

Abstract

We report a class of potent and selective dopamine D3 receptor antagonists based upon tranylcypromine. Although tranylcypromine has a low affinity for the rat D3 receptor (Ki = 12.8 μM), our efforts have yielded (1R,2S)-11 (CJ-1882), which has Ki values of 2.7 and 2.8 nM at the rat and human dopamine D3 receptors, respectively, and displays respective selectivities of >10000-fold and 223-fold over the rat and human D2 receptors. Evaluation in a β-arrestin functional assay showed that (1R,2S)-11 is a potent and competitive antagonist at the human D3 receptor.
Introduction
The dopamine-3 (D3) receptor subtype has been identified as an important target for agents currently in clinical use for the treatment of a variety of neurological diseases, including schizophrenia, Parkinson’s disease, and depression. However, all of the clinically approved drugs targeting the D3 receptor have a very limited selectivity over D2 receptors and other off-targets.1,2 Considerable effort has been devoted in the past decade to the design of potent and selective D3 ligands.1−16 Although it was initially challenging to design highly selective D3 ligands, due to the high degree of sequence homology between the D2 and D3 receptors, recent SAR studies have demonstrated the feasibility of such a design. For example, upon the basis of pramipexole (1), a potent D3 agonist with only modest selectivity over the D2 receptor, we designed and prepared compounds CJ-1638 (2) and CJ-1639 (3), which are potent and selective D3 agonists (Figure 1).14 Compounds 2 and 3 bind to the D3 receptor, with Ki values <1 nM and display >1000-fold selectivity over both the D1 and D2 receptors. Further, with the determination of the D3 receptor crystal structure and derived computational models, the small molecule SARs have been fortified.17 Not only have highly D3 receptor selective ligands been discovered, but the roles of the orthosteric site and a secondary binding pocket have further defined the drug–protein interactions responsible for affinity, selectivity, and efficacy.18,19
Figure 1.

Chemical structures of pramipexole and two previously reported selective D3 agonists.
Potent and selective D3 antagonists may have a therapeutic potential for the treatment of drug addictions and relate disorders.2 Herein we report the design and SAR study of a new class of D3 antagonists. Analysis of several classes of known D3 antagonists shows that their structures can be divided into three regions as shown in Figure 2: the headgroup (in blue), the linker (in black), and the tail (in red). The headgroup consists of a basic amine tethered to an aromatic group (typically phenyl), substituted with a hydrogen bonding acceptor (a nitrile in 4(3) or a methoxyl group in 6(20)), or one or two small hydrophobic groups in 5(21,22) and 7.13 We first selected a new “head” group with these features for the design and development of a new class of D3 antagonists.
Figure 2.
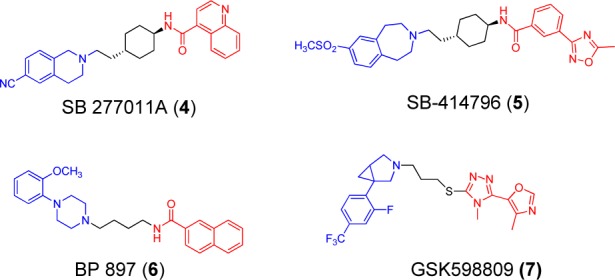
Chemical structures of previous dopamine D3 receptor antagonists.
Among potential “head” groups under consideration, we found that tranylcypromine (8, Figure 3) was attractive because, like pramipexole (1) and 7, it contains a structurally rigid phenethylamine moiety. Because the affinities of tranylcypromine for the dopamine receptors are not known, we first tested the commercially available, racemic tranylcypromine in our dopamine receptor binding assays using rat brain.23,24 Our data showed that tranylcypromine has a weak affinity for the D3 receptor with a Ki value = 12.8 μM and displays 4-fold selectivity over the D2 receptor (Table 1). Although tranylcypromine is a weak D3 ligand and has a very limited selectivity over the D2 receptor, it was used as a starting point for our SAR study, which has ultimately yielded a class of potent and selective D3 antagonists.
Figure 3.

Chemical structures and absolute configurations of the novel compounds.
Table 1. Binding Affinities at Rat Dopamine D1-Like, D2-Like, and D3 Receptors.
|
K ± SEM (nM) |
selectivity |
||||
|---|---|---|---|---|---|
| compd | D3 | D2-like | D1-like | D3/D2 | D3/D1 |
| 2 | 0.40 ± 0.087 | 725 ± 45 | 1616 ± 167 | 1827 | 4074 |
| 6 | 1.2 ± 0.10 | 468 ± 51 | 1042 ± 101 | 390 | 868 |
| 7 | 46 ± 5.6 | 18417 ± 2251 | 38807 ± 3038 | 400 | 484 |
| (±)-8 | 12793 ± 1870 | 58737 ± 5908 | 116250 ± 15103 | 4.3 | 9.1 |
| (1S,2R)-9 | 1171 ± 91 | 95593 ± 5877 | 122700 ± 10120 | 82 | 105 |
| (1R,2S)-9 | 1195 ± 71 | 96648 ± 9225 | 90587 ± 3531 | 81 | 76 |
| (1S,2R)-10 | 108 ± 7.5 | 2137 ± 247 | 3088 ± 33 | 20 | 29 |
| (1R,2S)-10 | 44 ± 5.8 | 1312 ± 170 | 2825 ± 297 | 30 | 64 |
| (±)-11 | 4.6 ± 0.40 | >100000 | 2201 ± 150 | >21000 | 479 |
| (±)-12 | 19 ± 1.8 | 503 ± 63 | 743 ± 71 | 26 | 39 |
| (±)-13 | 28 ± 2.1 | >300000 | 1804 ± 126 | >29000 | 64 |
| (1S,2R)-11 | 457 ± 34 | >100000 | 20653 ± 2035 | >218 | 45 |
| 1R,2S)-11 | 2.7 ± 0.30 | >300000 | 25810 ± 1867 | >100000 | 9559 |
Results and Discussion
Our previous study showed that the propyl substituent on the amine group in pramipexole enhances the binding affinity to the D3 receptors by at least 1 order of magnitude.14 We therefore investigated whether substitution of a propyl group on the primary amine in tranylcypromine would improve the binding affinity to the D3 receptor. Chiral resolution of racemic 2-phenyl-cyclopropanamine according to the reported method,25 followed by addition of the propyl group afforded two stereoisomers (1S,2R)-9 and (1R,2S)-9. Binding data showed that both (1S,2R)-9 and (1R,2S)-9 bind to the rat D3 receptor with Ki values of 1.2 μM and display approximately 80-fold selectivity over the D2 receptor (Table 1). Thus, addition of a propyl group to the primary amine in tranylcypromine indeed improves the binding affinity for the D3 receptor, as well as the selectivity over the D2 receptor.
Our previous study showed that introduction of appropriate linker and tail groups in compounds 2 and 3 onto the amine group in pramipexole significantly enhanced its selectivity for the rat D3 receptor over that for the D2 receptor.14 Accordingly, we synthesized (1S,2R)-10 and (1R,2S)-10 with the same linker and tail groups used in compound 2 appended onto the amine group in compounds (1S,2R)-9 and (1R,2S)-9. Compounds (1S,2R)-10 and (1R,2S)-10 have Ki values of 108 and 44 nM to the rat D3 receptor, respectively, representing a 10–20-fold improvement over (1S,2R)-9 and (1R,2S)-9. However, in contrast to the marked improved selectivity of compound 2 over pramipexole for the D3 receptor over the D2 receptor observed in our previous study,14 both (1S,2R)-10 and (1R,2S)-10 only have modest 20–30-fold selectivity over the rat D2 receptor.
Addition of a hydrophobic group such as Cl to the phenyl ring has been shown to enhance the binding affinity of antagonists to the D3 receptor. We have therefore synthesized three compounds (11, 12, and 13) in which a Cl substitution was installed in the para-, ortho-, or meta-position of the phenyl ring of 10. Because (1S,2R)-10 and (1R,2S)-10 do not differ markedly in their binding affinity to the rat D3 receptor or their selectivity over the rat D2 receptor, we first synthesized and evaluated the racemic forms of 11–13. Compounds (±)-11, (±)-12, and (±)-13 have Ki values of 4.6, 19, and 28 nM, respectively, to the rat D3 receptor. (±)-11 and (±)-13 also display high (>10000 times) selectivity over the rat D2 receptor, neither compound showing measurable binding to the rat D2 receptor at 100 μM. However, both (±)-11 and (±)-13 have significant affinity for the rat D1-like receptors with Ki values of 2.2 and 1.8 μM, respectively.
Because (±)-11 showed high affinity for the rat D3 receptor, we resolved the stereoisomers, (1S,2R)-11 and (1R,2S)-11). (1R,2S)-11 has a Ki value of 2.7 nM to the rat D3 receptor and is >100 times more potent than (1S,2R)-11 (Ki = 457 nM). Furthermore, (1R,2S)-11 has no appreciable binding to the rat D2 receptor at concentrations as high as 300 μM and consequently has >100,000-fold selectivity for the rat D3 receptor over the rat D2 receptor. (1R,2S)-11 shows weak binding affinity to the D1-like receptors, with a Ki value of 25.8 μM, and has >9000-fold selectivity for the rat D3 receptor over the rat D1-like receptors.
We next assessed the binding affinities of (±)-11, (±)-12, (±)-13, and (1R,2S)-11 to the human D2 and D3 receptors using transfected cell lines and included several previously reported D2/D3 antagonists as controls. The data are provided in Table 2.
Table 2. Binding Affinities at Human D2 and D3 Receptors.
|
Ki ± SEM (nM) |
selectivity | ||
|---|---|---|---|
| compd | D3 | D2 | D3/D2 |
| (±)-11 | 2.61 ± 0.19 | 667 ± 230 | 256 |
| (±)-12 | 22.1 ± 1.87 | 923 ± 213 | 42 |
| (±)-13 | 37.9 ± 3.57 | 1,220 ± 33.7 | 32 |
| (1R,2S)-11 | 2.80 ± 0.556 | 623 ± 105 | 223 |
| N-methylspiperone | 0.265 ± 0.008 | 0.133 ± 0.009 | 0.5 |
| eticlopride | 0.134 ± 0.004 | 0.086 ± 0.001 | 0.6 |
| raclopride | 13.4 ± 0.695 | 12.7 ± 1.21 | 1 |
| butaclamol | 6.39 ± 0.58 | 2.58 ± 0.473 | 0.4 |
| PG619 | 6.70 ± 0.77 | 1,090 ± 21 | 163 |
| PG648 | 1.88 ± 0.11 | 746 ± 123 | 397 |
Racemic compounds (±)-11, (±)-12, (±)-13, and the pure enantiomer (1R,2S)-11 have Ki values to the human D3 receptor of 2.61, 22.1, 37.9, and 2.80 nM, respectively. These values are similar to their respective Ki values of 4.6, 19, 26, and 2.7 nM to the rat D3 receptor. Compounds (±)-11, (±)-12, (±)-13, and (1R,2S)-11 have Ki values, respectively, of 667, 923, 1220, and 623 nM, to the human D2 receptor. Thus, with the exception of (±)-12, these compounds have higher binding affinities to the human D2 receptor than to the rat D2 receptor. The selectivities of (±)-11 and (±)-13 and the pure enantiomer (1R,2S)-11 for the human D3 receptor over the human D2 receptor are 256-, 32-, and 223-fold, respectively. These are lower than the selectivities observed for these compounds for the rat D3 receptor over the rat D2 receptor. The known D3 antagonists N-methylspiperone,26 eticlopride,27 raclopride,28 and butaclamol,29 all bind to the human D3 receptor with high affinities but show no selectivity between the human D2 and D3 receptors (Table 2). In comparison, PG61922 and PG648,12 two previously reported selective D3 antagonists, bind to the human D3 receptor with Ki values of 6.70 and 1.88 nM, respectively, displaying selectivities of 163- and 397-fold respectively, for the human D3 receptor over the human D2 receptor.
To assess the functional activity of (1R,2S)-11 at the D3 receptor, we tested it in a DiscoveRX D3 functional assay using the U2OS cell line transfected with human dopamine D3 receptor.30 In this assay, a D3 agonist, such as pramipexole, stimulates β-arrestin binding to the D3 receptor, while a D3 antagonist, such as 6 (BP 897),20 blocks the association of β-arrestin induced by a D3 agonist. Assessed in this manner, pramipexole has an agonist activity with an EC50 value of 3.7 ± 0.65 nM. (1R,2S)-11 has no agonist activity (EC50 > 100 μM), but it dose-dependently inhibits the binding of β-arrestin to the D3 receptor induced by 100 nM of pramipexole and has an IC50 value of 327 ± 126 nM (Table 3). In comparison, 6 shows no agonist activity but has potent antagonist activity, with an IC50 value of 13 ± 1.7 nM (Table 3). Hence, (1R,2S)-11 is a potent D3 antagonist, albeit less potent than 6. We performed a Schild regression analysis for (1R,2S)-11 in the human D3 functional assay (Figure 4). In these experiments, D3 activity, measured on the basis of β-arrestin binding to the receptor, was stimulated by pramipexole in the presence of three concentrations of (1R,2S)-11. The data were analyzed according to the methods of Kenakin.31 In the Schild plot (the inset in Figure 4), it can be seen that increasing concentrations of (1R,2S)-11 shift the pramipexole dose–response curve to the right, consistent with antagonist activity, with a KB value of 20 nM. The slope of the Schild plot was −0.83, close to unity, indicating that (1R,2S)-11 is a competitive D3 antagonist.
Table 3. Efficacy at the Human D3 Receptor in the DiscoveRx PathHunter eXpress β-Arrestin Assaya.
| agonist activity |
Antagonist Activity | ||
|---|---|---|---|
| compd | ED50 (nM) | Emax | IC50 (nM) |
| 1 | 3.7 ± 0.65 | 100 | ND |
| 6 | >100000 | 13 ± 1.7 | |
| (1R,2S)-11 | >100000 | 327 ± 126 | |
Data are the mean ± SEM of 3–6 independent determinations (2 determinations if inactive). ND = not determined.
Figure 4.

Schild analysis of (1R,2S)-11 at the human D3 receptor in the DiscoveRx PathHunter eXpress β-arrestin assay. D3 activity was stimulated by pramipexole. Schild transformation (insert) indicated a pA2 of −7.96, corresponding to a KB value of 20 nM. The slope was −0.83. Data shown are representative of two independent determinations.
The synthesis of compounds 9–13 is shown in Scheme 1. Enantiomerically pure (1R,2S)-2-phenylcyclopropanamine and (1S,2R)-2-phenylcyclo-propanamine were obtained by resolution of commercially available (±)-trans-2-phenylcyclopropylamine (8) according to the reported method.25 Reductive amination of (1R,2S)-8 and (1S,2R)-8 using propionaldehyde and NaBH4 gave (1R,2S)-9 and (1S,2R)-9 in good yield. Reductive amination of (1R,2S)-9 and (1S,2R)-9 with N-(cis-3-hydroxy-3-(2-oxoethyl)cyclobutyl)-2-naphthamide using sodium triacetoxyborohydride as a reductant gave (1R,2S)-10 and (1S,2R)-10. Compounds (±)-11–(±)-13 were prepared similarly. Reductive amination of the appropriate commercially available racemic chloro-substituted trans-2-phenylcyclopropanamine (±)-14–16 afforded (±)-17–19. The final compounds (±)-11–13 were prepared by reductive amination of (±)-17–19 with N-(cis-3-hydroxy-3-(2-oxoethyl)cyclobutyl)-2-naphthamide. N-(cis-3-Hydroxy-3-(2-oxo-ethyl)cyclobutyl)-2-naphthamide was prepared as described previously.14
Scheme 1. Synthesis of 9–13.

Reagents and conditions: (a) propionaldehyde, NaBH4, MeOH, RT; (b) N-(cis-3-hydroxy-3-(2-oxoethyl)cyclobutyl)-2-naphthamide, NaBH(OAc)3, HOAc, DCM, RT, 4 h.
The synthesis of (1S,2R)-11 is shown in Scheme 2. The optically pure key intermediate (1S,2S)-2-(4-chlorophenyl)cyclopropane-carboxylic acid 24 was prepared according to a reported method.32 Treatment of the acid chloride 20 with sodium (1R)-(+)-2,10-camphorsultam afforded the enoyl sultams 21 in excellent yield (>93%). Reaction of 21 with diazomethane in the presence of a catalytic amount of palladium acetate gave a cyclopropanated diastereomeric mixture of (1R)-(+)-2,10-camphorsultam-(1S,2S)-2-phenylcyclopropane-carboxamide 22 and (1R)-(+)-2,10-camphorsultam-(1R,2R)-2-phenyl-cyclopropane-carboxamide 23 in a ratio of 7:1. Recrystallization from ethanol afforded pure cyclopropanoyl sultams 22. Treatment of 22 with titanium isopropoxide in benzyl alcohol, followed by lithium hydroxide hydrolysis and acidification, afforded the cyclopropanecarboxylic acid (1S,2S)-24. Boc-protected (1S,2R)-2-phenylcyclopropylamine 25 was obtained from the carboxylic acid using a Curtius rearrangement of the corresponding acyl azide followed by addition of t-butanol to the isocyanate intermediate. Removal of the Boc group, followed by reductive amination, gave (1S,2R)-17. The final compound (1S,2R)-11 was obtained by reductive-amination of (1S,2R)-17 with N-(cis-3-hydroxy-3-(2-oxoethyl)cyclo-butyl)-2-naphthamide. (1R,2S)-11 was made by a method similar to that used for (1S,2R)-11, except that (1S)-(−)-2,10-camphorsultam was used as a chiral auxiliary.
Scheme 2. Synthesis of (1S,2R)-11.

Reagents and conditions: (a) (1R)-(+)-2,10-camphorsultam, NaH, THF, 0 °C, 30 min, then RT overnight; (b) CH2N2, Pd(OAc)2, DCM, RT, 10 h; (c) (i)Ti(iOPr)4, BzOH, 150 °C, 30 min, (ii) 2 M LiOH, MeOH, RT, 2 h, (iii) 4 M HCI; (d) (i) ethyl chloroformate. Et3N, acetone, 0 °C, 2 h, (ii) NaN3,1 h, (iii) 90 °C, toluene, 3 h, (iv) ButOH, reflux, 16 h. (e) TFA, DCM, RT, 12 h; (f) propionaldehyde, NaBH4, MeOH, RT; (g) N-(cis-3-hydroxy-3-(2-oxoethyl)cyclobutyl)-2-naprittiamlde, NaBH(OAc)3, HOAc, DCM, RT, 4 h.
Summary
Starting from tranylcypromine, we have designed and synthesized a class of potent and selective dopamine D3 receptor ligands. The best compound, (1R,2S)-11, had a Ki value of 2.7 nM to the rat D3 receptor and displayed a selectivity of >100000-fold over the rat D2 receptor and >9000-fold over the rat D1-like receptors. (1R,2S)-11 binds to the human D3 receptor with a Ki value of 2.8 nM and displays 223-fold selectivity over the human D2 receptor. Functional data and Schild analysis showed that (1R,2S)-11 is a potent and competitive antagonist at the human D3 receptor. (1R,2S)-11 (CJ-1882) is being further evaluated for its pharmacokinetics, brain bioavailability, and therapeutic potential for the treatment of drug abuse.
Experimental Section
General Methods
Solvents and reagents were obtained commercially and used without further purification. Reactions were monitored by TLC carried out on 250 μm silica gel plates 60F-254 (E. Merck) using UV light as visualizing agent. Silica gel 60, particle size 15–40 μm (E. Merck), was used for flash column chromatography. NMR spectra were recorded on a Bruker Avance 300 spectrometer (300 MHz). Chemical shifts (δ) are reported as δ values (ppm) downfield relative to TMS as an internal standard, with multiplicities reported in the standard manner. All final compounds have purities >95%, as determined by HPLC (UV detection at 254 nm).
(1S,2R)-2-Phenyl-N-propylcyclopropanamine ((1S,2R)-9)
Propionaldehyde (82 mg, 1.41 mmol) was added to a solution of (1S,2R)-2-phenylcyclopropanamine (188 mg, 1.41 mmol) in MeOH (10 mL), and the reaction mixture was stirred at room temperature for 2 h. Sodium borohydride (79 mg, 2.12 mmol) was then added, and the mixture was stirred at room temperature for 1 h. The reaction was quenched with H2O (30 mL) and extracted with ethyl acetate (40 mL). The residue was chromatographed (hexane:EtOAc = 50:50) to give (1S,2R)-9 (178 mg, 72% yield) as a colorless oil. 1H NMR (CD3OD, 300 MHz) δ 7.37–7.12 (m, 5H), 3.20–3.08 (m, 2H), 2.97–2.90 (m, 1H), 2.56–2.45 (m, 1H), 1.82–1.70 (m, 2H), 1.57–1.45 (m, 1H), 1.36 (dd, J = 6.7, 14.4 Hz, 1H), 1.04 (t, J = 7.4 Hz). 13C NMR (CD3OD, 75 MHz) δ 139.42, 129.68, 127.91, 127.56, 50.88, 39.04, 22.32, 20.55, 13.35, 11.19.
(1R,2S)-2-Phenyl-N-propylcyclopropanamine ((1R,2S)-9)
Propionaldehyde (82 mg, 1.41 mmol) was added to a solution of (1R,2S)-2-phenylcyclopropanamine (188 mg, 1.41 mmol) in MeOH (10 mL), and the reaction mixture was stirred at room temperature for 2 h. Sodium borohydride (79 mg, 2.12 mmol) was then added, and the mixture was stirred at room temperature for 1 h. The reaction was quenched with H2O (30 mL) and extracted with EtOAc (40 mL). The residue was chromatographed (hexane:EtOAc = 50:50) to give (1R,2S)-9 (171 mg, 69% yield) as a colorless oil. 1H NMR (CD3OD, 300 MHz) δ 7.37–7.12 (m, 5H), 3.20–3.08 (m, 2H), 2.97–2.90 (m, 1H), 2.56–2.45 (m, 1H), 1.82–1.70 (m, 2H), 1.57–1.45 (m, 1H), 1.36 (dd, J = 6.7, 14.4 Hz, 1H), 1.04 (t, J = 7.4 Hz). 13C NMR (CD3OD, 75 MHz) δ 139.42, 129.68, 127.91, 127.56, 50.88, 39.04, 22.32, 20.55, 13.35, 11.19.
N-(cis-3-Hydroxy-3-(2-(((1S,2R)-2-phenylcyclopropyl)(propyl)-amino)ethyl)cyclobutyl)-2-naphthamide ((1S,2R)-10)
N-(cis-3-Hydroxy-3-(2-oxoethyl)cyclobutyl)-2-naphthamide (80 mg, 0.28 mmol), sodium triacetoxyborohydride (90 mg, 0.43 mmol), and AcOH (26 mg, 0.43 mmol) were added to a solution of (1S,2R)-9 (50 mg, 0.28 mmol) in CH2Cl2 (20 mL). The mixture was stirred at room temperature for 4 h and then quenched by addition of H2O (30 mL). The mixture was extracted with CH2Cl2 (30 mL × 3). The organic solvent was removed under vacuum, and the residue was chromatographed (MeOH:EtOAc = 10:90) to give (1S,2R)-10 (43 mg, 34% yield) as a colorless oil. 1H NMR (CD3OD, 300 MHz) δ 8.39 (s, 1H), 8.00–7.80 (m, 4H), 7.60–7.50 (m, 2H), 7.40–7.20 (m, 5H), 4.25–4.00 (m, 1H), 3.60–3.25 (m, 4H), 3.20–3.02 (m, 1H), 2.75–2.60 (m, 3H), 2.40–2.09 (m, 4H), 1.90–1.65 (m, 3H), 1.53 (dd, J = 7.1, 14.4 Hz, 1H), 1.05 (t, J = 7.4 Hz, 3H). 13C NMR (CD3OD, 75 MHz) δ 169.90, 164.83, 138.89, 136.25, 133.99, 132.65, 129.99, 129.91, 129.31, 128.85, 128.76, 128.21, 127.86, 127.18, 124.91, 68.81, 58.31, 53.29, 46.48, 44.19, 44.09, 38.24, 23.66, 18.84, 14.49, 11.23.
N-(cis-3-Hydroxy-3-(2-(((1R,2S)-2-phenylcyclopropyl)(propyl)amino)-ethyl)cyclobutyl)-2-naphthamide ((1R,2S)-10)
N-(cis-3-Hydroxy-3-(2-oxoethyl)cyclobutyl)-2-naphthamide (80 mg, 0.28 mmol), sodium triacetoxyborohydride (90 mg, 0.43 mmol), and AcOH (26 mg, 0.43 mmol) were added to a solution of (1R,2S)-9 (50 mg, 0.28 mmol) in CH2Cl2 (20 mL), and the mixture was stirred at room temperature for 4 h. The reaction was quenched by addition of H2O (30 mL), and the mixture was extracted with CH2Cl2 (30 mL × 3). The organic solvent was removed under vacuum, and the residue was chromatographed (MeOH:EtOAc = 10:90) to give (1R,2S)-10 (36 mg, 29% yield) as a colorless oil. 1H NMR (CD3OD, 300 MHz) δ 8.39 (s, 1H), 8.00–7.80 (m, 4H), 7.60–7.50 (m, 2H), 7.40–7.20 (m, 5H), 4.25–4.00 (m, 1H), 3.60–3.25 (m, 4H), 3.20–3.02 (m, 1H), 2.75–2.60 (m, 3H), 2.40–2.09 (m, 4H), 1.90–1.65 (m, 3H), 1.53 (dd, J = 7.1, 14.4 Hz, 1H), 1.05 (t, J = 7.4 Hz, 3H). 13C NMR (CD3OD, 75 MHz) δ 169.90, 164.83, 138.89, 136.25, 133.99, 132.65, 129.99, 129.91, 129.31, 128.85, 128.76, 128.21, 127.86, 127.18, 124.91, 68.81, 58.31, 53.29, 46.48, 44.19, 44.09, 38.24, 23.66, 18.84, 14.49, 11.23.
N-(cis-3-(2-((((±)-trans)-2-(2-Chlorophenyl)cyclopropyl)(propyl)-amino)ethyl)-3-hydroxycyclobutyl)-2-naphthamide ((±)-12)
N-(cis-3-Hydroxy-3-(2-oxoethyl)cyclobutyl)-2-naphthamide (82 mg, 0.29 mmol), sodium triacetoxyborohydride (91 mg, 0.43 mmol), and AcOH (26 mg, 0.43 mmol) were added to a solution of (±)-18 (60 mg, 0.29 mmol) in CH2Cl2 (20 mL), and the mixture was stirred at room temperature for 4 h. The reaction was quenched by addition of H2O (30 mL), and the mixture was extracted with CH2Cl2 (30 mL × 3). The organic solvent was removed under vacuum, and the residue was chromatographed (MeOH:EtOAc = 10:90) to give (±)-12 (49 mg, 36% yield) as a colorless oil. 1H NMR (CD3OD, 300 MHz) δ 8.39 (s, 1H), 8.00–7.85 (m, 4H), 7.70–7.52 (m, 2H), 7.47 (dd, J = 1.6, 7.4 Hz, 1H), 7.40–7.20 (m, 2H), 7.09 (dd, J = 2.0, 7.4 Hz, 1H), 4.25–4.02 (m, 1H), 3.70–3.30 (m, 4H), 3.30–3.20 (m, 1H), 2.75–2.60 (m, 2H), 2.40–2.10 (m, 4H), 2.00–1.50 (m, 4H), 1.07 (t, J = 7.3 Hz, 3H). 13C NMR (CD3OD, 75 MHz) δ 170.16, 136.48, 136.40, 135.76, 134.23, 132.87, 130.91, 130.19, 129.95, 129.52, 129.05, 128.97, 128.91, 128.06, 127.59, 125.09, 69.09, 58.35, 53.41, 46.50, 44.48, 44.29, 38.48, 34.56, 21.52, 18.85, 14.44, 11.42.
N-(cis-3-(2-((((±)-trans)-2-(4-Chlorophenyl)cyclopropyl)(propyl)-amino)ethyl)-3-hydroxycyclobutyl)-2-naphthamide ((±)-11)
(±)-11 was prepared in a manner similar to that used for (±)-12 in 35% yield. 1H NMR (CDCl3, 300 MHz) δ 8.28 (s, 1H), 7.90–7.75 (m, 4H), 7.60–7.50 (m, 2H), 7.23 (d, J = 8.5 Hz, 2H), 6.94 (d, J = 8.5 Hz, 2H), 6.53 (d, J = 7.7 Hz, 1H), 4.40–4.20 (m, 1H), 3.00–2.40 (m, 6H), 2.25–1.70 (m, 6H), 1.60–1.50 (m, 2H), 1.25–1.00 (m, 2H), 0.89 (t, J = 7.3 Hz, 3H). 13C NMR (CDCl3, 75 MHz) δ 167.21, 140.12, 134.93, 132.82, 131.83, 131.72, 129.14, 128.71, 128.67, 127.95, 127.84, 127.58, 127.25, 126.96, 123.75, 71.19, 57.69, 53.27, 48.76, 44.73, 44.57, 37.51, 34.44, 24.94, 20.15, 17.02, 12.23.
N-(cis-3-(2-((((±)-trans)-2-(3-Chlorophenyl)cyclopropyl)(propyl)-amino)ethyl)-3-hydroxycyclobutyl)-2-naphthamide ((±)-13)
(±)-13 was prepared in a manner similar to that used for (±)-12 in 32% yield. 1H NMR (CD3OD, 300 MHz) δ 8.39 (s, 1H), 8.00–7.80 (m, 4H), 7.70–7.50 (m, 2H), 7.40–7.10 (m, 4H), 4.25–4.10 (m, 1H), 3.60–3.10 (m, 5H), 2.75–2.65 (m, 3H), 2.35–2.10 (m, 4H), 1.90–1.50 (m, 4H), 1.05 (t, J = 7.3 Hz, 3H). 13C NMR (CD3OD, 75 MHz) δ 170.14, 141.61, 136.45, 136.01, 134.20, 132.86, 131.60, 130.18, 129.50, 129.04, 128.95, 128.51, 128.04, 127.45, 126.01, 125.09, 69.05, 58.49, 53.47, 46.64, 44.42, 44.27, 38.47, 34.50, 23.42, 19.02, 14.99, 11.41.
N-(cis-3-(2-(((1S,2R)-2-(4-Chlorophenyl)cyclopropyl)(propyl)-amino)ethyl)-3-hydroxycyclobutyl)-2-naphthamide ((1S,2R)-11)
N-(cis-3-Hydroxy-3-(2-oxoethyl)cyclobutyl)-2-naphthamide (191 mg, 0.67 mmol), sodium triacetoxyborohydride (212 mg, 1.01 mmol), and AcOH (60 mg, 1.01 mmol) were added to a solution of (1S,2R)-17 (140 mg, 0.67 mmol) in CH2Cl2 (20 mL), and the mixture was stirred at room temperature for 4 h. The reaction was quenched by addition of H2O (30 mL), and the mixture was extracted with CH2Cl2 (30 mL × 3). The organic solvent was removed under vacuum, and the residue was chromatographed (MeOH:EtOAc = 10:90) to give (1S,2R)-11 (79 mg, 25% yield) as a colorless oil. 1H NMR (CDCl3, 300 MHz) δ 8.28 (s, 1H), 7.90–7.75 (m, 4H), 7.60–7.50 (m, 2H), 7.23 (d, J = 8.5 Hz, 2H), 6.94 (d, J = 8.5 Hz, 2H), 6.53 (d, J = 7.7 Hz, 1H), 4.40–4.20 (m, 1H), 3.00–2.40 (m, 6H), 2.25–1.70 (m, 6H), 1.60–1.50 (m, 2H), 1.25–1.00 (m, 2H), 0.89 (t, J = 7.3 Hz, 3H). 13C NMR (CDCl3, 75 MHz) δ 167.21, 140.12, 134.93, 132.82, 131.83, 131.72, 129.14, 128.71, 128.67, 127.95, 127.84, 127.58, 127.25, 126.96, 123.75, 71.19, 57.69, 53.27, 48.76, 44.73, 44.57, 37.51, 34.44, 24.94, 20.15, 17.02, 12.23.
N-(cis-3-(2-(((1R,2S)-2-(4-Chlorophenyl)cyclopropyl)(propyl)-amino)ethyl)-3-hydroxycyclobutyl)-2-naphthamide ((1R,2S)-11)
(1R,2S)-11 was prepared in 35% yield in a manner similar to that used for (1S,2R)-11, except that (1S)-(−)-2,10-camphorsultam was used as a chiral auxiliary. 1H NMR (CDCl3, 300 MHz) δ 8.28 (s, 1H), 7.90–7.75 (m, 4H), 7.60–7.50 (m, 2H), 7.23 (d, J = 8.5 Hz, 2H), 6.94 (d, J = 8.5 Hz, 2H), 6.53 (d, J = 7.7 Hz, 1H), 4.40–4.20 (m, 1H), 3.00–2.40 (m, 6H), 2.25–1.70 (m, 6H), 1.60–1.50 (m, 2H), 1.25–1.00 (m, 2H), 0.89 (t, J = 7.3 Hz, 3H). 13C NMR (CDCl3, 75 MHz) δ 167.21, 140.12, 134.93, 132.82, 131.83, 131.72, 129.14, 128.71, 128.67, 127.95, 127.84, 127.58, 127.25, 126.96, 123.75, 71.19, 57.69, 53.27, 48.76, 44.73, 44.57, 37.51, 34.44, 24.94, 20.15, 17.02, 12.23.
In Vitro Dopamine Receptor Binding Assays at the Rat Dopamine Receptors
The binding affinities of all the synthetic compounds were determined at the D3, D2, and D1-like receptors in membranes prepared from the brains of adult, male Sprague–Dawley rats (Pel-Freez, Rogers, AR). All compounds were dissolved in 100% EtOH at a concentration of 5 mM.
[3H]R(+)-7-OH-DPAT Binding Assay
The [3H]R-(+)-7-OH-DPAT binding assay for the rat D3 dopamine receptors was performed as described.23 A rat ventral striatum (nucleus accumbens and olfactory tubercles) was prepared in assay buffer (50 mM Tris, 1 mM EDTA; pH 7.4 at 23 °C) to yield a final concentration of 10 mg original wet weight (oww)/mL. Membranes were incubated with [3H]R-(+)-7-OH-DPAT (0.15 nM, SA = 163 Ci/mmol; GE Healthcare) and different concentrations of the test compounds (10–10 to 10–4 M). Nonspecific binding was defined by 1 μM spiperone (Sigma-Aldrich). Assay tubes were incubated at 23 °C for 90 min. The reaction was terminated by rapid vacuum filtration. Data were analyzed using SigmaPlot 10. Ki values were calculated using KD = 0.15 nM for [3H]7-OH-DPAT23 and are expressed as the mean ± SEM of 3–5 independent determinations.
[3H]Spiperone Binding Assay
[3H]Spiperone binding assays for rat D2-like receptors were performed as described24 for [3H]R-(+)-7-OH-DPAT with the following modifications. Assays were performed using membranes prepared from rat caudate-putamen, which expresses D2 receptors in high density but with very low levels of D3 receptors, and the final membrane homogenate concentration was 1.5 mg oww/mL. The assay buffer was 50 mM Tris-HCl, 5 mM KCl, 2 mM MgCl2, and 2 mM CaCl2, pH 7.4 at 23 °C; the concentration of [3H]spiperone (60–96 Ci/mmol; GE Healthcare, PerkinElmer, or American Radiolabeled Chemicals) was 0.2 nM, and the incubation time was 90 min at 23 °C. Nonspecific binding was defined in the presence of 1 μM (+)-butaclamol (Sigma-Aldrich). Ki values were calculated using the experimentally determined KD value for [3H]spiperone of 0.4 nM.
[3H]SCH 23390 Binding Assay
[3H] SCH 23390 binding assay for rat D1-like dopamine receptors was performed as described33 for [3H]spiperone binding except the concentration of [3H]SCH 23390 (60 Ci/mmol; American Radiolabeled Chemicals) was 0.3 nM. Ki values were calculated using the KD value for [3H]SCH 23390 of 0.3 nM.
In Vitro Dopamine Receptor Binding Assays at the Human Dopamine Receptors
HEK293 cells stably expressing human dopamine D2 and D3 receptors were grown in a 1:1 mixture of DMEM and Ham’s F12 culture media, supplemented with 20 mM HEPES, 2 mM l-glutamine, 0.1 mM nonessential amino acids, 1× antibiotic/antimycotic, 10% heat-inactivated fetal bovine serum, and 200 μg/mL hygromycin (Life Technologies, Grand Island, NY) and kept in an incubator at 37 °C and 5% CO2. Upon reaching 80–90% confluence, cells were harvested using premixed Earle’s Balanced Salt Solution (EBSS) with 5 μM EDTA (Life Technologies) and centrifuged at 3000 rpm for 10 min at 21 °C. The supernatant was removed, and the pellet was resuspended in 10 mL of hypotonic lysis buffer (5 mM MgCl2·6H2O, 5 mM Tris, pH 7.4 at 4 °C) and centrifuged at 20000 rpm for 30 min at 4 °C. The pellet was then resuspended in fresh EBSS buffer made from 8.7 g/L Earle’s Balanced Salts without phenol red (US Biological, Salem, MA), 2.2 g/L sodium bicarbonate, pH to 7.4. A Bradford protein assay (Bio-Rad, Hercules, CA) was used to determine the protein concentration, and membranes were diluted to 500 μg/mL and stored in a −80 °C freezer for later use.
Immediately prior to testing, all test compounds were freshly dissolved in 30% DMSO and 70% H2O to a stock concentration of 100 μM. To assist the solubilization of free-base compounds, 10 μL of glacial acetic acid was added along with the DMSO. Each test compound was then diluted into 13 half-log serial dilutions using 30% DMSO vehicle; final test concentrations ranged from 10 μM to 10 pM. Previously frozen membranes were diluted in fresh EBSS to a 100 μg/mL stock for binding. Radioligand competition experiments were conducted in glass tubes containing 300 μL of fresh EBSS buffer with 0.2 mM sodium metabisulfite, 50 μL of diluted test compound, 100 μL of membranes (10 μg total protein), and 50 μL of [3H]N-methylspiperone (0.4 nM final concentration; PerkinElmer). Nonspecific binding was determined using 10 μM butaclamol (Sigma-Aldrich, St. Louis, MO), and total binding was determined with 30% DMSO vehicle. All compound dilutions were tested in triplicate and the reaction incubated for 1 h at room temperature. The reaction was terminated by filtration through Whatman GF/B filters, presoaked for 1 h in 0.5% polyethylenimine, using a Brandel R48 filtering manifold (Brandel Instruments, Gaithersburg, MD). The filters were washed 3 times with 3 mL of ice-cold EBSS buffer and transferred to scintillation vials. Then 3 mL of CytoScint liquid scintillation cocktail (MP Biomedicals, Solon, OH) was added and vials were counted using a PerkinElmer Tri-Carb 2910 TR liquid scintillation counter (Waltham, MA). IC50 values for each compound were determined from dose–response curves, and Ki values were calculated using the Cheng–Prusoff equation.34 These analyses were performed using GraphPad Prism version 5.00 for Windows (GraphPad Software, San Diego, CA). Reported Ki values were determined from least three independent experiments.
Dopamine D3 β-Arrestin Functional Assay
Functional activity at the D3 receptor was determined using a PathHunter eXpress β-arrestin assay kit (DiscoveRx, Fremont, CA) for human D3 receptors transfected in U20S cells.30 Compounds were screened for agonist and antagonist activity according to the manufacturer’s instructions. Pramipexole (100 nM) was used as the reference agonist. Data were analyzed using SigmaPlot 10 and are presented as the mean ± SEM of 4–6 independent determinations. For the Schild analysis, D3 activity was stimulated by pramipexole in the presence of 3 concentrations of (1R,2S)-11 and analyzed according to the methods of Kenakin.31
Acknowledgments
This work was supported by a grant from the National Institute on Drug Abuse, National Institutes of Health (R01 DA032943 to S.W.) and the NIDA–Intramural Research Program (A.H.N, T.M.K). We thank Dr. David R. Sibley at NINDS/NIH for providing HEK293 cells stably expressing human dopamine D2 and D3 receptors, Heather Spaulding at the University of Kansas Medical Center, and Catherine Schweppe and Caitlin Burzynski at NIDA/NIH for their technical assistance.
Supporting Information Available
Experimental details of chemical synthesis and chemical data. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Newman A. H.; Grundt P.; Nader M. A. Dopamine D3 receptor partial agonists and antagonists as potential drug abuse therapeutic agents. J. Med. Chem. 2005, 48, 3663–79. [DOI] [PubMed] [Google Scholar]
- Heidbreder C. A.; Newman A. H. Current perspectives on selective dopamine D(3) receptor antagonists as pharmacotherapeutics for addictions and related disorders. Ann. N. Y. Acad. Sci. 2010, 1187, 4–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reavill C.; Taylor S. G.; Wood M. D.; Ashmeade T.; Austin N. E.; Avenell K. Y.; Boyfield I.; Branch C. L.; Cilia J.; Coldwell M. C.; Hadley M. S.; Hunter A. J.; Jeffrey P.; Jewitt F.; Johnson C. N.; Jones D. N.; Medhurst A. D.; Middlemiss D. N.; Nash D. J.; Riley G. J.; Routledge C.; Stemp G.; Thewlis K. M.; Trail B.; Vong A. K.; Hagan J. J. Pharmacological actions of a novel, high-affinity, and selective human dopamine D(3) receptor antagonist, SB-277011-A. J. Pharmacol. Exp. Ther. 2000, 294, 1154–65. [PubMed] [Google Scholar]
- Hackling A.; Ghosh R.; Perachon S.; Mann A.; Holtje H. D.; Wermuth C. G.; Schwartz J. C.; Sippl W.; Sokoloff P.; Stark H. N-(omega-(4-(2-Methoxyphenyl)piperazin-1-yl)alkyl)carboxamides as dopamine D2 and D3 receptor ligands. J. Med. Chem. 2003, 46, 3883–99. [DOI] [PubMed] [Google Scholar]
- Macdonald G. J.; Branch C. L.; Hadley M. S.; Johnson C. N.; Nash D. J.; Smith A. B.; Stemp G.; Thewlis K. M.; Vong A. K.; Austin N. E.; Jeffrey P.; Winborn K. Y.; Boyfield I.; Hagan J. J.; Middlemiss D. N.; Reavill C.; Riley G. J.; Watson J. M.; Wood M.; Parker S. G.; Ashby C. R. Jr. Design and synthesis of trans-3-(2-(4-((3-(3-(5-methyl-1,2,4-oxadiazolyl))- phenyl)carboxamido)cyclohexyl)ethyl)-7-methylsulfonyl-2,3,4,5-tetrahydro-1H-3-benzazepine (SB-414796): a potent and selective dopamine D3 receptor antagonist. J. Med. Chem. 2003, 46, 4952–64. [DOI] [PubMed] [Google Scholar]
- Campiani G.; Butini S.; Trotta F.; Fattorusso C.; Catalanotti B.; Aiello F.; Gemma S.; Nacci V.; Novellino E.; Stark J. A.; Cagnotto A.; Fumagalli E.; Carnovali F.; Cervo L.; Mennini T. Synthesis and pharmacological evaluation of potent and highly selective D3 receptor ligands: inhibition of cocaine-seeking behavior and the role of dopamine D3/D2 receptors. J. Med. Chem. 2003, 46, 3822–39. [DOI] [PubMed] [Google Scholar]
- Bettinetti L.; Schlotter K.; Hubner H.; Gmeiner P. Interactive SAR studies: rational discovery of super-potent and highly selective dopamine D3 receptor antagonists and partial agonists. J. Med. Chem. 2002, 45, 4594–7. [DOI] [PubMed] [Google Scholar]
- Chen J.; Ding K.; Levant B.; Wang S. Design of novel hexahydropyrazinoquinolines as potent and selective dopamine D3 receptor ligands with improved solubility. Bioorg. Med. Chem. Lett. 2006, 16, 443–6. [DOI] [PubMed] [Google Scholar]
- Ji M.; Chen J.; Ding K.; Wu X.; Varady J.; Levant B.; Wang S. Design, synthesis and structure–activity relationship studies of hexahydropyrazinoquinolines as a novel class of potent and selective dopamine receptor 3 (D3) ligands. Bioorg. Med. Chem. Lett. 2005, 15, 1701–5. [DOI] [PubMed] [Google Scholar]
- Varady J.; Wu X.; Fang X.; Min J.; Hu Z.; Levant B.; Wang S. Molecular modeling of the three-dimensional structure of dopamine 3 (D3) subtype receptor: discovery of novel and potent D3 ligands through a hybrid pharmacophore- and structure-based database searching approach. J. Med. Chem. 2003, 46, 4377–92. [DOI] [PubMed] [Google Scholar]
- Ehrlich K.; Gotz A.; Bollinger S.; Tschammer N.; Bettinetti L.; Harterich S.; Hubner H.; Lanig H.; Gmeiner P. Dopamine D2, D3, and D4 selective phenylpiperazines as molecular probes to explore the origins of subtype specific receptor binding. J. Med. Chem. 2009, 52, 4923–35. [DOI] [PubMed] [Google Scholar]
- Newman A. H.; Grundt P.; Cyriac G.; Deschamps J. R.; Taylor M.; Kumar R.; Ho D.; Luedtke R. R. N-(4-(4-(2,3-Dichloro- or 2-methoxyphenyl)piperazin-1-yl)butyl)heterobiarylcarboxamides with functionalized linking chains as high affinity and enantioselective D3 receptor antagonists. J. Med. Chem. 2009, 52, 2559–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Micheli F.; Arista L.; Bertani B.; Braggio S.; Capelli A. M.; Cremonesi S.; Di-Fabio R.; Gelardi G.; Gentile G.; Marchioro C.; Pasquarello A.; Provera S.; Tedesco G.; Tarsi L.; Terreni S.; Worby A.; Heidbreder C. Exploration of the amine terminus in a novel series of 1,2,4-triazolo-3-yl-azabicyclo[3.1.0]hexanes as selective dopamine D3 receptor antagonists. J. Med. Chem. 2010, 53, 7129–39. [DOI] [PubMed] [Google Scholar]
- Chen J.; Collins G. T.; Levant B.; Woods J.; Deschamps J. R.; Wang S. CJ-1639: A Potent and Highly Selective Dopamine D3 Receptor Full Agonist. ACS Med. Chem. Lett. 2011, 2, 620–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song R.; Yang R. F.; Wu N.; Su R. B.; Li J.; Peng X. Q.; Li X.; Gaal J.; Xi Z. X.; Gardner E. L. YQA14: a novel dopamine D3 receptor antagonist that inhibits cocaine self-administration in rats and mice, but not in D3 receptor-knockout mice. Addict. Biol. 2012, 17, 259–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Micheli F.; Heidbreder C. Dopamine D3 receptor antagonists: a patent review (2007–2012). Expert Opin. Ther. Pat. 2013, 23, 363–81. [DOI] [PubMed] [Google Scholar]
- Chien E. Y.; Liu W.; Zhao Q.; Katritch V.; Han G. W.; Hanson M. A.; Shi L.; Newman A. H.; Javitch J. A.; Cherezov V.; Stevens R. C. Structure of the human dopamine D3 receptor in complex with a D2/D3 selective antagonist. Science 2010, 330, 1091–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman A. H.; Beuming T.; Banala A. K.; Donthamsetti P.; Pongetti K.; LaBounty A.; Levy B.; Cao J.; Michino M.; Luedtke R. R.; Javitch J. A.; Shi L. Molecular determinants of selectivity and efficacy at the dopamine D3 receptor. J. Med. Chem. 2012, 55, 6689–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keck T. M.; Burzynski C.; Shi L.; Newman A. H. Beyond small-molecule SAR: using the dopamine D3 receptor crystal structure to guide drug design. Adv. Pharmacol. 2014, 69, 267–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pilla M.; Perachon S.; Sautel F.; Garrido F.; Mann A.; Wermuth C. G.; Schwartz J. C.; Everitt B. J.; Sokoloff P. Selective inhibition of cocaine-seeking behaviour by a partial dopamine D3 receptor agonist. Nature 1999, 400, 371–5. [DOI] [PubMed] [Google Scholar]
- Grundt P.; Carlson E. E.; Cao J.; Bennett C. J.; McElveen E.; Taylor M.; Luedtke R. R.; Newman A. H. Novel heterocyclic trans olefin analogues of N-{4-[4-(2,3-dichlorophenyl)piperazin-1-yl]butyl}arylcarboxamides as selective probes with high affinity for the dopamine D3 receptor. J. Med. Chem. 2005, 48, 839–848. [DOI] [PubMed] [Google Scholar]
- Grundt P.; Prevatt K. M.; Cao J.; Taylor M.; Floresca C. Z.; Choi J. K.; Jenkins B. G.; Luedtke R. R.; Newman A. H. Heterocyclic analogues of N-(4-(4-(2,3-dichlorophenyl)piperazin-1-yl)butyl)arylcarboxamides with functionalized linking chains as novel dopamine D3 receptor ligands: potential substance abuse therapeutic agents. J. Med. Chem. 2007, 50, 4135–46. [DOI] [PubMed] [Google Scholar]
- Bancroft G. N.; Morgan K. A.; Flietstra R. J.; Levant B. Binding of [3H]PD 128907, a putatively selective ligand for the D3 dopamine receptor, in rat brain: a receptor binding and quantitative autoradiographic study. Neuropsychopharmacology 1998, 18, 305–16. [DOI] [PubMed] [Google Scholar]
- Levant B.; Grigoriadis D. E.; DeSouza E. B. Characterization of [3H]quinpirole binding to D2-like dopamine receptors in rat brain. J. Pharmacol. Exp. Ther. 1992, 262, 929–35. [PubMed] [Google Scholar]
- Kinzel O.; Alfieri A.; Altamura S.; Brunetti M.; Bufali S.; Colaceci F.; Ferrigno F.; Filocamo G.; Fonsi M.; Gallinari P.; Malancona S.; Hernando J. I.; Monteagudo E.; Orsale M. V.; Palumbi M. C.; Pucci V.; Rowley M.; Sasso R.; Scarpelli R.; Steinkuhler C.; Jones P. Identification of MK-5710 ((8aS)-8a-methyl-1,3-dioxo-2-[(1S,2R)-2-phenylcyclo- propyl]-N-(1-phenyl-1H-pyrazol-5-yl)hexahydro-imidazo[1,5-a]pyrazine-7(1H)-carbox amide), a potent smoothened antagonist for use in Hedgehog pathway dependent malignancies, part 2. Bioorg. Med. Chem. Lett. 2011, 21, 4429–35. [DOI] [PubMed] [Google Scholar]
- Andree B.; Nyberg S.; Ito H.; Ginovart N.; Brunner F.; Jaquet F.; Halldin C.; Farde L. Positron emission tomographic analysis of dose-dependent MDL 100,907 binding to 5-hydroxytryptamine-2A receptors in the human brain. J. Clin. Psychopharmacol. 1998, 18, 317–23. [DOI] [PubMed] [Google Scholar]
- Claytor R.; Lile J. A.; Nader M. A. The effects of eticlopride and the selective D3-antagonist PNU 99194-A on food- and cocaine-maintained responding in rhesus monkeys. Pharmacol., Biochem. Behav. 2006, 83, 456–64. [DOI] [PubMed] [Google Scholar]
- Kohler C.; Hall H.; Ogren S. O.; Gawell L. Specific in vitro and in vivo binding of 3H-raclopride. A potent substituted benzamide drug with high affinity for dopamine D2 receptors in the rat brain. Biochem. Pharmacol. 1985, 34, 2251–9. [DOI] [PubMed] [Google Scholar]
- Chrzanowski F. A.; McGrogan B. A.; Maryanoff B. E. The pKa of butaclamol and the mode of butaclamol binding to central dopamine receptors. J. Med. Chem. 1985, 28, 399–400. [DOI] [PubMed] [Google Scholar]
- Olson K. R.; Eglen R. M. Beta-galactosidase complementation: a cell-based luminescent assay platform for drug discovery. Assay Drug Dev. Technol. 2007, 5, 137–44. [DOI] [PubMed] [Google Scholar]
- Kenakin T. P.Pharmacologic Analysis of Drug–Receptor Interaction; Raven Press: New York, 1987. [Google Scholar]
- Vallgarda J.; Appelberg U.; Csoregh I.; Hacksell U. Stereoselectivity and Generality of the Palladium-Catalyzed Cyclopropanation of Alpha,Beta-Unsaturated Carboxylic Acids Derivatized with Oppolzers Sultam. J. Chem. Soc., Perkin Trans. 1 1994, 461–70. [Google Scholar]
- Levant B.Characterization of Dopamine Receptors. In Current Protocols in Pharmacology; John Wiley & Sons, Inc.: New York, 2001. [Google Scholar]
- Cheng Y.; Prusoff W. H. Relationship between the inhibition constant (Ki) and the concentration of inhibitor which causes 50% inhibition (IC50) of an enzymatic reaction. Biochem. Pharmacol. 1973, 22, 3099–3108. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.