitor-1 (4EGI-1), which inhibits protein–protein interaction.22 The high throughput screening (HTS) campaign of small molecule libraries employed a cell free fluorescence polarization assay (FP). In vitro, 1 inhibits expression of regulatory proteins such as cyclins D1 and E, C-myc, and Bcl-2 without affecting the expression of housekeeping proteins such as actin and α-tubulin and enhances the dissociation of eIF4G from eIF4E. Moreover, we reported significantly lower IC50 for the inhibition of proliferation of transformed malignant Ph+, which is transformed by the bcr-abl, cells than that for nonmalignant nontransformed Ph– cells by 1.22 Similarly, Tamburini et al. reported that 1 dramatically reduces the clonogenic growth of AML progenitors with a moderate impairment of normal CD34+ hematopoietic progenitor cologenicity.23 Together, these studies support the proposition that inhibition of cap-dependent translation initiation will affect predominantly cells whose growth is fast or unregulated rather than normal cells.8 In vivo, it effectively inhibits xenograft tumor growth in a mouse model of human cancer.24 Moreover, 1 proved to be an important molecular probe in understanding the role of eIF4F in memory formation and consolidation,25 autism spectrum disorders,26 and viral infection.27
Titration of GB1-eIF4E, a fusion protein tagged with a solubility enhancing domain, with 1 and measuring the 15N-HSQC spectrum suggests that it is binding to residues present on the eIF4G-binding surface of eIF4E.22 In the absence of high resolution structure of the complex of 1 with eIF4E we carried out extensive structure–activity relationship studies to identify the essential pharmacophore and understand the structural latitude presented by this prototypical inhibitor of eIF4E/eIF4G interaction.28,29 Structural rigidification that introduces conformational constraint around rotatable bonds was practiced advantageously in many systems and was reported to contribute to higher specificity and potency, greater metabolic stability, and improved bioavailability.30−36
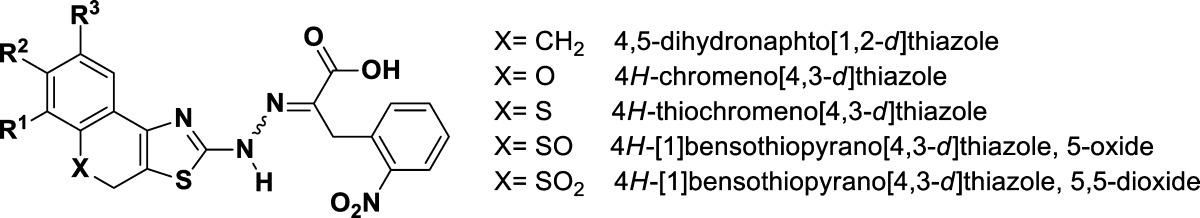
In light of the importance of the phenyl substitution on position 4 of the thiazolidine ring, we sought to delineate the spatial relationship between these two aromatic moieties by restricting the rotation around the bond connecting the two rings. Ring closure between the 1- or 6-position of the 3,4-dichlorophenyl ring (C) and the 5-position of the thiazolidine ring (B) in 1 will generate nearly coplanar, condensed, and rigid tricyclic systems that are formally formed by rotamers representing dihedral angles of 0° and 180° around the bond connecting the 4-thiazolyl and the phenyl ring (Scheme 1, structures type P1 and type P2). Specifically, the resultant rigid tricyclic scaffolds generated by bridging position 5 of the thiazolidine with the ortho position of the phenyl ring (substituting position 4 of the same thiazolidine ring) via one of the following linkers, ethylene, methylene oxide, methylenesulfide, methylenesulfoxide, and methylenesulfone, will fuse the thiazolidine ring to 3,4-dihydrotetralin, chromene, thiachromene, oxothiachromene, and thiodioxochromene, respectively. Evidently, these fused tricyclic scaffolds are found in many biological active compounds. For example: 4,5-dihydronaphtho[1,2-d]thiazole scaffold is found in a ligand that has significant 5-HT3 receptor affinity37 and in ligands that act as allosteric enhancers of A1 adenosine receptors;38 4H-chromeno[4,3-d]thiazole scaffold is present in some antibacterial agents;39 4H-thiochromeno[4,3-d]thiazole scaffold has been reported to constitute agents with antimicrobial, analgesic, anti-inflammatory, and anesthetic activities,40,41 and 4H-sulfoxo- and 4H-sulfonochromeno[4,3-d]thiazole scaffolds constitute some antifungal agents.42
Scheme 1. Strategy for the Rigidification of 1.

With the intention to characterize the impact of partial molecular rigidification, we connected rings B and C forming either the 3,4-dihydrotetralin or chromene ring systems. We have focused our study on the previously reported prototypic 1 and kept the o-NO2-substitution on ring A and the dichloro-substitution on ring C. In addition, we substituted ring C with the more polar dimethoxy-substitutions. Our study reported herein explored the role of conformational rigidification of the 4-[3′,4′-dichlorophenyl]thiazolyl part of 1 on its potency to inhibit eIF4E/eIF4G interaction employing a fluorescence polarization assay and proliferation of human cancer cells.
Results and Discussion
Chemistry
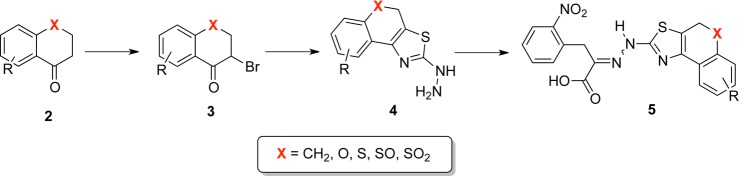
Conceptually, there are two possible modes for connecting rings B and C of 1 by forming a new six-membered ring that is part of the fused tricyclic system. In the first mode (P1), position C5 of the 2-thiazolyl (ring B) and the position C2′ of 3′,4′-dichlorphenyl (ring C) are connected to form the new six-membered ring (Scheme 1, system I). In the second mode (P2), the same position of the 2-thiazolyl (ring B) is connected to C6′ of 3′,4′-dichlorphenyl (ring C), again forming another variant of a fused tricyclic system (Scheme 1, system II). In addition, the availability of (E)- and (Z)-configurations around the carbimino bond of the hydrazono function enables formation of two geometrical isomers for each of the two cyclization modes described above. While the mode of cyclization is dictated by the choice of starting material, isomers (E) and (Z) are generated simultaneously as a mixture that requires chromatographic separation. The purity of the final rigidified 1 mimetic was established by analytical RP-HPLC and exceeded 95%, and their structural integrity and identity was established by 1H- and 13C NMR and HR-MS.
Our stepwise synthetic strategy followed the general pathway used in the synthesis of (E/Z)-1 (Scheme 2).43 Accordingly, α-bromination of the bicyclic ketone 2 generated the corresponding phenacyl bromide 3. Reaction of the bromide 3 with the thiosemicarbazide led to the formation of the 2-hydrazinyl-thiazole 4 that was then condensed with the o-nitro-phenylpyruvic acid to generate the partially rigidified (E/Z)-1 mimetic 5 (Scheme 2).
Scheme 2. Synthetic Strategy of Partially Rigidified 1 Mimetic.
Synthesis of the 4,5-dihydronaphtho[1,2-d]thiazolyl-containing 1 mimetic 13a and 13b is outlined in Scheme 3. Friedel–Crafts acylation of o-dichlorobenzene, 6, with succinic anhydride in the presence of AlCl3 or n-BuLi44 led to the formation of the respective isomers 4-(3,4-dichlorophenyl)- and 4-(2,3-dichlorophenyl)-4-oxobutanoic acid (7a and 7b, respectively). Clemmensen reduction of the ketones45,46 was followed by intramolecular polyphosphoric acid-mediated acylation,47 generating the corresponding α-tetralone derivatives 9a(45) and 9b(46) that were then α-brominated to the corresponding 2-bromo-6,7-dichloro-3,4-dihydronaphthalen-1(2H)-one, 10a,46,48 and 2-bromo-5,6-dichloro-3,4-dihydronaphthalen-1(2H)-one, 10b. Hantzsch-type reaction49 between thiosemicabazide and these α-bromoketones generated predominantly the corresponding hydrazines 11a and 11b in moderate yields side-by-side with lesser amounts of the side-product amines 12a and 12b. Finally, condensation between 4,5-dihydronaphtho[1,2-d]thiazol-2-yl)hydrazines, 11a and 11b, with 2-(o-nitrophenyl)pyruvic acid in the presence of 5% acetic acid afforded the isomeric mixture of the desired rigidified 1 mimetic, (E/Z)-13a (type P1) and (E/Z)-13b (type P2) in 35–40% yield. The individual (E)- and (Z)-isomers were separated by reverse phase column chromatography. In a separate reaction, when a mixture of 11a and 12a treated with 2-(o-nitrophenyl)pyruvic acid under similar reaction conditions only the formation (E/Z)-13a was observed with the unreacted diazine 12a, presumably due to its weak nucleophilicity relative to 11a. And also considering the tedious chromatographic process required for the isolation of the hydrazine (11a) and the thiadiazine (12a), it was decided to use the crude amine mixtures in the syntheses of other analogical derivatives of 1 (compounds (E/Z)-23a–b, (E/Z)-29a–b, (E/Z)-32a–b, (E/Z)-35a–b, and (E/Z)-46a–c, Schemes 4–7). The respective yields of the hydrazines were based on the LCMS analyses of the mixtures.
Scheme 3. Synthesis of 4,5-Dihydronaphtho[1,2-d]thiazolyl-Containing 1 Mimetic.

(i) Succinic anhydride, AlCl3, 65 °C, 80%; (ii) succinic anhydride, n-BuLi, −78 C, 30%; (iii) Zn(Hg), concd HCl, toluene, reflux, 36 h, 30–60%; (iv) polyphosphoric acid, 130 °C, 12 h, 30%; (v) Br2, ether, 30 min, 88–90%; (vi) thiosemicarbazide, dioxane, 48 h, 40–50%; (vii) 2-(o-nitrophenyl)pyruvic acid, 5% AcOH in EtOH (1:2, v/v), reflux, 1 h, 38–45%.
Scheme 4. Synthesis of Chromene Derived 1 Mimetic.

(i) β-Propiolactone, NaH, DMF, 100 °C, 42%; (ii) β-propiolactone, NaOH, H2O, 100 °C, 50%; (iii) HFliq, −78 °C, 12 h, 76%; (iv) Eaton’s Reagent, 70 °C, 75%; (v) pyridinium bromide perbromide, CHCl3–EtOH, 50 °C, 74–76%; (vi) thiosemicarbazide, dioxane, 40–50%; (vii) 2-(o-nitrophenyl)pyruvic acid, 5% AcOH–ethanol (1:2, v/v), reflux, 1 h, 35–40%.
Scheme 7. Synthesis of Dimethoxy Derivatives of Tetralin and Chromene Based 1 Mimetic.

(i) 3-Chloro propanoyl chloride, BF3·etherate, 60 °C, 3 h; (ii) K2CO3, RT, 20 h; (iii) succinic anhydride, AlCl3, nitrobenzene; (iv) Zn–Hg, concd HCl, reflux, 40%; (v) P2O5, benzene, reflux, 4 h, 57–79%; (vi) oxetan-2-one, NaH, DMF (vii) P2O5, benzene, 90 °C, 6 h, 57%; (viii) pyridinium bromide perbromide, CHCl3–EtOH, 50 °C, 30 min, 48–75%; (ix) thiosemicarbazide, dioxane; (x) 2-(o-nitrophenyl)pyruvic acid, 5% AcOH–ethanol (1:2, v/v), 35–41%.
Synthesis of rigidified oxymethylene-bridged 1 mimetic (E/Z)-23a (type P1) and (E/Z)-23b (type P2) started with O-alkylation of 2,3-dichloro- and 3,4-dichlorophenol, respectively, with β-propiolacone in the presence of sodium hydroxide or sodium hydride. This was followed by acid catalyzed cyclization of the 3-phenoxypropionic acids 15 and 18 into the corresponding chroman-4-one derivatives 16, 19, and 20.50 While the cyclization of 3-(2,3-dichlorophenoxy)propanoic acid (15) in the presence of either HFliq or Eaton’s reagent51 afforded 7,8-dichlorochroman-4-one (16) in good yield, cyclization of 3-(3,4-dichlorophenoxy)propanoic acid (18) in the presence of HFliq afforded 7,8-dichlorochroman-4-one (19) but in the presence of Eaton’s reagent it yielded 5,6-dichlorochroman-4-one (20). Because of excessive steric hindrance considerations, we shelved ketone 20 and carried α-bromination of 16 and 19 successfully by employing pyridinium bromide perbromide (PBPB).52 In contrast, attempt to use Br2 under conditions similar to those used for the preparation of 10a and 10b generated multiple products and mixtures difficult to resolve. Conversion of the α-bromo ketones 21a and 21b followed to the hydrazines 22a and 22b and the subsequent condensation with 2-(o-nitrophenyl)pyruvic acid followed the same procedures as described above for the 4,5-dihydronaphtho[1,2-d]thiazolyl-containing 1 mimetic 13a and 13b and generated the anticipated oxymethylene-bridged 1 mimetic (E/Z)-23a and (E/Z)-23b (Scheme 4).
As a logical extension of oxymethylene to thiomethylene rigidified analogues of 1, the synthesis of a set of rigidified mimetic of 1, 29a, and 29b was performed. The synthetic protocol started from the S-alkylation of 2,3- and 3,4-dichlorothiophenol with 3-bromopropionic acid in the presence of sodium hydroxide at elevated temperature, yielding respective thioaryl propionic acids, 25a and 25b, which on H2SO4 cyclization afforded 4-thiochroman-1-ones, 26a and 26b, in excellent yield.53 The 2-bromo-4-thiochroman-4-ones,5427a and 27b, obtained from the thiachromanones 25a and 25b, respectively, gave thiomethylene rigidified mimetic of 1, (E/Z)-29a and (E/Z)-29b in 35–40% yields (Scheme 5) by following the general synthetic steps described in Scheme 2.
Scheme 5. Synthesis of Thio-chromene Derived 1 Mimetic.

(i) 3-Bromopropionic acid, NaOH, 100 °C, 88–94%; (ii) concd H2SO4, −10 °C–RT, 90%; (iii) Br2, 60–69%; (iv) thiosemicarbazide, dioxane, 40–50%; (vi) 2-(o-nitrophenyl)pyruvic acid, 5% AcOH–ethanol (1:2, v/v), reflux, 1 h, 35–40%.
With a view to functionalize the thiochromene derived mimetic on the sulfur atom, α-bromo thiochromenes, 27a and 27b, were subjected to oxidation at different conditions55 to afford 33a and 33b, which on treatment with thiosemicarbazide followed by 2-(o-nitrophenyl)pyruvic acid gave the methylene sulfoxide and methylene sulfone bridged 1 mimetic (E/Z)-32a, (E/Z)-32b, (E/Z)-35a, and (E/Z)-35b, respectively (Scheme 6).
Scheme 6. Synthesis of Methylene Sulfoxide and Methylene Sulfone Derived 1 Mimetic.

(i) m-Chloroperbenzoic acid (1 equiv), CHCl3, RT, 70–76%; (ii) m-chloroperbenzoic acid (4 equiv), CHCl3, reflux, 72–83%; (iii) thiosemicarbazide, dioxane, 40–50%; (iv) 2-(o-nitrophenyl)pyruvic acid, acetic acid, 2 h, RT, 40–42%; (v) 2-(o-nitrophenyl)pyruvic acid, acetic acid, 30 min, RT, 35–45%.
To examine the effect of a dimethoxy substituent on the ring A replacing a dichloro substituent, we synthesized three dimethoxy substituted rigidified analogues of 1 starting from the respective tetralone and chromanones. Thus, 6,7-dimethoxy-4-chromanone (43a)56−58 afforded (E/Z)-46a, while 7,8-dimethoxy-4-chromanone (43b)59,60 and 6,7-dimethoxy tetralone (43c) obtained from 4-oxo-4-(3,4-dimethoxyphenyl)butanoic acid (39)61 yielded (E/Z)-46b and (E/Z)-46c, respectively, in analogy to Scheme 2 (Scheme 7).
Biology
Our previously described fluorescence polarization (FP) assay was used to measure the binding efficiency of new 1 mimetic to eIF4E by competing with a fluorogenic eIF4G derived peptide containing the conserved eIF4E-binding motif (KRYDREFLLGF).22 Displacement of the Nα-fluorescein tagged eIF4G-drived peptide by a competing ligand causes a measurable decrease in FP. The nontagged version of the same eIF4G-derived peptide and DMSO were used as positive and negative controls, respectively. The binding affinity of the new rigidified mimetic of 1 in which the 3,4-dichlorphenylthiazol-2-yl system was replaced with either 4,5-dihydronaphthol[1,2-d]thiazol-3-yl or 4H-chromeno[4,3-d]thiazol-3-yl moieties or 4H-thiochromeno[4,3-d]thiazol-3-yl or 4H-sulfoxo- and sulfonochromeno[4,3-d]thiazol-3-yl moieties was tested in the FP assay and were compared to that of (Z)-isomer of parent 1. The results were presented as a ratio between the IC50 of (Z)-1; the concentration of (Z)-1 needed to displace 50% of the fluorescent-tagged peptide, and the IC50 of the new 1 analogue, and the concentration of the new analogue needed to displace 50% of the fluorescent-tagged peptide when measured in the same 384-well plate (Figure 1).
Figure 1.

Dose Dependent Inhibition of eIF4E/eIF4G interaction by rigidified 1 mimetic.
The simplest of the rigidified 1 mimetic, (E/Z)-13a, containing an ethylene bridge between C-6′-(2′,3′-dichlorophenyl) ring (C) and C-5 of thiazolyl ring (B), and (E/Z)-13b, containing an ethylene bridge between C-2′-(4′,5′-dichlorophenyl) ring (C) and C-5 of thiazolyl ring (B), display a 1.5- to 2-fold increase in binding affinity as compared to parent (Z)-1 (entries 2–5, Table 1). Linking thiazolyl ring B with the phenyl ring C via an oxymethylene (entries 6–9, Table 1) contribute to further increase in binding affinity, reaching in (Z)-23a a 3-fold increase (IC50 = 15.5 μM) relative to the parent (Z)-1. However, rigidification via bridges containing thiomethylene (entries 10–13, Table 1), methylenesulfoxide (entries 14–17, Table 1), or methylenesulfone (entries 18–21, Table 1) reverse the trend and do not contribute to binding affinity enhancement relative to the nonrigidified parent 1. Interestingly, the most potent binders to eIF4E in this series are 1 mimics containing the dihydronaphtho- and the chromeno-ring systems carrying dimethoxy substituents (IC50 = 10.5 μM for both (E)-46c and (E)-46a, respectively). A plausible explanation for the decrease in binding affinity of the polar methylenesulfoxide- and methylenesulfone-containing 1 mimetic relative to the less polar thiomethylene- and oxymethylene-containing analogues is the marked decrease in the calculated partition coefficient (CLogP) associated with the increased polarity. Comparison of CLogPs of (Z)-isomers of 13a (5.21), 23a (4.65), 29a (4.96), 32a (3.15), and 35a (2.88) that are constrained by the respective ethylene-, oxymethylene-, thiomethylene-, methylenesolfoxide-, and methylenesulfone-bridges supports the above-mentioned proposition.
Table 1. Binding of the 1 Rigidified Mimetic to eIF4E as Measured by Fluorescence Polarization Assay (FP) and Their Potency to Inhibit Cancer Cells Proliferation As Measured by Sulforhodamine B (SRB) Assay.
| SRB assay
IC50 (μM) |
||||||||
|---|---|---|---|---|---|---|---|---|
| entry | compd | X | R1 | R2 | R3 | FP assay IC50 (μM) | CRL-2813 | CRL-2351 |
| 1 | (Z)-1 | Cl | Cl | H | 43.5 ± 1.52 | 15.3 ± 2.5 | 11.6 ± 0.2 | |
| 2 | (E)-13a | CH2 | H | Cl | Cl | 21.5 ± 0.70 | 3.8 ± 0.3 | 4.1 ± 0.7 |
| 3 | (Z)-13a | CH2 | H | Cl | Cl | 34 ± 4.24 | 12.0 ± 0.6 | >20 (NA)a |
| 4 | (E)-13b | CH2 | Cl | Cl | H | 32 ± 1.41 | 3.6 ± 0.3 | 4.4 ± 0.6 |
| 5 | (Z)-13b | CH2 | Cl | Cl | H | 43.5 ± 2.12 | 10.6 ± 1.4 | 11.2 ± 0.6 |
| 6 | (E)-23a | O | Cl | Cl | H | 25 ± 1.41 | 5.8 ± 0.0 | 16.6 ± 4.2 |
| 7 | (Z)-23a | O | Cl | Cl | H | 15.5 ± 0.70 | 12.3 ± 0.4 | >20 (NA) |
| 8 | (E)-23b | O | H | Cl | Cl | 18.5 ± 0.70 | 3.1 ± 0.1 | 5.2 ± 0.6 |
| 9 | (Z)-23b | O | H | Cl | Cl | 27 ± 1.41 | 8.7 ± 4.1 | 15.4 ± 2.8 |
| 10 | (E)-29a | S | H | Cl | Cl | 15.5 ± 0.70 | 5.1 ± 0.1 | 17.6 ± 2.8 |
| 11 | (Z)-29a | S | H | Cl | Cl | 14 ± 0.0 | 10.5 ± 0.1 | 17.7 ± 2.7 |
| 12 | (E)-29b | S | Cl | Cl | H | 28 ± 1.41 | 4.5 ± 0.1 | 9.1 ± 1.3 |
| 13 | (Z)-29b | S | Cl | Cl | H | 11.5 ± 0.70 | 7.0 ± 2.2 | 16.7 ± 0.1 |
| 14 | (E)-32a | SO | H | Cl | Cl | 30.5 ± 2.12 | 7.5 ± 0.7 | >20 (NA) |
| 15 | (Z)-32a | SO | H | Cl | Cl | 30 ± 1.41 | 4.1 ± 0.1 | 10.3 ± 0.4 |
| 16 | (E)-32b | SO | Cl | Cl | H | 48.5 ± 6.36 | 11.2 ± 1.7 | >20 (NA) |
| 17 | (Z)-32b | SO | Cl | Cl | H | 32.5 ± 2.12 | 11.4 ± 0.3 | 19.8 ± 0.3 |
| 18 | (E)-35a | SO2 | H | Cl | Cl | 40.5 ± 6.36 | >20 (NA) | >20 (NA) |
| 19 | (Z)-35a | SO2 | H | Cl | Cl | 39 ± 5.65 | >20 (NA) | >20 (NA) |
| 20 | (E)-35b | SO2 | Cl | Cl | H | 49 ± 2.82 | >20 (NA) | >20 (NA) |
| 21 | (Z)-35b | SO2 | Cl | Cl | H | 49 ± 2.82 | >20 (NA) | >20 (NA) |
| 22 | (E)-46c | CH2 | H | OMe | OMe | 10.5 ± 0.70 | 12.1 ± 0.7 | >20 (NA) |
| 23 | (Z)-46c | CH2 | H | OMe | OMe | 45 ± 2.82 | >20 (NA) | >20 (NA) |
| 24 | (E)-46b | O | OMe | OMe | H | 16.5 ± 3.53 | >20 (NA) | >20 (NA) |
| 25 | (Z)-46b | O | OMe | OMe | H | 55.5 ± 4.94 | >20 (NA) | >20 (NA) |
| 26 | (E)-46a | O | H | OMe | OMe | 10.5 ± 0.70 | >20 (NA) | >20 (NA) |
| 27 | (Z)-46a | O | H | OMe | OMe | 51.5 ± 4.94 | >20 (NA) | >20 (NA) |
NA: not active.
Evidently, regardless of the substitution on ring C (dichloro or dimethoxy), we do not observe a significant difference in binding affinity trends between type P1 (a series) and type P2 (b series) for the rigidified 1 mimetic (e.g., cf. IC50 in the FP assay of (E)-13a and (Z)-13a with (E)-13b and (Z)-13b, 21.5 and 34 with 32 and 43.5 μM, respectively, or (E)-46a and (Z)-46a with (E)-46b and (Z)-46b, 10.5, 30, and 51.5 with 16.5 and 55.5 μM, respectively).
The inhibition of cell proliferation data obtained in the SRB (sulforhodamine B) cell proliferation assay with human cancer cell lines CRL-2351 breast and CRL-2813 melanoma indicated that the rigidified 1-derived mimetic inhibited cell proliferation with IC50 values around 1–20 μM. With few exceptions that might relate to solubility and/or cell penetration issues, almost all compounds displaying higher affinity to eIF4E than (Z)-1 in the FP assay were also more potent inhibitors in the SRB cell proliferation assay, suggesting that the compounds inhibit cell proliferation through inhibition of translation initiation. In comparison with the parent nonconstrained 1 that inhibits equally the breast cancer and melanoma cells, the rigidified 1 mimetic compounds were, in general, more potent in inhibiting proliferation of CRL-2813 melanoma cells vs the CRL-2351 breast cancer cells. While (Z)-13a, (Z)-23a, (E)-32a, and (E)-32b (IC50= 12.0, 12.3, 7.5, and 11.2 μM, respectively) were slightly more potent than (Z)-1 (IC50= 15.3 μM) in inhibiting proliferation of the melanoma cells, they did not inhibit the proliferation of breast cancer cells up to 20 μM. Interestingly, the more polar rigidified 1 mimetic compounds, those that are composed of 4H-[1]benzothiopyrano[4,3-d]thiazole, 5,5-dioxide (35a and 35b, CLogP = 2.88), and dimethoxy-substituted dihydronaphtho[1,2-d]thiazole (Z)-46c, CLogP = 3.63) and 4H-chromeno[4,3-d]thiazole (46a and 46b, CLogP = 2.98), in spite of having inhibitory binding affinity comparable to that of (Z)-1 (CLogP = 4.76) were devoid of cell proliferation inhibitory activity up to concentrations of 20 μM.
Evidently, we think of the cancer cell proliferation inhibitory activities as reporting of global activity that combine on- and off-target effects. The high sensitivity of the adherent human melanoma CRL-2813 relative to the human breast cancer CRL-2351 to inhibition of proliferation by the rigidified 1 mimetic compounds may be attributed to the presence of a BRAF mutation that makes them significantly more dependent on highly efficient cap-dependent translation initiation.62
Disruption of eIF4E/eIF4G Interaction
We chose (E)-29a, a rigidified 1 mimetic that showed enhanced potency in the FP-assay and the SRB-cell proliferation assay, and (E)-35b, another 1 mimetic with very low activity in both assays, and tested their ability to disrupt the eIF4E/eIF4G complex formation in CRL-2813 melanoma cells (Figure 2). The state of association of eIF4E with eIF4G and 4E-BP1 was determined by pull-down experiment on m7GDP agarose resin. This demonstrated that full-length eIF4G is displaced from eIF4E by (E)-29a that was not the case with the FP-less active derivative (E)-35b, in which no effect was observed. The disruption of the eIF4E/eIF4G, however, increased the 4E-BP1 binding to eIF4E, which is consistent with our previous finding that 1 inhibits eIF4E/eIF4G interaction independently of 4E-BP1 binding to eIF4E.24 We speculated that this increase in the amount of eIF4E/4E-BP1 complex is likely due to the dissociation of eIF4G that exposes of a larger 4E-BP1 binding footprint that is present on eIF4E and is partially obscured by the bound eIF4G.22
Figure 2.

eIF4F complex formation disruption by (E)-29a: (a) (E)-29a displaces eIF4G from eIF4E and enhances 4E-BP1 binding in melanoma CRL-2813 cell lysate. After incubation of CRL-2813 cells with 30 μM of each compound for 3 h, the cells lysate was then used. A cap-affinity chromatography and SDS-PAGE immunoblotting were used to detect eIF4E, eIF4G, and 4E-BP1. The eIF4E lanes shown come from the same gel and Western blot. (b) Quantitative analysis of the effect of (E)-29a and (E)-35b on complex protein levels relative to eIF4E.
Conclusions
In an effort to optimize 1, the hit compound that was found to inhibit eIF4E/eIF4G protein–protein interaction and translation initiation both in vitro and in vivo, as a molecular probe and potential drug candidate, we focused on developing a rigidified 1 mimetic. Rigidification of the 2-(4-(3,4-dichlorophenyl)thiazol-2-yl) moiety by introduction of a ethylene, methylene oxide, methylenesulfide, methylenesulfoxide, or methylenesulfone bridges locking condensed tricyclic systems yielded some very potent 1 mimics. One of these is (E)-29a, which carries a methylenesulfide bridge, is 3-fold more potent than the parent 1 in competing for the binding to eIF4E in the cell-free FP assay (Figure 1) with an IC50 = 15.5 μM, disrupts very effectively eIF4E/eIF4G protein–protein interaction and concomitantly increases very effectively binding of 4E-BP1 to eIF4E (Figure 2), and last but not least is a potent inhibitor of human melanoma CRL-2813 cells proliferation IC50 = 5.1 μM (Table 1). Taken together, these results suggest that the binding site on eIF4E for the 2-(4-(3,4-dichlorophenyl)thiazol-2-yl) moiety in 1 accommodates very nicely the fused nearly coplanar tricyclic system, which may mimic very closely a rotamer population in 1 that binds to the macromolecular target. Recently, NMR mapping of a solution of N-terminal fused eIF4E and 1 and high resolution X-ray analysis of co-crystal structures of eIF4E and 1 or some of its analogues suggest that dissociation of eIF4E–eIF4G complex in the presence of these inhibitors is through an allosteric mechanism (unpublished data). It is very likely that the similar activity profiles of 1 and the rigidified fused tricyclic 1 mimetic reported here strongly suggests that the latter also share a similar molecular mechanism. For now, we conclude that the orientation of the phenyl in position 4 relative to the thiazolidine ring depends on the nature of the bridge in the fused tricyclic system, the substituents on that phenyl ring, and interplay between the former two and the configuration around the hydrazone function. Our ongoing efforts will continue to look at optimization through rigidification modes introducing conformational and configurational constraints to develop novel 1 mimetics as effective molecular probes. We are currently utilizing these optimized 1 mimetics for the direct confirmation of their allosteric mechanism of action.
Experimental Section
Chemistry
General
All the starting materials were obtained from commercial sources and used as purchased. Chromatography solvents were HPLC grade and were used without further purification. Thin layer chromatography (TLC) analysis was performed using Merck silica gel 60 F-254 thin layer plates. LC-MS analyses were performed on Waters 2695 separator module with (APCI mode) (XTerra C8 30 mm × 100 mm column) micromass ZQ employing a flow rate of 0.5 mL/min and a solvent system that includes A, 0.1% v/v formic acid in water, and B, 0.1% v/v formic acid in acetonitrile. The scan range was m/z 100–1000. HRMS analyses were performed on Agilent Technologies, 6120 time-of-flight-LC/MS instrument employing a linear gradient of A, 0.1% v/v FA in water, and B, 0.1% v/v TFA in acetonitrile. Melting points were measured in open Pyrex capillaries in MEL TEMP “Electronthermal” apparatus and are uncorrected. The purity of tested compounds was >95% as determined by RP-HPLC on a C18 Xbridge column (4.6 mm × 100 mm, 1 mL/min) with effluents monitored at 254 nm in Waters 2695 separator module. Solvent system employed included a linear gradient of A, 0.1% v/v TFA in water, and B, 0.1% v/v TFA in acetonitrile. NMR spectra were recorded on a Varian 400 or 500 MHz spectrometers. The signal of the deuterated solvent was used as internal reference. Chemical shifts (δ) are given in ppm and are referenced to residual not fully deuterated solvent signal. Coupling constants (J) are given in Hz.
Synthesis of 4-(3,4-Dichlorophenyl-1-oxo-butyric Acid (7a)
AlCl3 (1.99 g, 15 mmol) was added to a solution of succinic anhydride (5 g, 50 mmol) in 1,2-dichlorobenzene (44.1 g, 3 mmol) at RT. The reaction mixture was heated to 60 °C for 2.5 h and then inverse quenched onto cold water (120 mL), maintaining the temperature below 50 °C, and stirred for 30 min. Then hexane (60 mL) was added, and the stirring continued for 2 h to afford 7a as an off-white solid, which was filtered and dried under vacuum.
7a
White powder, yield 5.92 g (80%); mp 181–182 °C. 1H NMR (DMSO-d6, 400 MHz) in ppm: δ 2.54–2.57 (t, 2H), 3.24 (t, 2H), 7.79 (d, J = 8 Hz, 1H), 7.90–7.93 (dd, J = 8 and 4 Hz, 1H), 8.12 (d, J =J = 4 Hz, 1H). 13C NMR (DMSO-d6, 100 MHz) in ppm: δ 28.4, 33.9, 128.6, 130.4, 131.7, 136.6, 137.2, 174.3, 197.5. Purity of 100% as determined by RP-HPLC, tR = 13.55 min (linear gradient system of 0–100% B in A for 26 min). ESI-MS calcd MW for C10H8Cl2O3, 247.07; found, m/z = 247.01 (M – H)−.
Synthesis of 4-(3,4-Dichlorophenyl)butyric Acid (8a)
Pure Zn dust (98%) (2.6 g, 40 mmol) and HgCl2, (0.18 g, 0.66 mmol) were stirred with concentrated HCl (0.25 mL) and water (0.5 mL) for 10 min. The aqueous solution was then syringed out and the amalgamated zinc was suspended in a mixture of water (4 mL) and concd HCl (8 mL). To this suspension were added 7a (1.0 g, 4 mmol) followed by toluene (8 mL) and refluxed with stirring for 36 h with the addition of concd HCl (4 mL) every 5 h. After cooling to RT, the reaction mixture was filtered and the filtrate was extracted with ethyl acetate (100 mL), dried over anhydrous Na2SO4, and concentrated under vacuum to give the crude butyric acid (8a) as oil. It was then chromatographed on a silica gel column using hexane–ethyl acetate mixture (9:1 v/v) as eluent.
8a
White solid, yield 0.3 g (30%), Rf 0.4 (methanol–dichloromethane, 1:9, v/v); mp 61–63 °C. 1H NMR (CDCl3, 400 MHz) in ppm: δ 1.91–1.97 (m, 2H), 2.35–2.39 (t, 2H), 2.61–2.64 (t, 2H), 7.00–7.02 (dd, J = 8 and 4 Hz, 1H), 7.26 (d, J = 4 Hz, 1H), 7.34 (d, J = 8 Hz, 1H). 13C NMR (CDCl3, 100 MHz) in ppm: δ 26.0, 33.3, 34.2, 123.2, 125.7, 128.1, 130.5, 131.8, 141.6, 179.9. Purity of 98.4% as determined by RP-HPLC, tR = 15.16 min (linear gradient system of 0–100% B in A for 26 min). ESI-MS calcd MW for C10H10Cl2O2, 232.98; found, m/z = 231.02 (M – H)−.
Synthesis of 6,7-Dichloro-3,4-dihydronaphthalen-1(2H)-one (9a)
Polyphosphoric acid (35 g) was heated to melt at 120 °C for 30 min. To this was added 8a (1.2 g, 5.1 mmol) and heated further with stirring for 10 h at 130 °C. After cooling to RT, dilution with water (100 mL) and extraction with ethyl acetate (100 mL) yielded an organic phase that was washed with a saturated solution of NaHCO3 (50 mL), dried over anhydrous Na2SO4, and evaporated under vacuum. The oily residue was chromatographed on silica gel column with hexane–ethyl acetate (98:2, v/v) to obtain 9a.
9a
White solid, yield 0.32 g (30%); mp 110–111 °C. 1H NMR (CDCl3, 500 MHz) in ppm: δ 2.09–2.14 (m, 2H), 2.62 (t, J = 6.0 Hz, 2H), 2.89 (t, J = 6.0 Hz, 2H), 7.34 (s, 1H), 8.03 (s, 1H). 13C NMR (CDCl3, 125 MHz) in ppm: δ 23.8, 29.0, 38.7, 129.1, 130.8, 131.5, 132.3, 137.7, 143.9, 196.3. Purity of 100% as determined by RP-HPLC, tR = 16.79 min (linear gradient system of 0–100% of B in A for 26 min). ESI-MS calcd MW for C10H8Cl2O, 215.08; found, m/z = 214.97 [M + H]+.
Synthesis of 2-Bromo-6,7-dichloro-3,4-dihydronaphthalen-1(2H)-one (10a)
To a solution of 9a (0.1 g, 0.464 mmol) in dry diethyl ether (5 mL) was added a solution of bromine (0.074 g, 0.464 mmol) in ether (1 mL) and stirred at RT for 30 min. The residue obtained after the removal of the solvent under vacuum was treated with an aqueous solution of NaHCO3 (5% w/v, 10 mL) and extracted with dichloromethane (50 mL). The organic layer was dried over anhydrous Na2SO4, concentrated under vacuum, and chromatographed on a silica gel column with ethyl acetate–hexane mixture (5:95, v/v) to afford the pure bromide 10a.
10a
White powder, yield 0.120 g (88%); mp 128–130 °C. 1H NMR (CDCl3, 400 MHz) in ppm: δ 2.83–2.88 (m, 2H), 3.22–3.29 (m, 2H), 4.69 (t, 1H), 7.40 (s, 1H), 8.12 (s, 1H). 13C NMR (CDCl3, 100 MHz) in ppm: δ 25.4, 31.4, 49.2, 129.6, 130.5, 130.8, 132.1, 138.8, 142.3, 188.8. Purity of 98.9% as determined by RP-HPLC, tR = 18.53 min (linear gradient system of 0–100% B in A for 26 min). ESI-MS calcd MW for C10H7BrCl2O, 293.97; found, m/z = 292.75 (M – H)−.
Synthesis 1-(7,8-Dichloro-4,5-dihydronaphtho[1,2-d]thiazol-2-yl)hydrazine (11a) and 8,9-Dichloro-5,6-dihydro-4aH-naphtho[1,2-e][1,3,4]thiadiazin-3-amine (12a)
A solution of 10a (0.4 g, 1.36 mmol) and thiosemicarbazide (0.124 g, 1.36 mmol) in anhydrous dioxane (20 mL) was heated to 80 °C for 1 h and then stirred at RT for 48 h. The resulting precipitate was filtered, washed with dioxane (10 mL), and suspended in 2 M Na2CO3 (15 mL). The pale greenish-yellow solid of 11a and 12a was filtered, washed with water, and separated by preparative RP-HPLC using a linear gradient system of 10–50% B in A for 25 min.
11a
Off-white solid, yield 0.150 g (39%); mp 160–162 °C. 1H NMR (DMSO-d6, 400 MHz) in ppm: δ 2.86–2.95 (m, 4H), 7.49 (s, 1H), 7.67 (s, 1H), 9.27 (bs, 2H). 13C NMR (DMSO-d6, 100 MHz) in ppm: δ 21.3, 27.8, 123.9, 129.0, 129.6, 130.5, 131.9, 132.0, 135.9. Purity of 65.5% as determined by RP-HPLC, tR = 13.20 min (linear gradient system of 0–100% B in A for 26 min). ESI-MS calcd MW for C11H9Cl2N3S, 286.18; found, m/z = 285.95 [M + H]+.
12a
Off-white solid, yield 0.077 g (20%); mp 214–216 °C. 1H NMR (DMSO-d6, 500 MHz) in ppm: δ 1.73–1.84 (m, 2H), 2.77–2.95 (m, 2H), 4.31–4.35 (m, 1H), 7.66 (s, 1H), 8.05 (s, 1H). 13C NMR (DMSO-d6, 125 MHz) in ppm: δ 25.8, 27.1, 39.6, 127.0, 129.2, 129.7, 130.5, 131.4, 141.6, 148.0, 164.4. Purity of 96.8% as determined by RP-HPLC, tR = 12.44 min (linear gradient system of 0–100% B in A for 26 min). ESI-MS calcd MW for C11H9Cl2N3S, 286.18; found, m/z = 285.95 [M + H]+.
Synthesis of (E/Z)-2-(2-(7,8-Dichloro-4,5-dihydronaphtho[1,2-d]thiazol-2-yl)hydrazono)-3-(2-nitrophenyl)propanoic Acid [(E/Z)-13a]
A suspension of 11a (0.186 g, 0.649 mmol) in 5% (v/v) acetic acid (7 mL) was added to 2-(o-nitrophenyl)pyruvic acid (0.135 g, 0.649 mmol) in ethanol (14 mL) and was heated at 90–100 °C for 1 h. The yellow precipitate that formed upon cooling to RT was filtered, washed with water, and dried. The crude mixture containing the two isomers, (E)-13a and (Z)-13a, was purified on a RP-C18 FCC (100 g cartridge, flow rate = 40 mL/min) using a solvent system consisting of A, triethylammonium bicarbonate buffer (50 mM, pH = 8.5), and B, methanol. The purified isomers were precipitated from their respective fractions following acidification with 10% HCl and separated by centrifugation. Repeated washes of the pellets with 5% HCl followed by thorough washes with water and drying under vacuum yielded the following:
(E)-13a
Yellow powder; yield 0.06 g (20%); mp 255–256 °C. 1H NMR (DMSO-d6, 400 MHz) in ppm: δ 2.85–2.89 (m, 2H), 2.93–2.97 (m, 2H), 4.27 (s, 2H), 7.05 (d, J = 8.0 Hz, 1H), 7.47–7.51 (m, 2H), 7.55 (s, 1H), 7.60–7.65 (m, 1H), 8.04–8.06 (m, 1H), 12.44 (bs, 1H). 13C NMR (DMSO-d6, 100 MHz) in ppm: δ 21.3, 27.7, 29.8, 123.6, 125.7, 128.5, 129.2, 129.5, 129.7, 130.5, 131.7, 134.5, 136.0, 149.6, 166.0. Purity of 100% as determined by RP-HPLC, tR = 8.37 min (linear gradient system of 50–100% B in A for 20 min). HRMS(ESI) m/z calcd for C20H14Cl2N4O4S [M + H]+, 477.01128; found 477.01929.
(Z)-13a
Yellow powder; yield 0.055 g (18%); mp 254–255 °C. 1H NMR (DMSO-d6, 400 MHz) in ppm: δ 2.73–2.80 (m, 2H), 2.88–2.92 (m, 2H), 4.15 (s, 2H), 7.46–7.55 (m, 3H), 7.59 (s, 1H), 7.66–7.70 (m, 1H), 8.02–8.05 (m, 1H), 12.72 (bs, 1H). 13C NMR (DMSO-d6, 100 MHz) in ppm: δ 21.2, 27.7, 36.7, 123.9, 125.2, 128.9, 129.3, 129.8, 130.4, 132.5, 133.60, 134.2, 135.9, 149.7, 164.7. Purity of 99.2% as determined by RP-HPLC, tR = 10.79 min (linear gradient system of 50–100% B in A for 20 min). HRMS(ESI) m/z calcd for C20H14Cl2N4O4S [M+H]+, 477.01128; found 477.01923.
Synthesis of 4-(2,3-Dichlorophenyl)butyric Acid (7b)
A solution of 1,2-dichlorobenzene (1.47 g, 10 mmol) in dry THF (10 mL) was added dropwise to n-butyl lithium (4 mL, 1.8 M in hexanes) at −78 °C under N2, and the mixture was stirred for half an hour. To the pale-yellow reaction mixture, a solution of succinic anhydride (1.0 g, 10 mmol) in dry THF (10 mL) was added slowly (15 min) and stirred for 1 h at −78 °C. The reaction was quenched by water (20 mL) and acidified with 5 N HCl. The organic phase obtained following extraction with DCM (100 mL) was dried over anhydrous Na2SO4 and concentrated under vacuum to yield an oily residue which after recrystallization in toluene afforded 7b.
7b
White crystals; yield 0.74 g (30%); mp 116–118 °C. 1H NMR (CDCl3, 400 MHz) in ppm: δ 2.81 (t, 2H), 3.20 (t, 2H), 7.25–7.29 (m, 1H), 7.34–7.36 (m, 1H), 7.53–7.55 (m, 1H). 13C NMR (CDCl3, 100 MHz) in ppm: δ 28.4, 37.6, 126.9, 127.9, 132.4, 134.3, 141.4, 178.7, 200.8. Purity of 99.4% as determined by RP-HPLC, tR = 12.76 min (linear gradient system of 0–100% B in A for 26 min). ESI-MS calcd MW for C10H8Cl2O3, 247.07; found, m/z = 244.96 [M + H]+.
Synthesis of 5,6-Dichloro-3,4-dihydronaphthalen-1(2H)-one (9b)
Step A
Pure Zn dust (98%) (4.0 g, 61 mmol) and HgCl2, (0.4 g, 1.4 mmol) were stirred with concd HCl (0.25 mL) and water (6.6 mL) for 10 min. The aqueous solution was removed, and the zinc amalgam was then suspended in water (2.5 mL) and concd HCl (6 mL). The stirred suspension was treated with 7b (2.3 g, 9.3 mmol) and toluene (3.5 mL) and refluxed for 24 h with the addition of concd HCl (2 mL) in every 6 h. The reaction mixture was cooled to RT and filtered, and the filtrate was extracted with ethyl acetate (100 mL). The ethyl acetate fraction was dried over anhydrous Na2SO4 and concentrated under vacuum to give 8b as white solid.
8b
White solid; yield 1.2 g (57%). Purity of 99.2% as determined by RP-HPLC, tR = 15.01 min (linear gradient system of 0–100% B in A for 26 min). ESI-MS calcd MW for C10H10Cl2O2, 233.09; found, m/z = 230.74 (M – H)−.
Step B
8b (1.2 g, 5.1 mmol) was added to a polyphosphoric acid melt (35 g) at 130 °C and stirred for 10 h. The reaction mixture was cooled to RT, and water (100 mL) was added. This mixture was extracted with ethyl acetate (100 mL) and washed with saturated solution of NaHCO3 (50 mL). The organic phase was dried over anhydrous Na2SO4 and concentrated in vacuum. The oily residue was subjected to silica gel column chromatography with hexane–ethyl acetate mixture (98:2 v/v) to obtain pure 9b.
9b
Pale-yellow solid; yield 0.32 g (30%); mp 95–96 °C. 1H NMR (CDCl3, 400 MHz) in ppm: δ 2.12–2.17 (m, 2H), 2.59–2.62 (t, 2H), 3.01–3.04 (t, 2H), 7.38 (d, J = 8.0 Hz, 1H), 7.86 (d, J = 8.0 Hz, 1H). 13C NMR (CDCl3, 100 MHz) in ppm: δ 22.3, 28.1, 38.0, 126.4, 128.4, 128.5, 132.6, 138.5, 143.9, 196.7. Purity of 98.3% as determined by RP-HPLC, tR = 16.90 min (linear gradient system of 0–100% B in A for 26 min). ESI-MS calcd MW for C10H8Cl2O, 215.08; found, m/z = 214.91 [M + H]+.
Synthesis of 2-Bromo-5,6-dichloro-3,4-dihydronaphthalen-1(2H)-one (10b)
Synthesis of 10b followed the identical procedure as described for 10a.
10b
White powder; yield 0.13 g (90%); mp 102–103 °C. 1H NMR (CDCl3, 400 MHz) in ppm: δ 2.49–2.59 (m, 2H), 3.16–3.19 (m, 2H), 4.67–4.69 (m, 1H), 7.47 (d, J = 8.0 Hz, 1H), 7.95 (d, J = 8.0 Hz, 1H). 13C NMR (CDCl3, 100 MHz) in ppm: δ 24.9, 30.6, 48.6, 127.7, 129.0, 132.5, 139.5, 142.5, 189.3. Purity of 98.4% as determined by RP-HPLC, tR = 18.58 min (linear gradient system of 0–100% B in A for 26 min). ESI-MS calcd MW for C10H7BrCl2O, 293.37; found, m/z = 294.84 (M + H)+.
Synthesis of 1-(8,9-Dichloro-4,5-dihydronaphtho[1,2-d]thiazol-2-yl)hydrazine (11b) and 9,10-Dichloro-5,6-dihydro-4aH-naphtho[1,2-e][1,3,4]thiadiazin-3-amine (12b)
Synthesis of 11b and 12b followed the identical procedure as described for 11a and 12a adjusted to 0.085 mmol scale for the 10b and thiosemicarbazide. Both 11b and 12b were purified on a RP-C18 FCC (100 g cartridge, flow rate = 40 mL/min) using 0–40% B in A in 2 h.
11b
Off-white solid; yield 0.092 g (38%); mp 197–199 °C. 1H NMR (DMSO-d6, 400 MHz) in ppm: δ 2.88–2.95 (m, 2H), 3.09–3.15 (m, 2H), 7.53 (d, J = 8.0 Hz, 1H), 7.61 (d, J = 8.0 Hz, 1H), 9.74 (bs, 2H). 13C NMR (DMSO-d6, 100 MHz) in ppm: δ 20.9, 26.7, 122.6, 123.0, 129.1, 130.3, 131.0, 131.9, 134.9, 143.1, 168.2. Purity of 85.7% as determined by RP-HPLC, tR = 13.39 min (linear gradient system of 0–100% B in A for 26 min). ESI-MS calcd MW for C11H9Cl2N3S, 286.18; found, m/z = 283.94 (M – H)−.
12b
Off-white solid; yield 0.051 g (21%); mp 213-215 °C. 1H NMR (DMSO-d6, 500 MHz) in ppm: δ 1.83–1.90 (m, 2H), 2.79–2.85 (m, 2H), 3.19–3.23 (m, 2H), 4.31–4.35 (m, 1H), 7.62 (d, J = 5.0 Hz, 1H), 7.99 (d, J = 5.0 Hz, 1H). 13C NMR (DMSO-d6, 125 MHz) in ppm: δ 25.5, 26.1, 34.0, 125.6, 129.5, 129.7, 131.6, 135.1, 140.6, 148.5. Purity of 98.7% as determined by RP-HPLC, tR = 12.7 min (linear gradient system of 0–100% B in A for 26 min). ESI-MS calcd MW for C11H9Cl2N3S, 286.18; found, m/z = 283.94 (M – H)−.
Synthesis of (E/Z)-2-(2-(6,7-Dichloro-4,5-dihydronaphtho[1,2-d]thiazol-2-yl)hydrazono)-3-(2-nitrophenyl)propanoic Acid [(E/Z)-13b]
Synthesis of (E)-13b and (Z)-13b followed the identical procedure as described for (E)-13a and (Z)-13a adjusted to 0.0454 mmol scale for the 11b and 2-(o-nitrophenyl)pyruvic acid.
(E)-13b
Yellow powder; yield 0.061 g (28%); mp 254–255 °C. 1H NMR (DMSO-d6, 500 MHz) in ppm: δ 2.92–2.97(m, 2H), 3.11–3.15 (m, 2H), 4.20 (s, 2H), 7.05–7.08 (m, 1H), 7.45–7.50 (m, 3H), 7.62–7.68 (m, 1H), 8.04–8.07 (m, 1H), 12.3 (bs, 1H). 13C NMR (DMSO-d6, 125 MHz) in ppm: δ 20.9, 26.6, 29.8, 122.2, 125.7, 128.4, 129.1, 129.5, 130.3, 131.0, 131.8, 134.5, 135.0, 149.7, 166.1. Purity of 98.7% as determined by RP-HPLC, tR = 8.69 min (linear gradient system of 50–100% B in A for 20 min). HRMS(ESI) m/z calcd for C20H14Cl2N4O4S [M+H]+, 477.01128; found 477.01951.
(Z)-13b
Yellow powder; yield 0.036 g (17%); mp 261–262 °C. 1H NMR (DMSO-d6, 500 MHz) in ppm: δ 2.84 (t, 2H), 3.06 (t, 2H), 4.15 (s, 2H), 7.44 (d, J = 5 Hz, 1H), 7.49–7.55 (m, 3H), 7.67–7.70 (m, 1H), 8.03–8.05 (m, 1H), 12.70 (bs, 1H). 13C NMR (DMSO-d6, 125 MHz) in ppm: δ 20.9, 26.5, 36.7, 122.5, 125.2, 128.8, 129.0, 130.4, 130.9, 132.5, 134.1, 134.8, 149.7, 164.7. Purity of 100% as determined by RP-HPLC, tR = 11.39 min (linear gradient system of 50–100% B in A for 20 min). HRMS(ESI) m/z calcd for C20H14Cl2N4O4S [M+H]+, 477.01128; found 477.01961.
Synthesis of 3-(2,3-Dichlorophenoxy)propanoic Acid (15)
Oxetan-2-one (0.72 g, 10 mmol) was added dropwise (5 min) to a solution of 2,3-dichlorophenol, 14 (1.63 g, 10 mmol) in 0.25 M NaOH (4 mL) and stirred overnight at 100 °C. After cooling to RT, the reaction mixture was diluted with water (10 mL), acidified with concd HCl (2 mL), and extracted with diethyl ether (2 × 20 mL). The combined organic phases were washed with 10% (w/v) NaHCO3 (100 mL). The solid 15 obtained upon acidification of the aqueous layer to pH = 2 with concd HCl was filtered, washed thoroughly with water, and dried under vacuum.
15
White solid; yield 1.0 g (42%); mp 147–150 °C. 1H NMR (CD3OD, 400 MHz) in ppm: δ 2.00 (t, 2H), 4.28 (t, 2H), 6.98–7.00 (m, 1H), 7.06–7.08 (m, 1H), 7.17–7.21 (m, 1H). 13C NMR (CD3OD, 100 MHz) in ppm: δ 33.9, 65.2, 111.6, 121.4, 122.2, 127.7, 133.3, 155.8, 173.3. Purity of 91.1% as determined by RP-HPLC, tR = 13.74 min (linear gradient system of 0–100% B in A for 26 min). ESI-MS calcd MW for C9H8Cl2O3, 235.06; found, m/z = 232.84 (M – H)−.
Synthesis of 3-(3,4-Dichlorophenoxy)propanoic Acid (18)
A solution of 3,4-dichlorophenol, 17 (1.63 g, 10 mmol) in DMF (5 mL) was added dropwise (5 min) to a stirred suspension of NaH (0.4 g of 60% suspension in mineral oil, 10 mmol) in dry DMF (10 mL) and stirred for 30 min at RT and heated for 1 h at 100 °C. Then oxetan-2-one (0.72 g, 10 mmol) was added dropwise (5 min) and stirred overnight. After cooling to RT, it was diluted with water (10 mL), acidified with concd HCl (2 mL), and extracted with diethyl ether (2 × 20 mL). The combined organic phases were extracted with 10% (w/v) NaHCO3 (100 mL). The solid 18 obtained upon acidification of the aqueous phase to pH = 2 with (10 mL) was filtered, washed thoroughly with water, and dried under vacuum.
18
White solid; yield 1.2 g (50%); mp 114–116 °C. 1H NMR (CD3OD, 400 MHz) in ppm: δ 2.74 (t, 2H), 4.19 (t, 2H), 6.83–6.86 (m, 1H), 7.06 (d, J = 4.0 Hz, 1H), 7.35 (d, J = 8.0 Hz, 1H). 13C NMR (CD3OD, 100 MHz) in ppm: δ 33.9, 64.3, 114.7, 116.3, 123.6, 130.6, 132.5, 158.2, 173.4. Purity of 88.6% as determined by RP-HPLC, tR = 14.29 min (linear gradient system of 0–100% B in A for 26 min). ESI-MS calcd MW for C9H8Cl2O3, 235.06; found, m/z = 232.87 (M – H)−.
Synthesis of 7,8-Dichloro-2,3-dihydrochromen-4-one (16) and 6,7-Dichloro-2,3-dihydrochromen-4-one (19)
Method A
Warning! This reaction was carried out in a dedicated HF-reaction apparatus type I following strict adherence to the manufacturer’s instruction. A suspension of 15 or 18 (0.5 g, 2.12 mmol) in liquid HF (50 mL) was allowed to stir overnight at RT. The residue obtained after removing the HF under vacuum was dissolved in ether (50 mL) and washed with 10% (w/v) NaHCO3 (50 mL), and the separated organic phase was dried over anhydrous MgSO4 to yield 16 or 19.
Method B
15 or 18 (0.5 g, 2.12 mmol) was stirred in Eaton’s Reagent (20 mL) at RT for 1 h followed by 5 h at 70 °C. The dark-red reaction mixture was cooled to RT and quenched into ice-cold water (100 mL) and left for 30 min. The precipitate obtained from 15 was filtered and dried under vacuum to afford 16. The reaction of 18 yielded a precipitate containing a mixture of 19 and 20, was dried under vacuum and purified by FCC on a silica gel column employing ethyl acetate–hexane mixture (2:8, v/v) to obtain pure 19 and 20.
16
Off-white solid; yield 0.350 g (76%, method A), 0.4 g (80%, method B); mp 90–92 °C. 1H NMR (CDCl3, 400 MHz) in ppm: δ 2.82 (t, 2H), 4.65 (t, 2H), 7.10 (d, J = 8.0 Hz, 1H), 7.12 (d, J = 8.0 Hz, 1H). 13C NMR (CDCl3, 100 MHz) in ppm: δ 37.2, 68.2, 120.8, 122.0, 122.9, 125.6, 140.5, 158.5, 190.2. Purity of 99.8% as determined by RP-HPLC, tR = 14.99 min (linear gradient system of 0–100% B in A for 26 min).
19
Off-white solid; yield 0.350 g (76%, method A), 0.125 g (27%, method B); mp 131–133 °C. 1H NMR (CDCl3, 400 MHz) in ppm: δ 2.79 (t, 2H), 4.52 (t, 2H), 7.10 (s, 1H), 7.90 (s, 1H). 13C NMR (CDCl3, 100 MHz) in ppm: δ 37.4, 67.6, 118.0, 120.2, 120.9, 126.0, 128.3, 140.0, 160.3, 189.9. Purity of 94.6% as determined by RP-HPLC, tR = 15.69 min (linear gradient system of 0–100% B in A for 26 min).
20
Off-white solid; yield 0.175 g (38%, method B); mp 111–114 °C. 1H NMR (CDCl3, 500 MHz) in ppm: δ 2.82–2.84 (m, 2H), 4.50–4.52 (m, 2H), 6.87 (d, 1H, J = 8.5 Hz), 7.47 (d, 1H, J = 8.5 Hz). 13C NMR (CDCl3, 125 MHz) in ppm: δ 38.8, 67.0, 118.0, 119.9, 127.7, 132.3, 135.6, 161.9, 189.1. Purity of 99.5% as determined by RP-HPLC, tR = 15.54 min (linear gradient system of 0–100% B in A for 26 min).
Synthesis of 3-Bromo-7,8-dichloro-2,3-dihydrochromen-4-one (21a) and 3-Bromo-6,7-dichloro-2,3-dihydrochromen-4-one (21b)
Pyridinium bromide perbromide (0.147 g, 0.46 mmol) was added to a solution of 16 or 19 (0.1 g, 0.46 mmol) in a mixture of anhydrous ethanol and chloroform (1:1 v/v, 10 mL) during 10 min. The reddish-brown mixture was stirred at 50 °C for 30 min and cooled to RT. The residue obtained after removal of solvent under vacuum was suspended in water (20 mL) and extracted with dichloromethane (20 mL). The organic phase was washed with 5% (w/v) NaHCO3 (20 mL) followed by water (20 mL), dried over anhydrous Na2SO4, and concentrated under vacuum. The crude mixture was purified by FCC using hexane–ethyl acetate (9:1, v/v) solvent mixtures to yield pure 21a and 21b.
21a
Pale-yellow solid; yield 0.1 g (74%); mp 134–136 °C. 1H NMR (CDCl3, 400 MHz) in ppm: δ 4.62–4.64 (m, 1H), 4.75–4.80 (m, 2H), 7.20 (d, J = 8.8 Hz, 1H), 7.79 (d, J = 8.8 Hz, 1H). 13C NMR (CDCl3, 100 MHz) in ppm: δ 44.0, 72.2, 118.2, 122.2, 123.9, 126.6, 141.5, 157.3, 184.0. Purity of 97.5% as determined by RP-HPLC, tR = 12.29 min (linear gradient system of 30–100% B in A for 26 min). ESI-MS calcd MW for C9H5BrCl2O2, 295.94; found, m/z = 294.84 (M – H)−.
21b
White powder; yield 0.104 g, (76%); mp 100–107 °C. 1H NMR (CDCl3, 400 MHz) in ppm: δ 4.59–4.66 (m, 3H), 7.20 (s, 1H), 7.98 (s, 1H). 13C NMR (CDCl3, 100 MHz) in ppm: δ 44.3, 71.7, 118.4, 120.2, 127.1, 129.3, 141.1, 159.0, 183.5. Purity of 98.4% as determined by RP-HPLC, tR = 11.61 min (linear gradient system of 30–100% B in A for 26 min). ESI-MS calcd MW for C9H5BrCl2O2, 295.94; found, m/z = 294.02 (M – H)−.
Synthesis of (E/Z)-2-(2-(6,7-Dichloro-4H-chromeno[4,3-d]thiazol-2-yl)hydrazono)-3-(2-nitrophenyl)propanoic Acid [(E/Z)-23a]
Step A
A solution of 21a (0.2 g, 0.67 mmol) and thiosemicarbazide (0.07 g, 0.67 mmol) in anhydrous dioxane (20 mL) was stirred at 60 °C for 24 h. The precipitate formed after cooling to RT was filtered, washed with dioxane (10 mL), and suspended in 2 M Na2CO3 (20 mL). The brown product was filtered, washed with water, and dried to yield crude 22a and used as such in the next step.
22a
Yield 0.08 g (39%). ESI-MS calcd MW for C10H7Cl2N3OS, 288.15; found, m/z = 291.94 (M + H)+.
Step B
Synthesis of (E)-23a and (Z)-23a followed the identical procedure as described for (E)-13a and (Z)-13a adjusted to 0.27 mmol scale for the 22a and 2-(o-nitrophenyl)pyruvic acid.
(E)-23a
Pale-brown fluffy solid; yield 0.028 g (22%); mp 252–253 °C. 1H NMR (DMSO-d6, 400 MHz) in ppm: δ 4.27 (s, 2H), 5.55 (s, 2H), 7.05 (d, 1H, J = 4 Hz), 7.19 (d, 1H, J = 8 Hz), 7.33 (d, 1H, J = 8 Hz), 7.49–7.51 (m, 1H), 7.61–7.65 (m, 1H), 8.05 (d, 1H, J = 8 Hz),12.40 (bs, 1H). 13C NMR (DMSO-d6, 100 MHz) in ppm: δ 29.9, 66.3, 119.8, 121.4, 123.5, 125.7, 128.5, 129.5, 131.4, 131.6, 134.5, 149.6, 150.7, 165.9. Purity of 99.5% as determined by RP-HPLC, tR = 11.60 min (linear gradient system of 30–100% B in A for 20 min). HRMS(ESI) m/z calcd for C19H12Cl2N4O5S [M+H]+, 478.99054; found 479.00179.
(Z)-23a
Bright-yellow fluffy solid; yield 0.016 g (12%); mp 253–254 °C. 1H NMR (DMSO-d6, 500 MHz) in ppm: δ 4.17 (s, 2H), 5.47 (s, 2H), 7.18–7.20 (m, 1H), 7.40 (d, 1H, J = 5 Hz), 7.51–7.56 (m, 2H), 7.68–7.71 (m, 1H), 8.05 (d, 1H, J = 5 Hz), 12.72 (bs, 1H). 13C NMR (DMSO-d6, 125 MHz) in ppm: δ 36.8, 66.2, 119.7, 121.8, 123.5, 125.3, 128.9, 131.5, 132.4, 133.7, 134.2, 149.6, 150.6, 164.7. Purity of 100% as determined by RP-HPLC, tR = 13.60 min (linear gradient system of 30–100% B in A for 20 min). HRMS(ESI) m/z calcd for C19H12Cl2N4O5S [M+H]+, 478.99054; found 479.02029.
Synthesis of (E/Z)-2-(2-(7,8-Dichloro-4H-chromeno[4,3-d]thiazol-2-yl)hydrazono)-3-(2-nitrophenyl)propanoic Acid [(E/Z)-23b]
Step A
Synthesis of 22b followed the identical procedure as described for 22a.
22b
Yield 0.11 g (57%). ESI-MS calcd MW for C10H7Cl2N3OS, 288.15; found, m/z = 291.88 (M + H)+.
Step B
Synthesis of (E)-23b and (Z)-23b followed the identical procedure as described for (E)-13a and (Z)-13a adjusted to 0.48 mmol scale for 22b and 2-(o-nitrophenyl)pyruvic acid.
(E)-23b
Yellow powder; yield 0.055 g (24%); mp 260–261 °C. 1H NMR (DMSO-d6, 500 MHz) in ppm: δ 4.28 (s, 2H), 5.45 (s, 2H), 7.06 (d, J =5.0 Hz, 1H), 7.17 (s, 1H), 7.43 (s, 1H), 7.50 (t, 1H), 7.64 (t, 1H), 8.05 (d, J =10.0 Hz, 1H). 13C NMR (DMSO-d6, 125 MHz) in ppm: δ 30.0, 65.6, 118.8, 123.2, 124.2, 125.7, 128.5, 129.6, 130.6, 131.7, 134.5, 149.6, 152.9, 165.9. Purity of 98.0% as determined by RP-HPLC, tR = 12.77 min (linear gradient system of 30–100% B in A for 20 min). HRMS(ESI) m/z calcd for C19H12Cl2N4O5S [M+H]+, 478.99054; found 478.99796.
(Z)-23b
Yellow powder; yield 0.037 g (16%); mp 265–266 °C. 1H NMR (DMSO-d6, 500 MHz) in ppm: δ 4.17 (s, 2H), 5.38 (s, 2H), 7.16 (s, 1H), 7.51–7.56 (m, 3H), 7.69 (s, 1H), 8.04 (d, J =10.0 Hz, 1H). 13C NMR (DMSO-d6, 125 MHz) in ppm: δ 36.8, 65.5, 118.7, 123.6, 124.3, 125.2, 129.0, 130.7, 132.4, 133.7, 134.2, 149.6, 152.8, 164.6. Purity of 99.5% as determined by RP-HPLC, tR = 14.74 min (linear gradient system of 30–100% B in A for 20 min). HRMS(ESI) m/z calcd for C19H12Cl2N4O5S [M+H]+, 478.99054; found 478.99785.
Synthesis of 3-(3,4-Dichlorophenylthio)propanoic Acid (25a) and 3-(2,3-Dichlorophenylthio)propanoic Acid (25b)
An ice-cold mixture consisting of 3-bromopropionic acid (2.12 g, 13.6 mmol) and Na2CO3 (1.172 g, 13.6 mmol) dissolved in water (10 mL) was added dropwise to a solution of either 3,4-dichlorobenzenethiol or 2,3-dichlorobenzenethiol (2.5 g, 13.6 mmol) in aqueous 2N NaOH solution (6 mL) and ethanol (2 mL). This reaction mixture was heated at 100 °C for 4 h, cooled to RT, and extracted with ether. The precipitate formed after acidification of the aqueous portion with ice-cold 3 N HCl to pH = 2 followed by cooling to 0 °C for 30 min was filtered off and dried to give either 25a or 25b.
25a
White solid; yield 3.0 g (88%); mp 70–72 °C. 1H NMR (CDCl3, 400 MHz) in ppm: δ 2.68 (t, 2H, J = 8 Hz), 3.15 (t, 2H, J = 8 Hz), 7.16–7.18 (m, 1H), 7.35 (d, J = 8.0 Hz, 1H), 7.41–7.43 (m, 1H), 11.40 (bs, 1H). 13C NMR (CDCl3, 100 MHz) in ppm: δ 28.9, 34.2, 129.3, 130.9, 131.1, 131.4, 133.2, 135.6, 178.1. Purity of 99.3% as determined by RP-HPLC, tR = 15.33 min (linear gradient system of 0–100% B in A for 26 min). ESI-MS calcd MW for C9H8Cl2O2S, 251.13, found: m/z = 249.01 (M – H)−.
25b
Off-white solid; yield 2.8 g (82%); mp 139–142 °C. 1H NMR (CDCl3, 500 MHz) in ppm: δ 2.61 (t, 2H), 3.20 (t, 2H), 7.31–7.36 (m, 1H), 7.39–7.42 (m, 1H), 12.43 (s, 1H). 13C NMR (CDCl3, 125 MHz) in ppm: δ 27.4, 33.6, 125.8, 127.2, 129.0, 129.2, 132.8, 139.2, 173.1. Purity of 95.6% as determined by RP-HPLC, tR = 14.96 min (linear gradient system of 0–100% B in A for 26 min). ESI-MS calcd MW for C9H8Cl2O2S, 251.13; found, m/z = 250.57 (M – H)−.
Synthesis of 6,7-Dichlorothiochroman-4-one (26a) and 7,8-Dichlorothiochroman-4-one (26b)
Ketones 25a or 25b (2.8 g, 11.1 mmol) were added portionwise to stirred concd H2SO4 (50 mL), previously cooled to −10 °C, and warmed to RT and left stand for 2 h. The dark-red reaction mixture was poured slowly over ice-cold water (400 mL) and kept aside for 1 h to form a white precipitate. Pure 26a and 26b were obtained after recrystallization from ethanol.
26a
White crystals; yield 2.0 g (77%); mp 143–146 °C. 1H NMR (CDCl3, 400 MHz) in ppm: δ 2.92 (m, 2H), 3.21–3.25 (m, 2H), 7.36 (s, 1H), 8.10 (s, 1H). 13C NMR (CDCl3, 100 MHz) in ppm: δ 26.8, 39.1, 129.1, 129.8, 130.3, 130.7, 138.1, 141.8, 192.2. Purity of 100% as determined by RP-HPLC, tR = 17.33 min (linear gradient system of 0–100% B in A for 26 min). ESI-MS calcd MW for C9H6Cl2OS, 233.11; found, m/z = 232.81 and 234.83 (M + H)+.
26b
Off-white solid; yield 2.3 g (88%); mp 112–115 °C. 1H NMR (CDCl3, 500 MHz) in ppm: δ 2.87 (m, 2H), 3.37 (m, 2H), 7.42–7.45 (m, 1H), 7.87–7.89 (m, 1H). 13C NMR (CDCl3, 125 MHz) in ppm: δ 26.0, 37.8, 126.4, 128.5, 128.7, 131.4, 137.5, 144.3, 192.9. Purity of 99.1% as determined by RP-HPLC, tR = 16.88 min (linear gradient system of 0–100% B in A for 26 min). ESI-MS calcd MW for C9H6Cl2OS, 233.11; found, m/z = 232.81 and 234.83 (M + H)+.
Synthesis of 3-Bromo-6,7-dichloro-2,3-dihydrothiochromen-4-one (27a) and 3-Bromo-7,8-dichloro-2,3-dihydrothiochromen-4-one (27b)
To a solution of (0.5 g, 2.14 mmol) 26a or 26b dissolved in dry chloroform (20 mL) was added dropwise a solution of bromine (0.342 g, 2.14 mmol) in chloroform (5 mL). The reaction mixture was stirred at RT for 2 h and then at 60 °C for 1 h. It was then cooled to RT and extracted with 10% (w/v) Na2S2O3 solution (25 mL). The organic phase was washed with water (50 mL), dried over anhydrous Na2SO4, filtered, and concentrated under vacuum to give crude bromide. It was subjected to silica gel FCC using hexane–ethyl acetate (9:1, v/v) solvent mixture to yield pure 27a and 27b.
27a
Off-white solid; yield 0.4 g (60%); mp 128–131 °C. 1H NMR (CDCl3, 500 MHz) in ppm: δ 3.44–3.48 (m, 1H), 3.67–3.70 (m, 1H), 4.90–4.92 (m, 1H), 7.40 (s, 1H), 8.18 (s, 1H). 13C NMR (CDCl3, 125 MHz) in ppm: δ 35.0, 48.2, 128.8, 130.6, 132.0, 139.0, 140.6, 185.4. Purity of 98.4% as determined by RP-HPLC, tR = 18.61 min (linear gradient system of 0–100% B in A for 26 min).
27b
Off-white solid; yield 0.461 g (69%); mp 104–106 °C. 1H NMR (CDCl3, 500 MHz) in ppm: δ 3.47–3.54 (m, 1H), 3.67–3.71 (m, 1H), 4.90–4.92 (m, 1H), 7.31 (d, 1H), 8.01 (d, 1H). 13C NMR (CDCl3, 125 MHz) in ppm: δ 34.6, 47.6, 126.7, 128.2, 129.5, 139.5, 142.9, 185.9. Purity of 99.1% as determined by RP-HPLC, tR = 18.23 min (linear gradient system of 0–100% B in A for 26 min).
Synthesis of (E/Z)-2-(2-(7,8-Dichloro-4H-thiachromeno[4,3-d]thiazol-2-yl)hydrazono)-3-(2-nitrophenyl)propanoic Acid [(E/Z)-29a]
Step A
A solution of 27a (0.5 g, 1.6 mmol) and thiosemicarbazide (0.145 g, 1.6 mmol) in anhydrous dioxane (20 mL) was stirred at RT for 24 h and then heated to 80 °C for 12 h. The reaction mixture was then cooled to RT, and the precipitate was filtered, washed with dioxane (20 mL), and dried. The solid was triturated with 2 M Na2CO3 (40 mL), filtered, thoroughly washed with water, and dried to yield 28a, which was used as such in the next step.
28a
Yield 0.304 g (62%). ESI-MS calcd MW for C10H7Cl2N3S2, 304.22; found, m/z = 303.92 and 305.94 (M + H)+.
Step B
A suspension of 28a (0.304 g, 1 mmol) in 5% (v/v) acetic acid (7 mL) was added to 2-(o-nitrophenyl)pyruvic acid (0.209 g, 1 mmol) in ethanol (14 mL) and was heated at 90–100 °C for 2 h. The yellow precipitate that formed upon cooling to RT was filtered, washed with water, and dried. The crude mixture containing the two isomers, (E)-29a and (Z)-29a, were purified on a RP-C18 FCC (100 g cartridge, flow rate = 40 mL/min) using a linear gradient of 0–60% B in A for 3 h (A, triethylammonium bicarbonate buffer (50 mM, pH = 8.5), and B, methanol). The purified isomers were precipitated from their respective pooled fractions following acidification with 10% (v/v) HCl to pH = 2 and separated by centrifugation. The pellets were washed consecutively with 5% (v/v) HCl and water and dried under vacuum to yield the following:
(E)-29a
Brown powder; yield 0.096 g (20%); mp 259–260 °C. 1H NMR (DMSO-d6, 500 MHz) in ppm: δ 4.26 (s, 2H), 4.28 (s, 2H), 7.06 (d, J =10.0 Hz, 1H), 7.48–7.52 (m, 1H), 7.51 (s, 1H), 7.58–7.66 (m, 1H), 7.69 (s, 1H), 8.06 (d, 1H), 12.10 (bs, 1H), 12.77 (bs, 1H). 13C NMR (DMSO-d6, 125 MHz) in ppm: δ 23.8, 29.9, 125.7, 128.5, 128.7, 128.8, 129.5, 130.2, 131.6, 132.1, 134.5, 149.6, 165.9. Purity of 99.2% as determined by RP-HPLC, tR = 10.33 min (linear gradient system of 50–100% B in A for 20 min). HRMS(ESI) m/z calcd for C19H12Cl2N4O4S2 [M+H]+, 494.96770; found 494.97625.
(Z)-29a
Brown powder; yield 0.035 g (7%); mp 230–231 °C. 1H NMR (DMSO-d6, 400 MHz) in ppm: δ 4.15 (s, 2H), 4.17 (s, 2H), 7.49–7.56 (m, 3H), 7.66–7.72 (m, 2H), 8.03 (d, J = 8 Hz, 1H), 12.66 (bs, 1H). 13C NMR (DMSO-d6, 100 MHz) in ppm: δ 23.7, 36.8, 125.2, 125.9, 128.6, 128.9, 130.3, 132.0, 132.4, 133.6, 134.2, 149.7, 164.7. Purity of 99.8% as determined by RP-HPLC, tR = 13.33 min (linear gradient system of 50–100% B in A for 20 min). HRMS(ESI) m/z calcd for C19H12Cl2N4O4S2 [M+H]+, 494.96770; found 494.97396.
Synthesis of (E/Z)-2-(2-(6,7-Dichloro-4H-thiachromeno[4,3-d]thiazol-2-yl)hydrazono)-3-(2-nitrophenyl)propanoic Acid [(E/Z)-29b]
Step A
Synthesis of 28b followed the identical procedure as described for 28a.
28b
Yield 0.280 g (57%). ESI-MS calcd MW for C10H7Cl2N3S2, 304.22; found, m/z = 303.92 and 305.94 (M + H)+.
Step B
Synthesis of (E)-29b and (Z)-29b followed the identical procedure as described for (E)-29a and (Z)-29a adjusted to 0.82 mmol scale for the 28a and 2-(o-nitrophenyl)pyruvic acid.
(E)-29b
Orange powder; yield 0.090 g (22%); mp 225–226 °C. 1H NMR (DMSO-d6, 400 MHz) in ppm: δ 4.27 (s, 2H), 4.32 (s, 2H), 7.05 (d, J = 8 Hz, 1H), 7.39 (d, J = 8 Hz, 1H), 7.47–7.52 (m, 1H), 7.59–7.68 (m, 2H), 8.05 (d, J = 8 Hz, 1H), 12.21 (s, 1H), 12.74 (s, 1H). 13C NMR (DMSO-d6, 100 MHz) in ppm: δ 24.2, 29.9, 124.2, 125.7, 127.5, 128.5, 129.5, 131.1, 131.7, 134.5, 148.5, 149.6, 166.0. Purity of 99.9% as determined by RP-HPLC, tR = 15.66 min (linear gradient system of 30–100% B in A for 20 min). HRMS(ESI) m/z calcd for C19H12Cl2N4O4S2 [M+H]+, 494.96770; found 494.97248.
(Z)-29b
Pale-brown powder; yield 0.035 g (9%); mp 229–230 °C. 1H NMR (DMSO-d6, 400 MHz) in ppm: δ 4.15 (s, 2H), 4.24 (s, 2H), 7.36–7.39 (m, 1H), 7.49–7.55 (m, 2H), 7.61–7.64 (m, 1H), 7.67–7.70 (m, 1H), 8.03 (d, J = 7.6 Hz, 1H), 12.65 (s, 1H). 13C NMR (DMSO-d6, 100 MHz) in ppm: δ 24.1, 36.8, 124.5, 125.2, 127.4, 128.3, 128.9, 130.4, 131.2, 132.4, 133.6, 134.2, 149.7, 164.7. Purity of 99.4% as determined by RP-HPLC, tR = 18.23 min (linear gradient system of 50–100% B in A for 20 min). HRMS(ESI) m/z calcd for C19H12Cl2N4O4S2 [M+H]+, 494.96770; found 494.97512.
Synthesis of 3-Bromo-6,7-dichlorothiochroman-4-one 1-oxide (30a) and 3-Bromo-7,8-dichloro-2,3-dihydrothiochromen-4-one-1-oxide (30b)
A solution of m-chloroperbenzoic acid (0.287 g, 1.72 mmol) in chloroform (10 mL) was added dropwise to a solution of 27a or 27b (0.4 g, 1.72 mmol) in chloroform (10 mL) over a period of 30 min. The resultant mixture was stirred at RT for 90 min and treated with saturated NaHCO3 solution (20 mL). The separated organic phase was washed with water (50 mL), dried over anhydrous Na2SO4, filtered, and evaporated under vacuum to yield 30a or 30b, respectively.
30a
White solid; yield 0.428 g (76%); mp 201–202 °C. Purity of 96.9% as determined by RP-HPLC, tR = 7.37 min (linear gradient system of 30–100% B in A for 26 min). ESI-MS calcd MW for C9H5BrCl2O2S, 328.01; found, m/z = 326.87 (M – H)−.
30b
White solid; yield 0.430 g (76%); mp 211–212 °C. 1H NMR (DMSO-d6, 500 MHz) in ppm: δ 4.19 (dd, 1H, J1 = 4 Hz, J2 = 14 Hz), 4.31 (t, 1H, J = 14 Hz), 6.11 (dd, 1H, J1 = 4 Hz, J2 = 13 Hz), 8.06 (s, 2H). 13C NMR (DMSO-d6, 100 MHz) in ppm: δ 44.8, 52.2, 129.8, 130.3, 133.8, 135.0, 139.3, 142.8, 186.5. Purity of 92.2% as determined by RP-HPLC, tR = 11.74 min (linear gradient system of 0–100% B in A for 26 min). ESI-MS calcd MW for C9H5BrCl2O2S, 328.01; found, m/z = 326.74 (M – H)−.
Synthesis of (E/Z)-2-(2-(7,8-Dichloro-5-oxido-4H-thiochromeno[4,3-d]thiazol-2-yl)hydrazono)-3-(2-nitrophenyl)propanoic Acid [(E/Z)-32a]
Step A
A solution of 30a (0.380 g, 1.15 mmol) and thiosemicarbazide (0.105 g, 1.15 mmol) in anhydrous dioxane (20 mL) was stirred at RT for 24 h and then heated at 70 °C for 24 h. The mixture was cooled to RT, and the resulting precipitate was filtered, washed with dioxane (20 mL), and dried. The solid was triturated with 2 M Na2CO3 (40 mL), filtered, washed with water, and dried under vacuum to yield the crude 31a.
31a
Yield 0.240 g (65%). ESI-MS calcd MW for C10H7Cl2N3OS2, 320.22; found, m/z = 321.83 (M + H)+.
Step B
To a suspension of 31a (0.186 g, 0.581 mmol) in acetic acid (10 mL) was added 2-(o-nitrophenyl)pyruvic acid (0.121 g, 0.581 mmol). The reaction mixture was stirred at RT for 2 h and diluted with water (20 mL), forming a brown precipitate that was filtered, washed with water (40 mL), and dried. The crude mixture of the two isomers, (E)-32a and (Z)-32a, were purified on a RP-C18 FCC (100 g cartridge, flow rate = 40 mL/min) using a linear gradient system of 0–100% of B in A for 3h (A, water, and B, methanol).
(E)-32a
Brown fluffy solid; yield 0.044 g (17%) mp: 204–206 °C. 1H NMR (DMSO-d6, 400 MHz) in ppm: δ 4.30 (s, 2H), 4.35 (d, 1H, J = 16.0 Hz), 4.80 (d, 1H, J = 16.0 Hz), 7.08 (d, 1H, J = 8.0 Hz), 7.48–7.52 (m, 1H), 7.62–7.66 (m, 1H), 7.89 (s, 1H), 8.06 (d, 1H, J = 8.0 Hz), 8.14 (s, 1H), 12.26–12.95 (m, 2H). 13C NMR (DMSO-d6, 100 MHz) in ppm: δ 30.0, 45.1, 125.7, 126.4, 128.5, 129.7, 131.0, 131.9, 134.5, 136.1, 136.9, 149.7, 165.8. Purity of 96.5% as determined by RP-HPLC, tR = 9.82 min (linear gradient system of 30–100% B in A for 20 min). HRMS(ESI) m/z calcd for C19H12Cl2N4O5S2 [M+H]+, 510.96261; found 510.97205.
(Z)-32a
Brown fluffy solid; yield 0.02 g (8%); mp 200–202 °C. 1H NMR (DMSO-d6, 500 MHz) in ppm: δ 4.19 (s, 2H), 4.29 (d, 1H, J = 15.0 Hz), 4.73 (d, 1H, J = 20.0 Hz), 7.52–7.57 (m, 2H), 7.69–7.72 (m, 1H), 8.00 (s, 1H), 8.05 (d, 1H, 5.0 Hz), 8.13 (s, 1H), 12.71 (s, 1H). 13C NMR (DMSO-d6, 125 MHz) in ppm: δ 36.8, 45.1, 115.8, 125.2, 126.7, 128.9, 129.1, 131.1, 131.7, 132.3, 134.1, 149.8, 164.6, 167.4. Purity of 95.1% as determined by RP-HPLC, tR = 10.38 min (linear gradient system of 30–100% B in A for 20 min). HRMS(ESI) m/z calcd for C19H12Cl2N4O5S2 [M+H]+, 510.96261; found 510.97105.
Synthesis of (E/Z)-2-(2-(7,8-Dichloro-4H-5-oxothiochromeno[4,3-d]thiazol-2-yl)hydrazono)-3-(2-nitrophenyl)propanoic Acid [(E/Z)-32b]
Step A
Synthesis of 31b followed the identical procedure as described for 31a.
31b
Yield 0.186 g (50%). ESI-MS calcd MW for C10H7Cl2N3OS2, 320.22; found, m/z = 319.77 (M – H)−.
Step B
Synthesis of (E)-32b and (Z)-32b followed the identical procedure as described for (E)-29a and (Z)-29a adjusted to 0.58 mmol scale for the 31b and 2-(o-nitrophenyl)pyruvic acid.
(E)-32b
Brown fluffy solid; yield 0.056 g (18%); mp 190–192 °C (dec). 1H NMR (DMSO-d6, 500 MHz) in ppm: δ 4.27–4.31 (m, 3H), 4.95–5.01 (m, 1H), 7.06–7.07 (m, 1H), 7.48–7.51 (m, 1H), 7.62–7.65 (m, 1H), 7.86–7.94 (m, 1H), 8.05–8.08 (m, 1H), 12.24 (s, 1H). 13C NMR (DMSO-d6, 125 MHz) in ppm: δ 30.0, 40.4, 125.5, 125.7, 128.5, 129.5, 131.6, 133.4, 134.6, 134.9, 136.6, 149.6, 165.9. Purity of 98.2% as determined by RP-HPLC, tR = 8.74 min (linear gradient system of 30–100% B in A for 26 min). HRMS(ESI) m/z calcd for C19H12Cl2N4O5S2 [M+H]+, 510.96261; found 510.96985.
(Z)-32b
Brown fluffy solid; yield 0.015 g (5%); mp > 300 °C. 1H NMR (DMSO-d6, 400 MHz) in ppm: δ 4.17–4.29 (m, 3H), 4.90 (d, 1H, J = 17.6 Hz), 7.51–7.59 (m, 2H), 7.68–7.73 (m, 1H), 7.89–7.96 (m, 2H), 8.04 (d, 1H, J = 8 Hz), 13.03 (s, 1H). 13C NMR (DMSO-d6, 100 MHz) in ppm: δ 45.0, 125.2, 125.8, 128.9, 129.9, 131.6, 132.6, 133.3, 133.6, 134.2, 134.8, 136.6, 149.8. Purity of 96.0% as determined by RP-HPLC, tR = 10.12 min (linear gradient system of 30–100% B in A for 26 min). HRMS(ESI) m/z calcd for C19H12Cl2N4O5S2 [M+H]+, 510.96261; found 510.96978.
Synthesis of 1,1-Dioxo-3-bromo-6,7-dichloro-2,3-dihydrothiochromen-4-one (33a)
A solution of m-chloroperbenzoic acid (2.21 g, 12.8 mmol) in chloroform (10 mL) was added dropwise over 30 min to a stirred solution of 27a (1.0 g, 3.20 mmol) in chloroform (40 mL) at 60 °C. The resultant mixture was refluxed for 6 h and cooled to RT. The precipitated sulfone was filtered and dried. Additional product was obtained by consecutive washings of the filtrate with saturated NaHCO3 solution (100 mL) and water (100 mL). The separated organic phase was dried over anhydrous Na2SO4, filtered, and evaporated under vacuum and combined with the previously obtained precipitate to afford the sulfone 33a.
33a
Off-white solid; yield 0.8 g (72%); mp 255–256 °C. 1H NMR (DMSO-d6, 500 MHz) in ppm: δ 4.65–4.68 (m, 1H), 4.77–4.82 (m, 1H), 5.87–5.91 (m, 1H), 8.22 (d, 1H), 8.28 (d, 1H). 13C NMR (DMSO-d6, 125 MHz) in ppm: δ 46.4, 56.6, 126.2, 128.4, 131.37, 137.7, 139.2, 141.5, 184.2. Purity of 100% as determined by RP-HPLC, tR = 8.79 min (linear gradient system of 30–100% B in A for 20 min). ESI-MS calcd MW for C9H5BrCl2O3S, 344.01; found, m/z = 342.76 (M – H)−.
Synthesis of 1,1-Dioxo-3-bromo-7,8-dichloro-2,3-dihydrothiochromen-4-one (33b)
A solution of m-chloroperbenzoic acid (1.21 g, 6.9 mmol) in chloroform (15 mL) was added dropwise over 30 min to a solution of 27b (0.543 g 1.74 mmol) in chloroform (10 mL) that was kept at 60 °C. After refluxing the reaction mixture for 12 h, it was cooled to RT, diluted with dichloromethane (50 mL), and washed with saturated NaHCO3 solution (3 × 100 mL) and water (100 mL). The organic phase was dried over anhydrous Na2SO4, filtered, and evaporated under vacuum to yield an oily residue, which yielded 33b following trituration with ice-cold hexane.
33b
White solid; yield 0.5 g (83%); mp 254–255 °C (dec.). Purity of 98.1% as determined by RP-HPLC, tR = 14.55 min (linear gradient system of 0–100% B in A for 26 min). ESI-MS calcd MW for C9H5BrCl2O3S, 344.01; found, m/z = 342.82 [M – H]−.
Synthesis of (E/Z)-2-(2-(7,8-Dichloro-4H-thiodioxochromeno[4,3-d]thiazol-2-yl)hydrazono)-3-(2-nitrophenyl)propanoic Acid [(E/Z)-35a]
Step A
A solution of 33a (0.7 g, 2 mmol) and thiosemicarbazide (0.181 g, 2 mmol) in anhydrous dioxane (30 mL) was stirred at 90 °C for 2 d. Cooling the reaction mixture to RT resulted in a precipitate that was filtered, washed with dioxane (10 mL), and dried. The solid was triturated in 2 M Na2CO3 (40 mL), filtered, washed thoroughly with water, and dried to yield the respective crude 34a.
34a
Yield 0.230 g (33%). ESI-MS calcd MW for C10H7Cl2N3O2S2, 336.22; found, m/z = 337.84 (M + H)+.
Step B
The 2-(o-nitrophenyl)pyruvic acid (0.154 g, 0.74 mmol) was added to a suspension of 34a (0.25 g, 0.74 mmol) in acetic acid (20 mL), and the reaction mixture was stirred at RT for 4 h. The formed precipitate was filtered, washed thoroughly with glacial acetic acid (20 mL), and dried to give (E)-35a. Diluting the combined acetic acid filtrates with water (100 mL) generated a precipitate that was washed with methanol to afford (Z)-35a.
(E)-35a
Brown fluffy solid; yield 0.16 g, (41%). mp 240–241 °C. 1H NMR (DMSO-d6, 400 MHz) in ppm: δ 4.28 (s, 2H), 5.11 (s, 2H), 7.06 (d, 1H, J = 8 Hz), 7.48–7.52 (m, 1H), 7.61–7.65 (m, 1H), 7.91 (s, 1H), 8.02–8.07 (m, 2H), 12.32 (bs, 1H). 13C NMR (DMSO-d6, 100 MHz) in ppm: δ 30.1, 49.2, 125.7, 125.8, 126.0, 127.5, 128.5, 129.6, 131.4, 131.5, 131.6, 134.6, 135.3, 137.4, 149.6, 165.8. Purity of 98.7% as determined by RP-HPLC, tR = 12.41 min (linear gradient system of 30–100% B in A for 26 min). HRMS(ESI) m/z calcd for C19H12Cl2N4O6S2 [M+H]+, 526.95753; found 526.96875.
(Z)-35a
Gray fluffy solid; yield 0.065 g, (17%). mp 248–249 °C. 1H NMR (DMSO-d6, 400 MHz) in ppm: δ 4.18 (s, 2H), 5.04 (s, 2H), 7.51–7.71 (m, 3H), 8.02–8.05 (m, 3H), 12.74 (s, 1H). 13C NMR (DMSO-d6, 100 MHz) in ppm: δ 36.9, 49.17, 117.5, 125.2, 125.8, 129.0, 131.2, 131.7, 132.2, 133.6, 134.2, 135.2, 136.2, 137.5, 140.7, 149.6, 164.7, 167.5. Purity of 99.2% as determined by RP-HPLC, tR = 13.63 min (linear gradient system of 30–100% B in A for 26 min). HRMS(ESI) m/z calcd for C19H12Cl2N4O6S2 [M+H]+, 526.95753; found 526.96705.
Synthesis of (E/Z)-2-(2-(7,8-Dichloro-4H-thiodioxochromeno[4,3-d]thiazol-2-yl)hydrazono)-3-(2-nitrophenyl)propanoic Acid [(E/Z)-35b]
Step A: Synthesis of 7,8-Dichloro-2-hydrazinyl-4H-thiochromeno[4,3-d]thiazole 5,5-Dioxide (34b)
Synthesis of 34b followed the identical procedure as described for 34a.
34b
Yield 0.350 g (50%). ESI-MS calcd MW for C10H7Cl2N3O2S2, 336.22; found, m/z = 337.97 (M + H)+.
Step B
To a suspension of 34b (0.25 g, 0.74 mmol) in acetic acid (20 mL) was added 2-(o-nitrophenyl)pyruvic acid (0.155 g, 0.74 mmol). The reaction mixture was stirred at RT for 4 h and then diluted with water (100 mL). The precipitated solid was filtered and dried. The crude mixture was purified on a RP-C18 FCC (100 g cartridge, flow rate = 40 mL/min) using a solvent system consisting of A, triethylammonium bicarbonate buffer (50 mM, pH = 8.5), and B, methanol. The purified isomers (E)-35b and (Z)-35b were precipitated from their respective pooled fractions following addition of 10% HCl (pH = 2) and separated by centrifugation. The pellets were washed consecutively with 5% (v/v) HCl and water and dried under vacuum to yield the following:
(E)-35b
Off-white fluffy solid; yield 0.080 g (20%); mp 236–237 °C. 1H NMR (DMSO-d6, 500 MHz) in ppm: δ 4.29 (s, 2H), 5.19 (s, 2H), 7.07 (d, 1H, J = 5 Hz), 7.49–7.51 (m, 1H), 7.62–7.65 (m, 1H), 7.84–7.87 (m, 1H), 7.94–7.98 (m, 1H), 8.05–8.07 (m, 1H), 12.20 (s, 1H). 13C NMR (DMSO-d6, 125 MHz) in ppm: δ 30.1, 51.1, 125.7, 126.5, 128.5, 128.7, 129.6, 131.5, 133.4, 134.4, 134.5, 135.2, 149.6, 165.8. Purity of 99.9% as determined by RP-HPLC, tR = 11.48 min (linear gradient system of 30–100% B in A for 26 min). HRMS(ESI) m/z calcd for C19H12Cl2N4O6S2 [M + H]+, 526.95753; found 526.95913.
(Z)-35b
Pale-green fluffy solid; yield 0.045 g (11%); mp 247–248 °C. 1H NMR (DMSO-d6, 400 MHz) in ppm: δ 4.18 (s, 2H), 5.13 (s, 2H), 7.51–7.57 (m, 2H), 7.70 (t, 1H, J = 8 Hz), 7.90–7.95 (m, 2H), 8.04 (d, 1H, J = 8 Hz), 12.70 (s, 1H). 13C NMR (DMSO-d6, 100 MHz) in ppm: δ 36.9, 51.0, 115.9, 125.2, 126.9, 128.5, 129.0, 132.1, 132.3, 133.5, 133.6, 134.2, 134.3, 135.8, 136.1, 141.4, 149.6, 164.7, 167.3. Purity of 99.9% as determined by RP-HPLC, tR = 12.41 min (linear gradient system of 30–100% B in A for 26 min). HRMS(ESI) m/z calcd for C19H12Cl2N4O6S2 [M + H]+, 526.95753; found 526.96467.
Synthesis of 3-Chloro-1-(2-hydroxy-4,5-dimethoxyphenyl)propan-1-one (37)
The borontrifluoride etherate (1.25 mL, 10 mmol) was added dropwise over 3 min to a mixture of 3,4-dimethoxyphenol (1.5 g, 10 mmol) and 2-chloropropanoyl chloride (1.8 mL, 20 mmol) and kept at 60 °C for 3 h. The reaction was quenched by pouring the mixture into a mixture of ice-cold water (30 mL) and dichloromethane (30 mL) and stirred overnight at RT. The organic phase was separated, dried with anhydrous Na2SO4, and concentrated under vacuum. The crude product was subjected to silica gel FCC employing hexane–ethyl acetate mixture (9:1, v/v) as eluent followed by recrystallization from dichloromethane to obtain 37.
37
Yellow crystals; Rf 0.85 (dichloromethane, silica gel); yield 1.0 g (44%); mp 133–135 °C. 1H NMR (CDCl3, 500 MHz) in ppm: δ 3.36 (t, 2H), 3.83 (s, 3H), 3.88 (s, 3H), 3.87–3.90 (m, 2H), 6.42 (s, 1H), 6.98 (s, 1H). 13C NMR (CDCl3, 125 MHz) in ppm: δ 38.7, 40.7, 56.4, 56.8, 100.8, 110.6, 111.3, 142.3, 157.3, 160.5, 200.1. Purity of 96.7% as determined by RP-HPLC, tR = 13.98 min (linear gradient system of 0–100% B in A for 26 min).
Synthesis of 6,7-Dimethoxy-2,3-dihydrochromene-4-one (43a)
A mixture of 37 (0.2 g, 0.8 mmol) and K2CO3 (0.56 g, 40 mmol) in ethanol (10 mL) was stirred for 20 h at RT. The reaction mixture was filtered, and the filtrate was concentrated under vacuum to afford a yellow residue. The residue was diluted with ethyl acetate (20 mL) and water (20 mL). The organic phase was washed with 5% (w/v) NaHCO3 solution (20 mL), dried over anhydrous Na2SO4, and concentrated under vacuum. The resultant residue was purified on a silica gel FCC using cyclohexane–ethyl acetate mixture (7:3, v/v) as eluent to yield.
43a
Off-white solid; Rf 0.35 (cyclohexane–ethyl acetate, 7:3 (v/v), silica gel); yield 0.07 g (42%); mp 124–127 °C. 1H NMR (CDCl3, 400 MHz) in ppm: δ 2.70–2.73 (m, 2H), 3.82 (s, 3H), 3.88 (s, 3H), 4.45–4.49 (m, 2H), 6.39 (s, 1H), 7.26 (s, 1H). 13C NMR (CDCl3, 100 MHz) in ppm: δ 37.4, 56.3, 56.4, 67.7, 100.2, 106.9, 113.7, 114.6, 156.2, 158.5, 190.7. Purity of 98.9% as determined by RP-HPLC, tR = 9.75 min (linear gradient system of 0–100% B in A for 26 min). ESI-MS calcd MW for C11H12O4, 208.21; found, m/z = 206.73 (M – H)−.
Synthesis of 3-Bromo-6,7-dimethoxy-2,3-dihydrochromen-4-one (44a)
Synthesis of 44a followed the identical procedure as described for 21a adjusted to 2.8 mmol scale for the 43a and pyridinium bromide perbromide.
44a
Off-white solid; Rf 0.41 (cyclohexane–ethyl acetate, 7:3 (v/v), silica gel); yield 0.6 g (75%); mp 154–156 °C. 1H NMR (CDCl3, 400 MHz) in ppm: δ 3.89 (s, 3H), 3.90 (s, 3H), 4.53–4.62 (m, 3H), 6.46 (s, 1H), 7.27 (s, 1H). 13C NMR (CDCl3, 100 MHz) in ppm: δ 45.4, 56.4, 56.6, 72.1, 100.1, 107.5, 111.1, 145.5, 157.1, 157.5, 184.1. Purity of 97.1% as determined by RP-HPLC, tR = 5.77 min (linear gradient system of 30–100% B in A for 26 min). ESI-MS calcd MW for C11H11BrO4, 287.11; found, m/z = 288.95 [M + H]+.
Synthesis of (E/Z)-2-(2-(7,8-Dimethoxy-4H-chromeno[4,3-d]thiazol-2-yl)hydrazono)-3-(2-nitrophenyl)propanoic Acid [(E/Z)-46a]
Step A: Synthesis of 1-(7,8-Dimethoxy-4H-chromeno[4,3-d]thiazol-2-yl)hydrazine (45a)
A solution of 3-bromo-6,7-dimethoxy-2,3-dihydrochromen-4-one (0.5 g, 1.7 mmol) and thiosemicarbazide (0.157 g, 1.7 mmol) in anhydrous dioxane (20 mL) was stirred at 80 °C for 2 d. The resulting yellow precipitate was filtered, washed with dioxane (10 mL), triturated in 2 M Na2CO3 solution (30 mL), and filtered, and washed thoroughly with water and dried to afford the crude 45a.
45a
Off-white solid; yield 0.34 g (72%). ESI-MS calcd MW for C12H13N3O3S, 279.31; found, m/z = 279.73 (M – H)−.
Step B
Synthesis of (E)-46a, and (Z)-46a followed the identical procedure as described for (E)-13a and (Z)-13a adjusted to 0.7 mmol scale for the 45a and 2-(o-nitrophenyl)pyruvic acid.
(E)-46a
Yellow powder; yield 0.055 g (17%); mp 202–204 °C. 1H NMR (DMSO-d6, 500 MHz) in ppm: δ 3.67 (s, 3H), 3.73 (s, 3H), 4.29 (s, 2H), 5.29 (s, 2H), 6.61 (s, 1H), 6.98 (s, 1H), 7.08 (d, 1H, J = 7.5 Hz), 7.49–7.52 (m, 1H), 7.63–7.66 (m, 1H), 8.06 (d, 1H, J = 8.5 Hz), 12.58 (s, 2H). 13C NMR (DMSO-d6, 125 MHz) in ppm: δ 29.8, 56.3, 56.4, 64.7, 102.2, 106.1, 125.7, 128.5, 129.5, 131.7, 134.5, 144.3, 147.9, 149.7, 149.8, 166.1. Purity of 99.0% as determined by RP-HPLC, tR = 9.07 min (linear gradient system of 30–100% B in A for 26 min). HRMS(ESI) m/z calcd for C21H18N4O7S [M + H]+, 471.08961; found 471.09633.
(Z)-46a
Yellow powder; yield 0.06 g (18%); mp 238–240 °C. 1H NMR (DMSO-d6, 500 MHz) in ppm: δ 3.67 (s, 6H), 4.16 (s, 2H), 5.20 (s, 2H), 6.57 (s, 1H), 7.02 (s, 1H), 7.50–7.55 (m, 2H), 7.67–7.69 (m, 1H), 8.03–8.05 (m, 1H), 12.78 (s, 1H). 13C NMR (DMSO-d6, 125 MHz) in ppm: δ 36.7, 56.3, 56.5, 64.6, 102.1, 106.4, 125.2, 128.9, 132.5, 133.6, 134.2, 144.3, 147.8, 149.7, 149.9, 164.7. Purity of 99.5% as determined by RP-HPLC, tR = 10.90 min (linear gradient system of 30–100% B in A for 26 min). HRMS(ESI) m/z calcd for C21H18N4O7S [M + H]+, 471.08961; found 471.09668.
Synthesis of 3-(2,3-Dimethoxyphenoxy)propanoic Acid (42)
The oxetan-2-one (1.8 g, 2.5 mmol) was added dropwise during 5 min to a stirred solution of 2,3-dimethoxyphenol (4.0 g, 25 mmol) in 0.625 M NaOH (40 mL), and the mixture was kept at 100 °C for 3 h. After cooling to RT, the reaction mixture was diluted with water (50 mL) followed by acidification with concd HCl (10 mL). The product was extracted into diethyl ether (2 × 100 mL), and the combined ethereal phases were then washed with 10% (w/v) NaHCO3 solution (100 mL). The separated aqueous phase was acidified with concd HCl to pH = 2 and kept at 4 °C overnight to afford 42.
42
Off-white solid; yield 2.5 g (40%); mp 104–106 °C. 1H NMR (CDCl3, 400 MHz) in ppm: δ 2.89 (t, 2H, J = 6.4 Hz), 3.81 (s, 3H), 3.85 (s, 3H), 4.29 (t, 2H, J = 6.4 Hz), 6.59 (d, 2H, J = 8.8 Hz), 6.94–6.97 (m, 1H). 13C NMR (CDCl3, 100 MHz) in ppm: δ 34.6, 56.3, 61.0, 64.6, 106.1, 107.4, 123.8, 138.9, 152.4, 153.8, 177.0. Purity of 93.5% as determined by RP-HPLC, tR = 9.81 min (linear gradient system of 0–100% B in A for 26 min). ESI-MS calcd MW for C11H14O5, 226.23; found, m/z = 224.99 (M – H)−.
Synthesis of 7,8-Dimethoxy-2,3-dihydrochromen-4-one (43b)
To a solution of 42 (1.1 g, 4.8 mmol) dissolved in dry benzene (40 mL) was added phosphorus pentoxide (6 g) and refluxed for 4 h and then cooled to RT. Benzene was decanted off, and the residue was triturated with benzene (2 × 10 mL) and collected with the previously decanted solvent. Ice-cold water (20 mL) was added dropwise to the residual slurry of phosphorus pentoxide, and the mixture was extracted with benzene (20 mL). The combined benzene fractions were washed successively with 10% (v/v) NaHCO3 (40 mL), 1N NaOH (40 mL), and water (40 mL). The organic phase was dried over anhydrous Na2SO4, filtered, and evaporated under vacuum to yield 43b.
43b
Pale-yellow solid; yield 0.8 g (79%); mp 102–104 °C. 1H NMR (CDCl3, 400 MHz) in ppm: δ 2.72–2.75 (m, 2H), 3.83 (s, 3H), 3.89 (s, 3H), 4.53–4.56 (m, 2H), 6.60 (d, 1H, J = 8.0 Hz), 7.64 (d, 1H, J = 8.0 Hz). 13C NMR (CDCl3, 100 MHz) in ppm: δ 37.7, 56.3, 61.2, 67.8, 105.7, 116.7, 123.3, 136.9, 155.9, 158.7, 190.8. Purity of 95.81% as determined by RP-HPLC, tR = 9.44 min (linear gradient system of 0–100% B in A for 26 min). ESI-MS calcd MW for C11H12O4, 208.21; found, m/z = 208.91 (M + H)+.
Synthesis of 3-Bromo-7,8-dimethoxy-2,3-dihydrochromen-4-one (44b)
Synthesis of 44b followed the identical procedure as described for 21a adjusted to 2.4 mmol scale for ketone 43b and pyridinium bromide perbromide.
44b
Pale-yellow solid; yield 0.33 g (48%); mp 118–120 °C. 1H NMR (CDCl3, 400 MHz) in ppm: δ 3.86 (s, 3H), 3.92 (s, 3H), 4.57–4.60 (m, 1H), 4.63–4.68 (m, 2H), 6.69 (d, 1H, J = 8.0 Hz), 7.69 (d, 1H, J = 8.0 Hz). 13C NMR (CDCl3, 100 MHz) in ppm: δ 45.4, 56.5, 61.4, 71.9, 106.8, 113.9, 124.5, 136.8, 154.8, 159.4, 184.4. Purity of 97.3% as determined by RP-HPLC, tR = 5.84 min (linear gradient system of 30–100% B in A for 26 min). ESI-MS calcd MW for C11H11BrO4, 287.11; found, m/z = 286.84 [M + H]+.
Synthesis of (E/Z)-2-(2-(6,7-Dimethoxy-4H-chromeno[4,3-d]thiazol-2-yl)hydrazono)-3-(2-nitrophenyl)propanoic Acid [(E/Z)-46b]
Step A: Synthesis of 1-(6,7-Dimethoxy-4H-chromeno[4,3-d]thiazol-2-yl)hydrazine, (45b)
A solution of 44b (0.4 g, 1.36 mmol) and thiosemicarbazide (0.158 g, 1.36 mmol) in anhydrous dioxane (20 mL) was stirred at 50 °C for 24 h, and the resulting precipitate was filtered, washed with dioxane (10 mL), and triturated with 2 M Na2CO3 (15 mL) to afford the crude 45b, which was used as such in the next step.
45b
White powder; yield 0.18 g (38%). ESI-MS calcd MW for C12H13N3O3S, 279.31; found, m/z = 279.90 (M + H)+.
Step B
Synthesis of (E)-46b and (Z)-46b followed the identical procedure as described for (E)-13a and (Z)-13a adjusted to 0.5 mmol scale for the 45b and 2-(o-nitrophenyl)pyruvic acid.
(E)-46b
Yellow solid; yield 0.08 g (34%); mp 258–260 °C. 1H NMR (DMSO-d6, 400 MHz) in ppm: δ 3.67 (s, 3H), 3.74 (s, 3H), 4.27 (s, 2H), 5.34 (s, 2H), 6.62 (d, 1H, J = 8 Hz), 7.06 (d, 1H, J = 8 Hz), 7.12 (d, 1H, J = 8 Hz), 7.49–7.51 (m, 1H), 7.63–7.65 (m, 1H), 8.05 (d, 1H, J = 8 Hz), 12.70 (s, 1H). 13C NMR (DMSO-d6, 100 MHz) in ppm: δ 29.8, 56.4, 60.9, 64.9, 125.6, 128.4, 131.7, 134.5, 137.9, 147.1, 148.5, 149.6, 166.1. Purity of 100% as determined by RP-HPLC, tR = 8.78 min (linear gradient system of 30–100% B in A for 26 min). HRMS(ESI) m/z calcd for C21H18N4O7S [M + H]+, 471.08961; found 471.09530.
(Z)-46b
Yellow solid; yield 0.03 g (13%); mp 239–241 °C. 1H NMR (DMSO-d6, 400 MHz) in ppm: δ 3.66 (s, 3H), 3.75 (s, 3H), 4.16 (s, 2H), 5.27 (s, 2H), 6.63 (d, 1H, J = 8.8 Hz), 7.16 (d, 1H, J = 8.8 Hz), 7.50–7.56 (m, 2H), 7.67–7.71 (m, 1H), 8.03–8.05 (m, 1H), 12.78 (bs, 1H). 13C NMR (DMSO-d6, 100 MHz) in ppm: δ 36.8, 56.4, 60.9, 64.8, 106.0, 117.6, 125.2, 128.9, 132.5, 133.6, 134.2, 137.8, 147.0, 149.6, 153.8, 164.7. Purity of 97.9% as determined by RP-HPLC, tR = 10.38 min (linear gradient system of 30–100% B in A for 26 min). HRMS(ESI) m/z calcd for C21H18N4O7S [M + H]+, 471.08961; found 471.09987.
Synthesis of 4-(3,4-Dimethoxyphenyl)-4-oxobutanoic Acid (39)
Veratrole, 38 (7.0 g, 50 mmol), was added dropwise over 30 min to a stirred suspension of succinic anhydride (6.0 g, 60 mmol) and AlCl3 (16.0 g, 120 mmol) in nitrobenzene (40 mL) that was maintained at 10 °C. The temperature was then slowly raised to RT, and the stirring continued for 12 h. The reaction mixture was then poured into ice-cold water and acidified with concd HCl to pH = 2. The formed precipitate was filtered off, redissolved in 1N NaOH (50 mL), and extracted with ether (100 mL). The precipitate formed upon the acidification of the aqueous phase to pH = 2 with concd HCl was filtered, washed with water, and dried to yield 39.
39
Pale-yellow solid; yield 5.4 g (46%); mp 169–171 °C. 1H NMR (CDCl3, 400 MHz) in ppm: δ 2.52–2.55 (m, 2H), 3.17–3.20 (m, 2H), 3.79 (s, 3H), 3.82 (s, 3H), 7.04 (d, J = 8.0 Hz, 1H), 7.43 (d, J = 4.0 Hz, 1H), 7.62–7.64 (m, 1H). 13C NMR (CDCl3, 100 MHz) in ppm: δ 28.6, 33.3, 56.1, 56.3, 110.7, 11.5, 123.1, 130.0, 149.1, 153.6, 174.5, 197.4. Purity of 92.7% as determined by RP-HPLC, tR = 9.22 min (linear gradient system of 0–100% B in A for 26 min). ESI-MS calcd MW for C12H14O5, 238.24; found, m/z = 238.80 (M + H)+.
Synthesis of 6,7-Dimethoxy-3,4-dihydronaphthalen-1(2H)-one (43c)
Step 1
Synthesis of 40 followed the identical procedure as described for 8a adjusted to 18 mmol scale for 39 and used in the next step without purification.
40
Brownish oil; yield 3.0 g (70%). Purity of 60.5% as determined by RP-HPLC, tR = 10.64 min (linear gradient system of 0–100% B in A for 26 min). ESI-MS calcd MW for C12H16O4, 224.25; found, m/z = 224.93 (M + H)+.
Step 2
40 (3.0 g, 13.3 mmol) was added to a melt of polyphosphoric acid (50 g) and stirred for 6 h at 90 °C. The reaction mixture was cooled to RT and diluted with ice-cold water (100 mL). It was extracted with ethyl acetate (100 mL), washed with a saturated solution of NaHCO3 (100 mL), dried over anhydrous Na2SO4, and evaporated under vacuum. The oily residue was crystallized from hexane–ethyl acetate (1:1) followed by trituration with diethyl ether to afford 43c.
43c
Colorless solid; yield 2.0 g (57%); mp 98–100 °C. 1H NMR (CDCl3, 400 MHz) in ppm: δ 2.03–2.08 (m, 2H), 2.50–2.54 (m, 2H), 2.81–2.84 (m, 2H), 3.84 (s, 3H), 3.87 (s, 3H), 6.61 (s, 1H), 7.45 (s, 1H). 13C NMR (CDCl3, 100 MHz) in ppm: δ 23.8, 29.6, 30.7, 56.1, 56.2, 108.6, 110.3, 125.9, 139.5, 148.0, 153.6, 197.3. Purity of 98.99% as determined by RP-HPLC, tR = 10.82 min (linear gradient system of 0–100% B in A for 26 min). ESI-MS calcd MW for C12H14O3, 206.24; found, m/z = 207.97 (M + H)+.
Synthesis of 2-Bromo-6,7-dimethoxy-3,4-dihydronaphthalen-1(2H)-one (44c)
Synthesis of 44c followed the identical procedure as described for 21a adjusted to 2.4 mmol scale for the 43c, and the crude was purified by silica gel FCC using a mixture of hexane–ethyl acetate (8:2, v/v).
44c
Off-white solid; yield 0.5 g (74%); mp 110–112 °C. 1H NMR (CDCl3, 400 MHz) in ppm: δ 2.39–2.49 (m, 2H), 2.75–2.81 (m, 1H), 3.18–3.24 (m, 1H), 3.86 (s, 3H), 3.90 (s, 3H), 4.64–4.65 (m, 1H), 6.64 (s, 1H), 7.47 (s, 1H). 13C NMR (CDCl3, 100 MHz) in ppm: δ 26.0, 32.4, 56.2, 56.3, 109.7, 110.2, 123.1, 138.3, 148.5, 154.4, 189.7. Purity of 97.0% as determined by RP-HPLC, tR = 13.24 min (linear gradient system of 0–100% B in A for 26 min). ESI-MS calcd MW for C12H13BrO3, 285.13; found, m/z = 287.97 (M + H)+.
Synthesis of (E/Z)-2-(2-(7,8-Dimethoxy-4,5-dihydronaphtho[1,2-d]thiazol-2-yl)hydrazono)-3-(2-nitrophenyl)propanoic Acid [(E/Z)-46c]
Step A: Synthesis of 1-(7,8-Dimethoxy-4,5-dihydronaphtho[1,2-d]thiazol-2-yl)hydrazine (45c)
A solution of 44c (0.4 g, 1.4 mmol) and thiosemicarbazide (0.127 g, 1.4 mmol) in anhydrous dioxane (20 mL) was stirred at 80 °C for 2 d. Cooling to RT afforded a yellow precipitate that was filtered, washed with dioxane (10 mL), and dried. The solid was triturated with a 2 M Na2CO3 (40 mL), filtered, washed thoroughly with water, and dried to yield crude 45c. 45c: Off-white powder, yield 0.25 g (64%). ESI-MS calcd MW for C13H15N3O2S, 277.34; found, m/z = 278.01 (M + H)+.
Step B
Synthesis of (E)-46c and (Z)-46c followed the identical procedure as described for (E)-13a and (Z)-13a adjusted to 0.5 mmol scale for the 45c and 2-(o-nitrophenyl)pyruvic acid.
(E)-46c
Yellow powder; yield 0.058 g (25%); mp 183–184 °C. 1H NMR (DMSO-d6, 500 MHz) in ppm: δ 2.77–2.80 (m, 2H), 2.86–2.89 (m, 2H), 3.67 (s, 3H), 3.70 (s, 3H), 4.27 (s, 2H), 6.86 (s, 1H), 7.05 (d, 1H, J = 8.0 Hz), 7.13 (s, 1H), 7.47–7.50 (m, 1H), 7.61–7.65 (m, 1H), 8.04 (d, 1H, J = 8.5 Hz). 13C NMR (DMSO-d6, 125 MHz) in ppm: δ 21.9, 28.4, 29.7, 56.0, 56.2, 106.7, 113.0, 125.6, 127.4, 128.4, 129.6, 131.9, 134.4, 148.0, 148.2, 149.7, 166.2. Purity of 97.6% as determined by RP-HPLC, tR = 10.00 min (linear gradient system of 30–100% B in A for 26 min). HRMS(ESI) m/z calcd for C22H20N4O6S [M + H]+, 469.11035; found 469.11588.
(Z)-46c
Yellow powder; yield 0.037 g (16%); mp 259–260 °C. 1H NMR (DMSO-d6, 500 MHz) in ppm: δ 2.72–2.74 (m, 2H), 2.82–2.85 (m, 2H), 3.73 (s, 6H), 4.16 (s, 2H), 6.84 (s, 1H), 7.15 (s, 1H), 7.49–7.56 (m, 2H), 7.67–7.71 (m, 1H), 8.03–8.05 (m, 1H). 13C NMR (DMSO-d6, 125 MHz) in ppm: δ 21.8, 28.3, 36.6, 56.2, 125.2, 127.4, 128.8, 132.7, 133.5, 134.1, 148.0, 148.4, 149.7, 164.6. Purity of 99.3% as determined by RP-HPLC, tR = 11.22 min (linear gradient system of 30–100% B in A for 26 min). HRMS(ESI) m/z calcd for C22H20N4O6S [M + H]+, 469.11035; found 469.11944.
Biology
Fluorescent Polarization Assay
The C-terminal fluorescein-labeled peptide KKQYDREFLLDFQFK was synthesized by Research Genetics. This sequence contains the Y(X)4LΦ motif and was optimized for solubility and binding to eIF4E. A truncated eIF4E containing a deletion of N-terminal 26 amino acids was expressed as a GST fusion protein (GST-DN26-eIF4E). For the screening assay, a solution containing approximately 0.5 mM GST-DN26-eIF4E, 20 nM labeled peptide, and 2 mM DTT in a buffer composed of 50 mM sodium phosphate and 50 mM potassium chloride at pH 6.5 was used. Measurements of FP and fluorescent anisotrophy (FA) were made in black 384-well plates (Corning) using a Perkin Elmer Wallac EnVision 2100 Multilabel Reader.
Antiproliferation Activity
The antiproliferation activity of rigidified mimetic of 1 were investigated on CRL-2813 cell lines and CRL-2351 cell lines by sulforhodamine B (SRB) assay. Adherent human breast cancer and melanoma cells (CRL-2351 and CRL-2813) were plated in 96-well plates and maintained for 5 days in the presence of 0.54–20 μM compounds, and cell proliferation was measured by the SRB assay. Briefly, cells were fixed in 10% cold trichloroacetic acid at 4 °C overnight, extensively washed with double-distilled H2O, and air-dried. Plates were then incubated with 0.057% SRB in 1% acetic acid for 1 h at RT, washed with 1% acetic acid to remove the unbound dye, and air-dried. The bound dye was solubilized by addition of 10 mM Tris (pH 10), and the absorbance was determined in a Thermo Scientific Multiskan FC plate reader at 510 nm. The data calculations were carried out as described.64
Assessment of the Disruption of eIF4E/eIF4G Interaction by the m7GTP Pull-Down Assay
Melanoma CRL-2813 cell lines were incubated for 3 h in the presence of the 30 μM of each of the compounds and 0.4% DMSO, harvested by centrifugation, and lysed by multiple freeze–thaw cycles. Subsequently, the lysate was incubated with m7GTP–Sepharose beads for 1 h at 4 °C to pull-down eIF4E. The beads were separated, extensively washed, and the bound eIF4E eluted with free m7GTP, the supernatant was separated by SDS-PAGE, and Western blotted by polyclonal antibodies against 4E-BP1 and monoclonal antibodies against eIF4E and eIF4G.
Western Blot Analysis
Western blot analysis was carried out as described.65 Anti-eIF4E antibody (catalogue no. 610269) was from BD Transduction Laboratories (San Jose, CA). anti-4EBP-1 (catalogue no. 9452S) and anti-eIF4G (catalogue no. 2498S) antibodies were purchased from Cell Signaling Technology, Inc. (Danvers, MA).
Acknowledgments
This work was sponsored in part by NCI grant no. 5R01CA121357 to M.C. and Egenix, Inc.
Glossary
Abbreviations Used
- DTT
dithiothreitol
- SDS-PAGE
sodium dodecyl sulfate-polyacrylamide gel electrophoresis
- Tris
tris(hydroxymethyl)aminomethane)
- eIF4E
eukaryotic translation initiation factor 4E
- FP
fluorescence polarization
- PBPB
pyridinium bromide perbromide
- FCC
flash column chromatography
Supporting Information Available
Characterization data and NMR spectra of all new compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Sonenberg N.; Hinnebusch A. G. Regulation of translation initiation in eukaryotes: mechanisms and biological targets. Cell 2009, 136, 731–745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Rubeis S.; Bagni C. Regulation of Molecular Pathways in the Fragile X Syndrome: Insights into Autism Spectrum Disorders. J. Neurodev. Disord. 2011, 3, 257–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avdulov S.; Li S.; Michalek V.; Burrichter D.; Peterson M.; Perlman D. M.; Manivel J. C.; Sonenberg N.; Yee D.; Bitterman P. B.; Polunovsky V. A. Activation of translation complex eIF4F is essential for the genesis and maintenance of the malignant phenotype in human mammary epithelial cells. Cancer Cell 2004, 5, 553–563. [DOI] [PubMed] [Google Scholar]
- Gebauer F.; Hentze M. W. Molecular mechanisms of translational control. Nature Rev. Mol. Cell Biol. 2004, 5, 827–835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pestova T. V.; Lorsch J. R.; Hellen C. U. T.. The Mechanism of Translation Initiation in Eukaryotes; Cold Spring Harbor Laboratory Press: Woodbury, NY, 2007; Vol. 48. [Google Scholar]
- Poulin F.; Sonenberg N.. Mechanism of Translation Initiation in Eukaryotes; Landes Bioscience: Austin, TX, 2003. [Google Scholar]
- Sonenberg N. eIF4E, the mRNA cap-binding protein: from basic discovery to translational research. Biochem. Cell. Biol. 2008, 86, 178–183. [DOI] [PubMed] [Google Scholar]
- De Benedetti A.; Graff J. R. eIF-4E expression and its role in malignancies and metastases. Oncogene 2004, 23, 3189–3199. [DOI] [PubMed] [Google Scholar]
- Goodfellow I. G.; Roberts L. O. Eukaryotic initiation factor 4E. Int. J. Biochem. Cell Biol. 2008, 40, 2675–2680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raught B.; Gingras A. C. eIF4E activity is regulated at multiple levels. Int. J. Biochem. Cell Biol. 1999, 31, 43–57. [DOI] [PubMed] [Google Scholar]
- Gingras A. C.; Raught B.; Sonenberg N. eIF4 initiation factors: effectors of mRNA recruitment to ribosomes and regulators of translation. Annu. Rev. Biochem. 1999, 68, 913–963. [DOI] [PubMed] [Google Scholar]
- Koromilas A. E.; Lazaris-Karatzas A.; Sonenberg N. mRNAs containing extensive secondary structure in their 5′ non-coding region translate efficiently in cells overexpressing initiation factor eIF-4E. EMBO J. 1992, 11, 4153–4158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graff J. R.; Konicek B. W.; Carter J. H.; Marcusson E. G. Targeting the eukaryotic translation initiation factor 4E for cancer therapy. Cancer Res. 2008, 68, 631–634. [DOI] [PubMed] [Google Scholar]
- Gkogkas C. G.; Khoutorsky A.; Ran I.; Rampakakis E.; Nevarko T.; Weatherill D. B.; Vasuta C.; Yee S.; Truitt M.; Dallaire P.; Major F.; Lasko P.; Ruggero D.; Nader K.; Lacaille J. C.; Sonenberg N. Autism-related deficits via dysregulated eIF4E-dependent translational control. Nature 2013, 493, 371–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Napoli I.; Mercaldo V.; Boyl P. P.; Eleuteri B.; Zalfa F.; De Rubeis S.; Di Marino D.; Mohr E.; Massimi M.; Falconi M.; Witke W.; Costa-Mattioli M.; Sonenberg N.; Achsel T.; Bagni C. The fragile X syndrome protein represses activity-dependent translation through CYFIP1, a new 4E-BP. Cell 2007, 134, 1042–1054. [DOI] [PubMed] [Google Scholar]
- Tsai P. T.; Hull C.; Chu Y.; Greene-Colozzi E.; Sadowski A. R.; Leech J. M.; Steinberg J.; Crawley J. N.; Regehr W. G.; Sahin M. Autistic-like behaviour and cerebellar dysfunction in Purkinje cell Tsc1 mutant mice. Nature 2012, 488, 647–651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia Y.; Polunovsky V. A.; Bitterman P. B.; Wagner C. R. Cap-dependent translation initiation factor eIF4E: an emerging anticancer drug target. Med. Res. Rev. 2012, 32, 786–814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rousseau D.; Gingras A. C.; Pause A.; Sonenberg N. The eIF4E-binding proteins 1 and 2 are negative regulators of cell growth. Oncogene 1996, 13, 2415–2420. [PubMed] [Google Scholar]
- Meric F.; Hunt K. K. Translation initiation in cancer: a novel target for therapy. Mol. Cancer Ther. 2002, 1, 971–979. [PubMed] [Google Scholar]
- Aktas B. H.; Halperin J. A.; wagner G.; Chorev M.. Inhibition of Translation Initiation as a Novel Paradigm for Cancer Therapy. Elsevier Inc Academic Press: New York, 2011. [Google Scholar]
- Kong J.; Lasko P. Translational control in cellular and developmental processes. Nature Rev. Genet. 2012, 13, 383–394. [DOI] [PubMed] [Google Scholar]
- Moerke N. J.; Aktas H.; Chen H.; Cantel S.; Reibarkh M. Y.; Fahmy A.; Gross J. D.; Degterev A.; Yuan J.; Chorev M.; Halperin J. A.; Wagner G. Small-molecule inhibition of the interaction between the translation initiation factors eIF4E and eIF4G. Cell 2007, 128, 257–267. [DOI] [PubMed] [Google Scholar]
- Tamburini J.; Green A. S.; Bardet V.; Chapuis N.; Park S.; Willems L.; Uzunov M.; Ifrah N.; Dreyfus F.; Lacombe C.; Mayeux P.; Bouscary D. Protein synthesis is resistant to rapamycin and constitutes a promising therapeutic target in acute myeloid leukemia. Blood 2009, 114, 1618–1627. [DOI] [PubMed] [Google Scholar]
- Chen L.; Aktas B. H.; Wang Y.; He X.; Sahoo R.; Zhang N.; Denoyelle S.; Kabha E.; Yang H.; Freedman R. Tumor suppression by small molecule inhibitors of translation initiation. Oncotarget 2012, 3, 869–881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoeffer C. A.; Cowansage K. K.; Arnold E. C.; Banko J. L.; Moerke N. J.; Rodriguez R.; Schimdt E. K.; Klosi E.; Chorev M.; Lloyd R. E.; Pierre P.; Wagner G.; LeDoux J. E.; Klann E. Inhibition of the interactions between eukaryotic initiation factors 4E and 4G impairs long-term associative memory consolidation but not reconsolidation. Proc. Natl. Acad. Sci. U. S. A. 2011, 108, 3383–3388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santini E.; Huynh T. N.; MacAskill A. F.; Carter A. G.; Pierre P.; Ruggero D.; Kaphzan H.; Klann E. Exaggerated translation causes synaptic and behavioural aberrations associated with autism. Nature 2013, 493, 411–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMahon R.; Zaborowska I.; Walsh D. Noncytotoxic Inhibition of Viral Infection through eIF4F-Independent Suppression of Translation by 4EGI-1. J. Virol. 2011, 85, 853–864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabha E.; Wang Y.; Chen L.; Zhang N.; Rodriguez R.; Papadopoulos P.; Wagner G.; Aktas B. H.; Halperin J. A.; Chorev M.. Identification of the pharmacophore of 4EGI-1, an inhibitor of translation initiation with anti-cancer activity. In 240th ACS National Meeting & Exposition, Boston, MA, United States, August 22–26; American Chemical Society: Boston, 2010; pp MEDI-28.
- Takrouri K.; Chen T.; Papadopoulos E.; Sahoo R.; Kabha E.; Chen H.; Cantel S.; Wagner G.; Halperin J. A.; Aktas B. H.; Chorev M. Structure–activity relationship study of 4EGI-1, small molecule eIF4E/eIF4G protein–protein interaction inhibitors. Eur. J. Med. Chem. 2014, 77, 361–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caprathe B. W.; Jaen J. C.; Wise L. D.; Heffner T. G.; Pugsley T. A.; Meltzer L. T.; Parvez M. Dopamine autoreceptor agonists as potential antipsychotics. 3.6-Propyl-4,5,5a,6,7,8-hexahydrothiazolo[4,5-f]quinolin-2-amine. J. Med. Chem. 1991, 34, 2736–2746. [DOI] [PubMed] [Google Scholar]
- Hu Q.; Negri M.; Jahn-Hoffmann K.; Zhuang Y.; Olgen S.; Bartels M.; Müller-Vieira U.; Lauterbach T.; Hartmann R. W. Synthesis, biological evaluation, and molecular modeling studies of methylene imidazole substituted biaryls as inhibitors of human 17α-hydroxylase-17,20-lyase (CYP17)—Part II: Core rigidification and influence of substituents at the methylene bridge. Bioorg. Med. Chem. 2008, 16, 7715–7727. [DOI] [PubMed] [Google Scholar]
- Lucas S.; Negri M.; Heim R.; Zimmer C.; Hartmann R. W. Fine-tuning the selectivity of aldosterone synthase inhibitors: structure–activity and structure–selectivity insights from studies of heteroaryl substituted 1,2,5,6-tetrahydropyrrolo[3,2,1-ij]quinolin-4-one derivatives. J. Med. Chem. 2011, 54, 2307–2319. [DOI] [PubMed] [Google Scholar]
- Jenkins P. R.; Wilson J.; Emmerson D.; Garcia M. D.; Smith M. R.; Gray S. J.; Britton R. G.; Mahale S.; Chaudhuri B. Design, synthesis and biological evaluation of new tryptamine and tetrahydro-beta-carboline-based selective inhibitors of CDK4. Bioorg. Med. Chem. 2008, 16, 7728–773939. [DOI] [PubMed] [Google Scholar]
- Sander K.; Kottke T.; Tanrikulu Y.; Proschak E.; Weizel L.; Schneider E. H.; Seifert R.; Schneider G.; Stark H. 2,4-Diaminopyrimidines as histamine H4 receptor ligands—scaffold optimization and pharmacological characterization. Bioorg. Med. Chem. 2009, 17, 7186–7196. [DOI] [PubMed] [Google Scholar]
- Chowdhury S.; Chafeev M.; Liu S.; Sun J.; Raina V.; Chui R.; Young W.; Kwan R.; Fu J.; Cadieux J. A. Discovery of XEN907, a spirooxindole blocker of NaV1.7 for the treatment of pain. Bioorg. Med. Chem. Lett. 2011, 21, 3676–3681. [DOI] [PubMed] [Google Scholar]
- Spadaro A.; Negri M.; Marchais-Oberwinkler S.; Bey E.; Frotscher M. Hydroxybenzothiazoles as new nonsteroidal inhibitors of 17beta-hydroxysteroid dehydrogenase type 1 (17beta-HSD1). PloS One 2012, 7, e29252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perrone R.; Berardi F.; Colabufo N. A.; Tortorella V.; Lograno M. D.; Daniele E.; Govoni S. Conformationally restricted thiazole derivatives as novel class of 5-HT3 receptor ligands. Farmaco 1995, 50, 77–82. [Google Scholar]
- Chordia M. D.; Murphree L. J.; Macdonald T. L.; Linden J.; Olsson R. A. 2-Aminothiazoles: a new class of agonist allosteric enhancers of A1 adenosine receptors. Bioorg. Med. Chem. Lett. 2002, 12, 1563–1566. [DOI] [PubMed] [Google Scholar]
- Malviya N. J.; Kulkarni A. M.; Jaimini D.; Jadhav B. L.; Karnik A. V. Chemoselective synthesis of heterocyclic derivatives of 18-norequilenine, 16-substituted 12H-11-oxa-15-aza-17-thiacyclopenta[a]phenanthrenene and their in vitro evaluation of antibacterial and antifungal activity. Indian J. Chem., Sect. B 2006, 45B, 1499–1503. [Google Scholar]
- Khazi I. A. M.; Gadad A. K.; Mahajanshetti C. S. Synthesis and biological activity of some 2-amino/arylamino-4H-(1)benzothiopyrano[4,3-d]thiazoles. Indian J. Heterocycl. Chem. 1995, 4, 243–248. [Google Scholar]
- Kikuchi T.; Nishio M.; Ito T.; Sekizawa Y.. Anesthetics for black carp. Japanese Patent JP 46024259, 1971.
- Guo C.; Fang L.; Liu Y.; Su X.; Li C.; Sun L.; Luo W.; Liang L.; Huang Y.. Preparation of thiochromanone (thio)semicarbazone derivatives as antifungal agents. Canadian Patent CN 101434595, 2009.
- Wagner G.; Chorev M.; Moerke N. J.; Aktas H.; Halperin J. A.. Preparation of cyclic inhibitors of eukaryotic translation initiation factor interaction and use in treating cancer. World Patent WO 200678942 A2, July27, 2006.
- Quallich G. J.; Williams M. T.; Friedmann R. C. Friedel–Crafts synthesis of 4-(3,4-dichlorophenyl)-3,4-dihydro-1(2H)-naphthalenone, a key intermediate in the preparation of the antidepressant sertraline. J. Org. Chem. 1990, 55, 4971–4973. [Google Scholar]
- Sato Y.; Mizobuchi S.; Tanabe K.; Inoue H.. 2-Bromo-1-tetralone derivatives. European Patent EP 125695November21, 1984.
- Balsamo A.; Breschi M.; Giannaccinia G.; Lapuccil A.; Lucacchiniz A.; Macchial B.; Maneral C.; Martinottiz C. M. E.; Nencettil S.; Rossellol A.; Scatizziz R. Synthesis and P-adrenergic properties of tetrahydronaphthalene analogs of dichloroisoproterenol. Eur. J. Med. Chem. 1993, 28, 735–741. [Google Scholar]
- House H. O.; McCaully R. J. Polyphosphoric Acid-Catalyzed Reaction of Anisole with γ-Butyrolactone. J. Org. Chem. 1959, 24, 725–726. [Google Scholar]
- Baker B. R.; Hopkins S. E. Irreversible enzyme inhibitors. CLXVII. Thymidine phosphorylase. 10. On the nature and dimensions of the hydrophobic bonding region. A. J. Med. Chem. 1970, 13, 87–89. [DOI] [PubMed] [Google Scholar]
- Arndt F.; Franke W.; Klose W.; Lorenz J.; Schwarz K. Synthesen von Thiazolo- und [1,3]Thiazino[1,2,4]triazinonen. Liebigs Ann. Chem. 1984, 1302–1307. [Google Scholar]
- E.I. Du Pont de Nemours and Co., Novel substituted chromanone oxides and their preparations. UK Patent GB 1265212 (A) 19720301, 1972; ; Chem. Abstr. 1972, 76, 140531t. [Google Scholar]
- Eaton P. E.; Carlson G. R.; Lee J. T. Phosphorus pentoxide-methanesulfonic acid. Convenient alternative to polyphosphoric acid. J. Org. Chem. 1973, 38, 4071–4073. [Google Scholar]
- Habermann J.; Ley S. V.; Scicinski J. J.; Scott J. S.; Smits R.; Thomas A. W. Clean synthesis of .alpha.-bromo ketones and their utilisation in the synthesis of 2-alkoxy-2,3-dihydro-2-aryl-1,4-benzodioxanes, 2-amino-4-aryl-1,3-thiazoles and piperidino-2-amino-1,3-thiazoles using polymer-supported reagents. J. Chem. Soc., Perkin Trans. 1 1999, 17, 2425–2427. [Google Scholar]
- Sarges R.Hydantoin derivatives as therapeutic agents. U.S. Patent US 4147795, April3, 1979.
- Patonay T.; Juhasz-Toth E.; Benyei A. Base-induced coupling of a-azido ketones with aldehydes—an easy and efficient route to trifunctionalized synthons 2-azido-3-hydroxy ketones, 2-acylaziridines, and 2-acylspiroaziridines. Eur. J. Org. Chem. 2002, 2, 285–295. [Google Scholar]
- MacKenzie N. E.; Thomson R. H. Ring contractions of thiochroman-4-ones and thiochromen-4-ones. J. Chem. Soc., Perkin Trans. 1 1982, 2, 395–402. [Google Scholar]
- Birch H. F.; Robertson A.; Subramaniam T. S. Synthesis of rotenone and its derivatives. X. 6,7-Dimethoxychroman-4-one. J. Chem. Soc. 1936, 1832–1834. [Google Scholar]
- Mahapatra T.; Jana N.; Nanda S. Chemoenzymatic synthesis and resolution of compounds containing a quaternary stereocenters adjacent to a carbonyl group. Tetrahedron: Asymmetry 2008, 19, 1224–1232. [Google Scholar]
- Tanada Y.; Mori K. Synthesis and absolute configuration of (−)-neuchromenin, a neurotrophic metabolite of Eupenicillium javanicum var. meloforme, and its enantiomer. Eur. J. Org. Chem. 2001, 10, 1963–1966. [Google Scholar]
- Pfeiffer P.; Haack E.; Willems J. Brasilin and hematoxylin question. VI. Synthesis of tetramethylanhydrohematoxylin. Ber. Dtsch. Chem. Ges., Abt. B: Abhandlungen 1928, 61B, 294–299. [Google Scholar]
- Sugihara H.; Sanno Y. Stereoselective synthesis of cis- and trans-3-amino-4-chromanols. Chem. Pharm. Bull. 1977, 25, 859–866. [DOI] [PubMed] [Google Scholar]
- Bandaranayake W. M.; Riggs N. V. Anomalous acetoxylation of aromatic nuclei: some structural requirements in the substrate. Aust. J. Chem. 1981, 34, 115–129. [Google Scholar]
- Paraiso K. H. T.; Xiang Y.; Rebecca V. W.; Abel E. V.; Chen Y. A.; Munko A. C.; Wood E.; Fedorenko I. V.; Sondak V. K.; Anderson A. R. A.; Ribas A.; Dalla Palma M.; Nathanson K. L.; Koomen J. M.; Messina J. L.; Smalley K. S. M. PTEN Loss Confers BRAF Inhibitor Resistance to Melanoma Cells through the Suppression of BIM Expression. Cancer Res. 2011, 71, 2750–2760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palakurthi S. S.; Aktas H.; Grubissich L. M.; Mortensen R. M.; Halperin J. A. Anticancer effects of thiazolidinediones are independent of peroxisome proliferator-activated receptor gamma and mediated by inhibition of translation initiation. Cancer Res. 2001, 61, 6213–6218. [PubMed] [Google Scholar]
- Ziegeler G.; Ming J.; Koseki J. C.; Sevinc S.; Chen T.; Ergun S.; Qin X.; Aktas B. H. Embryonic lethal abnormal vision-like HuR-dependent mRNA stability regulates post-transcriptional expression of cyclin-dependent kinase inhibitor p27Kip1. J. Biol. Chem. 2010, 285, 15408–15419. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.