

Abstract

Translocator protein (18 kDa), known as TSPO, is a recognized biomarker of neuroinflammation. Radioligands with PET accurately quantify TSPO in neuroinflammatory conditions. However, the existence of three human TSPO genotypes that show differential affinity to almost all useful TSPO PET radioligands hampers such studies. There is an unmet need for genotype-insensitive, high-affinity, and moderately lipophilic TSPO ligands that may serve as leads for PET radioligand development. To address this need, we varied the known high-affinity TSPO ligand (l)-N,N-diethyl-2-methyl-3-(2-phenylquinolin-4-yl)propanamide in its aryl scaffold, side chain tether, and pendant substituted amido group while retaining an N-methyl group as a site for labeling with carbon-11. From this effort, oxygen-tethered N-methyl-aryloxypropanamides emerged as new high-affinity TSPO ligands with attenuated lipophilicity, including one example with attractive properties for PET radioligand development, namely N-methyl-N-phenyl-2-{[2-(pyridin-2-yl)quinolin-4-yl]oxy}propanamide (22a; rat Ki = 0.10 nM; human TSPO genotypes Ki = 1.4 nM; clogD = 4.18).
Introduction
Translocator protein 18 kDa (TSPO),1 formerly known as the peripheral benzodiazepine receptor,2 is located predominantly at the mitochondrial membrane3 in association4 with a voltage-dependent anion channel and an adenine nucleotide transporter. TSPO is present in several major organs, and is particularly dense in adrenal gland, heart, kidney, and testis.3 Low amounts are present in normal human brain,5 primarily in microglia where TSPO plays a crucial role in membrane biogenesis,6 and in steroid7 and heme8 biosynthesis. Activated microglia upregulate TSPO in instances of neuronal damage9−11 as seen in many neurological disorders including cerebral ischemia,12 Alzheimer’s disease,13,14 Parkinson’s disease,15 and multiple sclerosis.16 TSPO can therefore serve as an important biomarker for neuroinflammation.17
[11C]PK 11195 ([11C]1), first as racemate18 and then as the higher affinity (R)-enantiomer ([11C](R)-1),19 has long been used to detect human TSPO in vivo with PET.10,11 However, accurate quantification of TSPO density with [11C](R)-1 is confounded by limited brain uptake,20 a low ratio of specific to nonspecific binding,21 and an unfavorable metabolic profile.22 In view of these deficiencies, new TSPO radioligands have been developed from other structural classes with superior imaging characteristics (Chart 1).23−25 These include, for example, [11C]PBR28 ([11C]2), [11C]DAA1106 ([11C]3), [11C]DPA713 ([11C]4), [18F]FBR ([18F]5), PBR111 ([11C]6), and [18F]FEPPA ([18F]7).
Chart 1. Structures of Some PET TSPO Radioligands.

With the advent of the more sensitive radioligand, [11C]2,26 heterogeneity in human TSPO binding to PET radioligands has been discovered. Thus, [11C]2 fails to image TSPO in some human subjects.27−29 Furthermore, in a study of deceased individuals diagnosed with multiple sclerosis, 46% had brain TSPO showing high affinity (Ki ∼ 4 nM) for 2, 23% low affinity (Ki ∼ 200 nM), and 31% intermediate affinity.30 This heterogeneity in binding affinity was found to derive from a genetic polymorphism among individuals of European ancestry, namely an Ala147Thr31,32 mutation in TSPO. Three populations exist: those homozygous for Ala147, homozygous for Thr147, and heterozygous for Ala147/Thr147, now dubbed high-affinity binders (HABs), low-affinity binders (LABs), and mixed-affinity (MABs), respectively. Several other second-generation TSPO ligands are also genotype-sensitive to different extents.33−36 PET measurements of TSPO density in studies of neuroinflammation assume that radioligand binding affinity is the same in all subjects because the output measure in PET experiments is usually a function of binding potential (Bmax/KD), the product of TSPO density (Bmax) to radioligand affinity (1/KD). Therefore, PET radioligands with heterogeneous binding affinity may impair data interpretation, especially comparisons of binding potentials for patient populations with those of normal subjects, unless all subjects are characterized for TSPO genotype.37 However, genotyping is resource demanding and would be unnecessary if a genotype-insensitive TSPO radioligand could be employed.
Pharmuka Laboratories, who introduced the prototypical TSPO ligand 1, later reported N,N-diethyl-2-methyl-3-(2-phenylquinolin-4-yl)propanamide (±Q; 10a) as a potent inhibitor of [3H]1 in rat brain cortex (IC50 = 13.7 nM).38 TSPO strongly bound the l-enantiomer (−)-10a (PK 14067; IC50 = 5.4 nM), and not the d-enantiomer, (+)-10a (PK 14068; IC50 = 4000 nM). The binding affinity of (−)-10a was also found to be high in human cerebral cortex (Ki = 44 nM)5 although clearly much lower than in rat. Thus, the (2-arylquinolinyl-4-yl)propanamide (−)-10a represents a unique structural class of TSPO ligand that has so far been neglected for PET radioligand development. In this study, we explored this structural class for potential to generate high-affinity, acceptably lipophilic and human genotype-insensitive TSPO ligands to serve as leads for PET radioligand development.
Results and Discussion
Successful PET radioligands for imaging proteins in brain are required to display a wide array of properties.39−41 Among these properties are (i) high affinity and selectivity for the target protein, (ii) low molecular weight and intermediate polar surface area for blood–brain barrier penetration, (iii) moderate lipophilicity for adequate brain entry in the absence of excessive nonspecific binding, and (iv) amenability to labeling with a positron-emitter. In addition, PET radioligands for imaging TSPO in humans should ideally be insensitive to genotype. This study aimed to develop TSPO ligands as leads with a desirable combination of properties for PET radioligand development. Ligands were developed by modifying the 2-(arylquinolinyl-4-yl)propanamide 10a and initially assessed for binding affinity toward rat TSPO. Lipophilicities (clogD) were estimated by computation. The lipophilicity cost for high ligand affinity may be indexed as a lipophilicity efficiency parameter (LipE), defined42−44 as ligand pIC50 (or pKi) minus clogD. We sought ligands with improved LipE scores in addition to other desirable properties. Several ligands that we found to have appealing properties were also assayed against human HAB and LAB TSPOs to assess genotype sensitivity. Many new high-affinity TSPO ligands emerged from this effort and a few of these are promising new leads to PET radioligands.
Chemistry
Ligands were synthesized in one of three general ways, depending on the tether X in the general structure (Figure 1).
Figure 1.

Generalized structure of compounds tested in this study.
For ligands with X = CH2 (10a–w), a propargyl aniline (9a–t), prepared in situ by copper(I)-catalyzed addition of a terminal butynamide to an aldimine, was subjected to intramolecular cyclization to the dihydroquinoline. A second equivalent of aldimine (or adventitious oxygen) enabled oxidation of the dihydroquinoline to the desired quinoline, with the entire sequence conducted in one pot (method C).45 The requisite butynamides were prepared either by PyBroP-mediated amidation (9a–9k; method A)46 or by α-alkylation of amides with propargyl bromide (9a, 9l–9t; method B)47 (Scheme 1).
Scheme 1. Synthesis of Ligands 10a–w.

Reagents and conditions: (i) PyBroP, DIPA, DCM, rt; (ii) propargyl bromide, LDA, THF, −78 °C; (iii) 10% CuCl/AgOTf, DCA, 100 °C.
For a ligand analogous to 10m (X = CH2) with X = S (12), the 2-mercaptoamide (11) was prepared by treatment of thiolactic acid with N-methylaniline,48 followed by addition of the product to 4-chloro-2-phenylquinoline in the presence of base49 (Scheme 2).
Scheme 2. Synthesis of S-Tethered Ligand 12.

Reagents and conditions: (i) PhNHMe, 190 °C; (ii) t-BuOK, t-BuOH, DMF, 135 °C.
A similar reaction with palladium catalysis50 was used to prepare the analogous ligand with X = NH (14) (Scheme 3). The required amide 13 was readily prepared from 2-bromo-N-methyl-N-phenylpropanamide by conversion into the azide followed by reduction.51
Scheme 3. Synthesis of N-Tethered Ligand 14.

Reagents and conditions: (i) NaN3, DMF, 70 °C; (ii) Ph3P, H2O, THF, rt; (iii) HCl(g), toluene, rt; (iv) 4% Pd(OAc)2, 8% DPEPhos, K3PO4, dioxane, 85 °C.
For the analogous ligand with X = O (15a), 2-phenyl-4-quinolone was alkylated with 2-bromo-N-methyl-N-phenylpropanamide in the presence of base52 (Scheme 4). Two truncated versions of 15a in which either the phenyl group (15b) or the benzo fusion was absent (15c), were made similarly (Scheme 4). A series of 1,x-naphthyridine analogues of 15a (x = 5–8; 19a–d) and a quinazoline analogue (x = 3; 19e) were also prepared similarly (Scheme 5).
Scheme 4. Syntheses of O-Tethered Ligands 15a–c.

Reagents and conditions: (i) Cs2CO3, acetone, rt.
Scheme 5. Syntheses of Naphthyridine, Quinazoline, and 2-Pyridylquinoline Analogues of 15a.

Reagents and conditions: (i) PhCOCl, TEA, DCM, DMAP, rt; (ii) BuLi, THF, −78 °C then N-methoxy-N-methylacetamide, −30 °C; (iii) for 17c, PhCO2H, PyBroP, DIPA, DMAP, DCM, rt; (iv) NaOH, dioxane, 110 °C; (v) for 18e, NaOH, DMSO, 110 °C; (vi) for 19a–e, Cs2CO3, acetone, rt; (vii) PyBroP, DIPA, DCM, rt; (viii) NaOH, dioxane, 110 °C; (ix) for 22a, K2CO3, MeCN, 50 °C; (x) Cs2CO3, acetone, rt.
The o-benzamidoacetopyridines 17a,b,d were made by acylation of o-benzamidobromopyridines (16a,b,d) according to Weinreb’s method (Scheme 5),53 whereas 17c was made by benzoylation of 4-acetyl-3-aminopyridine (Scheme 5). 17a–d were then subjected to Camps cyclization54,55 to give the respective 1,x-naphthyridones (18a–d). The quinazoline 18e was prepared by hydrolysis of 4-chloro-2-phenylquinazoline. The naphthyridones and quinazolinone (18a–e) were then alkylated in the same manner (method D) used to make 15a to give the ligands 19a–e (Scheme 5). The 2-pyridinylquinolines 22a–c were synthesized similarly to round out the series of regioisomeric nitrogen-substituted ligands (Scheme 5).
Determination of Absolute Configuration of (−)-10a
Initially, the absolute configuration of (−)-10a was unknown. We considered that this information could be valuable in subsequent TSPO ligand design. Therefore, compound 10a was resolved by chiral HPLC and the optical rotations of the separate enantiomers were measured. We confirmed that (−)-10a was the higher affinity enantiomer (Table 1). (−)-10a is a thick syrup, and attempts to crystallize the picrate salt failed. This precluded X-ray crystallography for determination of absolute configuration, and so we resorted to VCD.56 The solvent-corrected IR and VCD spectra for (−)-10a were obtained (Supporting Information Figure S1). A conformational search of the R-enantiomer at the molecular mechanics level was performed on the entire molecule followed by optimizations using a B3LYP “functional” on a 6-31G(d) basis set with Gaussian 09. These calculations revealed 12 conformers that were all within 1.5 kcal/mol of the lowest-energy conformer (Supporting Information Figure S2). VCD and IR spectra were calculated on the optimized geometries of these conformers. Their Boltzmann summation was compared with the observed spectra of (−)-10a (Supporting Information Figure S3). Given the agreement between calculation and experiment, the absolute configuration of (−)-10a was assigned to be R.
Table 1. TSPO Ligands Based on 10a: Dependence of Binding Affinity, Lipophilicity, and Genotype Sensitivity on Side Chain Alkyl Substituents.
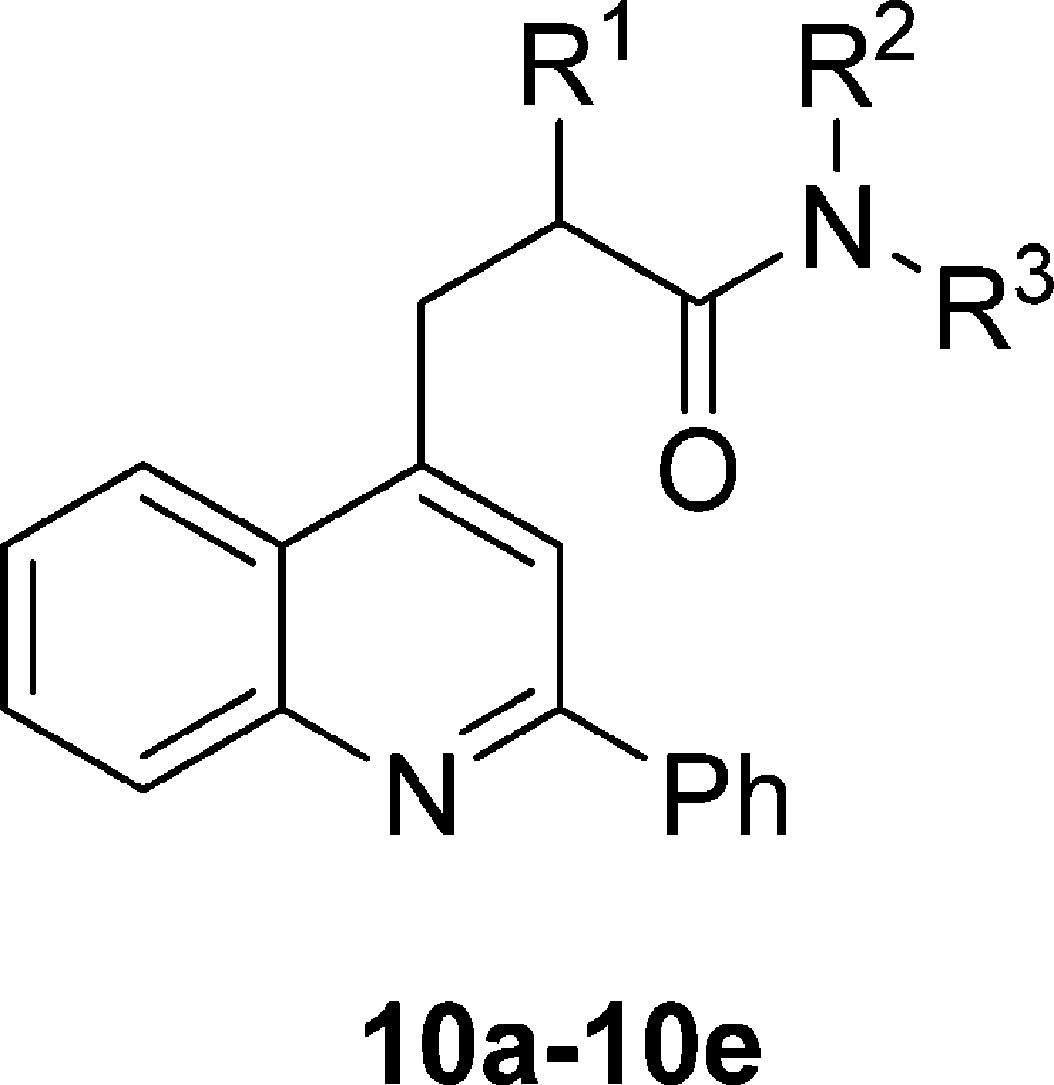
| ligand | R1 | R2 | R3 | rat Ki (nM)a | cLogD | LipE | LAB Ki (nM)b | HAB Ki (nM)c | (LAB Ki)/(HAB Ki) |
|---|---|---|---|---|---|---|---|---|---|
| 1 (PK 11195) | 0.5 ± 0.3 | 3.97 | 4.3 ± 0.2 | 4 ± 1 | 4 ± 1 | 1.0 ± 0.5 | |||
| 10a | Me | Et | Et | 2.1 ± 0.6 | 4.33 | 4.4 ± 0.1 | |||
| (R)-10a (PK 14067) | (R)-Me | Et | Et | 0.90 ± 0.09 | 4.33 | 4.72 ± 0.04 | 743 ± 190 | 10 ± 2 | 76 ± 27 |
| (S)-10a (PK 14068) | (S)-Me | Et | Et | 73 ± 36 | 4.33 | 2.9 ± 0.1 | |||
| 10b | H | Et | Et | 32 ± 11 | 4.84 | 2.7 ± 0.2 | |||
| 10c | Et | Et | Et | 14 ± 6 | 5.26 | 2.6 ± 0.2 | |||
| 10d | CH2-R2 | CH2-R1 | Me | 53 ± 22 | 3.69 | 3.6 ± 0.2 | |||
| 10e | Me | Me | Me | 40 ± 8 | 4.58 | 2.8 ± 0.1 |
Mean ± SD for n = 6, except for 1 (n = 60), and (R)-10a (n = 5).
Mean ± SD for n = 6, except for 1 (n = 12).
Mean ± SD for n = 6, except for 1 (n = 14).
Effect of Structural Changes to 10a on Rat TSPO Binding Affinity and LipE
The binding affinities (Ki) of all TSPO ligands, including 10a and its enantiomers, were determined on rat brain homogenates (Tables 1–5). Except for 10a, ligands were tested as racemates only. Assuming that all these ligands bind TSPO enantioselectively, as observed for 10a, the high-affinity enantiomer is expected to have about 2-fold higher affinity than that recorded for the racemate. Altering the chiral center of 10a by either removing the methyl group entirely (10b) or by lengthening this group from methyl to ethyl (10c) had a substantial detrimental effect on TSPO affinity. Therefore, a chiral center incorporating a methyl group appeared necessary for high TSPO affinity. With few exceptions, we retained this methyl group in all subsequently prepared ligands.
Table 5. Oxygen-Tethered TSPO Ligands Based on 15a: Dependence of Binding Affinity, Lipophilicity, and Genotype Sensitivity on Scaffold Aryl Groups.

| ligand | Ar | A | B | C | D | E | rat Ki (nM)a | cLogD | LipE | LAB Ki (nM)b | HAB Ki (nM)c | (LAB Ki)/(HAB Ki) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 15a | Ph | CH | CH | CH | CH | CH | 0.070 ± 0.004 | 4.73 | 5.42 ± 0.03 | 1.3 ± 0.2 | 0.5 ± 0.2 | 2.5 ± 0.9 |
| 15b | H | CH | CH | CH | CH | CH | 5.7 ± 0.5 | 2.96 | 5.29 ± 0.04 | 66 ± 22 | 3.3 ± 0.9 | 20 ± 9 |
| 15c | Ph | H | H | CH | 3.3 ± 0.6 | 3.52 | 5.3 ± 0.3 | 62 ± 11 | 2.0 ± 0.6 | 31 ± 11 | ||
| 19a | Ph | N | CH | CH | CH | CH | 1.4 ± 0.5 | 4.27 | 4.6 ± 0.1 | 23 ± 7 | 1.1 ± 0.3 | 21 ± 8 |
| 19b | Ph | CH | N | CH | CH | CH | 0.6 ± 0.1 | 4.15 | 5.0 ± 0.1 | 25 ± 8 | 1.3 ± 0.5 | 19 ± 9 |
| 19c | Ph | CH | CH | N | CH | CH | 0.3 ± 0.2 | 4.15 | 5.4 ± 0.3 | 47 ± 26 | 1.4 ± 0.4 | 33 ± 20 |
| 19d | Ph | CH | CH | CH | N | CH | 0.29 ± 0.03 | 3.91 | 5.63 ± 0.04 | 4.7 ± 0.7 | 0.9 ± 0.2 | 5 ± 2 |
| 19e | Ph | CH | CH | CH | CH | N | 0.13 ± 0.08 | 5.66 | 4.4 ± 0.5 | 2 ± 1 | 2 ± 1 | 1 ± 1 |
| 22a | o-Py | CH | CH | CH | CH | CH | 0.10 ± 0.05 | 4.18 | 5.9 ± 0.2 | 1.4 ± 0.5 | 1.4 ± 0.2 | 1.0 ± 0.4 |
| 22b | m-Py | CH | CH | CH | CH | CH | 0.20 ± 0.03 | 4.06 | 5.6 ± 0.1 | 12 ± 3 | 0.90 ± 0.09 | 13 ± 4 |
| 22c | p-Py | CH | CH | CH | CH | CH | 0.22 ± 0.02 | 4.06 | 5.60 ± 0.04 | 7 ± 3 | 1.0 ± 0.5 | 7 ± 5 |
Mean ± SD for n = 6, except for 19d, 22b (n = 5).
Mean ± SD for n = 6, except for 15a,b, 19c (n = 5), and 15c (n = 4).
Mean ± SD for n = 6, except for 15c (n = 5).
A tertiary amide is present in almost all high-affinity TSPO ligands (e.g., see Chart 1). Previous analogues of 1 that have had rotation about the amide locked or restricted have significantly reduced affinity for TSPO.57 Nonetheless, we found that complete restriction of amide bond rotation in the pyrrolidinyl ligand 10d had almost no adverse effect on affinity (cf. 10e; Table 1). The ability of the pyrrolidinyl ring to rotate with respect to the isoquinolinyl group perhaps compensates for absence of amide bond rotation. Creation of this pyrrolidinyl ring appreciably improved the LipE score (Table 1).
Methylation at a secondary amido nitrogen with [11C]methyl iodide has been a successful strategy for preparing useful PET radioligands.58 Therefore, we retained an N-methyl group in subsequently prepared ligands to provide a potential site for rapid labeling with carbon-11 (t1/2 = 20.4 min). We found that progressive lengthening of the remaining N-alkyl substituent from N-methyl to N-butyl (10e–h) dramatically increased TSPO affinity, yet this effect reached a plateau at N-butyl, as the N-pentyl compound (10i) offered no further improvement in affinity (Table 2). The N-propyl analogue offered the highest LipE score in this series but still with a quite high clogD value. Chain branching effects were not as predictable. Thus, TSPO affinity increased on replacing N-ethyl (10f) with N-isopropyl (10j) yet decreased on replacing N-propyl (10g) with N-sec-butyl (10k) or N-iso-butyl (10l). The N-isopropyl analogue (10j) gave the best LipE score among analogues with branched N-alkyl groups and the lowest clogD value among 10e–l.
Table 2. TSPO Ligands Based on 10a: Dependence of Binding Affinity, Lipophilicity, and Genotype Sensitivity on Amido Substituent.
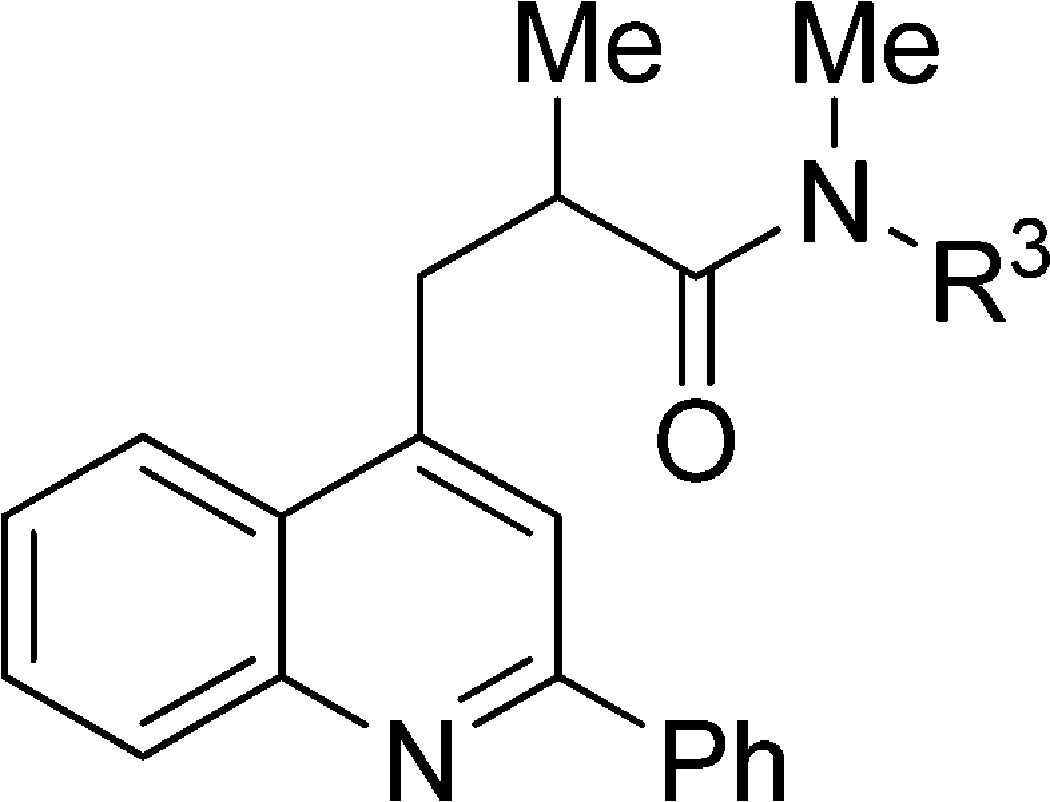
| ligand | R3 | rat Ki (nM)a | cLogD | LipE | LAB Ki (nM)b | HAB Ki (nM)c | (LAB Ki)/(HAB Ki) |
|---|---|---|---|---|---|---|---|
| 10e | Me | 40 ± 8 | 4.58 | 2.8 ± 0.1 | |||
| 10f | Et | 9 ± 2 | 4.28 | 3.8 ± 0.1 | |||
| 10g | Pr | 1.2 ± 0.4 | 4.74 | 4.2 ± 0.1 | |||
| 10h | Bu | 0.4 ± 0.2 | 5.53 | 3.9 ± 0.2 | 89 ± 3 | 1.0 ± 0.6 | 89 ± 50 |
| 10i | Pen | 0.6 ± 0.1 | 6.61 | 2.6 ± 0.1 | |||
| 10j | i-Pr | 2.9 ± 0.9 | 4.22 | 4.3 ± 0.1 | 973 ± 409 | 15 ± 2 | 64 ± 28 |
| 10kd | sec-Bu | 5 ± 3 | 5.20 | 2.9 ± 0.2 | |||
| 10l | i-Bu | 5 ± 2 | 5.00 | 3.3 ± 0.1 | 185 ± 81 | 6 ± 2 | 32 ± 18 |
| 10m | Ph | 0.9 ± 0.2 | 5.57 | 3.5 ± 0.1 | 53 ± 22 | 1.9 ± 0.5 | 27 ± 14 |
| 10n | Bn | 5 ± 3 | 6.53 | 1.8 ± 0.2 | |||
| 10o | o-Py | 1.6 ± 0.6 | 5.26 | 3.6 ± 0.2 | 119 ± 56 | 4 ± 1 | 30 ± 18 |
| 10p | m-Py | 10 ± 3 | 5.35 | 2.7 ± 0.1 | |||
| 10q | p-Py | 106 ± 24 | 5.33 | 1.7 ± 0.1 | |||
| 10r | o-F-Ph | 0.9 ± 0.2 | 5.54 | 3.5 ± 0.1 | |||
| 10s | m-F-Ph | 0.8 ± 0.2 | 5.64 | 3.5 ± 0.1 | |||
| 10t | p-F-Ph | 0.4 ± 0.1 | 5.32 | 4.1 ± 0.2 |
Mean ± SD for n = 6, except for 10h,l (n = 9).
Mean ± SD for n = 6, except for 10h,j (n = 3) and 10m (n = 5).
Mean ± SD for n = 6, except for 10m,o (n = 5).
10k is a pair of unresolved diastereomers.
An N-phenyl (10m) or N-benzyl (10n) group was well-tolerated, with 10m showing subnanomolar TSPO affinity (Table 2). TSPO showed variable sensitivity to substitution of the N-phenyl group in 10m with a pyridinyl or fluorophenyl group. Binding affinity varied with nature and position of the new heteroatom. Thus, replacement of a phenyl group with an o-pyridinyl group (10o) left binding affinity unchanged at subnanomolar. However, there was a 10-fold loss of affinity in the m-pyridinyl analogue (10p) and a further 10-fold loss in the p-pyridinyl analogue (10q). In contrast to the pyridinyl analogues, all three fluoro isomers (10r–t) showed almost identical subnanomolar affinity. The p-fluorophenyl ligand 10t had the highest LipE score but still possessed high computed lipophilicity.
Chlorophenyl groups have featured in examples of high-affinity TSPO ligands from other structural classes such as the isoquinoline carboxamide 1. Therefore, we also prepared the chlorophenyl isomers 10u–w of the N-methyl,N-isopropyl ligand 10j (Table 3). All three ligands showed lower TSPO affinity than the phenyl analogue 10j, with the o-isomer (10u) showing lowest affinity.
Table 3. TSPO Ligands Based on 10a: Dependence of Rat Binding Affinity and Lipophilicity on Pendant Aryl Substituent.

| ligand | Ar | rat Ki (nM)a | cLogD | LipE |
|---|---|---|---|---|
| 10j | Ph | 2.9 ± 0.9 | 4.22 | 4.3 ± 0.1 |
| 10u | o-Cl-Ph | 22 ± 6 | 4.50 | 3.2 ± 0.1 |
| 10v | m-Cl-Ph | 4 ± 1 | 4.71 | 3.7 ± 0.1 |
| 10w | p-Cl-Ph | 4 ± 1 | 4.83 | 3.6 ± 0.1 |
Mean SD for n = 6, except for 10u (n = 9).
Having explored how TSPO affinity was affected by structural change at the amido nitrogen, we next explored changes to the methylene group that bridges the amide to the quinolinyl ring. We replaced the methylene tether of 10m with an oxygen (15a), sulfur (12), or NH tether (14). In each case, rat TSPO binding affinity was greatly increased, and particularly so for the oxygen-tethered compound (15a) which showed a Ki value of 70 pM (Table 4). Increased affinity may be due to improved spatial relations of the amido group relative to the quinolinyl ring via altered bond lengths and dihedral angle. Further benefits of the heteroatom tethers were decreased lipophilicities, resulting in higher LipE scores.
Table 4. TSPO Ligands Based on 10a: Dependence of Rat Binding Affinity, Lipophilicity, and Genotype Sensitivity on Tether for Pendant Alkyl Carboxamido Group.
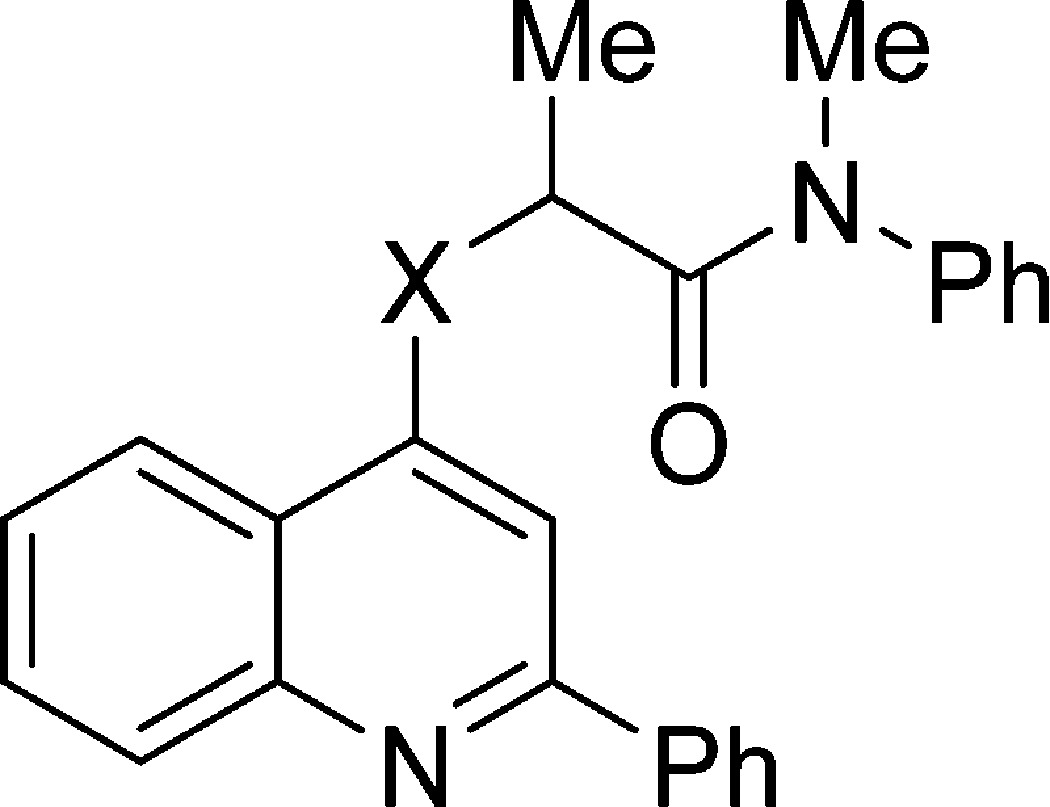
| ligand | X | rat Ki (nM)a | cLogD | LipE | LAB Ki (nM)b | HAB Ki (nM)c | (LAB Ki)/(HAB Ki) |
|---|---|---|---|---|---|---|---|
| 10m | CH2 | 0.9 ± 0.2 | 5.57 | 3.5 ± 0.1 | 53 ± 22 | 1.9 ± 0.5 | 27 ± 14 |
| 12 | S | 0.39 ± 0.04 | 5.31 | 4.10 ± 0.05 | 26 ± 5 | 2.5 ± 0.6 | 10 ± 3 |
| 14 | NH | 0.19 ± 0.01 | 4.11 | 5.62 ± 0.03 | 4 ± 1 | 0.44 ± 0.08 | 9 ± 3 |
| 15a | O | 0.070 ± 0.004 | 4.73 | 5.42 ± 0.03 | 1.3 ± 0.2 | 0.5 ± 0.2 | 2.5 ± 0.9 |
Mean SD for n = 6, except for 14 (n = 5).
Mean ± SD for n = 6, except for 10m (n = 5).
Mean ± SD for n = 6, except for 10m (n = 5).
We chose to focus on preparing further analogues of the very high affinity O-tethered lead (15a). We next considered whether high TSPO affinity might be retained in less lipophilic analogues in which the phenyl-quinoline scaffold was modified by removing an aromatic ring or by inserting a nitrogen atom. Compounds that lacked either the pendant 2-phenyl group (15b) or the benzo fusion (15c) illustrated that these two rings, and especially the phenyl group, were quite important for very high TSPO affinity (Table 5). Nonetheless, 15b and 15c still exhibited affinity in the low nanomolar range, with radically reduced clogD values and greatly enhanced LipE scores. All nitrogen substitutions in the quinoline core resulted in ligands that retained near- or subnanomolar affinity for TSPO, with affinities slightly increasing in the following order 19a < 19b < 19c ∼ 19d < 19e. Like the quinazoline (19e), the 2-(o-pyridyl)quinoline (22a) had very high affinity, approaching that of 15a, but with much lower computed lipophilicity and hence a much higher LipE score. The other pyridylquinolines (22b,c) showed virtually equal subnanomolar affinity.
Overall, the presence of a heteroatom tether strikingly increased LipE score, as may be readily appreciated from a plot of pKi versus clogD for ligands with each type of tether (Figure 2). In particular, ligands with oxygen tethers clearly cluster into a separate group to those with methylene tethers. Thus, introduction of a heteroatom tether was very effective in mitigating the demands35 of the TSPO binding site for high ligand lipophilicity.
Figure 2.

Plot of rat pKi versus clogD for ligands having different side chain tethers.
Assessment of Ligand Sensitivity to Human TSPO Genotype
Compound 1, the lead compound (R)-10a, examples of methylene-tethered ligands (10h,j,l,m,o), and the heteroatom-tethered compounds (12, 14, 15a–c, 19a–e, 22a–c) were selected to evaluate their human genotype sensitivities by measurement of their Ki values for binding to leukocytes from HABs and LABs. Our measurements confirmed that 1 has low genotype sensitivity (Table 1). (R)-10a showed about 10-fold lower affinity to HAB TSPO than to rat TSPO (Table 1). Affinity for LAB TSPO was about 76-fold lower than for HAB TSPO. All the methylene-tethered ligands had similar or lower affinity to HAB TSPO than to rat TSPO and had high genotype sensitivities (Table 2). Thus, overall, variation in amide substituents had little impact on genotype sensitivity.
For the group of ligands in which only the tether atom differed (10m, 12, 14, 15a) affinity for HAB TSPO was again somewhat lower than for rat TSPO, and genotype sensitivity reduced progressively across the tether series CH2, S, NH, and O (Table 4). The oxygen-tethered ligand 15a showed very low sensitivity (2.5) in addition to subnanomolar affinity and high LipE score. We surmise this improvement may relate to altered bond lengths and torsional angles at the oxygen atom.
Two truncated versions of 15a (15b,c) showed somewhat lower affinity for rat TSPO. Remarkably, however, they showed low nanomolar HAB binding affinities and LAB/HAB Ki ratios similar to those of 15a (Table 5). The LipE scores of 15b and 15c were similar to those of 15a.
Finally, we looked at the effects of introducing a second nitrogen into the 2-phenylquinoline scaffold (Table 5). Of the eight compounds tested, four (19a,b,d, 22b) displayed greater than 10-fold lower binding affinity to LAB TSPO than to HAB TSPO. The remaining four compounds were either much less sensitive (19d, 22c) or insensitive (19e, 22a) to TSPO genotype. Notably, compounds having the second nitrogen in nearest proximity to the quinolinyl nitrogen had the least sensitivity to genotype. Ligand 22a offered the most appealing combination of properties as a lead for PET radioligand development, including high HAB TSPO affinity, genotype insensitivity, and high LipE score for binding to rat and human TSPO, which are all improved over corresponding values for 10a.
An important consideration in attempts to develop genotype-insensitive PET radioligands for TSPO, is whether genotype sensitivity is likely to increase with ligand affinity. Generally, we observed that genotype sensitivity tended to decrease with HAB TSPO affinity among the tested ligands (Figure 3).
Figure 3.

Plot of genotype sensitivity versus HAB TSPO binding affinity for tested ligands.
Conclusions
Judicious structural modifications to 10a led to several TSPO ligands with affinity in the nanomolar or subnanomolar range plus an enhanced LipE score. Truncation of the phenyl-isoquinoline scaffold as in ligands 15b and 15c was particularly effective in reducing LipE score, and these ligands may serve as leads for further PET radioligand development. Introduction of an oxygen tether in place of the methylene tether was particularly effective in generating compounds with low genotype sensitivity as leads for PET radioligand development. Ligand 22a presents an especially appealing array of properties for this purpose.
Experimental Section
Materials and Methods
Reagents and solvents were purchased unless stated otherwise. Air-sensitive reagents were stored under N2 in a PureLab HE glovebox (Innovative Technology; Amesbury, MA). Melting points were determined on an SMP10 apparatus (Stuart; Staffordshire, UK). Boiling point vacuum pressures were determined on a DVR-200 apparatus (J-Kem Scientific Inc.; St. Louis, MO). Optical rotations were determined on a P-1010 instrument (JASCO Inc.; Easton, MD). IR-VCD spectra were recorded on a Chiral IR-2X instrument equipped with DualPEM (BioTools, Inc.; Jupiter, FL). Absolute configuration was determined with ComputeVOA (BioTools Inc.), employing a B3LYP “functional”, 6-31G(d) basis set on Gaussian 09 (Gaussian Inc.; Pittsburgh, PA). 1H (400 MHz), 13C NMR (100 MHz), and 19F NMR (376 MHz) spectra were recorded on an Avance 400 instrument (Bruker; Billerica, MA). Chemical shifts for 19F are reported relative to neat TFA in a coaxial insert (δ = −76.6). HRMS were obtained at the Mass Spectrometry Laboratory, School of Chemical Sciences, University of Illinois Urbana—Champaign using a Micromass Q-Tof Ultima instrument for ESI (Waters Corp.; Columbia, MD) or a GCT Premier instrument for EI (Waters Corp.). Preparative HPLC was performed with either a Luna PFP(2) (5 μm; 100 Å; 30 mm × 250 mm), Gemini C18 (10 μm; 110 Å; 30 mm × 250 mm), or a Lux Amylose-2 (3 μm; 10 mm × 250 mm) column. HPLC separation conditions are given in parentheses and refer to column type, flow rate (mL/min), and organic phase ‘O’/aq phase ‘A’ ratio, as follows: O1 = MeOH, O2 = MeCN, A1 = H2O, A2 = NH4CO3H (10 mM), A3 = Et2NH (0.1%). The chemical purities of all compounds were established by HPLC on either a Luna PFP(2) (5 μm; 100 Å; 4.6 mm × 250 mm), Gemini C18 (5 μm; 110 Å; 4.6 mm × 250 mm), or a Lux Amylose-2 (3 μm; 4.6 mm × 250 mm) column. Chemical purities were all >95% and typically >99%, as monitored by absorbance at 220 nm. cLogD was computed with Pallas for Windows software version 3.8 in default option (CompuDrug; Bal Harbor, FL).
Method A: N,N-Diethyl-2-methylpent-4-ynamide (9a)
DIPA (1.0 mL, 6.0 mmol) was added dropwise to a solution of 2-methylpent-4-ynoic acid59 (0.33 mL, 3.0 mmol), diethylamine (0.34 mL, 3.3 mmol), and PyBroP (1.4 g, 3.0 mmol) in DCM (3 mL) at rt. This mixture was stirred for 2 h and then the solvent removed. The residue was taken up in EtOAc (30 mL) and washed successively with 5% KHSO4 (30 mL × 3), brine (30 mL), 5% NaHCO3 (30 mL × 3), and brine (30 mL), and finally dried (Na2SO4). FC (hexanes/EtOAc, 1:1) of the residue gave 9a as a colorless oil (0.33 g, 66%). HRMS–ESI (m/z): [M + H]+ calcd for C10H18NO, 168.1388; found, 168.1383. 1H NMR (CDCl3): δ 3.41–3.33 (m, 4H), 2.87 (m, J = 2.4 Hz, 1H), 2.55–2.49 (ddd, J = 17, 7.2, 2.4 Hz, 1H), 2.34–2.28 (ddd, J = 17, 7.2, 2.4 Hz, 1H), 1.97 (t, J = 2.4 Hz, 1H), 1.21 (t, J = 6.8 Hz, 3H), 1.21 (t, J = 6.8 Hz, 3H), 1.12 (t, J = 6.8 Hz, 3H). 13C NMR (CDCl3): δ 174.1, 82.7, 69.0, 41.9, 40.4, 35.4, 23.3, 17.7, 14.9, 13.1.
As described in the Supporting Information, this method also gave: N,2-dimethyl-N-propylpent-4-ynamide (9b), N-butyl-N,2-dimethylpent-4-ynamide (9c), N,2-dimethyl-N-pentylpent-4-ynamide (9d), N-(butan-2-yl)-N,2-dimethylpent-4-ynamide (9e), N,2-dimethyl-N-(2-methylpropyl)pent-4-ynamide (9f), N-benzyl-N,2-dimethylpent-4-ynamide (9g), N,2-dimethyl-N-(pyridine-2-yl)pent-4-ynamide (9h), N,2-dimethyl-N-(pyridine-3-yl)pent-4-ynamide (9i), N,2-dimethyl-N-(pyridin-4-yl)pent-4-ynamide (9j), N-(4-fluorophenyl)-N,2-dimethylpent-4-ynamide (9k), N-(2-fluorophenyl)-N-methylpropanamide (9sa), N-(3-fluorophenyl)-N-methylpropanamide (9ta), N-(4-acetylpyridin-3-yl)benzamide (17c), N-(2-acetylphenyl)pyridine-2-carboxamide (20a), and N-(2-acetylphenyl)pyridine-4-carboxamide (20c).
Method B: N,N-Diethyl-2-methylpent-4-ynamide (9a)
n-Butyl lithium in hexanes (1.6 M, 28 mL, 44 mmol) was added dropwise to a solution of DIPA (6.1 mL, 43 mmol) in THF (130 mL) under Ar at −75 °C. This solution was slowly warmed to 0 °C and then cooled back to −75 °C. N,N-Diethylpropionamide (5.6 mL, 39 mmol) was added dropwise. This solution was slowly warmed to −20 °C and then cooled back to −75 °C. Propargyl bromide (87 mmol) in toluene (9.4 mL) was added dropwise. This solution slowly was warmed to rt and stirred for several hours. Brine (200 mL) was added, the organic layer separated, and the aqueous phase extracted with ether (100 mL × 2). The combined extracts were washed with brine (100 mL) and dried (MgSO4). Fractional distillation of the residue gave 9a as a colorless oil (bp 53–54 °C at 5.8 mmHg; 4.8 g, 71%). A small quantity was further purified by HPLC (Gemini, 30, O1/A1, 55:45); d 0.90 g/mL.
As described in the Supporting Information, this method also gave: N,N-diethylpent-4-ynamide (9l), N,N,2-triethylpent-4-ynamide (9m), 1-methyl-3-(prop-2-yn-1-yl)pyrrolidin-2-one (9n), N,N,2-trimethylpent-4-ynamide (9o), N-ethyl-N,2-dimethylpent-4-ynamide (9p), N,2-dimethyl-N-(propan-2-yl)pent-4-ynamide (9q), N,2-dimethyl-N-phenylpent-4-ynamide (9r), N-(2-fluorophenyl)-N,2-dimethylpent-4-ynamide (9s), and N-(3-fluorophenyl)-N,2-dimethylpent-4-ynamide (9t).
Method C: N,N-Diethyl-2-methyl-3-(2-phenylquinolin-4-yl)propanamide (10a)
Copper(I) chloride (10 mg, 0.10 mmol) and then silver triflate (26 mg, 0.10 mmol) were added to a solution of N-benzylideneaniline (362 mg, 2.00 mmol) and 9a (167 mg, 1.00 mmol) in DCA (1.0 mL) under Ar. The solution was heated to 100 °C for 17 h, cooled to rt, and filtered through diatomaceous earth. The mixture was taken up in EtOAc (10 mL) and washed with aqueous NH4Cl/NH4OH (pH 8, 10 mL). The aqueous phase was extracted with EtOAc (10 mL × 2). The combined extracts were washed with brine (15 mL) and dried (MgSO4). HPLC (PFP, 30, O2/A2, 65:35) of this material gave 10a as a dark-yellow syrup (0.12 g, 36%). HRMS–ESI (m/z): [M + H]+ calcd for C23H27N2O, 347.2123; found, 347.2114. 1H NMR (CDCl3): δ 8.20 (bs, 1H), 8.15 (m, 2H), 8.04 (dd, J = 8.4, 0.8 Hz, 1H), 7.74 (s, 1H), 7.70 (dt, J = 7.8, 1.2 Hz, 1H), 7.55 (dt, J = 7.8, 1.2 Hz, 1H), 7.53–7.48 (2H), 7.44 (tt, J = 7.2, 1.6 Hz, 1H), 3.52 (dd, J = 13.6, 9.2 Hz, 1H), 3.36–3.24 (m, 2H), 3.19–3.09 (m, 1H), 2.87 (dq, J = 7.2, 2.4 Hz, 2H), 1.31 (d, J = 6.4 Hz, 3H), 0.88 (t, J = 7.2 Hz, 3H), 0.75 (t, J = 7.2 Hz, 3H). 13C NMR (CDCl3): δ 174.4, 156.9, 148.4, 146.7, 139.5, 130.6, 129.3, 128.8, 127.5, 126.5, 126.2, 123.2, 120.0, 41.7, 40.6, 37.0, 36.7, 19.0, 14.5, 13.0.
As described in the Supporting Information, this method also gave: N,N-diethyl-3-(2-phenylquinolin-4-yl)propanamide (10b), N,N-diethyl-2-[(2-phenylquinolin-4-yl)methyl]butanamide (10c), 1-methyl-3-[(2-phenylquinolin-4-yl)methyl]pyrrolidin-2-one (10d), N,N,2-trimethyl-3-(2-phenylquinolin-4-yl)propanamide (10e), N-ethyl-N,2-dimethyl-3-(2-phenylquinolin-4-yl)propanamide (10f), N,2-dimethyl-3-(2-phenylquinolin-4-yl)-N-propylpropanamide (10g), N-butyl-N,2-dimethyl-3-(2-phenylquinolin-4-yl)propanamide (10h), N,2-dimethyl-N-pentyl-3-(2-phenylquinolin-4-yl)propanamide (10i), N,2-dimethyl-3-(2-phenylquinolin-4-yl)-N-(propan-2-yl)propanamide (10j), N-(butan-2-yl)-N,2-dimethyl-3-(2-phenylquinolin-4-ylpropanamide (10k), N,2-dimethyl-N-(2-methylpropyl)-3-(2-phenylquinolin-4-yl)propanamide (10l), N,2-dimethyl-N-phenyl-3-(2-phenylquinolin-4-yl)propanamide (10m), N-benzyl-N,2-dimethyl-3-(2-phenylquinolin-4-yl)propanamide (10n), N,2-dimethyl-3-(2-phenylquinolin-4-yl)-N-(pyridin-2-yl)propanamide (10o), N,2-dimethyl-3-(2-phenylquinolin-4-yl)-N-(pyridin-3-yl)propanamide (10p), N,2-dimethyl-3-(2-phenylquinolin-4-yl)-N-(pyridin-4-yl)propanamide (10q), N-(2-fluorophenyl)-N,2-dimethyl-3-(2-phenylquinolin-4-yl)propanamide (10r), N-(3-fluorophenyl)-N,2-dimethyl-3-(2-phenylquinolin-4-yl)propanamide (10s), N-(4-fluorophenyl)-N,2-dimethyl-3-(2-phenylquinolin-4-yl)propanamide (10t), 3-[2-(2-chlorophenyl)quinolin-4-yl)-N,2-dimethyl-N-(propan-2-yl)propanamide (10u), 3-[2-(3-chlorophenyl)quinolin-4-yl)-N,2-dimethyl-N-(propan-2-yl)propanamide (10v), and 3-[2-(4-chlorophenyl)quinolin-4-yl)-N,2-dimethyl-N-(propan-2-yl)propanamide (10w).
(R)-N,N-Diethyl-2-methyl-3-(2-phenylquinolin-4-yl)propanamide [(R)-10a]
Chiral HPLC (Lux, 12, O2/A3, 50:50) of 10a gave (R)-10a (tR 5.3 min; 99% ee; [α]18D −90° (c 2.86, EtOH).
(S)-N,N-Diethyl-2-methyl-3-(2-phenylquinolin-4-yl)propanamide [(S)-10a]
Chiral HPLC, as described above, also gave (S)-10a (tR 6.2 min; 96% ee; [α]18D +95° (c 3.35, EtOH).
N-Methyl-N-phenyl-2-sulfanylpropanamide (11)
N-Methylaniline (11 mL, 102 mmol) and thiolactic acid (9.0 mL, 102 mmol) were heated to 190 °C under Ar for 3 d. Water was collected in a Dean–Stark apparatus. The mixture was cooled to rt, taken up in CHCl3 (250 mL), washed successively with 10% HCl (100 mL × 2), saturated NaHCO3 (100 mL × 2), and brine (100 mL), and dried (MgSO4). The product was distilled (bp = 144 °C at 7.4 mmHg) to give 11 as a yellow oil (3.8 g, 19%). HRMS–ESI (m/z): [M + H]+ calcd for C10H14NOS, 196.0796; found, 196.0795. 1H NMR (CDCl3): δ 7.45 (t, J = 7.2 Hz, 2H), 7.38 (t, J = 7.6 Hz, 1H), 7.27 (d, J = 7.2 Hz, 2H), 3.37–3.31 (m, 1H), 3.29 (s, 3H), 2.13 (d, J = 10 Hz, 1H), 1.45 (d, J = 7.2 Hz, 3H). 13C NMR (CDCl3): δ 173.6, 143.5, 123.0, 128.2, 127.3, 37.8, 33.5, 22.9.
N-Methyl-N-phenyl-2-[(2-phenylquinolin-4-yl)sulfanyl]propanamide (12)
A mixture of 11 (115 mg, 0.59 mmol), 4-chloro-2-phenylquinoline (120 mg, 0.50 mmol), t-BuOK (61 mg, 0.55 mmol), and t-BuOH (53 μL, 0.55 mmol) in DMF (5.0 mL) was heated to 135 °C under Ar for 3 d. The mixture was cooled to rt, diluted with water (50 mL), and extracted into EtOAc (20 mL × 3). The combined extracts were washed with water (10 mL × 5) and brine (10 mL) and dried (MgSO4). Product was isolated by HPLC (Gemini, 43, O1/A2, 80:20) to give 12 as a light-yellow syrup (128 mg, 64%). HRMS–ESI (m/z): [M + H]+ calcd for C25H23N2OS, 399.1531; found, 399.1532. 1H NMR (CDCl3): δ 8.15 (dd, J = 8.0, 0.8 Hz, 2H), 7.98 (dd, J = 6.8, 1.6 Hz, 2H), 7.72 (dt, J = 7.4, 1.2 Hz, 1H), 7.54–7.46 (5H), 7.19 (t, J = 2.0 Hz, 3H), 6.94 (2H), 4.12 (q, J = 6.8 Hz, 1H), 3.23 (s, 3H), 1.58 (d, J = 6.8 Hz, 3H). 13C NMR (CDCl3): δ 171.5, 156.8, 148.3, 144.7, 143.2, 139.5, 130.4, 130.3, 130.0, 129.7, 129.0, 128.3, 127.8, 127.2, 126.8, 124.6, 119.7, 42.8, 38.0, 19.0.
2-Amino-N-methyl-N-phenylpropanamide hydrochloride (13)
NaN3 (2.58 g, 39.8 mmol) and 2-bromo-N-methyl-N-phenylpropanamide60 (2.42 g, 10.0 mmol) in DMF (50 mL) were heated to 70 °C under Ar for 2 h. The solution was cooled to rt, diluted with water (500 mL), and extracted into ether (200 mL × 3). The combined extracts were washed successively with water (100 mL × 5) and brine (100 mL) and dried (Na2SO4). The solvent was removed to yield the azide as a yellow oil (1.98 g, 97%), which was taken up in THF (100 mL). Ph3P (2.54 g, 9.70 mmol) and water (260 μL, 15 mmol) were added, and the mixture stirred overnight at rt. The solvent was removed, the residue taken up in benzene (or toluene), and HCl (g) bubbled through for 10 min. The hydrochloride salt was washed with ether to give 13 as a cream powder (1.78 g, 83%); mp 230–233 °C dec. HRMS–ESI (m/z): [M + H]+ calcd for C10H15N2O, 179.1184; found, 179.1182. 1H NMR (D2O): δ 7.72–7.63 (3H), 7.55–7.52 (2H), 4.83 (q, J = 7.2 Hz, 1H), 4.21 (q, J = 7.2 Hz, 1H), 3.57 (s, 3H), 3.43 (s, 3H), 1.79 (d, J = 6.8 Hz, 3H), 1.41 (d, J = 6.8 Hz, 3H). 13C NMR (D2O): δ 170.4, 141.0, 130.4, 129.3, 127.2, 47.3, 37.9, 15.8.
N-Methyl-N-phenyl-2-[(2-phenylquinolin-4-yl)amino]propanamide (14)
A mixture of (13) (322 mg, 1.50 mmol), 4-chloro-2-phenylquinoline (240 mg, 1.00 mmol), Pd(OAc)2 (9 mg, 0.04 mmol), DPEPhos (43 mg, 0.080 mmol), and K3PO4 (742 mg, 3.50 mmol) in dioxane (4.0 mL) was heated to 85 °C under Ar for 2 d. The mixture was cooled to rt and filtered through diatomaceous earth, which was then rinsed with EtOAc (20 mL). The organic phase was washed with water (20 mL) and the water extracted with EtOAc (20 mL × 2). The combined extracts were washed with brine (20 mL) and then dried (MgSO4). The product was isolated by FC (CHCl3/MeOH/TEA, 30:1:1%) and recrystallized (cyclohexane/EtOAc) to give 14 as pale-yellow chunks (0.13 g, 34%); mp 190–191 °C. HRMS–ESI (m/z): [M + H]+ calcd for C25H24N3O, 382.1919; found, 382.1917. 1H NMR (CDCl3): δ 8.05 (d, J = 8.0 Hz, 1H), 7.91 (d, J = 6.8 Hz, 2H), 7.85 (dd, J = 8.4, 0.8 Hz, 1H), 7.65 (dt, J = 6.8, 1.2 Hz, 1H), 7.52–7.41 (7H), 7.29–7.27 (2H), 6.41 (s, 1H), 5.92 (d, J = 8.0 Hz, 1H), 4.46 (dquint, J = 6.8, 1.6 Hz, 1H), 3.33 (s, 3H), 1.42 (d, J = 6.8 Hz, 3H). 13C NMR (CDCl3): δ 173.0, 158.3, 148.8, 148.5, 142.7, 141.0, 130.3, 130.2, 129.4, 128.8, 128.7, 128.5, 127.6, 127.4, 124.6, 119.7, 118.1, 96.8, 48.4, 38.0, 18.6.
Method D: N-Methyl-N-phenyl-2-[(2-phenylquinolin-4-yl)oxy]propanamide (15a)
2-Phenylquinolin-4(1H)-one hydrochloride (144 mg, 0.56 mmol), 2-bromo-N-methyl-N-phenylpropanamide60 (203 mg, 0.84 mmol), and Cs2CO3 (736 mg, 2.26 mmol) were stirred in acetone (5.0 mL) at rt for 19 h. Solid was filtered off and washed with a little acetone. Solvent was then removed, and the residue was recrystallized (DCM/ether) to give 15a as a colorless solid (123 mg, 57%); mp 158–160 °C. HRMS–ESI (m/z): [M + H]+ calcd for C25H23N2O2, 383.1760; found, 383.1761. 1H NMR (CDCl3): δ 8.15 (d, J = 8.4 Hz, 1H), 8.07 (d, J = 8.0 Hz, 1H), 7.96 (d, J = 7.2 Hz, 2H), 7.69 (dt, J = 7.8, 1.2 Hz, 1H), 7.53 (tt, J = 6.8, 1.6 Hz, 2H), 7.47 (m, 2H), 7.34–7.31 (3H), 7.21–7.19 (2H), 5.09 (q, J = 6.4 Hz, 1H), 3.31 (s, 3H), 1.68 (d, J = 6.4 Hz, 3H). 13C NMR (CDCl3): δ 169.8, 160.6, 158.5, 149.4, 142.4, 140.4, 130.1, 130.0, 129.2, 129.0, 128.7, 128.5, 127.6, 127.3, 125.4, 122.2, 120.4, 98.9, 71.4, 38.2, 18.1.
As described in the Supporting Information, this method also gave: N-methyl-N-phenyl-2-(quinolin-4-yloxy)propanamide (15b), N-methyl-N-phenyl-2-[(2-phenylpyridin-4-yl)oxy]propanamide (15c), N-methyl-N-phenyl-2-[(2-phenyl-1,5-naphthyridin-4-yl)oxy)]propanamide (19a), N-methyl-N-phenyl-2-[(2-phenyl-1,6-naphthyridin-4-yl)oxy)]propanamide (19b), N-methyl-N-phenyl-2-[(2-phenyl-1,7-naphthyridin-4-yl)oxy)]propanamide (19c), N-methyl-N-phenyl-2-[(2-phenyl-1,8-naphthyridin-4-yl)oxy)]propanamide (19d), N-methyl-N-phenyl-2-[(2-phenylquinazolin-4-yl)oxy)]propanamide (19e), N-methyl-N-phenyl-2-{[2-(pyridin-3-yl)quinolin-4-yl]oxy}propanamide (22b), and N-methyl-N-phenyl-2-{[2-(pyridin-4-yl)quinolin-4-yl]oxy}propanamide (22c).
N-(3-Bromopyridin-4-yl)benzamide (16b)
Benzoyl chloride (3.7 mL, 32 mmol) was added dropwise to a solution of 4-amino-3-bromopyridine (5.01 g, 29.0 mmol) and TEA (10 mL) in DCM (25 mL) at rt under Ar. DMAP (325 mg, 2.90 mmol) was then added in one portion. This mixture was stirred for 30 min and then quenched by addition of water (250 mL). The organic layer was separated off and the aqueous phase extracted with DCM (100 mL × 2). The combined extracts were washed with brine (100 mL) and then dried (MgSO4). The solvent was removed and the product twice recrystallized (EtOAc/hexanes) to give 16b as cream needles (1.98 g, 24%); mp 99–101 °C. HRMS–ESI (m/z): [M + H]+ calcd for C12H10N2OBr, 276.9976; found, 276.9971. 1H NMR (CDCl3): δ 8.69 (s, 1H), 8.62 (br s, 1H), 8.57 (d, J = 5.6 Hz, 1H), 8.50 (d, J = 5.6 Hz, 1H), 7.95–7.92 (m, 2H), 7.63 (tt, J = 7.6, 1.2 Hz, 1H), 7.56 (tt, J = 7.6, 1.2 Hz, 2H). 13C NMR (CDCl3): δ 165.5, 151.6, 149.9, 142.5, 133.6, 132.9, 129.2, 127.2, 144.8, 111.0.
N-(3-Bromopyridin-2-yl)benzamide (16d)
Benzoyl chloride (3.9 mL, 33.3 mmol) was added dropwise to a solution of 2-amino-3-bromopyridine (4.98 g, 28.8 mmol) and TEA (8.0 mL, 57.9 mmol) in THF (25 mL) at −5 °C under Ar. This mixture was warmed to rt and stirred overnight. The precipitate was filtered off and washed several times with THF. Solvent was then removed, MeOH added, and the precipitate filtered off. This step was repeated with ether. The ether was removed and the residue twice recrystallized (cyclohexane/toluene, then cyclohexane/DME) to give 16d as fluffy cream needles (1.95 g, 24%); mp 109–111 °C. HRMS–ESI (m/z): [M + H]+ calcd for C12H10N2OBr, 276.9976; found, 276.9974. 1H NMR (CDCl3): δ 8.54 (br s, 1H), 8.49 (dd, J = 4.8, 1.6 Hz, 1H), 7.97–7.94 (3H), 7.60 (tt, J = 7.2, 1.2 Hz, 1H), 7.52 (tt, J = 7.6, 1.6 Hz, 2H), 7.05 (dd, J = 8.0, 4.8 Hz, 1H). 13C NMR (CDCl3): δ 165.0, 148.8, 147.5, 141.5, 134.2, 132.4, 128.9, 127.5, 121.6, 112.7.
Method E: N-(2-Acetylpyridin-3-yl)benzamide (17a)
n-BuLi in hexanes (24 mL, 38 mmol) was added dropwise to a solution of N-(2-bromopyridin-3-yl)benzamide61 (4.82 g, 17.4 mmol) in THF (280 mL) at −78 °C under Ar. This solution was stirred for another 30 min and then warmed to −30 °C for 15 min. N-Methoxy-N-methylacetamide (4.1 mL, 38 mmol) was added dropwise and the solution warmed to rt. The mixture was quenched with 0.5 M HCl (110 mL). Saturated NaHCO3 solution was added to neutralize the mixture, and the product was extracted into DCM (180 mL × 2). The combined extracts were washed with brine (180 mL) and then dried (MgSO4). The product was purified by FC (hexanes/EtOAc/TEA, 10:1:1%) to yield a yellow solid that was further triturated with hexanes to give 17a as a white powder (1.22 g, 29%); mp 89–91 °C. HRMS–ESI (m/z): [M + H]+ calcd for C14H13N2O2, 241.0977; found, 241.0979. 1H NMR (CDCl3): δ 12.53 (s, 1H), 9.32 (dd, J = 8.4, 1.2 Hz, 1H), 8.42 (dd, J = 4.4, 1.6 Hz, 1H), 8.08–8.06 (m, 2H), 7.59–7.52 (4H), 2.85 (s, 3H). 13C NMR (CDCl3): δ 206.3, 166.5, 143.0, 138.4, 137.4, 134.1, 132.4, 128.9, 128.7, 128.2, 127.5, 27.8.
This method also gave N-(3-acetylpyridin-4-yl)benzamide (17b), and N-(3-acetylpyridin-2-yl)benzamide (17c) (see Supporting Information).
Method F: 2-Phenyl-1,4-dihydro-1,5-naphthyridin-4-one (18a)
17a (1.17 g, 4.88 mmol) and NaOH (585 mg, 14.6 mmol) in dioxane (50 mL) were heated in a pressure vessel to 110 °C under N2 for 3 h. The mixture was cooled to rt, the solvent removed, and the residue taken up in water (10 mL) and hexanes (100 mL). The aqueous phase was acidified with 1 M HCl and then neutralized with satd NaHCO3. A cream precipitate was collected and dried in vacuo with BaO for 3 d to give 17a as a hard, mustard-yellow solid (0.93 g, 86%); mp >300 °C. HRMS–ESI (m/z): [M + H]+ calcd for C14H11N2O, 223.0871; found, 223.0872. 1H NMR (DMSO-d6): δ 8.49 (d, J = 2.8 Hz, 1H), 8.09 (d, J = 8.4 Hz, 1H), 8.04 (d, J = 7.2 Hz, 2H), 7.52–7.40 (4H), 6.85 (s, 1H), 3.62 (br s, 1H). 13C NMR (DMSO): δ 173.5, 156.1, 144.7, 143.9, 142.1, 139.7, 135.0, 128.6, 128.3, 126.9, 124.0, 107.6.
This method also gave 2-phenyl-1,4-dihydro-1,6-naphthyridin-4-one (18b), 2-phenyl-1,4-dihydro-1,7-naphthyridin-4-one (18c), and 2-phenyl-1,4-dihydro-1,8-naphthyridin-4-one (18d) (see Supporting Information).
2-Phenyl-1,4-dihydroquinazolin-4-one (18e)
A slurry of 4-chloro-2-phenylquinazoline (5.18 g, 21.5 mmol) and NaOH (860 mg, 21.5 mmol) was heated to 110 °C in DMSO (200 mL) for 1 h. This slurry was then cooled to rt, whereupon fine colorless crystals appeared. These were collected and washed with water (500 mL × 2) to give 18e (1.28 g, 27%); mp 238–239 °C (lit. mp62 233–234 °C). HRMS–EI (m/z): [M + H]+ calcd for C14H11N2O, 222.0793; found, 222.0792. 1H NMR (HFIP–d2): δ 8.40 (d, J = 8.0 Hz, 1H), 8.24 (d, J = 8.8 Hz, 1H), 8.16 (dt, J = 7.2, 0.8 Hz, 1H), 7.95 (d, J = 7.6 Hz, 2H), 7.83 (t, J = 8.0 Hz, 1H), 7.60 (t, J = 7.6 Hz, 1H), 7.53 (t, J = 8.0 Hz, 2H), 4.95 (br s, 1H). 13C NMR (HFIP-d2): δ 166.3, 161.8, 151.6, 136.1, 135.6, 130.9, 128.6, 128.3, 127.6, 125.2, 122.7, 114.6. Addition of water to the mother liquor precipitated unreacted starting material (1.37 g).
Method G: N-Methyl-N-phenyl-2-{[2-(pyridin-2-yl)quinolin-4-yl]oxy}propanamide (22a)
2-(Pyridin-2-yl)-1,4-dihydroquinolin-4-one55 (222 mg, 1.00 mmol), 2-bromo-N-methyl-N-phenylpropanamide60 (270 mg, 1.12 mmol), and K2CO3 (834 mg, 6.03 mmol) in MeCN (35 mL) were heated to 55 °C (solution temp) under Ar for 16 h. The mixture was then cooled to rt and poured into stirring water (175 mL). After several min, the precipitate was collected and recrystallized (cyclohexane/EtOAc) to give 22a as colorless plates (267 mg, 70%); mp 185–187 °C. HRMS–ESI (m/z): [M + H]+ calcd for C24H22N3O2, 383.1708; found, 383.1712. 1H NMR (CDCl3): δ 8.77 (dd, J = 4.8, 0.8 Hz, 1H), 8.69 (d, J = 7.6 Hz, 1H), 8.28 (dd, J = 8.4, 0.8 Hz, 1H), 8.07 (d, J = 8.4 Hz, 1H), 7.87 (dt, J = 7.6, 1.6 Hz, 1H), 7.86 (s, 1H), 7.69 (dt, J = 7.2, 1.6 Hz, 2H), 7.51–7.45 (3H), 7.37 (dt, J = 4.8, 0.8 Hz, 2H), 5.06 (q, J = 6.8 Hz, 1H), 3.33 (s, 3H), 1.69 (d, J = 6.4 Hz, 3H). 13C NMR (CDCl3): δ 170.2, 160.8, 157.0, 156.3, 149.1, 148.7, 142.5, 136.8, 130.2, 129.9, 129.0, 128.6, 127.9, 125.8, 124.1, 122.4, 121.7, 121.4, 98.7, 71.1, 38.1, 18.5.
This method also gave N-phenyl-2-{[2-(pyridin-2-yl)quinolin-4-yl]oxy}propanamide (22d) (see Supporting Information).
Determination of Absolute Configuration of Enantiomer of (−)-10a
The solvent-corrected IR and VCD spectra of a 0.2 M solution of (−)-10a in CDCl3 in a BaF2 cell (100 μm path length) were recorded with 4 cm–1 resolution over the span of 22 h. PEM was optimized at 1400 cm–1.
Determination of Ligand Binding Affinities for Rat Brain TSPO
Binding assays were performed as previously described,26 except that crude rat brain homogenates were used instead of mitochondrial fractions. Data were analyzed with nonlinear regression curve-fitting software (GraphPad Prism 5; GraphPad Prism, San Diego, CA, USA). Briefly, whole rat brains from Sprague–Dawley rats were homogenized in cold HEPES buffer (50 mM; pH = 7.4) with a Teflon pestle and Glas-Col homogenizing system. The homogenates were centrifuged at 20000g for 15 min at 4 °C. The pellets were then resuspended, aliquotted into various vials, and stored at −80 °C. A self-displacement assay on 1 was used as a control along with each assay of test ligand with [3H]1 as reference radioligand. The individually calculated control KD values for 1 were compared to the reported value of 0.707 nM26 as an assurance of the correctness of results obtained on test ligands. The KD value of 0.707 nM for 1 was used as the dissociation constant to calculate Ki values for test ligands.
Determination of Ligand Binding Affinity to Human Leukocyte HAB and LAB TSPO
Assays on human leukocyte homogenates were performed as described previously32 Data were analyzed with nonlinear regression curve-fitting software (GraphPad Prism 5; GraphPad Prism). Ki values for the test TSPO ligands were measured in triplicate in two HAB and two low LAB tissues whose genotype had been predetermined. As a control in each assay, a self-displacement assay on 1 was performed to calculate KD. A mean KD of 4.7 nM29 was used to calculate test ligand Ki. All data were fitted to a one-site model to determine the ratio of Kis for test ligands between HABs and LABs.
Acknowledgments
This study was supported by the Intramural Research Program of the National Institutes of Health (NIH), specifically the National Institute of Mental Health. We thank the NIH PET Department for radioisotope production.
Glossary
Abbreviations Used
- DCA
1,2-dichloroethane
- DIPA
diisopropylethylamine
- DPEPhos
bis-[(2-diphenylphosphino)phenyl]ether
- HAB
high-affinity binder
- LAB
low-affinity binder
- MAB
mixed-affinity binder
- HFIP
hexafluoroisopropanol
- PFP
pentafluorophenyl
- PyBroP
bromotripyrrolidinophosphonium hexafluorophosphate
- TEA
triethylamine
- TSPO
translocator protein (18 kDa)
Supporting Information Available
Syntheses of compounds not described in main text and determination of absolute configuration of (−)-10a. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
This paper was written with contributions from all authors. All authors approved the final version of the manuscript.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Anholt R. R. H.; De Souza E. B.; Oster-Granite M. L.; Snyder S. H. Peripheral-type benzodiazepine receptors: autoradiographic localization in whole-body sections of neonatal rats. J. Pharmacol. Exp. Ther. 1985, 233, 517–526. [PubMed] [Google Scholar]
- Papadopoulos V.; Baraldi M.; Guilarte T. R.; Knudsen T. B.; Lacapère J. J.; Lindemann P.; Norenberg M. D.; Nutt D.; Weizman A.; Zhang M.-R.; Gavish M. Translocator protein (18 kDa): new nomenclature for the peripheral-type benzodiazepine receptor based on its structure and molecular function. Trends Pharmacol. Sci. 2006, 27, 402–409. [DOI] [PubMed] [Google Scholar]
- Syapin P. J.; Skolnick P. Characterization of benzodiazepine binding sites in cultured cells of neural origin. J. Neurochem. 1979, 32, 1047–1051. [DOI] [PubMed] [Google Scholar]
- McEnery M. W.; Snowman A. M.; Trifiletti R. R.; Snyder S. H. Isolation of the mitochondrial benzodiazepine receptor: Association with the voltage-dependent anion channel and the adenine nucleotide carrier. Proc. Natl. Acad. Sci. U. S. A. 1992, 89, 3170–3174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doble A.; Malgouris C.; Daniel M.; Daniel N.; Imbault F.; Basbaum A.; Uzan A.; Guérémy C.; Le Fur G. Labelling of peripheral-type benzodiazepine binding sites in human brain with [3H]PK 11195: anatomical and subcellular distribution. Brain Res. Bull. 1987, 18, 49–61. [DOI] [PubMed] [Google Scholar]
- Wright G.; Reichenbecher V. The effects of superoxide and the peripheral benzodiazepine receptor ligands on the mitochondrial processing of manganese-dependent superoxide dismutase. Exp. Cell Res. 1999, 246, 443–450. [DOI] [PubMed] [Google Scholar]
- Papadopoulos V.; Lecanu L.; Brown R. C.; Han Z.; Yao Z.-X. Peripheral-type benzodiazepine receptor in neurosteroid biosynthesis, neuropathology and neurological disorders. Neuroscience 2006, 138, 749–756. [DOI] [PubMed] [Google Scholar]
- Taketani S.; Kohno H.; Okuda M.; Furukawa T.; Tokunaga R. Induction of peripheral-type benzodiazepine receptors during differentiation of mouse erythroleukemia cells—a possible involvement of these receptors in heme biosynthesis. J. Biol. Chem. 1994, 269, 7527–7531. [PubMed] [Google Scholar]
- Benavides J.; Fage D.; Carter C.; Scatton B. Peripheral type benzodiazepine binding sites are a sensitive indirect index of neuronal damage. Brain Res. 1987, 421, 167–172. [DOI] [PubMed] [Google Scholar]
- Cagnin A.; Gerhard A.; Banati R. B. In vivo imaging of neuroinflammation. Eur. Neuropsychopharmacol. 2002, 12, 581–586. [DOI] [PubMed] [Google Scholar]
- Banati R. B. Visualising microglial activation in vivo. Glia 2002, 40, 206–217. [DOI] [PubMed] [Google Scholar]
- Imaizumi M.; Kim H.-J.; Zoghbi S. S.; Briard E.; Hong J.; Musachio J. L.; Ruetzler C.; Chuang D.-M.; Pike V. W.; Innis R. B.; Fujita M. PET imaging with [11C]PBR28 can localize and quantify upregulated peripheral benzodiazepine receptors associated with cerebral ischemia in rat. Neurosci. Lett. 2007, 411, 200–205. [DOI] [PubMed] [Google Scholar]
- Cagnin A.; Brooks D. J.; Kennedy A. M.; Gunn R. N.; Myers R.; Turkheimer F. E.; Jones T.; Banati R. B. In vivo measurement of activated microglia in dementia. Lancet 2001, 358, 461–467. [DOI] [PubMed] [Google Scholar]
- Kreisl W. C.; Lyoo C. H.; McGwier M.; Snow J.; Jenko K. J.; Kimura N.; Corona W.; Morse C. L.; Zoghbi S. S.; Pike V. W.; McMahon F. J.; Turner R. S.; Innis R. B. Biomarkers Consortium PET Radioligand Project Team. In vivo radioligand binding to translocator protein correlates with severity of Alzheimer’s disease. Brain 2013, 136, 2228–2238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ouchi Y.; Yoshikawa E.; Sekine Y.; Futatsubashi M.; Kanno T.; Ogusu T.; Torizuka T. Microglial activation and dopamine terminal loss in early Parkinson’s disease. Ann. Neurol. 2005, 57, 168–175. [DOI] [PubMed] [Google Scholar]
- Debruyne J. C.; Versijpt J.; Van Laere K. J.; De Vos F.; Keppens J.; Strijckmans K.; Achten E.; Slegers G.; Dierckx R. A.; Korf J.; De Reuck J. L. PET visualization of microglia in multiple sclerosis patients using [11C]PK11195. Eur. J. Neurol. 2003, 10, 257–264. [DOI] [PubMed] [Google Scholar]
- Owen D. R. J.; Matthews P. M.. Imaging brain microglial activation using positron emission tomography and translocator protein-specific radioligands. In Biomarkers of Neurological and Psychiatric Disease; Guest P. C.; Bahn S., Eds.; Academic Press: Waltham, MA, 2011; Vol. 101, pp 19–39. [DOI] [PubMed] [Google Scholar]
- Camsonne R.; Moulin M. A.; Crouzel C.; Syrota A.; Maziere M.; Comar D. C-11 Labeling of PK11195 and visualization of peripheral receptors of benzodiazepines by positron-emission tomography. J. Pharmacol. 1986, 17, 383. [Google Scholar]
- Shah F.; Hume S. P.; Pike V. W.; Ashworth S.; McDermott J. Synthesis of the enantiomers of [N-methyl-11C]PK 11195 and comparison of their behaviors as radioligands for PK binding sites in rats. Nucl. Med. Biol. 1994, 21, 573–581. [DOI] [PubMed] [Google Scholar]
- Debruyne J. C.; Van Laere K. J.; Versijpt J.; De Vos F.; Eng J. K.; Strijckmans K.; Santens P.; Achten E.; Slegers G.; Korf J.; Dierckx R. A.; De Reuck J. L. Semiquantification of the peripheral-type benzodiazepine ligand [11C]PK11195 in normal human brain and application in multiple sclerosis patients. Acta Neurol. Belg. 2002, 102, 127–135. [PubMed] [Google Scholar]
- Kropholler M. A.; Boellaard R.; Schuitemaker A.; Folkersma H.; van Berckel B. N. M.; Lammertsma A. A. Evaluation of reference tissue models for the analysis of [11C](R)-PK11195 studies. J. Cereb. Blood Flow Metab. 2006, 26, 1431–1441. [DOI] [PubMed] [Google Scholar]
- Greuter H. N. J. M.; van Ophemert P. L. B.; Luurtsema G.; van Berckel B. N. M.; Franssen E. J. F.; Windhorst B. D.; Lammertsma A. A. Optimizing an online SPE–HPLC method for analysis of (R)-[11C]1-(2-chlorophenyl)-N-methyl-N-(1-methylpropyl)-3-isoquinolinecarboxamide [(R)-[11C]PK11195] and its metabolites in humans. Nucl. Med. Biol. 2005, 32, 307–312. [DOI] [PubMed] [Google Scholar]
- Chauveau F.; Boutin H.; Van Camp N.; Dollé F.; Tavitian B. Nuclear imaging of neuroinflammation: a comprehensive review of [11C]PK11195 challengers. Eur. J. Nucl. Med. Mol. Imaging 2008, 35, 2304–2319. [DOI] [PubMed] [Google Scholar]
- Schweitzer P. J.; Fallon B. A.; Mann J. J.; Kumar J. S. D. PET tracers for the peripheral benzodiazepine receptor and uses thereof. Drug Discovery Today 2010, 15, 933–942. [DOI] [PubMed] [Google Scholar]
- Luus C.; Hanani R.; Reynolds A.; Kassiou M. The development of PET radioligands for imaging the translocator protein (18 kDa): What have we learned?. J. Labelled Compd. Radiopharm. 2010, 53, 501–510. [Google Scholar]
- Briard E.; Zoghbi S. S.; Imaizumi M.; Gourley J. P.; Shetty H. U.; Hong J.; Cropley V.; Fujita M.; Innis R. B.; Pike V. W. Synthesis and evaluation in monkey of two sensitive 11C-labeled aryloxyanilide ligands for imaging brain peripheral benzodiazepine receptors in vivo. J. Med. Chem. 2008, 51, 17–30. [DOI] [PubMed] [Google Scholar]
- Brown A. K.; Fujita M.; Fujimura Y.; Liow J. S.; Stabin M.; Ryu Y. H.; Imaizumi M.; Hong J.; Pike V. W.; Innis R. B. Radiation dosimetry and biodistribution in monkey and man of [11C]PBR28: a PET radioligand to image inflammation. J. Nucl. Med. 2007, 48, 2072–2079. [DOI] [PubMed] [Google Scholar]
- Fujita M.; Imaizumi M.; Zoghbi S. S.; Fujimura Y.; Farris A. G.; Suhara T.; Hong J.; Pike V. W.; Innis R. B. Kinetic analysis in healthy humans of a novel positron emission tomography radioligand to image the peripheral benzodiazepine receptor, a potential biomarker for inflammation. Neuroimage 2008, 40, 43–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreisl W. C.; Fujita M.; Fujimura Y.; Kimura N.; Jenko K. J.; Kannan P.; Hong J.; Morse C. L.; Zoghbi S. S.; Gladding R. L.; Jacobson S.; Oh U.; Pike V. W.; Innis R. B. Comparison of [11C](R)-PK 11195 and [11C]PBR28, two radioligands for translocator protein (18 kDa) in human and monkey: Implications for positron emission tomographic imaging of this inflammation biomarker. Neuroimage 2010, 49, 2924–2932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owen D. R.; Howell O. W.; Tang S.-P.; Wells L. A.; Bennacef I.; Bergstrom M.; Gunn R. N.; Rabiner E. A.; Wilkins M. R.; Reynolds R.; Matthews P. M.; Parker C. A. Two binding sites for [3H]PBR28 in human brain: implications for TSPO PET imaging of neuroinflammation. J. Cereb. Blood Flow Metab. 2010, 30, 1608–1618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owen D. R.; Yeo A. J.; Gunn R. N.; Song K.; Wadsworth G.; Lewis A.; Rhodes C.; Pulford D. J.; Bennacef I.; Parker C. A.; StJean P. L.; Cardon L. R.; Mooser V. E.; Matthews P. M.; Rabiner E. A.; Rubio J. P. An 18-kDa Translocator protein (TSPO) polymorphism explains differences in binding affinity of the PET radioligand PBR28. J. Cereb. Blood Flow Metab. 2012, 32, 1–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreisl W. C.; Jenko K. J.; Hines C. S.; Lyoo C. H.; Corona W.; Morse C. L.; Zoghbi S. S.; Hyde T.; Kleinman J. E.; Pike V. W.; McMahon F. J.; Innis R. B.; Biomarkers Consortium PET Radioligand Project Team. A genetic polymorphism for translocator protein 18 kDa affects both in vitro and in vivo radioligand binding in human brain to this putative biomarker of neuroinflammation. J. Cereb. Blood Flow Metab. 2013, 33, 53–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owen D. R. J.; Gunn R. N.; Rabiner E. A.; Bennacef I.; Fujita M.; Kreisl W. C.; Innis R. B.; Pike V. W.; Reynolds R.; Matthews P. M.; Parker C. A. Mixed-affinity binding in humans with 18-kDa translocator protein ligands. J. Nucl. Med. 2011, 52, 24–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizrahi R.; Rusjan P. M.; Kennedy J.; Pollock B.; Mulsant B.; Suridjan I.; De Luca V.; Wilson A. A.; Houle S. Translocator protein (18 kDa) polymorphism (rs6971) explains in vivo brain binding affinity of the PET radioligand [18F]-FEPPA. J. Cereb. Blood Flow Metab. 2012, 32, 968–972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pike V. W.; Taliani S.; Lohith T. G.; Owen D. R. J.; Pugliesi I.; Da Pozzo E.; Hong J.; Zoghbi S. S.; Gunn R. N.; Parker C. A.; Rabiner E. A.; Fujita M.; Innis R. B.; Martini C.; Da Settimo F. Evaluation of novel N1-methyl-2-phenylindol-3-ylglyoxylamides as a new chemotype of 18 kDa translocator protein-selective ligand suitable for the development of positron emission tomography radioligands. J. Med. Chem. 2011, 54, 366–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo Q.; Colasanti A.; Owen D. R.; Onega M.; Kamalakaran A.; Bennacef I.; Matthews P. M.; Rabiner E. A.; Turkheimer F. E.; Gunn R. N. Quantification of the specific translocator protein signal of 18F-PBR111 in healthy humans: a genetic polymorphism effect on in vivo binding. J. Nucl. Med. 2013, 54, 1915–1923. [DOI] [PubMed] [Google Scholar]
- Guo Q.; Owen D. R.; Rabiner E. A.; Turkheimer F. E.; Gunn R. N. Identifying improved TSPO PET imaging probes through biomathematics: the impact of multiple TSPO binding sites in vivo. NeuroImage 2012, 60, 902–910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubroeucq M.-C.; Bénavidès J.; Doble A.; Guilloux F.; Allam D.; Vaucher N.; Bertrand P.; Guérémy C.; Renault C.; Uzan A.; Le Fur G. Stereoselective inhibition of the binding of [3H]PK 11195 to peripheral-type benzodiazepine binding sites by a quinolinepropanamide derivative. Eur. J. Pharmacol. 1986, 128, 269–272. [DOI] [PubMed] [Google Scholar]
- Pike V. W. Positron-emitting radioligands for studies in vivo—probes for human psychopharmacology. J. Psychopharmacol. 1993, 7, 139–158. [DOI] [PubMed] [Google Scholar]
- Laruelle M.; Slifstein M.; Huang Y. Relationships between radiotracer properties and image quality in molecular imaging of the brain with positron emission tomography. Mol. Imaging Biol. 2003, 5, 363–375. [DOI] [PubMed] [Google Scholar]
- Patel S.; Gibson R. In vivo site-directed radiotracers: a mini-review. Nucl. Med. Biol. 2008, 35, 805–815. [DOI] [PubMed] [Google Scholar]
- Leeson P. D.; Springthorpe B. The influence of drug-like concepts on decision-making in medicinal chemistry. Nature Rev. Drug Discovery 2007, 6, 881–890. [DOI] [PubMed] [Google Scholar]
- Ryckmans T.; Edwards M. P.; Horne V. A.; Correia A. M.; Owen D. R.; Thompson L. R.; Tran I.; Tutt M. F.; Young T. Rapid assessment of a novel series of selective CB2 agonists using parallel synthesis protocols: a lipophilic efficiency (LipE) analysis. Bioorg. Med. Chem. Lett. 2009, 19, 4406–4409. [DOI] [PubMed] [Google Scholar]
- Shultz M. D. Setting expectations in molecular optimizations: strengths and limitations of commonly used composite parameters. Bioorg. Med. Chem. Lett. 2013, 23, 5980–5991. [DOI] [PubMed] [Google Scholar]
- Kuninobu Y.; Inoue Y.; Takai K. Copper(I)- and gold(I)-catalyzed synthesis of 2,4-disubstituted quinoline derivatives from N-aryl-2-propynylamines. Chem. Lett. 2007, 36, 1422–1423. [Google Scholar]
- Frérot E.; Coste J.; Pantaloni A.; Dufour M.-N.; Jouin P. PyBOP and PyBroP: two reagents for the difficult coupling of the α,α-dialkyl amino acid, Aib. Tetrahedron 1991, 47, 259–270. [Google Scholar]
- Woodbury R. P.; Rathke M. W. Isolation and reactions of α-lithio N,N-dimethylacetamide. J. Org. Chem. 1977, 42, 1688–1690. [Google Scholar]
- VanAllan J. A. A new process for the preparation of thioglycolylamides. J. Am. Chem. Soc. 1947, 69, 2914. [Google Scholar]
- Wolf C.; Lerebours R. Use of highly active palladium–phosphinous acid catalysts in Stille, Heck, amination, and thiation reactions of chloroquinolines. J. Org. Chem. 2003, 68, 7077–7084. [DOI] [PubMed] [Google Scholar]
- Margolis B. J.; Long K. A.; Laird D. L. T.; Ruble J. C.; Pulley S. R. Assembly of 4-aminoquinolines via palladium catalysis: a mild and convenient alternative to SNAr methodology. J. Org. Chem. 2007, 72, 2232–2235. [DOI] [PubMed] [Google Scholar]
- Knouzi N.; Vaultier M.; Carrié R. Réduction d’azides par la triphénylphosphine en présence d’eau: une méthode générale et chimiosélective d’accès aux amines primaires. Bull. Soc. Chim. Fr. 1985, 122, 815–819. [Google Scholar]
- Winters R. T.; Sercel A. D.; Showalter H. D. H. Efficient synthesis of peri-hydroxylated 9,10-anthracenedione ethers via alkylation of cesium phenolates. Synthesis 1988, 712–714. [Google Scholar]
- Nahm S.; Weinreb S. M. N-Methoxy-N-methylamides as effective acylating agents. Tetrahedron Lett. 1981, 22, 3815–3818. [Google Scholar]
- Camps R. Synthese von α- und γ-oxychinolinen. Ber. Dtsch. Chem. Ges. 1899, 32, 3228–3234. [Google Scholar]
- Jones C. P.; Anderson K. W.; Buchwald S. L. Sequential Cu-catalyzed amidation-base-mediated Camps cyclization: a two-step synthesis of 2-aryl-4-quinolones from o-halophenones. J. Org. Chem. 2007, 72, 7968–7973. [DOI] [PubMed] [Google Scholar]
- He Y.; Wang B.; Dukor R. K.; Nafie L. A. Determination of absolute configuration of chiral molecules using vibrational optical activity: a review. Appl. Spectrosc. 2011, 65, 699–723. [DOI] [PubMed] [Google Scholar]
- Cappelli A.; Anzini M.; Vomero S.; De Benedetti P. G.; Menziani M. C.; Giorgi G.; Manzoni C. Mapping the peripheral benzodiazepine receptor binding site by conformationally restrained derivatives of 1-(2-chlorophenyl)-N-methyl-N-(1-methylpropyl)-3-isoquinolinecarboxamide (PK11195). J. Med. Chem. 1997, 40, 2910–2921. [DOI] [PubMed] [Google Scholar]
- Pike V. W.; Halldin C.; Crouzel C.; Barré L.; Nutt D. J.; Osman S.; Shah F.; Turton D. R.; Waters S. L. Radioligands for PET studies of central benzodiazepine receptors and PK (peripheral benzodiazepine) binding sites—current status. Nucl. Med. Biol. 1993, 20, 503–525. [DOI] [PubMed] [Google Scholar]
- Pettigrew J. D.; Freeman R. P.; Wilson P. D. Total synthesis of (−)-xyloketal D and its enantiomer—confirmation of absolute stereochemistry. Can. J. Chem. 2004, 82, 1640–1648. [Google Scholar]
- Bischoff C. A. Studien über Verkettungen. XXVI. Das Aethylanilin. Ber. Dtsch. Chem. Ges. 1897, 30, 3178–3180. [Google Scholar]
- Garnier E.; Blanchard S.; Rodriguez I.; Jarry C.; Léger J.-M.; Caubère P.; Guillaumet G. New access to oxazolopyridines via hydroxyamidine derivatives; application to quinolines. Synthesis 2003, 2033–2040. [Google Scholar]
- Körner M. Ueber das Benzoyl-o-Amidobenzamid. J. Prakt. Chem. (Leipzig) 1887, 36, 155–165. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.