

Abstract

Compounds active at neurotensin receptors (NTS1 and NTS2) exert analgesic effects on different types of nociceptive modalities, including thermal, mechanical, and chemical stimuli. The NTS2 preferring peptide JMV-431 (2) and the NTS2 selective nonpeptide compound levocabastine (6) have been shown to be effective in relieving the pain associated with peripheral neuropathies. With the aim of identifying novel nonpeptide compounds selective for NTS2, we examined analogues of SR48692 (5a) using a FLIPR calcium assay in CHO cells stably expressing rat NTS2. This led to the discovery of the NTS2 selective nonpeptide compound 1-({[1-(4-fluorophenyl)-5-(2-methoxyphenyl)-1H-pyrazol-3-yl]carbonyl}amino)cyclohexane carboxylic acid (NTRC-739, 7b) starting from the nonselective compound 5a.
Introduction
Neurotensin (NT) is a tridecapeptide (Glu-Leu-Tyr-Glu-Asn-Lys-Pro-Arg-Arg-Pro-Tyr-Ile-Leu) that was identified 40 years ago from bovine hypothalamus.1 NT functions as a neurotransmitter and neuromodulator demonstrating a range of biological actions.2 In the CNS, NT is colocalized with and regulates the action of mesolimbic and nigrastriatal dopamine3−7 as well as mediating nonopioid analgesia and hypothermia.8,9 It is believed that NT operates primarily through interaction with two G-protein coupled receptors NTS1 and NTS2 (also referred to as NTR1, NTR2, NTRH, NTRL) to regulate these activities. A third receptor, NTS3, also known as sortilin, is a type I receptor bearing a single transmembrane domain.10−14 The ability of the NT receptor system to regulate CNS dopamine led researchers to postulate that NT might be an endogenous neuroleptic15 and that drugs acting via the NT system might therefore be useful as antipsychotic agents.16,17 Methamphetamine also produces profound disruption of dopamine flow in the mesolimbic and nigrastriatal dopamine networks with chronic methamphetamine exposure, eliciting behaviors resembling schizophrenia.18,19 It is thus no surprise that compounds acting via the NT network are now being investigated as potential treatments for methamphetamine abuse.20,21
The NT system also plays an important role in the pain processing network.8,22 The nonopioid analgesia mediated by the NT system has also generated much interest over the years as a potential means of circumventing the side effects produced by opioid analgesics including addiction, constipation, and respiratory depression. NT-mediated analgesia has been demonstrated with compounds selective for both the NTS1 and NTS2 receptors as well as nonselective compounds.23−28 Beyond this, there is now substantial evidence that both NTS1 and NTS2 can mediate relief from chronic or neuropathic pain.29 This type of pain is difficult to treat with current drugs and does not always respond well to opioid therapy.30 Taken together with their neuroleptic activity, it is easy to understand why the development of compounds acting via the NT network has engendered so much interest.
The NTS1 receptor has received the most attention since the discovery of the NT system as it binds NT with high affinity, hence the name NTRH (NT receptor high affinity), and is well behaved in heterologous expression systems. The NTS2 receptor, on the other hand, has received less attention by comparison, and as a consequence fewer compounds have thus far been identified that interact with this receptor. As is true for NTS1, the most potent and selective NTS2 compounds discovered to date have been obtained via modification of the NT(8–13) (1) fragment.31 Discovery of potent and selective nonpeptide compounds however has proven elusive and remains as a significant challenge.
Several important milestone compounds from NT receptor research are depicted in Chart 1. The 8–13 fragment of NT, compound 1, is as potent as the full length peptide, and several hexapeptide variants of 1 have been reported over the years that either favor NTS2 over NTS1 or are selective for NTS2 versus NTS1. The first of these is JMV-431 (2) that was produced via reduction of the tyrosine 11 amide bond and shows a clear preference for NTS2.32,33 While it is not bioavailable, it is active in several models of chronic pain when delivered intrathecally.28,29 The peptide NT79 (3), reported more recently,34 is selective for NTS2 (>100-fold) and has provided a wealth of information regarding the role of NTS2 in animal models of pain, antipsychotic activity, thermoregulation, and regulation of blood pressure.34 Like peptide 2, this compound attained NTS2 selectivity via modification of the tyrosine 11 residue. The most recent additions to the rolls of NTS2-selective compounds are the potent peptide–peptoid hybrids 4a and 4b.35,36 These compounds are ultraselective for NTS2, with selectivity ratios reaching 12000- and 22000-fold, respectively, with 4b also demonstrating excellent plasma stability.
Chart 1.

AlThough few in number, there are three prominent nonpeptide compounds that interact with NTS2 that have been used extensively in the characterization of the NT receptors. These include the two pyrazole-based compounds 5a (SR48692) and SR142948a (5b) and the histamine blocker levocabastine (6).37−39 Pyrazole compound 5a prefers NTS1 to NTS2 while 5b is nonselective, but they both antagonize the activity of NT at NTS1. Compound 6 is selective for NTS2 versus NTS1, but it is also a potent antagonist at histamine receptor 1 (H1). These compounds (5a,b and 6) highlight the fact that while NTS2-selective peptides exist, selective nonpeptide compounds are rare.
The SAR of pyrazole compounds 5a and 5b has been described at NTS1 but, to our knowledge, no systematic survey of SAR has ever been described at NTS2. With the goal of discovering novel NTS2 selective ligands, we undertook this endeavor by adapting a previously described calcium mobilization assay to our high throughput FLIPR Tetra system and confirmed the functional results of active compounds using a radioligand binding assay.40 This effort led to the identification of a nonpeptide compound that is selective for NTS2 versus NTS1, 1-({[1-(4-fluorophenyl)-5-(2-methoxyphenyl)-1H-pyrazol-3-yl]carbonyl}amino)cyclohexane carboxylic acid (7b), starting from the nonselective compound 5a. The newly discovered compound (7b) displays neither calcium nor binding activity at NTS1, but is a potent partial agonist at NTS2 (EC50 of 12 ± 6 nM and an Emax of 7% relative to 5b) that gave a Ki of 153 ± 10 nM in competition with [125I]NT. The details of this effort are presented herein.
Chemistry
The target compounds, depicted in Tables 2 and 3, were synthesized as described in Schemes 1–2. Commercially available materials were used when available. Those not commercially available were prepared according to literature precedent. Scheme 1 illustrates the synthesis of the key intermediate pyrazole carboxylic acids (11a–j). These were prepared as described by Labeeuw,41 although improved methods were recently described by Jiang et al.42 and also Baxendale et al.43 This employed a Knorr [3 + 2]-cyclization reaction between 4-aryl-2,4-diketoesters (8a–f) and arylhydrazines 9a–c in acetic acid at reflux. The resulting esters 10a–j were hydrolyzed using LiOH and dioxane to give 11a–j. The 4-aryl-2,4-diketoesters were commercially available with the exception of 8b. The 2,6-dimethoxy-butyrophenone used to synthesize compound 8b was prepared exactly as described by Lindh.44
Table 2. Antagonism of NT Induced Calcium Release at NTS1 Compared to Target Compound Induced Calcium Mobilization and Binding Affinity at NTS2 for 7-Chloroquinolyl (A), Naphthyl (B), and 4-F-Phenyl (C) Substituted Pyrazole Carboxamides at the NTS1 and NTS2 Receptors.
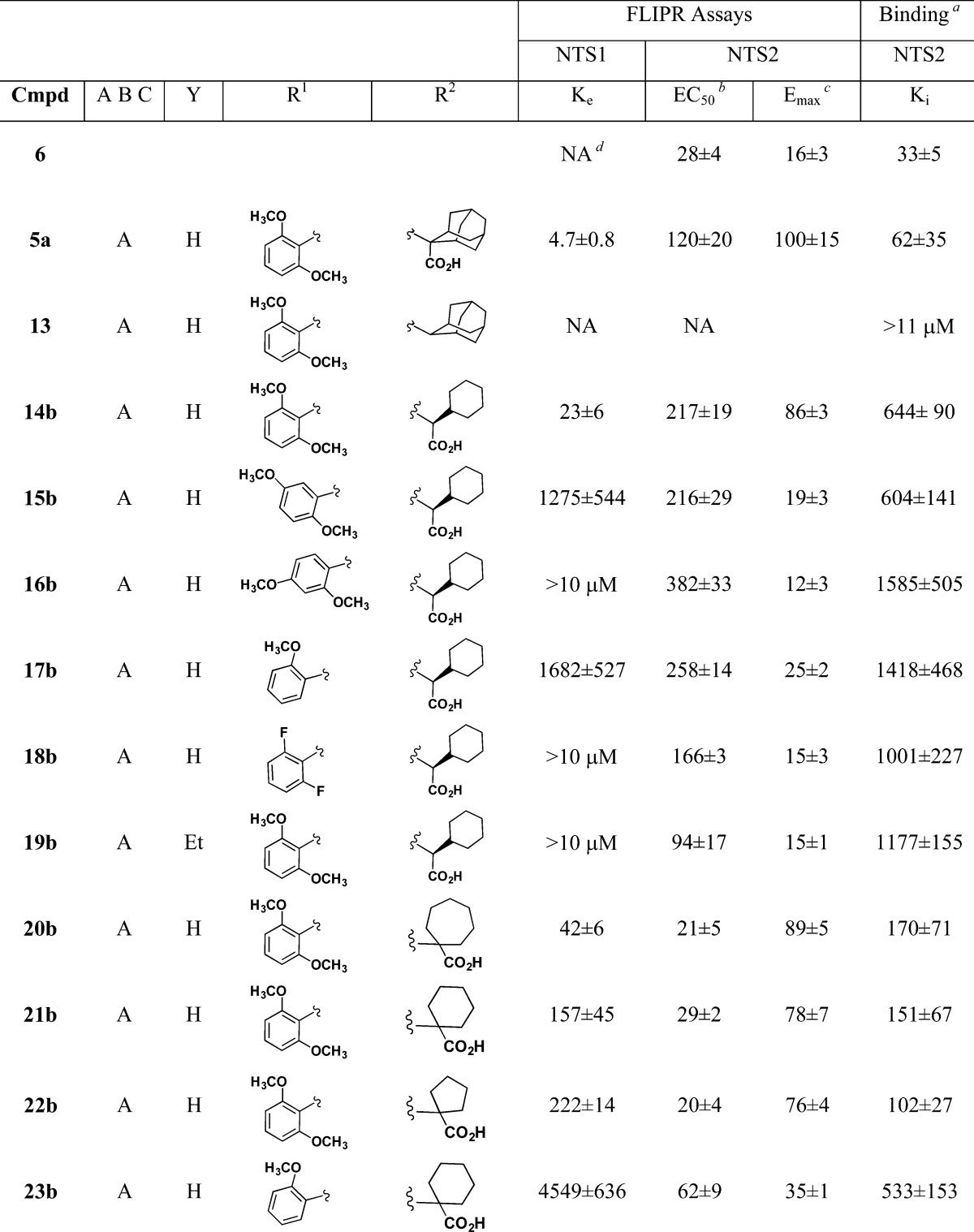
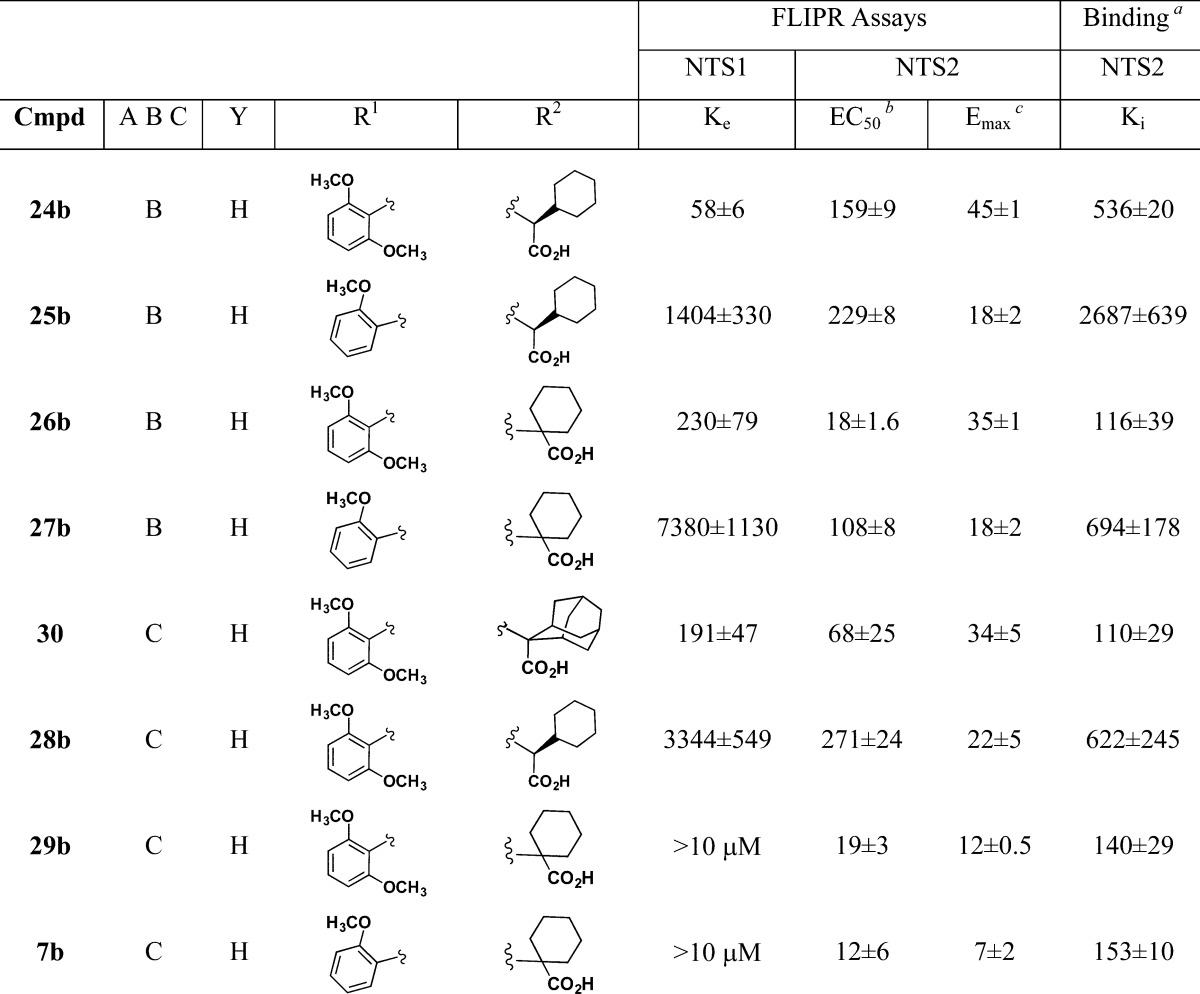
[125I]NT.
EC50, Ke, IC50, and Ki values are all nM ± SEM.
Emax value is % 5b.
Not active.
Table 3. Selectivity Ratios for 29b and 7b at the NTS1 and NTS2 Receptors Determined Using [125I]NT Radioligand Binding.
| compd | NTS1 Ki nM ± SEM | NTS2 Ki nM ± SEM | NTS2/NTS1 |
|---|---|---|---|
| 29b | 3210 ± 879 | 140 ± 29 | 23 |
| 7b | >25 μM | 153 ± 10 | 161 |
Scheme 1. Synthesis of Key Pyrazole Intermediates 11a–j.

Reagents and conditions: (i) HOAc, HCl, and 9a (7-chloroquinolin-4-yl)hydrazine·HCl)) or 9b (1-naphthylhydrazine·HCl) or 9c (4-fluorophenylhydrazine·HCl), reflux 4 h; (ii) LiOH 3 equiv, dioxane, RT 16 h.
Scheme 2. Synthesis of Target Compounds 7b, 13, 14b–29b, and 30.

Reagents and conditions: (i) HBTU, Et3N, CH2Cl2, 2-aminoadamantane·HCl, RT, 16 h (12e); (ii) SOCl2, toluene, reflux, 3 h; (iii) NaOH, THF, 12f, RT, 16 h; (iv) HBTU, Et3N, CH2Cl2, amino acid ester 12d, RT, 16 h; (v) TFA, CH2Cl2, RT, 16 h; (vi) HBTU, Et3N, CH2Cl2, amino acid ester 12a–c, RT, 16 h; (vii) LiOH, dioxane, RT, 16 h.
In Scheme 2, the synthetic methods used to produce target compounds 7b, 13, 14b–29b, and 30 are described. Thus, a given pyrazole carboxylic acid (11a–j) and amino acid ester (12a–d) or amine (12e) from Chart 2 were coupled using O-benzotriazol-1-yl-N,N,N′,N′-tetramethyluronium hexafluorophosphate (HBTU) and triethylamine to give ester intermediates 7a, and 14a–29a and the descarboxy target compound 13. The ester intermediates 7a, and 14a–29a were hydrolyzed to give final products using either basic (LiOH and dioxane) or acidic (trifluoroacetic acid (TFA) in CH2Cl2) conditions as shown. We used the tert-butyl rather than the methyl esters of l-cyclohexylglycine (12d) to avoid racemization of the chiral amino acid. We used the basic conditions for hydrolysis of achiral amino acid esters. The synthesis of target compound 30 was accomplished according to the method of Quére,´45 wherein pyrazole acid 11i was converted to its acid chloride using SOCl2 in toluene, followed by coupling of the acid chloride intermediate with the adamantyl amino acid 12f under Schotten–Bauman conditions. The adamantyl amino acid 12f was prepared as described by Nagasawa.46
Chart 2. Amines, Amino Acids, and Amino Acid Esters Used to Prepare Target Compounds 7b, 13, and 14b–29b.

Biological Results
The binding affinities of the test compounds for the rNTS1 and rNTS2 receptors listed in Tables 1 through 3 were determined using previously reported competitive binding assays.40 The NTS1 and 2 receptors were labeled using [125I]NT. The cells used in the binding assay were CHO-k1 cells (American Type Culture Collection) engineered to overexpress either the rNTS1 or rNTS2 receptor. Measures of functional agonist and antagonist activity were obtained by measuring changes in intensity of a calcium-sensitive fluorescent dye as an indirect measure of changes in internal calcium concentrations. These measurements were performed using a FLIPR Tetra (NTS2) or a FlexStation II plate reader for NTS1 (Molecular Devices) and were analyzed using GraphPad Prism software. Antagonist activity was measured as inhibition of NT-induced calcium release (NTS1) or inhibition of 5b-induced calcium release (NTS2).
Table 1. Functional and Radioligand Binding Data Obtained for NT, 1, 5a, 5b, and 6 at the NTS1 and NTS2 Receptors.
| FLIPR assay |
bindinga | ||||||
|---|---|---|---|---|---|---|---|
| NTS1 |
NTS2 |
NTS2 | |||||
| compd | EC50b | Emaxc | Ke | EC50 | Emaxd | IC50 | Ki |
| NT | 0.04 ± 0.012 | 100 ± 3 | NAe | 18.5 ± 1.2 | 18.9 ± 3 | ||
| 1 | 0.01 ± 0.002 | 114 ± 7 | NA | 5.4 ± 0.6 | 33 ± 11 | ||
| 5a | 4.7 ± 0.8 | 120 ± 20 | 100 ± 3 | 62 ± 35 | |||
| 5b | 1.5 ± 0.6 | 20 ± 5 | 100 ± 5 | 6 ± 2 | |||
| 6 | NA | NA | 28 ± 4 | 16 ± 3 | 33 ± 5 | ||
[125I]NT,
EC50, Ke, IC50, and Ki values are nM ± SEM.
Emax value is % NT.
Emax value is % 5b.
Not active.
Results and Discussion
In this article, we provide the details of our effort to identify nonpeptide compounds that are selective for NTS2 starting from the nonselective compound 5a. This included developing an assay capable of detecting activity at the NTS2 receptor based upon literature reports that compound 5a is an agonist at NTS2 (induced calcium mobilization at the NTS2 receptor). A summary of the key SAR elements of 5a discovered using the NTS2 calcium mobilization assay that led to the identification of the potent partial agonist 7b (NTRC-739) are provided in Table 2 and includes the NTS2 EC50 as well as the NTS1 Ke and the NTS2 binding affinity (Ki). Each of the appended molecular regions of 5a is discussed herein and includes: the dimethoxyphenyl ring, the amino acid side chain, the 7-chloroquinoline ring, and the 4-position of the pyrazole ring. The data obtained from the testing of NT reference compounds in this assay, including NT, 1, 5a,b and levocabastine (6), are provided in Table 1.
In attempting to develop a method of screening for NTS2 activity, we observed that the literature reports dealing with functional assays of NTS2 receptors yielded contradictory data, exhibiting cell-type expression- and species-dependent pharmacological properties with opposing patterns. Indeed, NT has been reported to be an agonist, an inverse agonist, and a neutral antagonist depending upon the cell expression system.13,47−49 Similar findings were reported for compounds 5a, 5b, and 6 as well when tested alongside NT in these systems. Specifically, the potent NTS1 antagonists 5a and 5b were found to be agonists at NTS2 as they mobilized calcium release while levocabastine (6) was found to behave either as an agonist, antagonist, or even as a partial inverse agonist.47−49
While the information above illustrated that there was much inconsistency between cell lines and expression systems for NTS2, we believed that each system would produce internally consistent (reproducible) data. We moved forward with the working hypothesis that the calcium mobilization described by these various laboratories was indeed NTS2-mediated and chose one system to study in greater detail. As we already had the CHO-k1-rNTS2 cell line available from previous work, this became the obvious choice.40 We studied the calcium release produced by analogues of the nonpeptide compound 5a by measuring their ability to either stimulate release of calcium or to block the calcium release stimulated by 5b and found that it responded to changes in structure (SAR), Table 2. This information was compared with that obtained for (NT and 1) as well as levocabastine, which revealed that this assay was capable of identifying a range of activities from potent agonist (5a and 5b) to partial agonist (6) to antagonist (NT and 1), Table 1 below, which implied that the structure of the compound made a direct impact on the resulting mobilization of calcium. This indicated that our hypothesis was correct and that we could identify NTS2 agonists directly and NTS2 antagonists indirectly against the agonist activity of 5b.
Further support for our hypothesis was obtained by collecting the binding affinity data in competition with [125I]NT for our study set of analogues of 5a,Table 2. These data revealed that compounds that modulated calcium release also competed with NT for binding at NTS2. Involvement of NTS2 was implicated further by the observation that compounds that mobilized calcium release in the CHO-k1-rNTS2 cell line did not produce calcium mobilization in the parent CHO line. Taken together, these data indicated that this assay method could be used to steer SAR studies in search of novel NTS2 selective compounds.
As described above, the baseline for our study was established using the compounds common to NT receptor research. In Table 1, we have summarized the calcium release and binding data obtained for NT, 1, 5a, and 5b, and the NTS2-selective compound levocabastine (6) in both our rNTS1 and rNTS2 cell lines. In our CHO-k1-NTS2 cell line, neither NT nor 1 showed any calcium release in our FLIPR assay as previously reported.49 Furthermore, we did not observe constitutive activity in this expression system as has been reported by other investigators using alternate expression systems.35,50 On the other hand, both 5a and 5b stimulated calcium release with EC50 values of 120 and 20 nM, respectively. Compound 5b was more potent than compound 5a in the calcium mobilization assay but equally efficacious, and was designated as our NTS2 full agonist standard (the use of the term agonist here refers to a compound that stimulates calcium release in this assay). Levocabastine (6), a compound used for years to distinguish NTS1 from NTS2, was also tested and found to be a potent partial agonist with an EC50 of 28 nM and an Emax of 16% relative to 5b. The activity of 5b and 6 are illustrated in Figure 1.
Figure 1.

Dose–response curves for 5b and 6 in CHO-k1-rNTS2 cells.
Neurotensin and 1 did not stimulate calcium release, but they both blocked the calcium release stimulated by compound 5b in an insurmountable manner as they showed a rightward shift in the dose–response curve with an accompanying dose-dependent drop in the Emax value of 5b.51,52 This is illustrated for NT in Figure 2. We thus determined and report here the IC50 for both NT and 1 in competition with 5b and found these to be 18.9 and 5.4 nM, respectively. The IC50 curve generated for NT against 5b is depicted in Figure 3.
Figure 2.

NT is an insurmountable antagonist of 5b mediated calcium mobilization.
Figure 3.

IC50 curve for NT versus 5b.
Corroboration of the functional data was obtained in the binding assay. We found that compound 5a gave a Ki of 62 nM, while 5b showed a 10-fold higher affinity with a Ki of 6 nM. As in the FLIPR assay, compound 5b has a higher affinity than 5a. NT gave a Ki of 18.5 nM. Compound 1 showed similar affinity to NT, while levocabastine (6) gave a Ki of 33 nM. The binding affinities found for NT, 1, and levocabastine are slightly higher than those found in other laboratories, while those for 5a and b are similar. We attribute this to our use of an in-plate whole cell binding method40 as opposed to the use of membrane preparations.
Relating the functional behaviors observed in vitro for these reference compounds to their demonstrated in vivo behaviors is instructive from a drug discovery perspective. NT possesses antipsychotic activity and mediates nonopioid analgesia, while both NT and levocabastine (6) are active in models of persistent pain.29,34 The pyrazole compounds 5a and 5b, on the other hand, are not analgesics in vivo but instead block the analgesic activity of NT in animal models of pain.28,53,54 This is also observed in animal models based on runaway mesolimbic dopamine where the neuroleptic action of NT is blocked by compounds 5a and 5b.21,38,55,56 According to the functional data in Table 1, the proven analgesic and antipsychotic compounds appeared as either antagonists of calcium mobilization (NT and peptide 1) or potent partial agonists of calcium mobilization (levocabastine, 6), while the compounds that possess neither analgesic nor antipsychotic activity in vivo, compounds 5a and 5b, appeared as potent agonists. This point of reference provided a means of parsing in vitro active compounds into two categories, those that might possess desirable behaviors in animal models and those that might not. As this data set is small, we set out to buttress these relationships using our functional assay to identify additional examples of novel NTS2-selective antagonists and potent partial agonists. This article focuses attention on the latter.
The results obtained in our SAR study to find a potent partial agonist based on compound 5a were centered on the three pyrazole scaffolds A–C, Table 2. We began by examining the effect of deleting the carboxylic acid group, compound 13, as the carboxyl group is known to be a primary attachment point for ligands of the NT receptors.57−59 We found that the descarboxy derivative 13 was inactive in both NTS1 and NTS2 FLIPR assays and gave a Ki > 11 μM in the radioligand binding assay. This behavior stands in stark contrast to the activity found for 5a and provided additional evidence that the calcium release observed for 5a (and 5b) results from these ligands interacting with NTS2.
We chose to use compound 14b as a surrogate for compound 5a because it was easier to produce compound libraries using the l-cyclohexyl glycine amino acid compared with the adamantyl amino acid. Compound 14b, like 5a, was previously reported to be a potent NTS1 antagonist by Quéré60 and thus was expected to be an agonist in the NTS2. The l-isomer was chosen as it is known to be more active than the R isomer.61 The data from 14b indicated that it would be a suitable stand in for 5a as it provided comparable agonist potency and efficacy for NTS2 (EC50 of 217 nM and Emax 86% of 5b) and NTS1 antagonist activity (Ke of 23 nM). The 10-fold difference in its relative binding affinity at NTS2 illustrated to us that the structure of the amino acid side chain significantly impacts binding affinity.
A comparison of the data obtained for compounds 14b–18b (Table 2) illustrates the SAR realized from changing the position of the 6-methoxy group (14b, 15b, and 16b), elimination of the 6-methoxy group (17b), and replacing the 2,6-methoxy groups with fluorine atoms (18b). We found that the NTS1 antagonist activity decreased across the series of positional isomers 14b–16b with Ke values of 23 nM, 1275 nM, and >10 μM, respectively. This trend continued for 17b and the difluoro derivative (18b), with Ke values of 1682 nM and >10 μM. Their NTS2 efficacy (Emax) also fell across this series of compounds while the functional potency (EC50) was little varied. The binding affinities showed surprisingly little variation compared with the change seen going between 5a and 14b and reinforced the notion that the structure of the amino acid side chain has a strong impact on binding affinity and appeared to set the range of observed activity within the study group.
Taken together, we found that the potency of antagonist activity for 14b at the NTS1 receptor relied heavily upon the 2,6-dimethoxyphenyl ring for its activity. At NTS2, however, it was the efficacy of calcium release that was most significantly affected by alteration of the 2,6-dimethoxyphenyl ring. The binding affinity data and NTS2 potency were much less affected by changes to this molecular region. This in turn suggested that an NTS2 potent partial agonist would not possess the 2,6-dimethoxyphenyl ring.
The data from compound 19b illustrated the effect on in vitro activity produced by alkylation of the 4-position of the pyrazole ring. The changes seen here resembled the previous set of compounds as 19b showed lower NTS1 antagonist relative to 14b and decreased efficacy at NTS2 with less effect on NTS2 potency and binding affinity. Overall, this example illustrated that alkylation of this position provided compounds that favored NTS2 activity over NTS1 but with only modest levels of affinity at NTS2. Collectively, the data suggested that this was not a substitution that would lead in the right direction.
The amino acid side chain is known to have a strong influence on ligand behavior, presumably due to its proximity to the carboxyl group, the primary anchoring point of the ligand to the receptor. The data obtained for compounds 5a, 14b, and 20b–23b in Table 2 illustrate the SAR of the large amino acid side chains. This group of compounds demonstrated that large cyclic (cycloheptyl, 20b), large bicyclic (adamantyl, 5a), and large pendant alicyclic (14b) structures are potent NTS1 antagonists with Ke values of 42, 4.7, and 23 nM, respectively. Compounds with smaller cyclic rings were somewhat less potent at NTS1, Ke = 222 nM for the five-membered cyclic ring (22b) and 157 nM for the six-membered cyclic ring (21b). According to these data then, the compounds with smaller cyclic rings (21b, 22b) trended toward NTS2 selectivity. Compounds 20b–22b also showed increased NTS2 agonist potency (lower EC50), but this was accompanied by full efficacy (high Emax relative to 5b) at NTS2, exactly opposite of that found in the potent partial agonist levocabastine (6).
In their favor, however, these same compounds (20b–22b) showed improved binding affinity at NTS2 versus 14b (Ki = 170, 151, and 102 nM versus 644 nM for 14b), more in line with 5a (Ki = 62 nM). For these compounds, the binding affinity and NTS2 potency improvement worked in concert with the lower NTS1 activity to provide enhanced NTS2 affinity and potency while maintaining low efficacy and lower NTS1 activity. This comparison of NTS2 agonist potency to the NTS1 antagonist potency for compounds 21b and 23b was an important clue to achieving selectivity in this series of compounds as this data reinforced the data from the previous series, which showed that the NTS1 receptor relied much more heavily upon the 2,6-dimethoxyphenyl ring for its activity compared with NTS2 and that the amino acid side chain could work in concert with the methoxyphenyl ring to retain these desired properties. This trend was further advanced in compounds that did not possess the chloroquinoline group.
Two key substitutions for the 7-chloroquinoline group (A) were the naphthyl (B) and 4-fluorophenyl (C) groups, Table 2. These substitutions were included in our compound libraries because the naphthyl compounds were known to yield NTS1 antagonists45 and the 4-fluorophenyl group was found in levocabastine (6). Like their 7-chloroquinolyl counterparts, the naphthyl-substituted 2,6-dimethoxyphenyl derivatives 24b and 26b possess potent NTS1 antagonist activity and also NTS2 agonist activity; however, distinct differences were observed that led us in the right direction.
Most apparent was that these compounds were inherently less potent at NTS1 and less efficacious at NTS2. The Ke values for NTS1 antagonist activity and the NTS2 Emax values were roughly half of that found for similarly substituted chloroquinoline-based compounds. This was observed for both the l-cyclohexyl glycine (24b) and the 1-aminocyclohexancarboxylic acid side chains (26b). Comparing the naphthyl (24b) and the 7-chloroquinolyl (14b) compounds, we found Ke values of 58 versus 23 nM and NTS2 Emax values of 45 and 86% of 5b, respectively. Comparing compounds 26b and 21b, we found Ke values of 230 versus 157 nM and NTS2 Emax values of 35 and 78%, respectively.
As seen in the 7-chloroquinolyl series (A), the naphthyl-substituted 2-methoxyphenyl derivatives 25b and 27b were less potent NTS1 antagonists than their 2,6-dimethoxyphenyl substituted counterparts. More surprising was the observation that the NTS2 efficacies for these compounds were nearly half again as low as that found in similarly substituted 7-chloroquinolyl compounds 17b and 23b. This is readily revealed in a comparison of the NTS2 efficacy data obtained for compound 21b, 23b, and the 2-methoxy naphthyl substituted compound 27b, with NTS2 Emax values of 78, 35, and 18%, respectively.
The naphthyl-substituted compounds with the 2,6-dimethoxyphenyl ring were more potent at NTS2 than their chloroquinoline-based counterparts (14b, 17b, 21b, and 23b), but naphthyl compounds bearing the 2-methoxyphenyl ring were equally potent. This was observed for compounds with the l-cyclohexyl glycine as well as those with 1-aminocyclohexancarboxylic acid as in 26b and 27b.
The NTS2 binding data observed for the naphthyl-substituted compounds 24b–27b was found to be in line with that seen for the similarly substituted 7-chloroquinolyl-substituted compounds, with the 2-methoxy derivatives showing lower affinity (higher Ki values) than the 2,6-dimethoxy counterparts. As before though, the compounds bearing the l-cyclohexyl glycine side chain (24b,25b) were less potent than those with the 1-aminocyclohexancarboxylic side chain (26b,27b).
Toward obtaining an NTS2 selective potent partial agonist, the calcium data profile for compound 26b looked promising as it moved closer to that of levocabastine (6). Compound 26b showed 2-fold greater NTS2 potency and comparable efficacy. The binding data was also similar, but in this case, compound 6 was more potent, showing a Ki 3.5-fold greater than that for 26b. The similarity between these two compounds ended, however, when comparing receptor selectivity, as compound 26b showed substantial NTS1 antagonist activity while levocabastine (6) displayed no activity at NTS1. The 2-methoxy naphthyl-substituted analogue 27b was not able to compensate for this deficit, for while its NTS1 antagonist activity was lowered by 20-fold, the NTS2 potency and binding affinity also suffered substantial losses.
Fortunately, the pyrazole scaffold (C) bearing the 4-fluorophenyl-substituent seen in compounds 30, 28b, 29b, and 7b overcame the deficiencies found in scaffold B to provide selective potent partial agonists. Comparison of the adamantyl-substituted compounds 5a and 30 highlights the contribution of the 4-fluorophenyl ring. These data show that the NTS1 antagonist activity was diminished by 40-fold for 30 versus 5a, with Ke values of 191 and 4.7 nM, respectively. The NTS2 potency, on the other hand, was doubled (EC50 values of 120 versus 68 nM) while the efficacy was less than half relative to 5a (Emax values of 100 versus 34% of 5b). This phenomenon was also observed in the data for compounds 28b, 29b, and 7b. These compounds are all far less active at NTS1 compared with their corresponding 7-chloroquinolyl (14b, 21b, and 23b) or naphthyl (24b, 26b, and 27b) analogues and thereby attain enhanced NTS2 selectivity with respect to calcium release. The 4-fluorophenyl-substituted compounds (29b and 7b), bearing the 1-aminocyclohexancarboxylic acid substituent, showed the most levocabastine-like profiles whether they had a 2,6-dimethoxyphenyl ring (29b) or a 2-methoxyphenyl ring (7b). In keeping with previous observations, the transition to the 2-methoxyphenyl ring (7b) from the 2,6-dimethoxyphenyl ring (29b) led to a nearly 50% reduction in NTS2 efficacy, with NTS2 Emax values of 15 and 8% of 5b, respectively.
The NTS2 binding affinity observed for 29b and 7b was consistent with similarly substituted 7-chloroquinolyl (21b, 23b) or naphthyl-substituted (26b, 27b) analogues bearing the 1-aminocyclohexanecarboxylic acid, with one important exception; for 7b, the binding affinity did not decrease in going from the 2,6-dimethoxyphenyl (29b) to the 2-methoxyphenyl ring substitution (7b) as seen in the Ki values of 140 and 153 nM, respectively. We imagine that this change in SAR for 29b and 7b could result from a change in binding mode at NTS2 that is permitted for 4-fluorophenyl but not 7-chloroquinolyl or naphthyl-substituted analogues. In the end, the 4-fluorophenyl-substituted pyrazole scaffold C was able to overcome the deficits found in scaffolds A and B to provide pyrazole-based compounds (29b and 7b) with enhanced NTS2 potency and binding affinity with significantly lower efficacy compared to 5a at the NTS2 receptor.
From the standpoint of calcium mobilization at NTS1 and NTS2, compounds (29b and 7b) appear to be selective for the NTS2 receptor. However, this was not a fair comparison, as one assay measures calcium mobilization (NTS2) while the other blockade of calcium mobilization (NTS1). We therefore acquired the radioligand binding data for both 29b and 7b at NTS1 in order to compare like measurements. As seen in Table 3, comparison of the relative binding affinities at NTS1 and NTS2 showed that compound 7b is 161-fold selective for NTS2 versus NTS1 while the dimethoxy analogue 29b shows only a 23-fold preference for NTS2 over NTS1. These data point out that while the calcium assay described herein is useful for identifying compounds that are active at NTS2, conclusions about selectivity require the use of the binding assay.
The identification of 7b as an NTS2 selective compound demonstrated that this calcium assay was useful for driving SAR studies in the pyrazole carboxamide series of compounds. However, the low efficacy of 7b, less than half of that found for levocabastine, prompted us to revisit mobilization of calcium in the parent CHO cell line as a negative control. We thus carried out a final study and examined all of the compounds reported herein that showed NTS2 Emax values less than 15% of 5b. This was done collectively by comparing all of the low efficacy compounds together in the same assay, under identical conditions, in our CHO-k1-NTS2 cells and simultaneously in the parent CHO line. The results of these experiments confirmed that the calcium mobilization observed was only found in the CHO cells stably expressing the NTS2 receptor.
Conclusions
In summary, we tested the compounds common to neurotensin research and have identified their associated calcium mobilization patterns at NTS1 and 2 using a FLIPR tetra using NT as our agonist standard for NTS1 and 5b as our agonist standard for NTS2. We found that compounds known to possess analgesic and antipsychotic activity in vivo appear as antagonists (NT and 1)) or potent partial agonists (levocabastine (6)) in this NTS2 in vitro assay. Using the assays described above in combination with binding data, we identified structural changes to compound 5a that led to the discovery of the NTS2 selective compound 7b, a pyrazole-based compound with in vitro properties similar to levocabastine in this assay, a compound known to be active against chronic pain. We are now working to identify the relevance of the functional data produced using this method by investigating compound 7b in recognized models of neuropathic pain in an attempt to establish an in vitro/in vivo correlation.
Experimental Section
Reactions were conducted under a nitrogen atmosphere using oven-dried glassware as required. All solvents and chemicals used were reagent grade. Anhydrous tetrahydrofuran (THF), dichloromethane (DCM), and N,N-dimethylformamide (DMF) were purchased from VWR and used without further purification. (7-Chloroquinolin-4-yl)hydrazine·HCl (9a) was purchased from Aldrich Chemical Corp. 1-Naphthylhydrazine·HCl (9b) was purchased from TCI America. 4-Fluorophenylhydrazine·HCl (9c) was purchased from Alfa Aesar. Most other reagents were obtained from either Aldrich Chemical Corp. or Fischer Scientific and used without further purification. Flash column chromatography was carried out using a Teledyne ISCO Combiflash Rf system and Redisep Rf gold prepacked HP silica columns. Evaporation of solvents was accomplished using rotary evaporators. The purity of target compounds was determined to be ≥95% by combustion analysis. Characterization of compounds was accomplished by a combination of TLC, combustion analysis, mass spectrometry (MS) as well as 1H and 13C NMR. NMR spectra were recorded on a Bruker Avance DPX-300 (300 MHz). Chemical shifts are reported in ppm relative to the tetramethylsilane, and coupling constant (J) values are reported in hertz (Hz). Low-resolution mass spectra were obtained using a Waters Alliance HT/Micromass ZQ system (ESI) in both positive and negative mode. Optical rotations were measured on an Auto Pol III polarimeter at the sodium D line. Thin layer chromatography (TLC) was performed on EMD precoated silica gel 60 F254 plates, and spots were visualized with UV light and I2 or phosphomolybdic acid stain. CHO-k1 cells were from the American Type Culture Collection, 125I–neurotensin was from PerkinElmer, Calcium 5 dye was from Molecular Devices, and cell culture reagents were from Life Technologies. The graphs provided in Figures 1–3 were obtained using Graphpad Prism Software.
General Methods
General Method for Preparation of Pyrazole Carboxylic Acid Esters (10a–j)
A magnetically stirred solution of a 4-aryl-2,4-diketoester sodium salt (8a–f, 5 mmol) and arylhydrazine hydrochloride (9a–c, 5 mmol) in glacial acetic acid (35 mL) and concd HCl (1.5 mL) was heated under reflux for 5 h and then cooled to rt. This was then poured into 300 mL of water and extracted five times with CH2Cl2. The extracts were washed carefully with satd NaHCO3 and then water and brine then dried over Na2SO4 and concentrated to give a crude material. This material was purified using flash chromatograph with a gradient of 0–100% EtOAc in hexanes over 30 min collecting peaks only. Flow rates are automatically set based on column size chosen for separation. Combination of desired fractions and evaporation provided methyl esters 10a–j as foamy solids.
General Method for Preparation of Pyrazole Carboxylic Acids (11a–j)
Pyrazole carboxylic acids 11a–j were prepared from the esters (10a–j) using the methyl ester hydrolysis method described below.
General Amide Coupling
To a magnetically stirred suspension of the appropriate pyrazole carboxylic acid (11a–j, 0.122 mmol) in anhydrous CH2Cl2 (20 mL) were added successively triethylamine (0.366 mmol), HBTU (0.146 mmol), and the appropriate amino acid ester hydrochloride salt (12a–e, 0.122 mmol). After stirring for 16 h, the resulting mixture was concentrated and the residue purified by flash chromatography using a gradient of 0–100% EtOAc in hexanes over 30 min collecting peaks only. Flow rates are automatically set based on column size chosen for separation. This gave an intermediate ester that was taken directly to the hydrolysis step.
General Methyl Ester Hydrolysis
To a magnetically stirred solution of methyl ester (7a,20–23a, 26a, 27a, and 29a) from the coupling reaction (0.1 mmol) in 1,4-dioxane (5 mL) was added 1N LiOH (2 mL), followed by stirring at room temperature overnight. The mixture was then concentrated, and the residue was taken up in water (2 mL) and extracted with ethyl acetate (15 mL). After this, 2 N HCl was added to the aqueous layer to precipitate the carboxylic acid final product. This was extracted twice with CH2Cl2, and the combined extracts were dried over Na2SO4 and concentrated to give solid final products.
General tert-Butyl Ester Hydrolysis
To a magnetically stirred solution of the tert-butyl ester intermediate (14–19a,24a,25a, and 28a) obtained from the coupling reaction (0.08 mmol) in CH2Cl2 (10 mL) was added excess TFA (10 mL) at room temp. After stirring for 16 h, the mixture was concentrated and then triturated with ethyl ether to give a solid product that was isolated by vacuum filtration, washed with ether, and then dried under high vacuum overnight.
Methyl 1-(1-(4-Fluorophenyl)-5-(2-methoxyphenyl)-1H-pyrazole-3-carboxamido)cyclohexanecarboxylate (7a)
Following the general amide coupling procedure, 7a was prepared from 11j and 12b as an amorphous off-white solid (55% yield). 1H NMR (CDCl3) δ 7.36 (t, J = 7.8 Hz, 1H) 7.23–7.31 (m, 3H), 7.18 (s, 1H), 6.94–7.04 (m, 3H), 6.80 (d, J = 8.5 Hz, 1H), 3.75 (s, 3H), 3.48 (s, 3H), 2.15–2.25 (m, 2H), 1.88–2.02 (m, 2H), 1.46–1.56 (m, 6H).
1-({[1-(4-Fluorophenyl)-5-(2-methoxyphenyl)-1H-pyrazol-3-yl]carbonyl}amino)cyclohexanecarboxylic Acid (7b)
Following the general methyl ester hydrolysis procedure, 7b was obtained from 7a as a white solid (85%). 1H NMR (300 MHz, CDCl3) δ 7.33–7.42 (m, 1H), 7.18–7.31 (m, 4H), 6.94–7.05 (m, 4H), 6.81 (d, J = 8.2 Hz, 1H), 3.44 (s, 3H), 2.28 (d, J = 13.9 Hz, 2H), 1.94–2.08 (m, 2H), 1.33–1.83 (m, 6H). 19F NMR (282 MHz, CDCl3) δ −113.42. 13C NMR (CDCl3) δ 175.00, 163.76, 163.45, 160.16, 156.36, 145.66, 142.48, 136.54, 136.50, 131.24, 131.16, 125.81, 125.70, 120.87, 118.66, 115.71, 115.41, 111.24, 109.45, 60.25, 54.94, 32.20, 25.13, 21.30. MS (ESI) m/z: 436.7 (M – H)−. Anal. (C24H24FN3O4) C, H, N.
Methyl 1-(7-Chloroquinolin-4-yl)-5-(2,6-dimethoxyphenyl)-1H-pyrazole-3-carboxylate (10a).45
Ester 10a was prepared via the general procedure starting from methyl 4-(2,6-dimethoxyphenyl)-2,4-dioxobutanoate sodium salt (8a) and 7-chloro-4-hydrazinylquinoline hydrochloride (9a) to give 10a as a yellow foam (68%). 1H NMR (CDCl3) δ 8.98 (d, J = 5.2 Hz, 1H), 8.88 (d, J = 1.8 Hz, 1H), 8.37 (d, J = 9.2 Hz, 1H), 7.82 (dd, J = 1.8, 9.2 Hz, 1H), 7.45 (d, J = 5.2 Hz, 1H), 7.34 (t, J = 8.4 Hz, 1H), 7.15–7.24 (m, 1H), 6.50 (d, J = 8.4 Hz, 2H), 4.01 (s, 3H), 3.48 (s, 6H).
Methyl 1-(7-Chloroquinolin-4-yl)-5-(2,6-dimethoxyphenyl)-4-ethyl-1H-pyrazole-3-carboxylate (10b)
Ester 10b was prepared from methyl 3-[(2,6-dimethoxyphenyl)carbonyl]-2-oxopentanoate, sodium salt (8b), and 9a according to the general method (pale-yellow solid, 45%). 1H NMR (CDCl3) δ 8.87 (d, J = 4.8 Hz, 1H), 8.18 (d, J = 1.9 Hz, 1H), 7.95 (d, J = 9.1 Hz, 1H), 7.50 (d, J = 2.0 Hz, 1H), 7.12–7.27 (m, 2H), 6.42 (d, J = 8.5 Hz, 2H), 3.76 (s, 3H), 3.50 (s, 6H), 2.69 (q, J = 7.4 Hz, 2H), 1.17 (t, J = 7.4 Hz, 3H).
Methyl 1-(7-Chloroquinolin-4-yl)-5-(2,5-dimethoxyphenyl)-1H-pyrazole-3-carboxylate (10c)
Ester 10c was prepared from methyl 4-(2,5-dimethoxyphenyl)-2,4-dioxobutanoate sodium salt (8c) and 9a according to the general method (pale-yellow solid, 57%). 1H NMR (CDCl3) δ 8.77 (d, J = 4.5 Hz, 1H), 8.14 (d, J = 2.1 Hz, 1H), 7.91 (d, J = 9.0 Hz, 1H), 7.54 (dd, J = 2.1, 8.95 Hz, 1H), 7.14 (s, 1H), 7.01 (d, J = 4.5 Hz, 1H), 6.78–6.91 (m, 2H), 6.57 (d, J = 8.9 Hz, 1H), 3.99 (s, 3H), 3.73 (s, 3H), 2.91 (s, 3H).
Methyl 1-(7-Chloroquinolin-4-yl)-5-(2,4-dimethoxyphenyl)-1H-pyrazole-3-carboxylate (10d)
Ester 10d was prepared from methyl 4-(2,4-dimethoxyphenyl)-2,4-dioxobutanoate sodium salt (8d) and 7-chloro-4-hydrazinylquinoline hydrochloride (9a) according to the general method (yellow amorphous solid, 45%). 1H NMR (CDCl3) δ 8.78 (d, J = 4.6 Hz, 1H), 8.14 (d, J = 1.8 Hz, 1H), 7.87 (d, J = 9.0 Hz, 1H), 7.52 (dd, J = 2.0, 9.0 Hz, 1H), 7.20 (d, J = 8.4 Hz, 1H), 7.09 (s, 1H), 7.02 (d, J = 4.6 Hz, 1H), 6.47 (dd, J = 2.2, 8.43 Hz, 1H), 6.19 (d, J = 2.1 Hz, 1H), 3.98 (s, 3H), 3.77 (s, 3H), 2.99 (s, 3H).
Methyl 1-(7-Chloroquinolin-4-yl)-5-(2,6-difluorophenyl)-1H-pyrazole-3-carboxylate (10e)
Ester 10e was prepared from methyl 4-(2,6-difluorophenyl)-2,4-dioxobutanoate sodium salt (8e) and 9a according to the general method (yellow amorphous solid, 52%). 1H NMR (CDCl3) δ 8.87 (d, J = 4.5 Hz, 1H), 8.14 (d, J = 1.9 Hz, 1H), 7.61–7.75 (m, 1H), 7.51 (dd, J = 1.9, 9.0 Hz, 1H), 7.41 (dd, J = 0.9, 8.7 Hz, 1H), 7.25–7.35 (m, 1H), 7.22 (d, J = 4.5 Hz, 1H), 6.77–6.89 (m, 2H), 4.01 (s, 3H).
Methyl 1-(7-Chloroquinolin-4-yl)-5-(2-methoxyphenyl)-1H-pyrazole-3-carboxylate (10f)
Ester 10f was prepared from methyl 4-(2-methoxyphenyl)-2,4-dioxo-butanoate sodium salt (8f) and 9a according to the general method (yellow solid, 43%). 1H NMR (CDCl3) δ 8.78 (d, J = 4.7 Hz, 1H), 8.15 (s, 1H), 7.91 (d, J = 9.0 Hz, 1H), 7.50 (td, J = 1.0, 9.0 Hz, 1H), 7.27–7.32 (m, 2H), 6.96–7.01 (m, 2H), 6.65 (d, J = 8.5 Hz, 2H), 4.0 (s, 3H), 4.0 (s, 3H).
Methyl 5-(2,6-Dimethoxyphenyl)-1-naphthalen-1-yl-1H-pyrazole-3-carboxylate (10g).62
Ester 10g was prepared from 8a and 1-naphthylhydrazine hydrochloride (9b) according to the general method (colorless foam, 55%). 1H NMR (CDCl3) δ 7.67–7.83 (m, 3H), 7.38–7.49 (m, 2H), 7.22–7.33 (m, 2H), 7.05–7.17 (m, 2H), 6.34 (d, J = 8.3 Hz, 2H), 3.96 (s, 3H), 3.41 (br s, 6H).
Methyl 5-(2-Methoxyphenyl)-1-naphthalen-1-yl-1H-pyrazole-3-carboxylate (10h).62
Ester 10h was prepared from methyl 4-(2-methoxyphenyl)-2,4-dioxobutanoate sodium salt (8f) and 9b according to the general method (off-white solid, 40%). 1H NMR (CDCl3) δ 7.78–7.88 (m, 2H), 7.62–7.71 (m, 1H), 7.44–7.53 (m, 2H), 7.29–7.36 (m, 1H), 7.24–7.29 (m, 1H), 7.10–7.24 (m, 3H), 6.81 (dt, J = 0.8, 7.5 Hz, 1H), 6.64 (d, J = 8.2 Hz, 1H), 3.97 (s, 3H), 3.13 (s, 3H).
Methyl 5-(2,6-Dimethoxyphenyl)-1-(4-fluorophenyl)-1H-pyrazole-3-carboxylate (10i)
Ester 10i was prepared from 8a and 4-fluorophenyl hydrazine hydrochloride (9c) according to the general method (white amorphous powder, 55%). 1H NMR (CDCl3) δ 7.22–7.33 (m, 3H), 6.89–7.00 (m, 3H), 6.51 (d, J = 8.3 Hz, 2H), 3.96 (s, 3H), 3.59 (s, 6H).
Methyl 1-(4-Fluorophenyl)-5-(2-methoxyphenyl)-1H-pyrazole-3-carboxylate (10j)
Ester 10j was prepared from 8f and 9c according to the general method (off-white powder, 45%). 1H NMR (CDCl3) δ 7.32–7.43 (m, 1H), 7.23–7.32 (m, 3H), 6.92–7.04 (m, 4H), 6.81 (d, J = 8.5 Hz, 1H), 3.97 (s, 3H), 3.44 (s, 3H).
1-(7-Chloroquinolin-4-yl)-5-(2,6-dimethoxyphenyl)-1H-pyrazole-3-carboxylic Acid (11a).45
Pyrazole acid 11a was prepared from ester 10a according to the general methyl ester hydrolysis method (pale-yellow powder, 78%). 1H NMR (300 MHz, DMSO-d6) δ 8.90 (d, J = 4.7 Hz, 1H), 8.17 (s, 1H), 7.73 (s, 2H), 7.26 (t, J = 8.4 Hz, 1H), 7.20 (d, J = 4.5 Hz, 1H), 6.99 (s, 1H), 6.54 (d, J = 8.5 Hz, 2H), 3.39 (s, 6H).
1-(7-Chloroquinolin-4-yl)-5-(2,6-dimethoxyphenyl)-4-ethyl-1H-pyrazole-3-carboxylic Acid (11b)
Pyrazole acid 11b was prepared from ester 10b according to the general methyl ester hydrolysis method (pale-yellow solid, 80%). 1H NMR (CDCl3) δ 10.36–11.51 (m, 2H), 8.87 (d, J = 4.8 Hz, 1H), 8.19 (d, J = 2.0 Hz, 1H), 7.94 (d, J = 9.0 Hz, 1H), 7.49 (dd, J = 2.0, 9.0 Hz, 1H), 7.19–7.32 (m, 1H), 7.17 (d, J = 4.7 Hz, 1H), 6.42 (d, J = 8.4 Hz, 2H), 3.50 (s, 6H), 2.68 (q, J = 7.4 Hz, 2H), 1.16 (t, J = 7.4 Hz, 3H). MS (ESI) m/z: 436.5 (M – H)−.
1-(7-Chloroquinolin-4-yl)-5-(2,5-dimethoxyphenyl)-1H-pyrazole-3-carboxylic Acid (11c)
Pyrazole acid 11c was prepared from ester 10c according to the general methyl ester hydrolysis method (amorphous, pale-yellow solid, 94%). 1H NMR (CDCl3) δ 8.80 (d, J = 4.7 Hz, 1H), 8.18 (d, J = 1.9 Hz, 1H), 7.91 (d, J = 9.2 Hz, 1H), 7.56 (dd, J = 1.9, 9.0 Hz, 1H), 7.19 (s, 1H), 7.02 (d, J = 4.7 Hz, 1H), 6.91–6.82 (m, 2H), 6.57 (d, J = 9.0 Hz, 1H), 3.75 (s, 3H), 2.92 (m, 3H).
1-(7-Chloroquinolin-4-yl)-5-(2,4-dimethoxyphenyl)-1H-pyrazole-3-carboxylic Acid (11d)
Pyrazole acid 11d was prepared from ester 10d according to the general methyl ester hydrolysis method (pale-yellow solid, 78%). 1H NMR (DMSO-d6) δ 8.91 (d, J = 4.7 Hz, 1H), 8.22 (d, J = 1.7 Hz, 1H), 7.78 (s, 1H), 7.76 (d, J = 2.0 Hz, 1H), 7.32 (d, J = 8.4 Hz, 1H), 7.24 (d, J = 4.7 Hz, 1H), 7.04 (s, 1H), 6.56 (dd, J = 2.2, 8.4 Hz, 1H), 6.34 (d, J = 2.2 Hz, 1H), 3.72 (s, 3H), 2.92 (s, 3H).
1-(7-Chloroquinolin-4-yl)-5-(2,6-difluorophenyl)-1H-pyrazole-3-carboxylic Acid (11e)
Pyrazole acid 11e was prepared from ester 10e according to the general methyl ester hydrolysis method (pale-yellow solid, 94%). 1H NMR (CD3OD) δ 8.90 (d, J = 4.7 Hz, 1H), 8.10 (s, 1H), 7.80 (d, J = 9.0 Hz, 1H), 7.62 (d, J = 9.0 Hz, 1H), 7.23–7.45 (m, 2H), 7.04 (s, 1H), 6.54 (d, J = 8.5 Hz, 2H).
1-(7-Chloroquinolin-4-yl)-5-(2-methoxyphenyl)-1H-pyrazole-3-carboxylic Acid (11f).63
Pyrazole acid 11f was prepared from ester 10f according to the general methyl ester hydrolysis method (pale-yellow solid, 92%). 1H NMR (DMSO-d6) δ 8.80 (d, J = 4.7 Hz, 1H), 8.15 (d, J = 2.1 Hz, 1H), 7.98 (d, J = 9.0 Hz, 1H), 7.69 (dd, J = 2.1, 9.0 Hz, 1H), 7.25–7.39 (m, 2H), 7.04 (d, J = 4.7 Hz, 1H), 6.97 (t, J = 7.4 Hz, 1H), 6.79 (d, J = 8.1 Hz, 1H), 6.70 (s, 1H), 2.91 (s, 3H).
5-(2,6-Dimethoxyphenyl)-1-naphthalen-1-yl-1H-pyrazole-3-carboxylic Acid (11g).62
Pyrazole acid 11g was prepared from ester 10g according to the general methyl ester hydrolysis method (off-white solid, 70%). 1H NMR (CDCl3) δ 7.78–7.86 (m, 2H), 7.72 (dd, J = 3.5, 6.3 Hz, 1H), 7.44–7.52 (m, 2H), 7.22–7.35 (m, 2H), 7.10–7.19 (m, 2H), 6.35 (d, J = 8.3 Hz, 2H), 3.41 (s, 6H).
5-(2-Methoxyphenyl)-1-naphthalen-1-yl-1H-pyrazole-3-carboxylic Acid (11h).62
Pyrazole acid 11h was prepared from ester 11h according to the general methyl ester hydrolysis method (off-white powder, 92%). 1H NMR (CDCl3) δ 7.81–7.91 (m, 2H), 7.68 (dd, J = 3.4, 6.2 Hz, 1H), 7.46–7.56 (m, 2H), 7.28–7.37 (m, 1H), 7.14–7.24 (m, 3H), 6.83 (t, J = 7.4 Hz, 1H), 6.65 (d, J = 8.3 Hz, 1H), 3.12 (s, 3H). MS (ESI) m/z: 343.2 (M – H)−.
5-(2,6-Dimethoxyphenyl)-1-(4-fluorophenyl)-1H-pyrazole-3-carboxylic Acid (11i)
Pyrazole acid 11i was prepared from ester 10i according to the general methyl ester hydrolysis method (off-white powder, 93%). 1H NMR (CDCl3) δ 7.23–7.36 (m, 3H), 6.91–7.03 (m, 3H), 6.52 (d, J = 8.5 Hz, 2H), 3.60 (s, 6H).
1-(4-Fluorophenyl)-5-(2-methoxyphenyl)-1H-pyrazole-3-carboxylic Acid (11j)
Pyrazole acid 11j was prepared from ester 10j according to the general methyl ester hydrolysis method (off-white powder, 91%). 1H NMR (CDCl3) δ 7.34–7.44 (m, 1H), 7.22–7.33 (m, 3H), 6.95–7.07 (m, 4H), 6.82 (d, J = 8.3 Hz, 1H), 3.44 (s, 3H).
1-(7-Chloroquinolin-4-yl)-5-(2,6-dimethoxyphenyl)-N-tricyclo[3.3.1.13,7]dec-2-yl-1H-pyrazole-3-carboxamide (13)
Compound 13 was prepared from 11a and 2-aminoadamantane hydrochloride (12e) following the general amide coupling method (off-white powder, 65%). 1H NMR δ (CDCl3) 8.77 (d, J = 4.7 Hz, 1H), 8.16 (d, J = 1.9 Hz, 1H), 7.96 (d, J = 9.0 Hz, 1H), 7.55 (dd, J = 1.9, 9.0 Hz, 1H), 7.24–7.37 (m, 2H), 7.13 (s, 1H), 6.92–7.02 (m, 2H), 6.64 (d, J = 8.3 Hz, 1H), 4.28 (d, J = 8.1 Hz, 1H), 2.98 (s, 6H), 2.02–2.11 (m, 2H), 1.80–1.95 (m, 9H), 1.70–1.79 (m, 2H), 1.59–1.70 (m, 2H). MS (ESI) m/z: 544.4 (M+H)+. Anal. (C31H31ClN4O3) C, H, N.
tert-Butyl (2S)-({[1-(7-Chloroquinolin-4-yl)-5-(2,6-dimethoxyphenyl)-1H-pyrazol-3-yl]carbonyl}amino)(cyclohexyl)ethanoate (14a)
Following the general amide coupling procedures, 14a was obtained from 11a and tert-butyl (2S)-amino(cyclohexyl)ethanoate hydrochloride (12d). The material was purified by flash chromatography (0–100% EtOAc/hexanes) to afford 14a (84%) as a pale-yellow film. 1H NMR (CDCl3) δ 8.78 (d, J = 4.7 Hz, 1H), 8.12 (d, J = 2.0 Hz, 1H), 7.97 (d, J = 9.0 Hz, 1H), 7.50 (dd, J = 2.1, 9.0 Hz, 1H), 7.42 (d, J = 9.0 Hz, 1H), 7.21 (t, J = 8.4 Hz, 1H), 7.01–7.12 (m, 2H), 6.39 (d, J = 8.2 Hz, 2H), 4.66 (dd, J = 5.1, 9.1 Hz, 1H), 3.40 (br s, 6H), 1.54–1.99 (m, 6H), 1.44–1.53 (m, 9H), 1.01–1.32 (m, 5H).
(2S)-({[1-(7-Chloroquinolin-4-yl)-5-(2,6-dimethoxyphenyl)-1H-pyrazol-3-yl]carbonyl}amino)(cyclohexyl)ethanoic Acid (14b).58
Following the general tert-butyl ester hydrolysis procedure, 14b was obtained as a pale-yellow solid in 91% yield from 14a. 1H NMR (CDCl3) δ 9.10 (br s, 1 H), 8.74 (br s, 1 H), 8.31 (d, J = 8.9 Hz, 1 H), 7.78 (d, J = 8.9 Hz, 1 H), 7.60–7.41 (m, 2 H), 7.39–7.25 (m, 1 H), 6.48 (d, J = 7.5 Hz, 2 H), 4.80 (dd, J = 4.7, 7.9 Hz, 1 H), 3.47 (br s, 6 H), 2.10–1.92 (m, 1 H), 1.90–1.60 (m, 5 H), 1.32–1.09 (m, 5 H). MS (ESI) m/z: 548.4 (M – H)−. [α]25D −18.1° (c 1.94, MeOH). Anal. (C29H29ClN4O5) C, H, N.
tert-Butyl (2S)-({[1-(7-Chloroquinolin-4-yl)-5-(2,5-dimethoxyphenyl)-1H-pyrazol-3-yl]carbonyl}amino)(cyclohexyl)ethanoate (15a)
Following the general amide coupling procedure, 15a was obtained from 11c and 12d as a pale-yellow film (70%). 1H NMR (CDCl3) δ 8.78 (d, J = 4.7 Hz, 1H), 8.16 (d, J = 1.9 Hz, 1H), 8.01 (d, J = 9.2 Hz, 1H), 7.55 (dd, J = 2.3, 9.0 Hz, 1H), 7.40 (d, J = 9.0 Hz, 1H), 7.14–7.11 (m, 1H), 7.00 (d, J = 4.7 Hz, 1H), 6.90 (d, J = 3.0 Hz, 1H), 6.83 (dd, J = 3.2, 9.0 Hz, 1H), 6.55 (d, J = 9.0 Hz, 1H), 4.69–4.62 (m, 1H), 3.72 (s, 3H), 2.89 (m, 3H), 2.05–1.60 (m, 6H), 1.50 (s, 9H), 1.39–1.11 (m, 5H).
(2S)-({[1-(7-Chloroquinolin-4-yl)-5-(2,5-dimethoxyphenyl)-1H-pyrazol-3-yl]carbonyl}amino)(cyclohexyl)ethanoic Acid (15b)
Following the general tert-butyl ester hydrolysis procedure, 15b was obtained (92%) as a white foam from 15a. 1H NMR (CDCl3) δ 9.00 (d, J = 5.3 Hz, 1H), 8.39 (d, J = 1.8 Hz, 1H), 8.17 (d, J = 9.2 Hz, 1H), 7.7 (dd, J = 1.9, 9.0 Hz, 1H), 7.30 (d, J = 9.0 Hz, 1H), 7.27–7.16 (m, 2H), 6.90 (d, J = 3.0 Hz, 1H), 6.83 (dd, J = 3.2, 9.0 Hz, 1H), 6.58 (d, J = 9.0 Hz, 1H), 4.81–4.72 (m, 1H), 3.78 (s, 3H), 2.90 (m, 3H), 2.05–1.60 (m, 6H), 1.37–1.03 (m, 5H). MS (ESI) m/z: 547.8 (M – H)−. [α]25D −2.6° (c 0.45, MeOH). Anal. (C29H29ClN4O5) C, H, N.
tert-Butyl (2S)-({[1-(7-Chloroquinolin-4-yl)-5-(2,4-dimethoxyphenyl)-1H-pyrazol-3-yl]carbonyl}amino)(cyclohexyl)ethanoate (16a)
Following the general amide coupling procedure, 16a was obtained as a pale-yellow film (86%) from 11d and 12d. 1H NMR (CDCl3) δ 8.79 (d, J = 4.7 Hz, 1H), 8.16 (d, J = 2.0 Hz, 1H), 7.96 (d, J = 9.0 Hz, 1H), 7.54 (dd, J = 2.1, 9.0 Hz, 1H), 7.40 (d, J = 9.0 Hz, 1H), 7.21 (d, J = 8.4 Hz, 1H), 7.07 (s, 1H), 7.01 (d, J = 4.6 Hz, 1H), 6.47 (dd, J = 2.3, 8.4 Hz, 1H), 6.18 (d, J = 2.3 Hz, 1H), 4.66 (dd, J = 5.1, 9.0 Hz, 1H), 3.77 (s, 3H), 2.96 (s, 3H), 1.56–1.81 (m, 6H), 1.48 (d, J = 4.5 Hz, 9H), 1.00–1.34 (m, 5H).
(2S)-({[1-(7-Chloroquinolin-4-yl)-5-(2,4-dimethoxyphenyl)-1H-pyrazol-3-yl]carbonyl}amino)(cyclohexyl)ethanoic Acid (16b)
Following the general tert-butyl ester hydrolysis procedure, 16b was obtained as a yellow solid in 86% yield from 16a. 1H NMR (CDCl3) δ 9.11 (br s, 1 H), 8.48 (s, 1 H), 8.27 (d, J = 9.1 Hz, 1 H), 7.80 (d, J = 8.9 Hz, 1 H), 7.46 (d, J = 8.4 Hz, 1 H), 7.42–7.35 (m, 1 H), 7.31 (d, J = 8.4 Hz, 1 H), 7.12 (s, 1 H), 6.59 (d, J = 8.4 Hz, 1 H), 6.23 (d, J = 1.7 Hz, 1 H), 4.73 (dd, J = 5.4, 8.4 Hz, 1 H), 3.81 (s, 3 H), 3.02 (s, 3 H), 1.87–1.61 (m, 5 H), 1.37–1.04 (m, 6 H). MS (ESI) m/z: 548.5 (M – H)−. [α]25D +3.1° (c 0.45, MeOH). Anal. (C29H29ClN4O5) C, H, N.
tert-Butyl (2S)-({[1-(7-Chloroquinolin-4-yl)-5-(2-methoxyphenyl)-1H-pyrazol-3-yl]carbonyl}amino)(cyclohexyl)ethanoate (17a)
Following the general amide coupling procedure, 17a was obtained from 11f and 12d in 94% yield as a pale-yellow solid. 1H NMR (CDCl3) δ 8.77 (d, J = 4.7 Hz, 1H), 8.16 (d, J = 1.9 Hz, 1H), 7.98 (d, J = 9.0 Hz, 1H), 7.55 (dd, J = 2.0, 9.1 Hz, 1H), 7.41 (d, J = 9.1 Hz, 1H), 7.23–7.35 (m, 2H), 7.09–7.16 (m, 1H), 6.89–7.03 (m, 2H), 6.64 (d, J = 8.1 Hz, 1H), 4.66 (dd, J = 5.0, 9.1 Hz, 1H), 2.98 (s, 3H), 1.53–1.99 (m, 7H), 1.49 (s, 9H), 1.01–1.35 (m, 6H).
(2S)-({[1-(7-Chloroquinolin-4-yl)-5-(2-methoxyphenyl)-1H-pyrazol-3-yl]carbonyl}amino)(cyclohexyl)ethanoic Acid (17b)
Following the general tert-butyl ester hydrolysis procedure, 17b was obtained as a pale-yellow film in 74% yield from 17a. 1H NMR (CDCl3) δ 9.09 (br s, 1 H), 8.76 (br s, 1 H), 8.33 (d, J = 8.9 Hz, 1 H), 7.82 (d, J = 8.9 Hz, 1 H), 7.57–7.32 (m, 4 H), 7.22 (s, 1 H), 7.10 (t, J = 6.9 Hz, 1 H), 6.70 (d, J = 7.7 Hz, 1 H), 4.78 (dd, J = 4.8, 8.2 Hz, 1 H), 3.04 (br s, 3 H), 2.10–1.90 (m, 1 H), 1.89–1.60 (m, 5 H), 1.25–1.07 (m, 5 H). MS (ESI) m/z: 518.0 (M – H)−. [α]25D −24.3° (c 2.04, MeOH). Anal. (C28H27ClN4O4) C, H, N.
tert-Butyl (2S)-({[1-(7-Chloroquinolin-4-yl)-5-(2,6-difluorophenyl)-1H-pyrazol-3-yl]carbonyl}-amino)(cyclohexyl)ethanoate (18a)
Following the general amide coupling procedure, 18a was obtained from 11e and 12d as a pale-yellow foam in 53% yield. 1H NMR (CDCl3) δ 8.89 (d, J = 4.7 Hz, 1H), 8.15 (d, J = 4.7 Hz, 1H), 7.75 (d, J = 9.0 Hz, 1H), 7.75 (d, J = 9.0 Hz, 1H), 7.52 (dd, J = 1.9, 9.0 Hz, 1H), 7.4 (d, J = 9.0 Hz, 1H), 7.32–7.25 (m, 3H), 7.2 (d, J = 9.0 Hz, 1H), 6.80 (t, J = 7.6 Hz, 1H), 4.69–4.63 (m, 1H), 1.90–1.85 (m, 1H), 1.84–1.60 (m, 5H), 1.50 (s, 9H), 1.32–1.06 (m, 5H).
(2S)-({[1-(7-Chloroquinolin-4-yl)-5-(2,6-difluorophenyl)-1H-pyrazol-3-yl]carbonyl}amino)(cyclohexyl)ethanoic Acid (18b)
Following the general tert-butyl ester hydrolysis procedure, 18b was obtained as a white foam (90%) from 18a. 1H NMR (CDCl3) δ 8.91 (d, J = 4.7 Hz, 1H), 8.18 (s, 1H), 8.17 (d, J = 9.2 Hz, 1H), 7.74 (d, J = 9.0 Hz, 1H), 7.53 (dd, J = 1.9, J = 9.0 Hz, 1H), 7.40 (d, J = 9.0 Hz, 1H), 7.20–7.05 (m, 4H), 6.80 (t, J = 7.6 Hz, 1H), 4.82–4.74 (m, 1H), 2.08–1.94 (m, 1H), 1.89–1.60 (m, 5H), 1.37–1.05 (m, 5H). MS (ESI) m/z: 523.6 (M – H)−. [α]25D −13.3° (c 0.44, MeOH). Anal. (C27H23ClF2N4O3) C, H, N.
tert-Butyl (2S)-({[1-(7-Chloroquinolin-4-yl)-5-(2,6-dimethoxyphenyl)-4-ethyl-1H-pyrazol-3-yl]carbonyl}amino)(cyclohexyl)ethanoate (19a)
Following the general amide coupling procedures, 19a was obtained as a pale-yellow film (86%) 11b and 12d. 1H NMR (CDCl3) δ 8.76 (d, J = 4.6 Hz, 1H), 8.09 (d, J = 2.1 Hz, 1H), 7.98 (d, J = 9.1 Hz, 1H), 7.48 (td, J = 2.2, 9.1 Hz, 2H), 7.21 (t, J = 8.4 Hz, 1H), 7.09 (d, J = 4.6 Hz, 1H), 6.40 (dd, J = 8.4, 15.6 Hz, 2H), 4.64 (dd, J = 4.9, 9.0 Hz, 1H), 3.53 (s, 3H), 3.42 (s, 3H), 2.54–2.82 (m, 2H), 1.61–1.95 (m, 6H), 1.49 (s, 9H), 1.17–1.36 (m, 5H), 1.06–1.15 (m, 3H). MS (ESI) m/z: 633.7 (M + H)+.
(2S)-({[1-(7-Chloroquinolin-4-yl)-5-(2,6-dimethoxyphenyl)-4-ethyl-1H-pyrazol-3-yl]carbonyl}amino)(cyclohexyl)ethanoic Acid (19b)
Following the general tert-butyl ester hydrolysis procedure, 19b was obtained as a pale-yellow solid in 96% yield from 19a. 1H NMR (CDCl3) δ 9.12 (d, J = 5.7 Hz, 1 H), 8.43 (s, 1 H), 8.38 (d, J = 9.2 Hz, 1 H), 7.77 (dd, J = 1.3, 9.3 Hz, 1 H), 7.56 (d, J = 8.7 Hz, 1 H), 7.48 (d, J = 5.7 Hz, 1 H), 7.34 (t, J = 8.4 Hz, 1 H), 6.51 (dd, J = 8.5, 15.5 Hz, 2 H), 4.77 (dd, J = 5.2, 8.7 Hz, 1 H), 3.67–3.45 (m, 6 H), 2.66 (dd, J = 3.2, 7.3 Hz, 2 H), 2.12–1.91 (m, 1 H), 1.90–1.60 (m, 6 H), 1.38–1.16 (m, 4 H), 1.15–1.01 (m, 3 H). MS (ESI) m/z: 575.8 (M – H)−. [α]25D +2.3° (c 0.98, MeOH). Anal. (C31H33ClN4O5) C, H, N.
Methyl 1-({[1-(7-Chloroquinolin-4-yl)-5-(2,6-dimethoxyphenyl)-1H-pyrazol-3-yl]carbonyl}amino)cycloheptanecarboxylate (20a)
Following the general amide coupling procedure, 20a was obtained from 11a and methyl 1-aminocycloheptanecarboxylate hydrochloride (12c) as an off-white powder in 65% yield. 1H NMR (CDCl3) δ 8.77 (d, J = 4.7 Hz, 1H), 8.16 (d, J = 1.9 Hz, 1H), 7.96 (d, J = 9.0 Hz, 1H), 7.55 (dd, J = 1.9, 9.0 Hz, 1H), 7.24–7.37 (m, 2H), 7.13 (s, 1H), 6.92–7.02 (m, 2H), 6.64 (d, J = 8.3 Hz, 1H), 4.28 (d, J = 8.1 Hz, 1H), 2.98 (s, 6H), 2.02–2.11 (m, 2H), 1.80–1.95 (m, 9H), 1.70–1.79 (m, 2H), 1.59–1.70 (m, 2H).
1-({[1-(7-Chloroquinolin-4-yl)-5-(2,6-dimethoxyphenyl)-1H-pyrazol-3-yl]carbonyl}amino)cycloheptanecarboxylic Acid (20b)
Following the general methyl ester hydrolysis procedure, 20b was obtained from 20a as a white powder in 86% yield. 1H NMR (CDCl3) δ 8.80 (d, J = 4.7 Hz, 1H), 8.14 (d, J = 2.1 Hz, 1H), 7.90 (d, J = 9.0 Hz, 1H), 7.50 (dd, J = 2.1, 9.0 Hz, 1H), 7.24–7.17 (m, 1H), 7.11 (s, 1H), 7.07 (d, J = 4.7 Hz, 1H) 6.40 (d, J = 8.7 Hz, 2H), 3.42 (s, 6H), 2.42–2.28 (m, 2H), 2.24–2.13 (m, 2H), 1.74–1.53 (m, 10H). MS (ESI) m/z: 519.5 (M – H)−. Anal. (C29H29ClN4O5) C, H, N.
Methyl 1-({[1-(7-Chloroquinolin-4-yl)-5-(2,6-dimethoxyphenyl)-1H-pyrazol-3-yl]carbonyl}amino)cyclohexanecarboxylate (21a)
Following the general amide coupling procedures, 21a was obtained from 11a and ethyl 1-aminocyclohexanecarboxylate hydrochloride (12b) as a white solid in 87% yield. 1H NMR (CDCl3) δ 8.74–8.84 (m, 1H), 8.14 (d, J = 2.1 Hz, 1H), 7.96 (d, J = 9.0 Hz, 1H), 7.52 (dd, J = 2.1, 9.0 Hz, 1H), 7.15–7.25 (m, 1H), 7.04–7.15 (m, 2H), 6.39 (d, J = 8.5 Hz, 2H), 4.25 (q, J = 7.2 Hz, 2H), 3.41 (s, 6H), 2.18 (d, J = 13.6 Hz, 2H), 1.86–2.02 (m, 2H), 1.43–1.74 (m, 6H), 1.23–1.34 (m, 3H).
1-({[1-(7-Chloroquinolin-4-yl)-5-(2,6-dimethoxyphenyl)-1H-pyrazol-3-yl]carbonyl}amino)cyclohexanecarboxylic Acid (21b)
Following the general methyl ester hydrolysis procedure, 21b was obtained from 21a in 87% yield as a white solid. 1H NMR (DMSO-d6) δ 8.92 (d, J = 4.7 Hz, 1H), 8.15 (d, J = 2.1 Hz, 1H), 7.86–7.75 (m, 2H), 7.69 (dd, J = 2.1, 9.0 Hz, 1H), 7.30–7.21 (m, 2H), 6.95 (s, 1H), 6.53 (d, J = 8.7 Hz, 1H), 3.43 (s, 6H), 2.17–2.06 (m, 2H), 1.85–1.72 (m, 2H), 1.62–1.25 (m, 6H). MS (ESI) m/z: 533.8 (M – H)−. Anal. (C28H27ClN4O5) C, H, N.
Methyl 1-({[1-(7-Chloroquinolin-4-yl)-5-(2,6-dimethoxyphenyl)-1H-pyrazol-3-yl]carbonyl}amino)cyclopentanecarboxylate (22a)
Following the general amide coupling procedures, 22a was obtained from 11a and methyl 1-aminocyclopentanecarboxylate hydrochloride (12a) as a white powder in 85% yield. 1H NMR (CDCl3) δ 8.78 (d, J = 4.7 Hz, 1H), 8.12 (d, J = 2.1 Hz, 1H), 7.94 (d, J = 9.0 Hz, 1H), 7.52 (dd, J = 2.1, 9.0 Hz, 1H), 7.29–7.22 (m, 1H), 7.21–7.16 (m, 1H), 7.11–7.05 (m, 1H), 6.39 (d, J = 8.5 Hz, 2H), 3.78 (s, 3H), 3.41 (s, 6H), 2.42–2.28 (m, 2H), 2.17–2.05 (m, 2H), 1.90–1.80 (m, 4H).
1-({[1-(7-Chloroquinolin-4-yl)-5-(2,6-dimethoxyphenyl)-1H-pyrazol-3-yl]carbonyl}amino)cyclopentanecarboxylic Acid (22b)
Following the general methyl ester hydrolysis procedure, 22b was obtained from 22a as a white powder in 72% yield. 1H NMR (CDCl3) δ 8.80 (d, J = 4.7 Hz, 1H), 8.14 (d, J = 2.1 Hz, 1H), 7.90 (d, J = 9.0 Hz, 1H), 7.53 (dd, J = 2.1, J = 9.0 Hz, 1H), 7.30–7.18 (m, 1H), 7.11 (s, 1H), 7.07 (d, J = 4.7 Hz, 1H) 6.40 (d, J = 8.7 Hz, 2H), 3.42 (s, 6H), 2.54–2.40 (m, 2H), 2.20–2.07 (m, 2H), 1.90–1.77 (m, 4H). MS (ESI) m/z: 547.8 (M – H)−. Anal. (C28H27ClN4O5) C, H, N.
Methyl 1-({[1-(7-Chloroquinolin-4-yl)-5-(2-methoxyphenyl)-1H-pyrazol-3-yl]carbonyl}amino)cyclohexanecarboxylate (23a)
Following the general amide coupling procedure, 23a was obtained from 11f and ethyl 1-aminocyclohexanecarboxylate hydrochloride (12b) as a white solid in 63% yield. 1H NMR (CDCl3) δ 8.78 (d, J = 4.7 Hz, 1H), 8.17 (d, J = 1.9 Hz, 1H), 7.96 (d, J = 9.0 Hz, 1H), 7.56 (dd, J = 2.2, 9.1 Hz, 1H), 7.24–7.34 (m, 2H), 7.10 (s, 1H), 6.90–7.01 (m, 1H), 6.64 (d, J = 8.7 Hz, 2H), 3.78 (s, 3H), 2.99 (s, 3H), 2.18 (d, J = 13.8 Hz, 2H), 1.88–2.01 (m, 2H), 1.63–1.74 (m, 6H).
1-({[1-(7-Chloroquinolin-4-yl)-5-(2-methoxyphenyl)-1H-pyrazol-3-yl]carbonyl}amino)cyclohexanecarboxylic Acid (23b)
Following the general methyl ester hydrolysis procedure, 23b was obtained from 23a as a white solid in 77% yield. 1H NMR (CDCl3) δ 8.80 (d, J = 4.7 Hz, 1H), 8.17 (d, J = 2.1 Hz, 1H), 7.90 (d, J = 9.0 Hz, 1H), 7.5 (dd, J = 2.1, J = 9.0 Hz, 1H), 7.35–7.21 (m, 1H), 7.20–7.11 (m, 2H), 6.63 (d, J = 8.7 Hz, 1H), 2.98 (s, 3H), 2.30–2.16 (m, 2H), 2.10–1.96 (m, 2H), 1.76–1.53 (m, 6H). MS (ESI) m/z: 503.6 (M – H)−. Anal. (C28H27ClN4O4) C, H, N.
tert-Butyl (2S)-Cyclohexyl({[5-(2,6-dimethoxyphenyl)-1-naphthalen-1-yl-1H-pyrazol-3-yl]carbonyl}amino)ethanoate (24a)
Following the general amide coupling procedures, 24a was obtained from 11g and 12d as a white solid in 53% yield. 1H NMR (CDCl3) δ 7.88–7.72 (m, 3H), 7.51–7.38 (m, 3H), 7.36–7.21 (m, 2H), 7.11 (t, J = 8.4 Hz, 1H), 7.07–7.03 (m, 1H), 6.33 (d, J = 5.9 Hz, 2H), 4.66 (dd, J = 5.4, 9.1 Hz, 1H), 3.40 (br s, 6H), 1.96–1.52 (m, 8H), 1.47 (s, 9H), 1.33–1.02 (m, 6H).
(2S)-Cyclohexyl({[5-(2,6-dimethoxyphenyl)-1-naphthalen-1-yl-1H-pyrazol-3-yl]carbonyl}amino)ethanoic Acid (24b).45
Following the general tert-butyl ester hydrolysis procedure 24b was obtained as a slightly yellow solid in 53% yield from 24a. 1H NMR (CDCl3) δ 7.82 (d, J = 6.8 Hz, 2 H), 7.73 (d, J = 6.4 Hz, 2 H), 7.48 (dd, J = 3.2, 6.2 Hz, 2 H), 7.38–7.21 (m, 2 H), 7.19–7.01 (m, 2 H), 6.33 (d, J = 8.1 Hz, 2 H), 4.73 (dd, J = 5.8, 8.3 Hz, 1 H), 3.40 (br s, 6 H), 1.93 (br s, 1 H), 1.83–1.55 (m, 5 H), 1.23–0.99 (m, 5 H). MS (ESI) m/z: 512.4 (M – H)−. [α]25D −26.7° (c 0.24, MeOH). Anal. (C30H31N3O5) C, H, N.
tert-Butyl (2S)-Cyclohexyl({[5-(2-methoxyphenyl)-1-naphthalen-1-yl-1H-pyrazol-3-yl]carbonyl}amino)ethanoate (25a)
Following the general amide coupling procedure, 25a was obtained from 11h and 12d in 81% yield as a colorless film. 1H NMR (CDCl3) δ 7.84 (s, 1H), 7.81 (s, 1H), 7.75 (d, J = 3.5 Hz, 1H), 7.55–7.46 (m, 2H), 7.43 (d, J = 9.1 Hz, 1H), 7.36–7.28 (m, 1H), 7.23–7.14 (m, 3H), 7.12 (s, 1H), 6.85–6.75 (m, 1H), 6.61 (d, J = 8.6 Hz, 1H), 4.66 (dd, J = 5.4, 9.1 Hz, 1H), 3.07 (s, 3H), 1.96–1.57 (m, 7H), 1.47 (s, 9H), 1.32–1.05 (m, 6H).
(2S)-Cyclohexyl({[5-(2-methoxyphenyl)-1-naphthalen-1-yl-1H-pyrazol-3-yl]carbonyl}amino)ethanoic Acid (25b)
Following the general tert-butyl ester hydrolysis procedure, 25b was obtained from 25a as a white foam (43%). 1H NMR (CDCl3) δ 7.91–7.81 (m, 2 H), 7.73–7.61 (m, 2 H), 7.56–7.47 (m, 2 H), 7.39–7.29 (m, 1 H), 7.25–7.18 (m, 2 H), 7.15 (s, 2 H), 6.80 (t, J = 7.4 Hz, 1 H), 6.63 (d, J = 8.3 Hz, 1 H), 4.74 (dd, J = 5.7, 8.8 Hz, 1 H), 3.12 (s, 3 H), 1.93 (br s, 1 H), 1.83–1.53 (m, 5 H), 1.24–1.02 (m, 5 H). MS (ESI) m/z: 482.3 (M – H)−. [α]25D −32.7° (c 0.56, MeOH). Anal. (C29H29N3O4) C, H, N.
Methyl 1-({[5-(2,6-Dimethoxyphenyl)-1-naphthalen-1-yl-1H-pyrazol-3-yl]carbonyl}amino)cyclohexanecarboxylate (26a)
Following the general amide coupling procedure, 26a was obtained from 11g and 12b as a white solid in 98% yield. 1H NMR (CDCl3) δ 7.73–7.89 (m, 3H), 7.45–7.52 (m, 2H), 7.27–7.37 (m, 2H), 7.06–7.16 (m, 2H), 7.02 (s, 1H), 6.32 (d, J = 8.3 Hz, 2H), 3.78 (s, 3H), 3.23–3.63 (m, 6H), 2.08–2.32 (m, 4H), 1.56 (br s, 8H).
1-({[5-(2,6-Dimethoxyphenyl)-1-naphthalen-1-yl-1H-pyrazol-3-yl]carbonyl}amino)cyclohexanecarboxylic Acid (26b).45
Following the general ethyl ester hydrolysis procedure, 26b was obtained from 26a as an off-white powder in 35% yield. 1H NMR (CDCl3) δ 7.88–7.78 (m, 2 H), 7.77–7.67 (m, 1 H), 7.54–7.45 (m, 2 H), 7.37–7.29 (m, 1 H), 7.28–7.22 (m, 4 H), 7.15 (t, J = 8.4 Hz, 1 H), 7.08 (s, 1 H), 6.34 (d, J = 8.5 Hz, 2 H), 3.41 (br s, 6 H), 2.24 (d, J = 14.1 Hz, 2 H), 2.10–1.94 (m, 2 H), 1.79–1.48 (m, 6 H). MS (ESI) m/z: 498.3 (M – H)−. Anal. (C30H31N3O5) C, H, N.
Methyl 1-({[5-(2-Methoxyphenyl)-1-naphthalen-1-yl-1H-pyrazol-3-yl]carbonyl}amino)cyclohexanecarboxylate (27a)
Following the general amide coupling procedures, 27a was obtained from 11h and 12b as a colorless film in 73% yield. 1H NMR (CDCl3) δ 7.93–7.80 (m, 2H), 7.80–7.71 (m, 1H), 7.59–7.46 (m, 2H), 7.40–7.29 (m, 1H), 7.25–7.12 (m, 4H), 7.10 (s, 1H), 6.87–6.74 (m, 1H), 6.62 (d, J = 8.2 Hz, 1H), 3.77 (s, 3H), 3.09 (s, 3H), 2.13 (br s, 2H), 1.96 (br s, 3H), 1.71–1.30 (m, 10H).
1-({[5-(2-Methoxyphenyl)-1-naphthalen-1-yl-1H-pyrazol-3-yl]carbonyl}amino)cyclohexanecarboxylic Acid (27b)
Following the general ethyl ester hydrolysis procedure, 27b was obtained from 27a as a white foam (66%). 1H NMR (CDCl3) δ 7.92–7.82 (m, 2 H), 7.68 (d, J = 3.4 Hz, 1 H), 7.53 (dd, J = 3.3, 6.3 Hz, 2 H), 7.38–7.30 (m, 1 H), 7.25–7.09 (m, 5 H), 6.82 (t, J = 7.4 Hz, 1 H), 6.63 (d, J = 8.2 Hz, 1 H), 3.10 (s, 3 H), 2.22 (d, J = 13.9 Hz, 2 H), 2.01 (t, J = 11.2 Hz, 2 H), 1.77–1.19 (m, 8 H), 0.97 (br s, 1 H). MS (ESI) m/z: 503.6 (M – H)−. Anal. (C28H27N3O4) C, H, N.
tert-Butyl (2S)-Cyclohexyl({[5-(2,6-dimethoxyphenyl)-1-(4-fluorophenyl)-1H-pyrazol-3-yl]carbonyl}amino)ethanoate (28a)
Following the general amide coupling procedure, 28a was obtained from 11i and 12d as an off-white powder in 94% yield. 1H NMR (CDCl3) δ 7.42–7.54 (m, 1H), 7.21–7.35 (m, 3H), 6.90–7.03 (m, 3H), 6.50 (dd, J = 8.5, 13.9 Hz, 2H), 4.67 (dd, J = 5.09, 9.14 Hz, 1H), 3.62 (s, 3H), 3.52 (s, 3H), 1.60–1.92 (m, 7H), 1.48–1.52 (m, 9H), 1.16–1.33 (m, 5H). MS (ESI) m/z: 536.5 (M – H)−.
(2S)-Cyclohexyl({[5-(2,6-Dimethoxyphenyl)-1-(4-fluorophenyl)-1H-pyrazol-3-yl]carbonyl}amino)ethanoic Acid (28b)
Following the general tert-butyl ester hydrolysis procedure, 28b was obtained as a white foam (83%) from 28a. 1H NMR (CD3OD) δ 7.23–7.42 (m, 3H), 6.99–7.15 (m, 2H), 6.76–6.86 (m, 1H), 6.62 (d, J = 8.29 Hz, 2H), 4.57 (d, J = 6.03 Hz, 1H), 3.60 (s, 6H), 1.74–2.10 (m, 7H), 1.21–1.44 (m, 4H). MS (ESI) m/z: 480.6 (M – H)−. [α]25D −19.7° (c 0.33, MeOH). Anal. (C26H26FN3O5) C, H, N.
Methyl 1-({[5-(2,6-Dimethoxyphenyl)-1-(4-fluorophenyl)-1H-pyrazol-3-yl]carbonyl}amino)cyclohexanecarboxylate (29a)
Following the general amide coupling procedure, 29a was prepared from 11i and 12b as a colorless solid in 50% yield. 1H NMR (CDCl3) δ 7.33–7.25 (m, 4H), 7.00–6.90 (m, 2H), 6.50 (d, J = 8.5 Hz, 2H), 3.75 (s, 3H), 3.58 (s, 6H), 2.25–2.13 (m, 2H), 2.01–1.89 (m, 2H), 1.74–1.52 (m, 6H).
1-({[5-(2,6-Dimethoxyphenyl)-1-(4-fluorophenyl)-1H-pyrazol-3-yl]carbonyl}amino)cyclohexanecarboxylic Acid (29b)
Following the general methyl ester hydrolysis procedure, 29b was obtained from 29a as a white solid (89%). 1H NMR (CDCl3) δ 7.36–7.21 (m, 4H), 7.03–6.95 (m, 2H), 6.50 (d, J = 8.5 Hz, 2H), 3.60 (s, 6H), 2.34–2.20 (m, 2H), 2.10–1.96 (m, 2H), 1.79–1.47 (m, 6H). MS (ESI) m/z: 466.4 (M – H)−. Anal. (C25H26FN3O5) C, H, N.
2-({[5-(2,6-Dimethoxyphenyl)-1-(4-fluorophenyl)-1H-pyrazol-3-yl]carbonyl}amino)tricycle-[3.3.1.13,7]decane-2-carboxylic Acid (30)
Following the method of Quéré,4530 was obtained from 11i and 9-aminobicyclo[3.3.1]nonane-9-carboxylic acid (12f). Briefly, 11i was heated under reflux in SOCl2 for 2 h, followed by cooling and concentration on a rotavap. The residue was then dissolved in toluene and evaporated 3× to remove HCl. The residue was then taken up in dry THF and added to a well agitated mixture of 5:1 THF:water (15 mL/g of acid chloride) containing 1.1 equiv of 2-aminoadamantane-carboxylic acid and 2.2 equiv of NaOH at 5 °C. Following the addition, the stirring was continued for 18 h at ambient temp. After this time, the mixture is made acidic with 1 N HCl and extracted 3× with CH2Cl2, dried over sodium sulfate, and evaporated. The residue was chromatographed using a 0–15% CH2Cl2:MeOH gradient. Evaporation provided the desired compound as an off-white solid (71%). 1H NMR (CDCl3) δ 7.20–7.36 (m, 4H), 6.90–7.04 (m, 3H), 6.51 (d, J = 8.5 Hz, 2H), 3.54–3.63 (m, 6H), 2.71–2.81 (m, 2H), 2.23 (d, J = 12.8 Hz, 2H), 2.07 (d, J = 12.2 Hz, 2H), 1.68–1.95 (m, 8H). MS (ESI) m/z: 518.9 (M – H)−. Anal. (C29H30FN3O5) C, H, N.
Pharmacological Methods
Calcium Mobilization Assay for NTS1 Receptor
CHO-k1-rNTS1 cells were maintained in DMEM/F12 medium supplemented with 10% fetal bovine serum (Gibco), 100 U/mL penicillin/100 μg/mL streptomycin, 100 μg/mL normocin (Invivogen), and 250 μg/mL geneticin. For calcium mobilization assays, cells were plated at 30000 cells/well in black, clear-bottom 96-well plates the day of the assay and incubated at 37 °C, 5% CO2. Prior to the assay, Calcium 5 dye (Molecular Devices) was reconstituted according to manufacturer instructions. The reconstituted dye was diluted 1:40 in assay buffer (1× HBSS, 20 mM HEPES, and 2.5 mM Probenicid (Sigma), pH 7.4). Growth media was removed, and 200 μL of this diluted dye was added to each well. Plates were incubated for 45 min at 37 °C, 5% CO2 after dye addition. For agonist assays, cells were pretreated with 1:10 addition (20 μL) of 10% DMSO in assay buffer, and the plates were returned to 37 °C, 5% CO2 for 15 min. Full-log serial dilutions of the test compounds were made at 10× the desired final concentration in 1% DMSO assay buffer and warmed to 37 °C. After the pretreatment incubation, fluorescence intensity was measured on a FlexStation II fluorometric imaging plate reader (Molecular Devices). Relative fluorescence units (RFU) were measured before (20 readings) and after (40 readings) the agonist compound addition for a total 60 s read time (excitation = 485 nm, emission = 525 nm, cutoff = 515 nm). For antagonist assays, cells were treated with Calcium 5 dye for 45 min at 37 °C, 5% CO2 as with the agonist assays. Then 1/10th volume of 10× final concentration of the test antagonist compounds were added in 1% DMSO in assay buffer, and the plate was incubated for 15 min at 37 °C, 5% CO2. For Ke assays, the cells were pretreated with a single concentration of test compound and the FlexStation II added serial dilutions of the control agonist NT. For IC50 assays, cells were pretreated with serial dilutions of test compound and the FlexStation II added 1 nM NT.
Calcium Mobilization Assay for NTS2 Receptor
CHO-k1-rNTS2 cells were maintained in DMEM/F12 supplemented with 10% FBS, pen/strep, 100 μg/mL normocin, and 400 μg/mL geneticin. For calcium mobilization assays, cells were plated at 25000 cells/well in black, clear-bottom 96-well plates the day before the assay and incubated at 37 °C, 5% CO2. Then 100 μL of reconstituted Calcium 5 dye (Molecular Devices, diluted 1:20 in assay buffer (1× HBSS, 20 mM HEPES, 2.5 mM Probenicid, pH 7.4)) was added per well and plates were incubated for 45 min at 37 °C, 5% CO2. For agonist assays, cells were pretreated with 1:10 addition of 10% DMSO and the plates were returned to 37 °C, 5% CO2. Full-log serial dilutions of the test compounds were made at 10× the desired final concentration in 1% DMSO assay buffer and warmed to 37 °C. After the pretreatment incubation, fluorescence intensity was measured on a FLIPR Tetra fluorometric imaging plate reader (Molecular Devices). Relative fluorescence units (RFUs) were measured every second for 100 s (14 readings before compound addition to establish baseline fluorescence, 85 after), exposure time 0.53 s, gain = 2000, excitation intensity = 30%, gate open = 10%. For antagonist assays, 1/10 volume of 10× concentration of the test compound dilutions in 10% DMSO in assay buffer was added to cells in lieu of the 10% DMSO only in assay buffer for the pretreatment. Ke assays were run against dose–response curves of the control agonist 5b, and IC50 assays were run against the EC80 of 5b (73 nM final concentration).
Competitive Binding Assays
Relative binding affinity was evaluated using 125I labeled neurotensin and CHO-k1 cell lines overexpressing either the rNTS1 or rNTS2 receptor essentially as described by Gendron.40 Briefly, cells were plated at 100000 cells/well in 24-well plates in complete DMEM/F12 medium supplemented with 10% fetal bovine serum, 100 units of penicillin/mL, 100 mg of streptomycin/mL, 0.1 mg/mL of normocin, and 250 mg/mL geneticin. Cells were incubated at 37 °C with 5% CO2 and 95% humidity for 48 h. Cells were then equilibrated for 10 min in Earle’s buffer (130 mM NaCl, 5 mM KCl, 1.8 mM CaCl2, 0.8 mM MgCl2, and 20 mM HEPES, pH 7.4) supplemented with 0.1% BSA and 0.1% glucose and then incubated with or without 10 nM test compound in 0.1 nM [125I]NT at 37 °C for 30 min. Cells were washed twice in Earle’s buffer, extracted in 1 mL of 0.1N NaOH and counted in a Packard Cobra II gamma counter for 1 min. Total binding was determined in the absence of test compound, and nonspecific binding was determined in the presence of 1.0 μM nonradiolabeled neurotensin.
Data Analysis
To determine EC50, IC50, and Ke values, data were fit to a three-parameter logistic equation using GraphPad Prism software. Ki values for radioligand binding assays were determined from IC50 values using the equation of Cheng and Prusoff. All data are from at least three independent experiments run in duplicate wells.
Acknowledgments
This research was supported by the National Institute on Drug Abuse, grant DA29961. We gratefully acknowledge the NIMH Chemical Synthesis and Drug Supply Program for providing us with the samples of 5b and 5a.
Glossary
Abbreviations Used
- NT
neurotensin
- NTS1
neurotensin receptor 1
- NTS2
neurotensin receptor 2
- CNS
central nervous system 5
- H1
histamine receptor 1
- FLIPR
fluorometric imaging plate reader
- CHO
Chinese hamster ovary
Supporting Information Available
Elemental analysis results for test compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Caraway R.; Leeman S. E. The isolation of a new hypotensive peptide, neurotensin, from bovine hypothalami. J. Biol. Chem. 1973, 248, 6854–6861. [PubMed] [Google Scholar]
- Boules M.; Li Z.; Smith K.; Fredrickson P.; Richelson E. Diverse roles of neurotensin agonists in the central nervous system. Front. Endocrinol. 2013, 4, 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hokfelt T.; Everitt B. J.; Theodorsson-Norheim E.; Goldstein M. Occurrence of neurotensinlike immunoreactivity in subpopulations of hypothalamic, mesencephalic, and medullary catecholamine neurons. J. Comp. Neurol. 1984, 222, 543–559. [DOI] [PubMed] [Google Scholar]
- Kalivas P. W.; Miller J. S. Neurotensin neurons in the ventral tegmental area project to the medial nucleus accumbens. Brain. Res. 1984, 300, 157–160. [DOI] [PubMed] [Google Scholar]
- Kalivas P. W.; Nemeroff C. B.; Prange A. J. Jr. Neuroanatomical site specific modulation of spontaneous motor activity by neurotensin. Eur. J. Pharmacol. 1982, 78, 471–474. [DOI] [PubMed] [Google Scholar]
- Ford A. P.; Marsden C. A. In vivo neurochemical and behavioural effects of intracerebrally administered neurotensin and d-Trp11-neurotensin on mesolimbic and nigrostriatal dopaminergic function in the rat. Brain Res. 1990, 534, 243–250. [DOI] [PubMed] [Google Scholar]
- Nemeroff C. B.; Hernandez D. E.; Luttinger D.; Kalivas P. W.; Prange A. J. Jr. Interactions of neurotensin with brain dopamine systems. Ann. N. Y. Acad. Sci. 1982, 400, 330–344. [DOI] [PubMed] [Google Scholar]
- Clineschmidt B. V.; McGuffin J. C. Neurotensin administered intracisternally inhibits responsiveness of mice to noxious stimuli. Eur. J. Pharmacol. 1977, 46, 395–396. [DOI] [PubMed] [Google Scholar]
- Nemeroff C. B.; Osbahr A. J. III; Manberg P. J.; Ervin G. N.; Prange A. J. Jr. Alterations in nociception and body temperature after intracisternal administration of neurotensin, beta-endorphin, other endogenous peptides, and morphine. Proc. Natl. Acad. Sci. U. S. A. 1979, 76, 5368–5371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka K.; Masu M.; Nakanishi S. Structure and functional expression of the cloned rat neurotensin receptor. Neuron 1990, 4, 847–854. [DOI] [PubMed] [Google Scholar]
- Vita N.; Laurent P.; Lefort S.; Chalon P.; Dumont X.; Kaghad M.; Gully D.; Le Fur G.; Ferrara P.; Caput D. Cloning and expression of a complementary DNA encoding a high affinity human neurotensin receptor. FEBS Lett. 1993, 317, 139–142. [DOI] [PubMed] [Google Scholar]
- Chalon P.; Vita N.; Kaghad M.; Guillemot M.; Bonnin J.; Delpech B.; Le Fur G.; Ferrara P.; Caput D. Molecular cloning of a levocabastine-sensitive neurotensin binding site. FEBS Lett. 1996, 386, 91–94. [DOI] [PubMed] [Google Scholar]
- Mazella J.; Botto J. M.; Guillemare E.; Coppola T.; Sarret P.; Vincent J. P. Structure, functional expression, and cerebral localization of the levocabastine-sensitive neurotensin/neuromedin N receptor from mouse brain. J. Neurosci. 1996, 16, 5613–5620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazella J.; Zsurger N.; Navarro V.; Chabry J.; Kaghad M.; Caput D.; Ferrara P.; Vita N.; Gully D.; Maffrand J. P.; Vincent J. P. The 100-kDa neurotensin receptor is gp95/sortilin, a non-G-protein-coupled receptor. J. Biol. Chem. 1998, 273, 26273–26276. [DOI] [PubMed] [Google Scholar]
- Nemeroff C. B. Neurotensin: perchance an endogenous neuroleptic?. Biol. Psychiatry 1980, 15, 283–302. [PubMed] [Google Scholar]
- Kitabgi P. Targeting neurotensin receptors with agonists and antagonists for therapeutic purposes. Curr. Opin. Drug Discovery Dev. 2002, 5, 764–776. [PubMed] [Google Scholar]
- Boules M.; Fredrickson P.; Richelson E. Neurotensin agonists as an alternative to antipsychotics. Expert Opin. Invest. Drugs 2005, 14, 359–369. [DOI] [PubMed] [Google Scholar]
- Tatetsu S.; Takaki M.; Miyagawa T.; Kozuma Y.; Kozuma Y. A study on the experimental production in the brain of histopathological changes resembling schizophrenia by chronic methamphetamine administration. Seishin Shinkeigaku Zasshi 1967, 69, 1363–1370. [PubMed] [Google Scholar]
- Machiyama Y. Chronic methamphetamine intoxication model of schizophrenia in animals. Schizophrenia Bull. 1992, 18, 107–113. [DOI] [PubMed] [Google Scholar]
- Frankel P. S.; Hoonakker A. J.; Alburges M. E.; McDougall J. W.; McFadden L. M.; Fleckenstein A. E.; Hanson G. R. Effect of methamphetamine self-administration on neurotensin systems of the basal ganglia. J. Pharmacol. Exp. Ther. 2011, 336, 809–815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanson G. R.; Hoonakker A. J.; Robson C. M.; McFadden L. M.; Frankel P. S.; Alburges M. E. Response of neurotensin basal ganglia systems during extinction of methamphetamine self-administration in rat. J. Pharmacol. Exp. Ther. 2013, 346, 173–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobner P. R. Neurotensin and pain modulation. Peptides 2006, 27, 2405–2414. [DOI] [PubMed] [Google Scholar]
- Boules M.; Johnston H.; Tozy J.; Smith K.; Li Z.; Richelson E. Analgesic synergy of neurotensin receptor subtype 2 agonist NT79 and morphine. Behav. Pharmacol. 2011, 22, 573–581. [DOI] [PubMed] [Google Scholar]
- Bredeloux P.; Cavelier F.; Dubuc I.; Vivet B.; Costentin J.; Martinez J. Synthesis and biological effects of c(Lys-Lys-Pro-Tyr-Ile-Leu-Lys-Lys-Pro-Tyr-Ile-Leu) (JMV2012), a new analogue of neurotensin that crosses the blood–brain barrier. J. Med. Chem. 2008, 51, 1610–1616. [DOI] [PubMed] [Google Scholar]
- Bredeloux P.; Costentin J.; Dubuc I. Interactions between NTS2 neurotensin and opioid receptors on two nociceptive responses assessed on the hot plate test in mice. Behav. Brain Res. 2006, 175, 399–407. [DOI] [PubMed] [Google Scholar]
- Smith K. E.; Boules M.; Williams K.; Richelson E. NTS1 and NTS2 mediate analgesia following neurotensin analog treatment in a mouse model for visceral pain. Behav. Brain Res. 2012, 232, 93–97. [DOI] [PubMed] [Google Scholar]
- Hughes F. M.; Shaner B. E.; May L. A.; Zotian L.; Brower J. O.; Woods R. J.; Cash M.; Morrow D.; Massa F.; Mazella J.; Dix T. A. Identification and functional characterization of a stable, centrally active derivative of the neurotensin (8–13) fragment as a potential first-in-class analgesic. J. Med. Chem. 2010, 53, 4623–4632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarret P.; Esdaile M. J.; Perron A.; Martinez J.; Stroh T.; Beaudet A. Potent spinal analgesia elicited through stimulation of NTS2 neurotensin receptors. J. Neurosci. 2005, 25, 8188–8196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tetreault P.; Beaudet N.; Perron A.; Belleville K.; Rene A.; Cavelier F.; Martinez J.; Stroh T.; Jacobi A. M.; Rose S. D.; Behlke M. A.; Sarret P. Spinal NTS2 receptor activation reverses signs of neuropathic pain. FASEB J. 2013, 27. [DOI] [PubMed] [Google Scholar]
- Finnerup N. B.; Sindrup S. H.; Jensen T. S. The evidence for pharmacological treatment of neuropathic pain. Pain 2010, 150, 573–581. [DOI] [PubMed] [Google Scholar]
- Carraway R. L., S E.. Structural requirements for the biological activity of neurotensin, a new vasoactive peptide. In Peptides: Chemistry, Structure and Biology; Walter R., Meienhofer J., Eds.; Ann Arbor Science: Ann Arbor, MI, 1975; pp 679–685. [Google Scholar]
- Dubuc I.; Sarret P.; Labbe-Jullie C.; Botto J. M.; Honore E.; Bourdel E.; Martinez J.; Costentin J.; Vincent J. P.; Kitabgi P.; Mazella J. Identification of the receptor subtype involved in the analgesic effect of neurotensin. J. Neurosci. 1999, 19, 503–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doulut S.; Rodriguez M.; Lugrin D.; Vecchini F.; Kitabgi P.; Aumelas A.; Martinez J. Reduced peptide bond pseudopeptide analogues of neurotensin. Pept. Res. 1992, 5, 30–38. [PubMed] [Google Scholar]
- Boules M.; Liang Y.; Briody S.; Miura T.; Fauq I.; Oliveros A.; Wilson M.; Khaniyev S.; Williams K.; Li Z.; Qi Y.; Katovich M.; Richelson E. NT79: A novel neurotensin analog with selective behavioral effects. Brain Res. 2010, 1308, 35–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Einsiedel J.; Held C.; Hervet M.; Plomer M.; Tschammer N.; Hubner H.; Gmeiner P. Discovery of highly potent and neurotensin receptor 2 selective neurotensin mimetics. J. Med. Chem. 2011, 54, 2915–2923. [DOI] [PubMed] [Google Scholar]
- Held C.; Plomer M.; Hubner H.; Meltretter J.; Pischetsrieder M.; Gmeiner P. Development of a metabolically stable neurotensin receptor 2 (NTS2) ligand. ChemMedChem 2013, 8, 75–81. [DOI] [PubMed] [Google Scholar]
- Gully D.; Canton M.; Boigegrain R.; Jeanjean F.; Molimard J. C.; Poncelet M.; Gueudet C.; Heaulme M.; Leyris R.; Brouard A. Biochemical and pharmacological profile of a potent and selective nonpeptide antagonist of the neurotensin receptor. Proc. Natl. Acad. Sci. U. S. A. 1993, 90, 65–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gully D.; Labeeuw B.; Boigegrain R.; Oury-Donat F.; Bachy A.; Poncelet M.; Steinberg R.; Suaud-Chagny M. F.; Santucci V.; Vita N.; Pecceu F.; Labbe-Jullie C.; Kitabgi P.; Soubrie P.; Le Fur G.; Maffrand J. P. Biochemical and pharmacological activities of SR 142948A, a new potent neurotensin receptor antagonist. J. Pharmacol. Exp. Ther. 1997, 280, 802–812. [PubMed] [Google Scholar]
- Dahl R.; Pedersen B.; Larsen B. Intranasal levocabastine for the treatment of seasonal allergic rhinitis: a multicentre, double-blind, placebo-controlled trial. Rhinology 1995, 33, 121–125. [PubMed] [Google Scholar]
- Gendron L.; Perron A.; Payet M. D.; Gallo-Payet N.; Sarret P.; Beaudet A. Low-affinity neurotensin receptor (NTS2) signaling: internalization-dependent activation of extracellular signal-regulated kinases 1/2. Mol. Pharmacol. 2004, 66, 1421–1430. [DOI] [PubMed] [Google Scholar]
- Labeeuw B.; Gully D.; Jeanjean F.; Molimard J.; Boigegrain R.. Substituted 1-Phenyl-3-pyrazolecarboxamides Active on the Neurotensin Receptors Their Preparation and Pharmaceutical Compositions Containing Them. U.S. Patent 5,723,483, 1998.
- Jiang J.; Huang W.; Zhai J.; Liu H.; Cai Q.; Xu L.; Wang W.; Ji Y. ‘One-pot’ synthesis of 4-substituted 1,5-diaryl-1H-pyrazole-3-carboxylates via lithium tert-butoxide-mediated sterically hindered Claisen condensation and Knorr reaction. Tetrahedron 2013, 69, 627–635. [Google Scholar]
- Baxendale I. R.; Cheung S.; Kitching M. O.; Ley S. V.; Shearman J. W. The synthesis of neurotensin antagonist SR 48692 for prostate cancer research. Biorg. Med. Chem. 2013, 21, 4378–4387. [DOI] [PubMed] [Google Scholar]
- Lindh J.; Sojberg P. J. R.; M L. Synthesis of aryl ketones by palladium(II)-catalyzed decarboxylative addition of benzoic acids to nitriles. Angew. Chem. Int. Ed. 2010, 49, 7733–7737. [DOI] [PubMed] [Google Scholar]
- Quéré L.Etude de l’interaction ligand-récepteur neurotensinergique. Ph.D. Thesis. University of Namur, Namur, Belgium, 1995. [Google Scholar]
- Nagasawa H. T.; Eberling J. A.; Shirota F. N. 2-Aminoadamantane-2-carboxylic acid, a rigid, achiral, tricyclic alpha-amino acid with transport inhibitory properties. J. Med. Chem. 1973, 16, 823–826. [DOI] [PubMed] [Google Scholar]
- Yamada M.; Yamada M.; Lombet A.; Forgez P.; Rostene W. Distinct functional characteristics of levocabastine sensitive rat neurotensin NT2 receptor expressed in Chinese hamster ovary cells. Life Sci. 1998, 62, 375–380. [DOI] [PubMed] [Google Scholar]
- Richard F.; Barroso S.; Martinez J.; Labbe-Jullie C.; Kitabgi P. Agonism, inverse agonism, and neutral antagonism at the constitutively active human neurotensin receptor 2. Mol. Pharmacol. 2001, 60, 1392–1398. [DOI] [PubMed] [Google Scholar]
- Vita N.; Oury-Donat F.; Chalon P.; Guillemot M.; Kaghad M.; Bachy A.; Thurneyssen O.; Garcia S.; Poinot-Chazel C.; Casellas P.; Keane P.; Le Fur G.; Maffrand J. P.; Soubrie P.; Caput D.; Ferrara P. Neurotensin is an antagonist of the human neurotensin NT2 receptor expressed in Chinese hamster ovary cells. Eur. J. Pharmacol. 1998, 360, 265–272. [DOI] [PubMed] [Google Scholar]
- Holst B.; Holliday N. D.; Bach A.; Elling C. E.; Cox H. M.; Schwartz T. W. Common structural basis for constitutive activity of the ghrelin receptor family. J. Biol. Chem. 2004, 279, 53806–53817. [DOI] [PubMed] [Google Scholar]
- Gaddum J. H. Theories of drug antagonism. Pharmacol. Rev. 1957, 9, 211–218. [PubMed] [Google Scholar]
- Kenakin T.; Jenkinson S.; Watson C. Determining the potency and molecular mechanism of action of insurmountable antagonists. J. Pharmacol. Exp. Ther. 2006, 319, 710–723. [DOI] [PubMed] [Google Scholar]
- Roussy G.; Dansereau M. A.; Baudisson S.; Ezzoubaa F.; Belleville K.; Beaudet N.; Martinez J.; Richelson E.; Sarret P. Evidence for a role of NTS2 receptors in the modulation of tonic pain sensitivity. Mol. Pain 2009, 5, 38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith D. J.; Hawranko A. A.; Monroe P. J.; Gully D.; Urban M. O.; Craig C. R.; Smith J. P.; Smith D. L. Dose-dependent pain-facilitatory and -inhibitory actions of neurotensin are revealed by SR 48692, a nonpeptide neurotensin antagonist: influence on the antinociceptive effect of morphine. J. Pharmacol. Exp. Ther. 1997, 282, 899–908. [PubMed] [Google Scholar]
- Costa F. G.; Frussa-Filho R.; Felicio L. F. The neurotensin receptor antagonist, SR48692, attenuates the expression of amphetamine-induced behavioural sensitisation in mice. Eur. J. Pharmacol. 2001, 428, 97–103. [DOI] [PubMed] [Google Scholar]
- Gully D.; Lespy L.; Canton M.; Rostene W.; Kitabgi P.; Le Fur G.; Maffrand J. P. Effect of the neurotensin receptor antagonist SR48692 on rat blood pressure modulation by neurotensin. Life Sci. 1996, 58, 665–674. [DOI] [PubMed] [Google Scholar]
- Labbe-Jullie C.; Barroso S.; Nicolas-Eteve D.; Reversat J. L.; Botto J. M.; Mazella J.; Bernassau J. M.; Kitabgi P. Mutagenesis and modeling of the neurotensin receptor NTR1. Identification of residues that are critical for binding SR 48692, a nonpeptide neurotensin antagonist. J. Biol. Chem. 1998, 273, 16351–16357. [DOI] [PubMed] [Google Scholar]
- Labbe-Jullie C.; Botto J. M.; Mas M. V.; Chabry J.; Mazella J.; Vincent J. P.; Gully D.; Maffrand J. P.; Kitabgi P. [3H]SR 48692, the first nonpeptide neurotensin antagonist radioligand: characterization of binding properties and evidence for distinct agonist and antagonist binding domains on the rat neurotensin receptor. Mol. Pharmacol. 1995, 47, 1050–1056. [PubMed] [Google Scholar]
- Henry J. A.; Horwell D. C.; Meecham K. G.; Rees D. C. A structure–affinity study of the amino acid side-chains in neurotensin: N and C terminal deletions and Ala-scan. Bioorg. Med. Chem. Lett. 1993, 3, 949–952. [Google Scholar]
- Quéré L.; Boigegrain R.; Jeanjean F.; Gully D.; Evrard G.; Durant F. Structural requirements of non-peptide neurotensin receptor antagonists. J. Chem. Soc., Perkin Trans. 1996, 2, 2639–2646. [Google Scholar]
- Labbe-Jullie C.; Deschaintres S.; Gully D.; Le Fur G.; Kitabgi P. Effect of the nonpeptide neurotensin antagonist, SR 48692, and two enantiomeric analogs, SR 48527 and SR 49711, on neurotensin binding and contractile responses in guinea pig ileum and colon. J. Pharmacol. Exp. Ther. 1994, 271, 267–276. [PubMed] [Google Scholar]
- Labeeuw B.; Gully D.; Jeanjean F.; Molimard J.; Boigegrain R.. Substituted 1-naphthyl-3-pyrazolecarboxamides which are active on neurotensin, their preparation and pharmaceutical compositions containing them. US005502059, 1996.
- Thomas J. B.; Navarro H.; Warner K. R.; Gilmour B. The identification of nonpeptide neurotensin receptor partial agonists from the potent antagonist SR48692 using a calcium mobilization assay. Bioorg. Med. Chem. Lett. 2009, 19, 1438–1441. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.