

Abstract
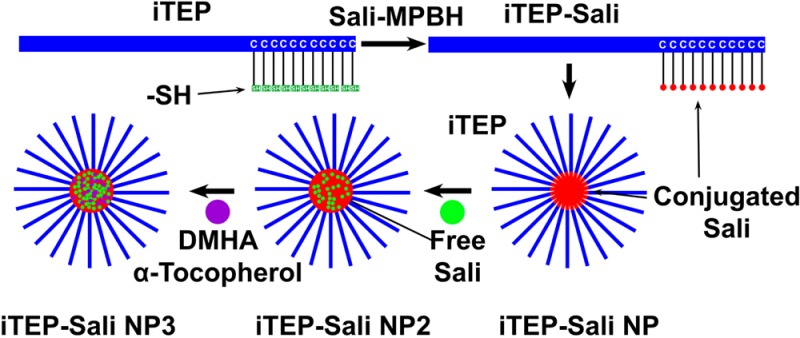
Salinomycin (Sali) has selective toxicity to cancer stem cells (CSCs), a subpopulation of cancer cells that have been recently linked with tumor multidrug resistance (MDR). To utilize its selective toxicity for cancer therapy, we sought to devise a nanoparticle (NP) carrier to deliver Sali to solid tumors through the enhanced permeability and retention effect and, hence, to increase its exposure to CSCs. First, hydrophobic Sali was conjugated to a hydrophilic, immune-tolerant, elastin-like polypeptide (iTEP); the amphiphilic iTEP–Sali conjugates self-assemble into NPs. Next, free Sali was encapsulated into the NPs alone or with two additives, N,N-dimethylhexylamine (DMHA) and α-tocopherol. The coencapsulation significantly improved the loading efficiency and release profile of Sali. The resulting NPs of the coencapsulation, termed as iTEP–Sali NP3s, have an in vitro release half-life of 4.1 h, four times longer than iTEP–Sali NP2s, the NPs that have encapsulated Sali only. Further, the NP3 formulation increases the plasma area under curve and the tumor accumulation of Sali by 10 and 2.4 times, respectively. Lastly, these improved pharmacokinetic and tumor accumulation profiles are consistent with a boost of CSC-elimination effect of Sali in vivo. In NP3-treated 4T1 orthotopic tumors, the mean CSC frequency is 55.62%, a significant reduction from the mean frequencies of untreated tumors, 75.00%, or free Sali-treated tumors, 64.32%. The CSC-elimination effect of the NP3 can further translate to a delay of tumor growth. Given the role of CSCs in driving tumor MDR and recurrence, it could be a promising strategy to add the NP3 to conventional cancer chemotherapies to prevent or reverse the MDR.
Keywords: cancer stem cells, salinomycin, iTEP nanoparticle, tumor multidrug resistance, coencapsulation additives
Introduction
Multidrug resistance (MDR) of cancer constitutes a significant challenge to clinical oncology and serves as one of the major reasons for tumor recurrence. Cancer stem cells (CSCs) were discovered in various types of cancer, including breast,1 colon,2 colorectal,3 lung,4 glioma,5 and pancreatic cancers.6 Recently, CSCs have been linked to the MDR since CSCs are often enriched in tumors after conventional chemotherapies;3,7,8 and CSCs were found more resistant to conventional chemotherapeutics than differentiated tumor cells.9,10 Mechanisms that CSCs utilize to achieve drug resistance include entering a quiescent cell cycle status,11 possessing a high DNA repair capacity,5 avoiding apoptosis,12 upregulating ATP-binding cassette transporter,12,13 and overexpressing of detoxification enzymes such as aldehyde dehydrogenase 1.14
The distinct susceptibility to conventional chemotherapeutics between CSCs and differentiated tumors leads to enrichment of the CSC populations in some tumors treated by conventional chemotherapeutics.3,7,8 What aggravates this alarming situation is CSCs’ ability to recapitulate heterogeneity and hierarchy.15,16 Therefore, CSCs not only are one important reason for MDR but also play a critical role in progression and metastasis of cancer.17 Consequently, a failure of eradicating CSCs in tumors could have a huge adverse impact on the prognosis of cancer. Due to this rationale, an effective way to elimination of CSCs becomes critical to prevent MDR development and results in a favored prognosis during cancer treatment by conventional chemotherapeutics.
Because of the role of CSCs in tumorigenesis and the MDR, there is an intensive, ongoing effort to discover new compounds that have selective toxicity to CSCs. Sali is one such compound and among the first group discovered.18 Proposed mechanisms behind the selective toxicity of Sali include p38 MAPK activation,19 accumulation of reactive oxygen species,20 inhibition of Wnt signaling pathways,21 and inhibition of p-glycoprotein activity by Sali.22 Consistent with its selective toxicity to CSCs and the role of CSCs in causing MDR, Sali can sensitize a wide spectrum of tumor cell lines that would be otherwise resistant to conventional cancer chemotherapeutics such as paclitaxel (PTX).23
While using Sali to eradicate CSCs may have immediate clinical benefits, such an application is currently hindered by poor solubility of Sali, as well as its toxicities to healthy tissues and cells in mammals.24−26 On the other hand, nanocarriers have in general been shown to increase tumor accumulation of small molecule drugs, redistribute their accumulation in organs, and/or resolve their solubility issues.27,28 Therefore, a nano delivery system is highly desired that can help Sali overcome the solubility issue and increase its accumulation in tumors while reducing its distribution in off-target organs. Recently, a hyaluronic acid based nanogel was shown in vitro to facilitate the uptake of Sali to CSCs, which are CD44 positive. The increased uptake compared to free Sali was attributed to interactions between hyaluronic acid and CD44.29 Meanwhile, another group used PEG-b-PCL polymeric micelles to deliver Sali as encapsulated drugs and found that the micelles improved CSC-specific toxicity of Sali in vivo using a mouse subcutaneous, xenograph model of human breast cancer. However, the improvement was marginal as the overall therapeutic outcomes resulted from micelle-delivered Sali and free Sali treatments were indifferent statistically.23
We speculated that one reason for the aforementioned suboptimal therapeutic outcomes of Sali may be that the micellar carrier failed to accumulate more Sali in tumors than the free form of Sali, and hence did not result in a stronger inhibition to tumor growth. The speculation cannot be answered as the report did not include pharmacokinetics and tumor accumulation evaluations.23 Here we seek to develop a novel and biocompatible nanoparticle (NP) carrier to deliver Sali to clinically and biologically relevant orthotopic breast tumors. We hypothesize that the NP using Sali as its hydrophobic, together with our new encapsulation method, can deliver satisfactory pharmacokinetics and tumor accumulation of Sali, which in turn lead to a better therapeutic outcome of Sali. To this end, we first obtained NPs self-assembled from the conjugates between Sali and iTEP, a biodegradable recombinant polypeptide we invented recently (M. Chen, personal communication); the immune tolerance nature of iTEP may prove an advantage when the carrier is administered repeatedly and tested in clinical trials, in which cases the immunogenicity of therapeutics becomes a focal point of consideration. Next, we exploited the iTEP–Sali NPs to encapsulate free Sali, which is a novel strategy for Sali delivery. Then, we found that encapsulated Sali was released in a controlled manner and maintained CSC-selective toxicity of Sali. We also discovered that coencapsulation of Sali with two additives, α-tocopherol and DMHA, noticeably improved the loading efficiency and drug release profile of the Sali. Another benefit of the resulting NPs is that they significantly retarded in vivo release of the Sali and therefore led to better pharmacokinetics and tumor accumulation profiles of the Sali. Through tumor regression study, we demonstrated that the encapsulated Sali was able to inhibit the growth of bulk tumors because of these desired properties conferred by the NPs. Lastly and most importantly, the NP-delivered Sali depleted CSCs in breast tumors more efficiently than free Sali.
Material and Methods
Materials
All chemicals, unless otherwise described, were purchased from Thermo Fisher Scientific Inc. (Waltham, MA, USA) at biological grade. Organic solvents including acetonitrile (ACN), dichloromethane (DCM), dimethylformamide (DMF), isopropanol, and methanol were purchased from Thermo Fisher Scientific Inc. (Waltham, MA, USA) at HPLC grade. The LB and TB media were prepared in our lab using standard formula.30 All the cell culture plates were purchased from Corning Inc. (Corning, NY, USA). The cell culture media and supplements including RPMI-1640 (with 2 mM l-glutamine), Media 199, and fetal bovine serum (FBS) were purchased from Life Technologies, Inc. (Carlsbad, CA, USA).
All antibodies were purchased from BioLegend (San Diego, CA, USA) including PE anti-mouse CD24 antibody (Cat. # 101808), APC anti-human CD44 antibody (Cat. # 338806), and their isotope control antibodies (Cat. # 400635, Cat. # 400119).
4T1, a highly metastatic murine cell line derived from a spontaneous syngeneic breast cancer of Balb/c mice, was purchased from American Type Culture Collection (Rockville, MD, USA). 4T1 cells were maintained in monolayer cultures within an RPMI 1640 medium supplemented with 10% FBS. Cells were maintained at 37 °C humidified atmosphere with 5% CO2.
Female Balb/c mice that were 24–28 days old (18–19 g) were purchased from Charles River Laboratories International, Inc. (USA). All the animal experiment protocols were approved by the Institutional Animal Care and Use Committee at the University of Utah.
iTEP Synthesis and iTEP–Sali Conjugate Synthesis and Characterization
These are described in the Supporting Information.
Size and Charge Measurements by Dynamic Light Scattering (DLS)
iTEPs or iTEP–Sali conjugates were measured using the Malvern Zetasizer Nano (Malvern, Chester County, PA, USA) at 25 μM in PBS at 37 °C using a low-volume disposable sizing cuvette or a clear disposable zeta cell. Before size measurements, all samples were reduced overnight in a 100 mM TCEP solution to reduce possible disulfide bonds. Each sample was measured in triplicate. The instrument settings include the following: material RI = 1.59, material absorption = 0.010, water dispersant RI = 1.330, viscosity = 0.6864 cP. The default value, 4.65 mm, was used as the measurement position. The instrument was allowed to automatically optimize the count rate, duration, and attenuator. Specifically, for zeta potential determination, the dispersant dielectric constant was set as 74.4.
Loading of Sali into iTEP–Sali NPs
5.0 mg of iTEP–Sali NPs and 3.0 mg of salinomycin were codissolved in 125 μL of DMF and mixed well. In some cases, 13.34 mg of DMHA (Sigma-Aldrich, St. Louis, MO, USA) and 0.87 mg of α-Tocopherol (Sigma-Aldrich, St. Louis, MO, USA) were also added into the mixture. The ratio of the above components was based on our independent trial experiments and previous reports.31,32 The mixture was then supplemented with 750 μL of Milli-Q water in a dropwise manner. The mixture was stirred for 0.5 h at room temperature before being dialyzed against 3 L of DI water for 3 h to remove DMF. The complete removal of DMF was critical and was monitored using a characteristic absorption of DMF at 210 nm. The product solution was filtered through 0.45 μm Acrodisc syringe filters with Supor Membrane (Pall Corporation, Port Washington, NY, USA) and concentrated by Amicon Ultra-15 centrifugal filter units (MW cutoff = 3000 Da). The encapsulated Sali is determined by HPLC after a precolumn derivatization by DNPH. The detailed procedure of this determination method is described in the Supporting Information. The loading efficiency was defined as in the following equation:
In Vitro Sali Release Assay
In vitro release profile of Sali from iTEP–Sali NPs was measured by a previously described dialysis method with minor modification.23 iTEP–Sali NPs with or without encapsulated Sali were diluted in 0.5 mL of Milli-Q water containing 4% BSA and kept in dialysis bags (Spectrum Laboratories, Inc., Rancho Dominguez, CA, USA, MW cutoff = 8000 Da). The bags were then immersed into 100 mL of PBS solution (pH = 7.4) in a beaker, respectively. The entire dialysis systems were shaken at 100 rpm at 37 °C. At predetermined time points, the 0.5 mL sample solutions in the bags were mixed and 10 μL of sample was collected from each sample. Sali inside the bags was presumed and measured as unreleased drug in the NPs. Concentrations of Sali were determined by HPLC after the precolumn derivatization with DNPH.
The relationship of the percentage of Sali release (F) with time (t) was fitted using the following equation and GraphPad V5.0.
K is the release rate constant:
Pharmacokinetics and Biodistribution Study
Pharmacokinetics studies were carried out according a protocol modified from a previous report.27 iTEP–Sali NPs or Sali were injected in a 100 μL solution into Balb/c mice via the tail vein at 5 mg/kg free Sali or encapsulated Sali dose. The Sali solution used for the injection contained 1% DMSO to dissolve Sali (Sigma-Aldrich, St. Louis, MO, USA). At predetermined time points, about 50 μL blood samples were collected by submandibular bleeding into tubes containing 150 μL of PBS with heparin at 1,000 U/mL. Blood plasma was collected from blood samples after samples were centrifuged to pellet blood cells. Next, 100 μL of isopropanol was added to each plasma sample to precipitate proteins. The precipitate was removed by centrifugation. The supernatants were collected; concentrations of Sali in the supernatant were determined by the precolumn derivitization with DNPH. The entire Sali abstraction procedure was preoptimized for a high Sali recovery rate. The changes of plasma Sali concentrations over time were fitted to a two-compartment model using GraphPad V5.0. The details are described below:
Using the GraphPad, we obtained the initial concentration (Cp(0)), the distribution rate constant (a), and elimination rate constant (b) directly. Other PK parameters were calculated from the known parameters using following equations:
For the biodistribution study, Balb/c mice were inoculated subcutaneously with 106 4T1 cells in 50 μL of PBS at #4 mammary fat pad on the right side of mouse abdomen. The inoculated tumors were allowed to grow to 200 mm3 before the study. iTEP–Sali NP3s or Sali was injected intravenously into the mice at 5 mg/kg Sali or an equivalent Sali dose, respectively. At 12 h postinjection, hearts, livers, spleen, lungs, kidneys, and tumors were harvested from the treated mice. The tissues were homogenized in 70% acetonitrile–water solution on ice for 3 min using a Brinkmann Polytron homogenizer (Brinkmann Instruments, Inc., Westbury, NY, USA). Next, 3 mL of the above abstract solution was used for liver tissues while 1 mL of the solution was used for heart, spleen, lungs, kidneys, and tumor tissues. After Sali was extracted from the homogenized tissues into the solution, the samples were centrifuged at 14,000 rpm for 10 min at 4 °C. Supernatants containing Sali were collected, and Sali quantity was determined by precolumn derivitization with DNPH. The final tissue distribution was normalized to percentage of initial dose averaged by weight of each organ (% ID/gram).
Cytotoxicity Studies
105 4T1 mammosphere cells or 103 regular 4T1 cells were seeded in wells of 96-well plates in 100 μL of RPMI-1640 medium, which contained serially diluted Sali, paclitaxel (PTX), or the NPs in tests for different studies. After the incubation with these drugs, live cells were quantified using the CellTiter 96 AQueous One Solution Cell Proliferation Assay (Promega, Madison, WI, USA). The plates were read with a SpectraMax M2 plate reader (Molecular Devices, Inc. , Sunnyvale, CA, USA). Quantities of live cells in each well were represented by the absorbance values at 490 nm of that well. Consequently, cell viabilities in each treated well were expressed as the absorbance values in percentage after the values were normalized with the mean absorbance value of the wells containing untreated cells. The viability data was fitted into a Sigmoidal dose–response curve using the GraphPad Prism 5.0 (GraphPad Software, Inc., La Jolla, CA, USA). The IC50 and 95% confidence index (CI) were obtained from the fitting. Mammosphere culture and cell collection are described in the Supporting Information.
Tumor Growth Study
Balb/c mice were inoculated subcutaneously with 4T1 cells as described earlier. At the seventh day after inoculation and when tumors all reached to or above 100 mm3, the mice were randomly assigned into three groups and treated with the control (1% DMSO PBS treatment), free Sali (5 mg/kg in 1% DMSO PBS), or iTEP–Sali NP3 (5 mg/kg encapsulated Sali equivalent in PBS), respectively. All dosing was administered intravenously. The dosing was repeated five times at a two-day interval. The length and width dimensions of tumors were measured by a caliper every other day. Tumor volumes were estimated using the following formula: tumor volume = (length × width2/2).23
The mice were sacrificed the day after the fifth treatment. The tumors were harvest to determine their weight and CSC populations. Pictures of the tumors were taken at the time of the tumor collection.
Quantification of CSC Frequencies in 4T1 Orthotopic Tumors
Single cell suspensions of cells were prepared from 4T1 tumor tissues that we collected from the tumor growth study according to a previously published protocol.32 An approximately 300 mg of tumor mass was incubated with 3 mL of medium 199 (Life Technologies, Inc.) with 250 U/mL ultrapure collagenase III (Worthington Biochemical, Inc., Lakewood, NJ, USA). The tissue digestion lasted for 2 h at 37 °C while shaken at 100 rpm. After digestion, 3 mL of serum-containing M-199 medium was added to inhibit the activity of collagenase, and cells were filtered through a 45 μm nylon mesh and washed with RPMI-1640 containing 10% FBS. Cells were counted, transferred to a 5 mL tube, and washed twice with DPBS with a 0.1% heat-inactivated FBS at 4 °C for 5 min at 1,000 rpm. Antibodies were then incubated with the cells for 30 min on ice at a 1:80 dilution as recommended by the manufacturer of the antibodies. After the incubations, cells were washed twice with the above washing medium and then were resuspended in 0.4 mL of DPBS/0.1% FBS containing 1 μg/mL DAPI per million cells. Flow cytometry analyses of the cells were performed on a Cytek DxP (Cytek Development, Inc., Fremont, CA, USA). Dead cells were eliminated by gating as they were stained by DAPI. Side scatter and forward scatter profiles were used to eliminate cell doublets. The gating for CSCs was set up based on the CD44 and CD24 expression profile of the cells collected from 4T1 mammospheres.
To study mammosphere-forming ability of tumors, the above-mentioned single tumor cells were seeded in wells of 6-well cell culture plate at a density of 500 cells/well. The culture medium was supplemented with B27 (1X, Invitrogen, CA, USA), 20 ng/mL EGF, 20 ng/mL bFGF (BD Biosciences, Franklin Lakes, NJ, USA), 4 μg/mL heparin (Sigma-Aldrich, MO, USA), and 0.5% methylcellulose (Stem Cell Technologies, Vancouver, BC, V5Z 1B3, Canada).33 After 7 days of culture, number of mammospheres in each well was counted and analyzed.
Results
Design and Generation of iTEP–Sali Nanoparticles
To design a nanocarrier to deliver Sali to solid tumors through the enhanced permeability and retention (EPR) effect,34,35 we took advantage of a self-assembling principle of amphiphiles that amphiphlies with strong, spatially separated amphiphilicity are able to assemble into micelle-like NPs.27,36 Since Sali is hydrophobic with a LogD value of 3.24 at pH 7.4, we paired it with a hydrophilic iTEP that we recently invented (Chen, personnel communication). We expected that attaching Sali to one terminus of the iTEP would introduce sufficient, segmented amphiphilicity and the amphiphilicity can drive the conjugates to form NPs. To this end, 32 cysteines were appended to the C-terminal of the iTEP as conjugation sites (Figure S1 in the Supporting Information). The resulting new iTEP (Figure S2 in the Supporting Information) maintains the hydrophilic nature of the parent iTEP (Figure S3 in the Supporting Information). To connect Sali to the new iTEP, Sali was first connected with the MPBH linker (Figures S4 and S5 in the Supporting Information); then the formed MPBH–Sali conjugates were connected with the new iTEP (see the Supporting Information). After iTEP–Sali conjugates were purified, we found that the conjugates displayed a hydrodynamics diameter of 109.7 ± 45.9 nm at 37 °C according to DLS measurements (Figure 1a). The size of the conjugates was much larger than that of the unconjugated iTEP, 7.9 ± 1.49 nm, which suggests a NP structure of iTEP–Sali conjugates. The sizes of iTEP–Sali NPs are narrowly dispersed with a low polydispersity index (PDI) value of 0.217. The NPs have a critical micelle concentration (CMC) of 15.32 μM and a neutral charge (Figures S6 and S7 in the Supporting Information). The NPs were not toxic, due presumably to the nonrelease of Sali from the NPs (Figure 1b). The nonrelease result is reasonable since Sali is connected with MPBH through a stable amide bond and the MPBH, in turn, is connected to the iTEPs through a stable thioester bond.
Figure 1.
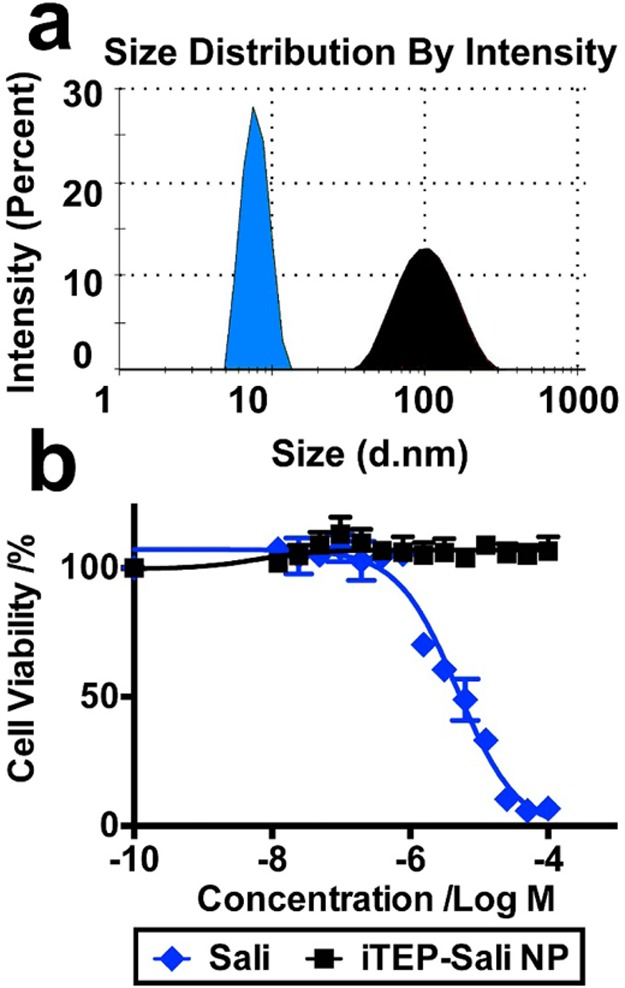
(a) Hydrodynamic diameters of the unconjugated iTEP which has a 32-cysteine appendix (blue filled area) and iTEP–Sali conjugate (black filled area). The measurements were performed using the DLS method at 37 °C. The sample concentrations were 25 μM for both. Before the measurement, the unconjugated iTEP was reduced in 100 mM TCEP solution overnight to cleave possible disulfide bonds. (b) Viability profiles of regular 4T1 cells after they were exposed to different concentrations of Sali or iTEP–Sali NPs for 48 h. The IC50 of free Sali is 4.8 μM. The NPs do not show any toxicity up to 100 μM.
Encapsulation of Sali in iTEP–Sali NPs
Since we have attained the iTEP–Sali NPs, next we tried to use the NPs to deliver Sali in an encapsulated form. Using a modified cosolvent method, we were able to load free Sali into the NPs. The loading efficiency for Sali is 25.0 ± 5.3%. We termed these NPs with encapsulated Sali as iTEP–Sali NP2 to differentiate them from the empty iTEP–Sali NPs. The iTEP–Sali NP2s have a mean diameter of 195.0 ± 95.7 nm and a PDI of 0.288 (Figure 2a). Next, through an in vitro release study, we found that the encapsulated Sali was released from iTEP–Sali NP2 at a fairly rapid release rate with a half-life of 1.0 h (Figure 2b and Table 1).
Figure 2.
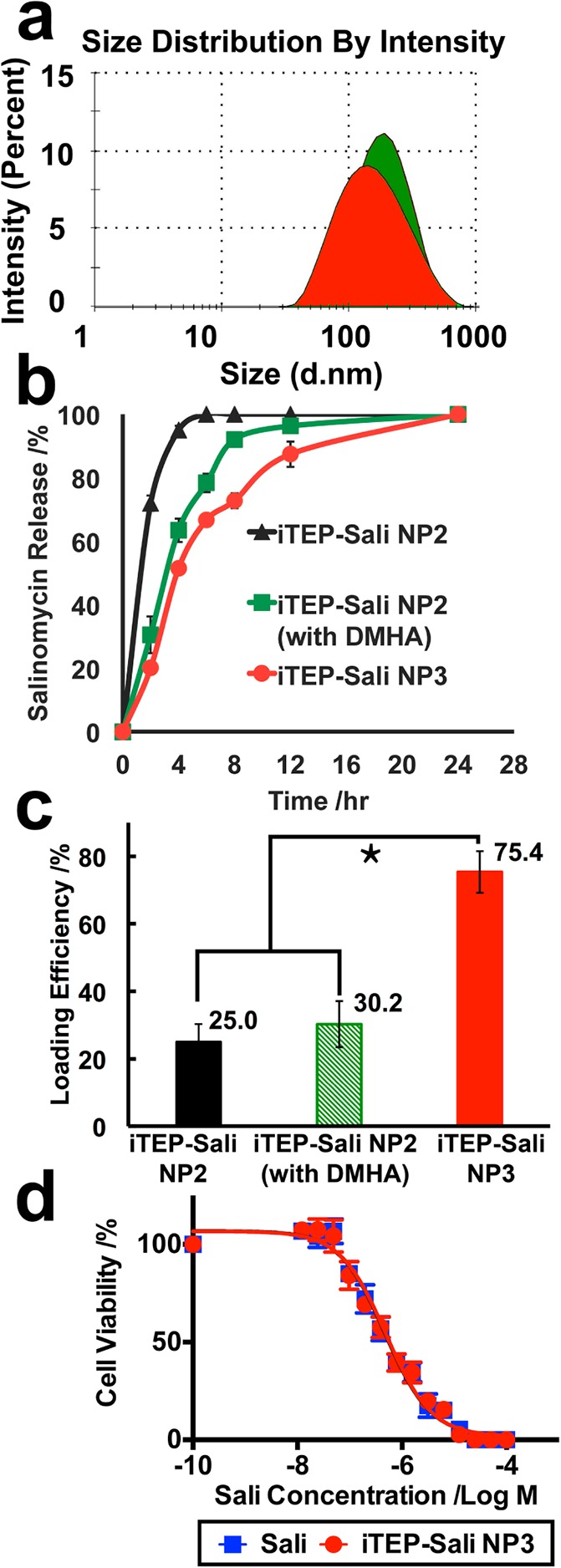
(a) Hydrodynamic diameters of iTEP–Sali NP2 (green filled area) and iTEP–Sali NP3 (red filled area) at 37 °C in PBS. The sample concentrations were 25 μM for both. The measurements were performed using DLS at 37 °C. (b) The release profile for Sali from TEP-Sali NP2, iTEP–Sali NP2 with DMHA, and iTEP–Sali NP3 respectively. The quantity of released Sali was determined by HPLC in combination with a precolumn derivatization with DNPH. The Sali release data were fit to a release model described in the Supporting Information. (c) The loading efficiency of Sali by NP2, NP2 plus DMHA, or NP3. The * indicates that the differences between the NP3 and NP2 or NP2 plus DMHA are statistically significant with a p < 0.0001 analyzed by one-way ANOVA. (d) Viability profiles of 4T1 mammosphere cells after they were exposed to different concentrations of Sali or iTEP–Sali NP3 for 48 h. The IC50 of free Sali is 0.49 μM with a 95% CI = 0.40–0.60 μM. The IC50 of the NP3 is 0.48 μM with a 95% CI = 0.35–0.59 μM.
Table 1. Release Kinetics of Sali from iTEP–Sali NP2, iTEP–Sali NP2 plus DMHA, or iTEP–Sali NP3.
| half-life |
|||
|---|---|---|---|
| iTEP–Sali NP2 | iTEP–Sali NP2 (with DMHA) | iTEP–Sali NP3 | |
| mean (h) | 1.04 | 2.93 | 4.13 |
| 95% CI | 0.99–1.09 | 2.73–3.15 | 3.86–4.43 |
| R2 | 0.99 | 0.98 | 0.98 |
Coencapsulation of Sali with DMHA and α-Tocopherol in iTEP–Sali NPs Together
Because Sali has an ionizable carboxyl group with a pKa around 4.0 that could confer negative charges to Sali at a neutral pH, we suspected the negative charges may have rendered instability to Sali encapsulation in hydrophobic cores of iTEP–Sali NP2s. The instability, in turn, may have led to the rapid release and low loading efficiency of Sali we observed. In order to test the conjecture and to attain a carrier having a slower release rate and higher loading efficiency of Sali, we coencapsulated Sali with DMHA, a positively charged hydrocarbon that has been used to neutralize encapsulated, negatively charged chemicals inside micelles.32 The results from our in vitro release study showed that coencapsulation of DMHA and Sali slowed down the release of Sali from the NP2s. The release half-life of Sali from the NP2 with DMHA was 2.9 h, which is significantly longer than that of the NP2s without DMHA (Figure 2b and Table 1). The coencapsulation, however, failed to boost the loading. The resulted loading efficiency of 30.2 ± 6.8% was same as that of the NP2s without DMHA (Figure 2c). One possible reason could be that DMHA is not hydrophobic enough. One early study pointed out that DMHA was not as effective as its more hydrophobic analogue, N,N-dimethyloctadecylamine (DMOA), in stabilizing the encapsulation of negatively charged chemicals.32 DMOA, however, was more toxic than both DMHA and Sali (Figure S8 in the Supporting Information), which excluded it from serving as an additive for the Sali coencapsulation.
We reasoned that, to exploit the coencapsulation system of DMHA and Sali, we might need to supplement more hydrophobicity to it. Therefore, we introduced a second additive, α-tocopherol (vitamin E), to the system. The α-tocopherol was able to double loading efficiency of rapamycin due to its ability to increase the hydrophobic core of micelles.31 We hypothesized that coencapsulation of α-tocopherol together with Sali and DMHA would not only provide a sufficiently hydrophobic environment for Sali but also neutralize the charge of Sali. This would mean that Sali can be trapped more stably inside of the hydrophobic core of the NPs. Consequently, we could see an improvement of both loading efficiency and release profiles of the coencapsulated Sali. The hypothesis was proven true as the coencapsulation led to a very high loading efficiency, 75.0 ± 6.17% (Figure 2c). In addition, the newly generated NPs, including Sali and two additives (termed iTEP–Sali NP3s), have a mean diameter of 179.9 ± 43.0 nm and share a size similar to that of NP2 (Figure 2a). Importantly, the release rate of Sali from the NP3s is remarkably slower than that of NP2s or NP2s with DMHA only (Figure 2b). The release half-life of NP3 was 4.1 h, which is significantly longer than 2.9 h of the NP2s with DMHA only or 1.0 h of the NP2s (Table 1). Together, these results showed that the paired additives, DMHA and α-tocopherol, improved both loading efficiency and release kinetics of encapsulated Sali in iTEP–Sali NPs. Moreover, iTEP–Sali NP3 solubilized Sali in aqueous solution, rendering a maximum solubility of 7,320 ± 230 μg/mL, which represents a 430-fold increase, given that the intrinsic solubility of Sali is only 17 μg/mL.37 Lastly, the results of the in vitro cytotoxicity study showed that iTEP–Sali NP3s possess the same level of selective toxicity to CSCs as free Sali (Figure 2d). Here, we used cells harvested from 4T1 tumor mammospheres as a model for CSCs because these cells carry characteristic phenotypes of CSCs, such as a CD24–/CD44+ phenotype and a mammosphere-forming capacity (Figure S9 in the Supporting Information). The IC50 of the NP3 to 4T1 mammosphere cells was 0.48 μM, which, statistically, was the same as the IC50 of free Sali, 0.49 μM (Figure 2d). This similarity is understandable as we expect that the encapsulated Sali is released from the NP3s during the incubation with the cells and exerts its toxicity the same as free Sali.
Pharmacokinetics and Tumor Accumulation of Coencapsulated Sali
After free Sali or iTEP–Sali NP3s was administered to mice at a dose of 5 mg/kg intravenously, plasma concentrations of free Sali or encapsulated Sali were sampled up to 12 h (Figure 3a). These concentration data fit best into a two-compartment pharmacokinetics model. According to the fitting results (Table 2), the AUC of the NP3-delivered Sali was 192.10 μM h, which was approximately 6 times greater than that of the free Sali, 30.70 μM h. Similarly, free Sali’s clearance was 6 times faster than that of the encapsulated Sali (CLsali, 216.87 μL/(h g) versus CLNP3, 34.66 μL/(h g)). The elimination half-life of the NP3-delivered Sali was approximately 5.2 times longer than that of the free Sali. Meanwhile, in summary, the results support a conclusion that the NP3s significantly retard the clearance of the encapsulated Sali from the systematic circulation, which, together with sizes of the NP3s (179.9 nm in diameter), might lead to greater tumor accumulation of Sali via the EPR effect. Indeed, there was a 2.4-fold increase of tumor Sali accumulation resulting from using the NP3s. At 12 h postinjection, the mean Sali concentration in tumors of those mice receiving free Sali was 1.17 ± 0.17% ID/g. In contrast, the mean concentration for the NP3-delivered Sali was 2.85 ± 0.12% ID/g (Figure 3b). At the same time, the NP3s reduced Sali accumulation in heart and lungs (Figure S10 in the Supporting Information).
Figure 3.
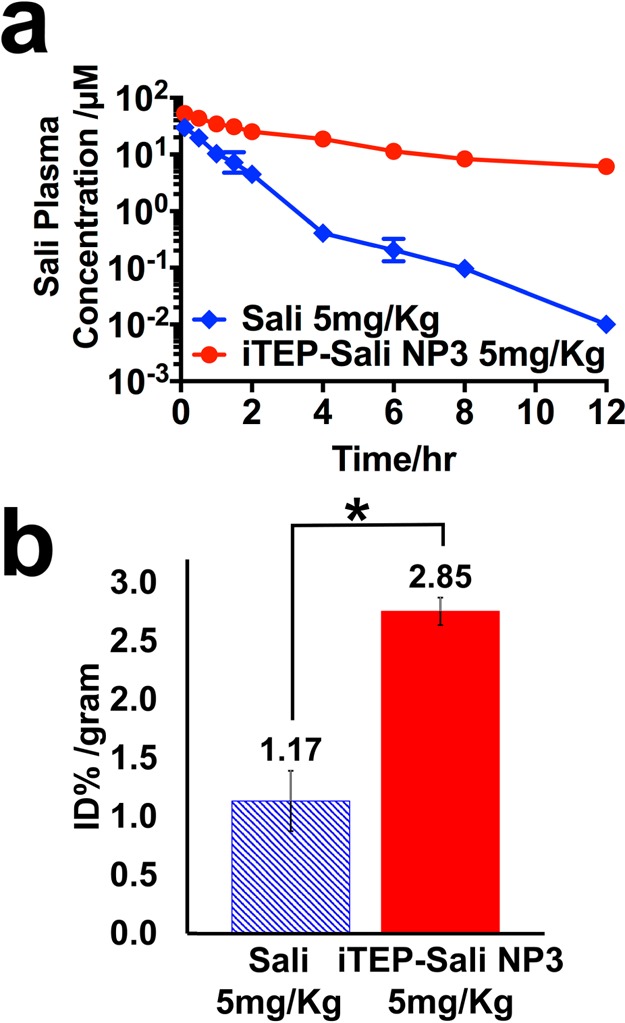
(a) Plasma Sali concentrations after they were administered as free form (blue line) or as encapsulated form in iTEP–Sali NP3 (red line) through intravenous injection. The concentrations were determined by HPLC in combination with a precolumn derivatization with DNPH. The Sali plasma concentration was plotted on the log scale based on 10 as a function of time postinjection. (b) Tumor accumulation of Sali after they were administered as free form (blue bar) or as encapsulated form in iTEP–Sali NP3 (red bar) through intravenous injection. The presented data are for tumor samples collected at 12 h post intravenous injection at 5 mg/kg. The quantities of Sali were expressed as percentage of initial dose normalized by weight of tumor, % ID/gram. The * indicates that the difference between Sali and iTEP–Sali-NP3 is statistically significant with a p = 0.0031 analyzed by one-way ANOVA.
Table 2. PK Parameters of Sali after It Was Delivered As a Free Form or an Encapsulated Form in the NP3.
| Sali | iTEP–Sali NP3 | |||
|---|---|---|---|---|
| mean | 95% CI | mean | 95% CI | |
| initial vol of distribn (V0, μL/g) | 197.44 | 166.82–241.84 | 118.78 | 107.57–132.63 |
| AUC (μM h) | 30.70 | 14.15–53.28 | 192.10 | 98.34–315.27 |
| clearance (CL, μL/(h g)) | 216.87 | 124.96–470.52 | 34.66 | 21.12–67.70 |
| elimination half-life (telimi, h) | 0.86 | 0.26–2.57 | 4.45 | 2.77–7.70 |
| distribn half-life (tdistribn, h) | 0.43 | 0.29–1.19 | 0.63 | 0.40–1.08 |
| R2 | 0.93 | 0.95 | ||
Reduction of CSCs in 4T1 Orthotopic Tumors by iTEP–Sali NP3s
Since iTEP–Sali NP3s extended the systematic circulation of Sali and increased tumor accumulation of Sali, it was intriguing to see whether such improvements could strengthen the selective inhibition of CSCs by Sali in vivo. To test this hypothesis, we inoculated murine 4T1 syngeneic tumor cells in mammary fat pads of Balb/c mice to establish an orthotopic tumor model. Once the tumors grew to desired sizes, we treated the mice with 5 doses of PBS as a control, 5 mg/kg free Sali, or iTEP–Sali NP3 loaded with 5 mg/kg Sali, respectively. After 5 treatments, we analyzed CSC frequencies in these tumors using CD24 and CD44 markers. The CSCs in tumors were defined operationally as those cells that have the same CD24 and CD44 expression profiles as those cells collected from 4T1 mammospheres (Figures S9 and S11 in the Supporting Information). For the control group, the mean CSC frequency was 75.00 ± 2.95%. Free Sali was able to lower the frequency significantly to 64.32 ± 5.72% (P < 0.05). More importantly, the NPs further reduced the value to 55.62 ± 3.73%, which was significantly lower than those of both the control group and free Sali-treated group (P < 0.05) (Figure 4a). These comparison results suggest that Sali is able to kill CSCs in vivo just as they are able to in vitro (Figure 2b), and, more significantly, iTEP–Sali NP3s are able to enhance the effect of Sali.
Figure 4.
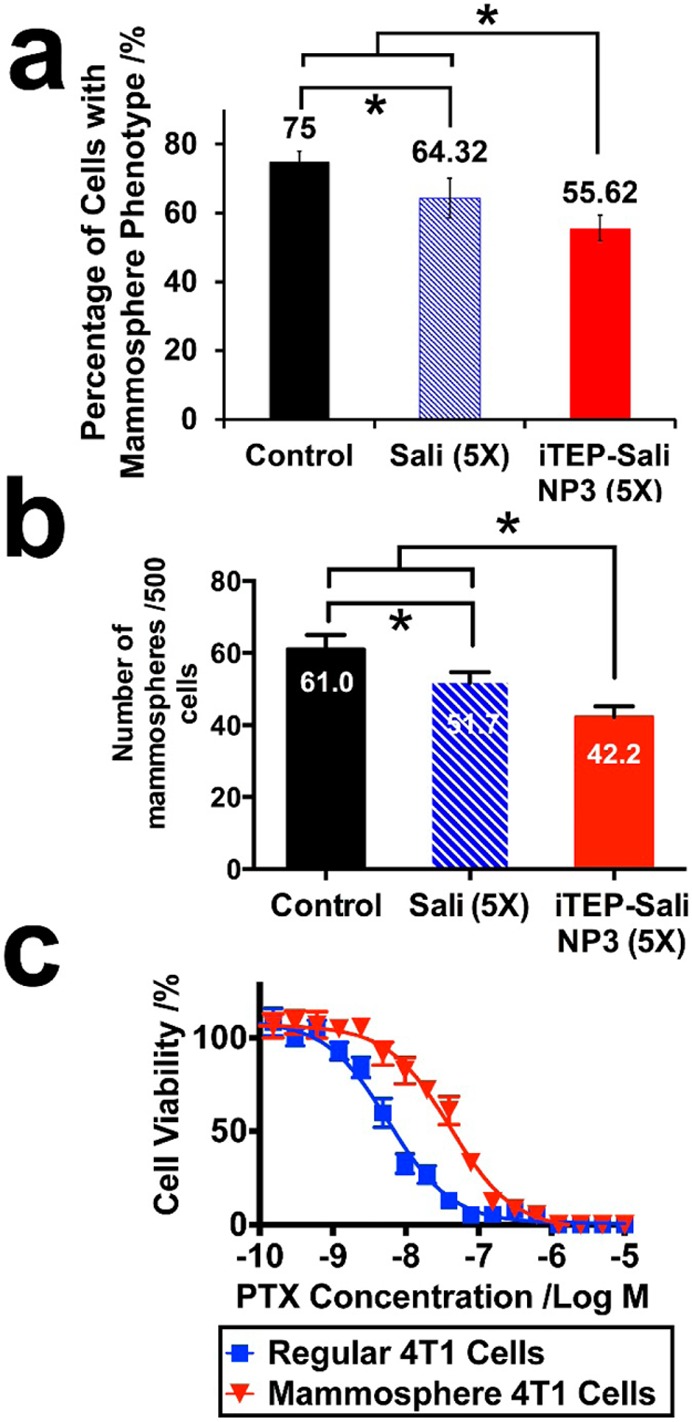
(a) The CSC frequencies of the 4T1 orthotopic tumors treated by controls (PBS), free Sali, or iTEP–Sali NP3. The * symbols indicate that the mean CSS frequency of the NP-treated tumor is significantly lower than that of control- or free Sali-treated tumors (p = 0.0004 and p = 0.0252, respectively), and that the mean frequency of the Sali-treated tumors is significantly lower than that of the control-treated tumors (p = 0.0133). The data were analyzed using one way ANOVA. (b) Mammospheres formed from 4T1 orthotropic tumors after these tumors were treated by controls (PBS), free Sali, or iTEP–Sali NP3. The * symbols indicate that the mammosphere-formation ability of the NP-treated tumor is significantly lower than that of control- or free Sali-treated tumors (p = 0.0035 and p = 0.0125, respectively), and that the mammosphere-formation ability of the Sali-treated tumors is significantly lower than that of the control-treated tumors (p = 0.0358). The data were analyzed using one way ANOVA. (c) The viability profile of regular 4T1 cells versus 4T1 mammosphere cells after they were exposed to different concentrations of PTX for 72 h. The IC50 of PTX to regular 4T1 cells 5.79 nM with a 95% CI = 4.90–6.86 nM. The IC50 of PTX to 4T1 mammosphere cells 40.48 nM with a 95% CI = 37.64–47.30 nM.
One functional feature of CSCs is their mammosphere-forming capacity.18,33,38 The capacity is relevant to the drug resistance of tumors,18,39,40 thus they are very significant to cancer therapy. To investigate functional significance of the NP3s’ effect in reducing CSCs in 4T1 orthotopic tumors, we examined mammosphere-forming ability of the cells collected from 4T1 tumors after these tumors were treated by the NP3, free Sali, and PBS. We found that the NP3-treated tumors had a significantly lower ability to generate mammospheres than the other two treatment groups (Figure 4b). There were on average 42.2 mammospheres formed per 500 NP3-treated tumor cells, which were significantly less than those formed from free Sali- or PBS-treated tumors (54.7 and 61.0 mammospheres per 500 tumor cells, respectively). Putting together, the superior effects of the NP3 over free Sali in eliminating CSCs and impeding the mammosphere-forming ability of 4T1 tumors are significant because they suggested that the NP3 was effective to alter cell composition of the tumors, and, more importantly, to diminish tumorigenesis and drug resistance potential of the tumors.
In a separate study, we found that the cells isolated from mammospheres were more resistant to PTX than general 4T1 cells cultured as monolayers (Figure 4c). The IC50 of PTX to the mammosphere cells was 7 times higher than the IC50 to regular 4T1 cells, 40.50 nM versus 5.79 nM (P < 0.05). The results are consistent with earlier reports that CSCs play a critical role in promoting the tumor MDR.18,39,40 Therefore, the aforementioned enhancing effect of iTEP–Sali NPs to the CSC-specific toxicity of Sali may have implications for overcoming the MDR.
Inhibition of 4T1 Tumor Growth by iTEP–Sali NP3s
Since CSCs play a role in tumorigenesis and maintain the hierarchy of tumors, depletion of CSCs in tumors may lead to a tumor regression or stabilization. Thus, we examined whether the superiority of the NP3-delivered Sali over free Sali in depleting CSCs can translate into an advantage of tumor growth inhibition or tumor regression. Our results showed that neither the NP3s nor free Sali were able to abolish tumors or to stop tumor growth (Figure 5a). Instead, both apparently slowed down the tumor growth. The growth-inhibition effect was especially prominent for the NPs as the mean tumor weight of the NP3-treated mice at day 16 was significantly smaller than that of the control group: 436.8 ± 137.9 mg versus 762.6 ± 193.8 mg (P < 0.05), respectively (Figures 5b and 5c). While it is clear the NP3-delivered Sali had an impact on overall tumor growth, the impact was not as potent as its impact on CSCs inside the 4T1 tumors. In contrast to the result that the NP3-treated 4T1 tumors had a significantly smaller CSC population than free Sali-treated 4T1 tumors after five treatments (Figure 4a), the mean weights of the NP3-treated and the free Sali-treated tumors were not statistically different (Figure 5c). One possible reason for the discrepancy is that cells in 4T1 tumors might, on average, be less sensitive to Sali than the CSC subpopulation in the tumors. Indeed, our results showed that Sali was 10 times less toxic to regular 4T1 cells than to cells isolated from 4T1 mammospheres (Figure 5d). Thus, it is possible that, as a result of lower toxicity of Sali to differentiated tumor cells and stromal cells, the increased Sali accumulation did not have the same degree of impact on the overall tumor growth as on CSCs. In addition, the NP3s did not completely eradicate the CSC subpopulation in the tumors. The surviving CSCs could reignite tumor growth that counteracted any tumor growth inhibition contributed by the NP3s. As a result, the therapeutic advantage of the NP3s was overshadowed.
Figure 5.
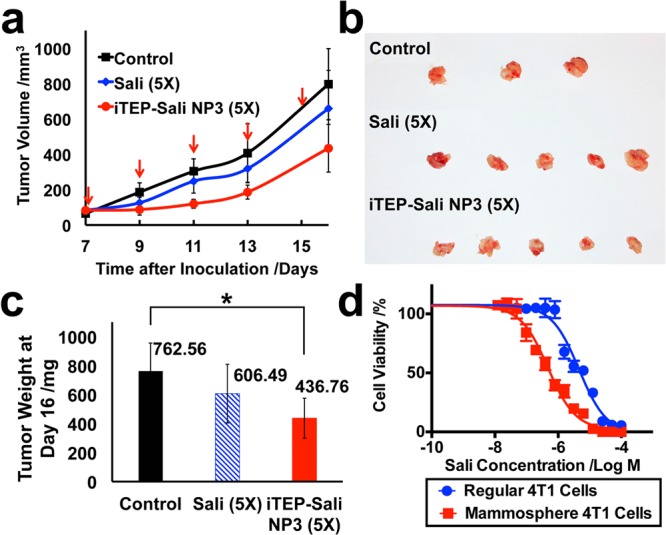
(a) The size changes of 4T1 orthotopic tumors after they were treated by five doses of controls (PBS), free Sali, or iTEP–Sali NP3. Both free Sali and the NP-encapsulated Sali were administered at 5 mg/kg. The five dosing dates are indicated as red arrows. The tumor dimensions were measured at the same day of dosing except for the last measurement, which was performed at day 16 post tumor inoculations. (b) Photos of the tumors collected at day 16 after the inoculations. (c) The mean tumor weights of each group of mice at the day 16 post tumor inoculations. The mean values were compared by one way ANNOVA. The * indicates a significant difference between the connected two means (p = 0.0307). (d) The viability profile of regular 4T1 cells versus 4T1 mammosphere cells after they were exposed to different concentrations of iTEP–Sali NP3 for 48 h. The IC50 of the NP3 to regular 4T1 cells 4.56 μM with a 95% CI = 3.49–5.95 μM. The IC50 of the NP3 to 4T1 mammosphere cells 0.48 μM with a 95% CI = 0.35–0.59 μM. The two IC50 are statistically different.
Discussion
In this study, we designed and characterized an iTEP-based Sali delivery system that improved the pharmacokinetics and tumor accumulation of Sali, as well as strengthened its specific CSC-inhibition effect of Sali. The results, in principle, validate the use of nanocarriers to boost the efficacy of CSC-specific drugs. Because CSCs were suggested to play a role in causing tumor MDR and recurrence, this enhancement effect of nanocarriers represented by iTEP–Sali NPs is significant to overcome the tumor and recurrence.
ITEPs were chosen as building blocks for the delivery system because they bring three primary advantages to the system. First, the nonimmunogenic nature of iTEPs could save the carrier from the risk of any carrier-specific immunogenicity and allow multiple dosing of the carrier. In contrast, if a pharmaceutical protein or polypeptide induces immune responses during its application, the responses could severely compromise its function.41,42 Second, recombinant polypeptides such as iTEPs are biodegradable so the carriers built upon the polypeptides are biodegradable and biocompatible as well. Indeed, iTEP carriers have not revealed any adverse effect in our in vivo studies. Finally, the sequences, length, and hydrophobicity of iTEPs can be definitely controlled and adjusted using genetic engineering approaches. This property of iTEPs becomes an advantage since it allows for the generation of a wide spectrum of new iTEPs or iTEP NPs to precisely meet a specific delivery need. The versatility of iTEP carriers offers the possibility to further optimize delivery systems not only for Sali but also for other CSC-targeting drugs. Examples of such optimization include appending iTEP with protein-based targeting ligands such as those antibodies recognizing CSC-specific antigens.
The Sali delivery system reported here represents a novel strategy to deliver and use Sali. Sali contributed the hydrophobicity to the iTEP–Sali conjugates and served as payloads at the same time. Previously, hydrophobic drugs were used as a hydrophobic component of micelle-forming amphiphilic conjugates and an encapsulated payload in the micelle.43 What we are reporting is the first application of this delivery strategy to Sali. A stable encapsulation represents the primary advantage of this strategy, which happens when a hydrophobic drug is packed inside a hydrophobic core contributed by the same drug.43
When charged hydrophobic drugs such as Sali are trapped inside a hydrophobic core of micelle-like NPs, the charges may destabilize the encapsulation, which was observed in both previous studies and this current study.23 To cancel out the charges, it is reasonable to coencapsulate an oppositely charged, hydrophobic molecule with the charged drugs. DMOA was believed to be a good candidate to serve as coencapsulate additives for negatively charged drugs such as Sali, because it is hydrophobic and positively charged. It also has acceptable toxicity according to a previous report.32 However, our data revealed strong toxicity of DMOA, especially in the comparison to Sali, which excluded DMOA for the coencapsulation with Sali. DMHA, a positively charged analogue of DMOH with no apparent toxicity, is a reasonable alternative. However, DMHA is not as hydrophobic as DMOA due to its short hydrocarbon chain. To address the insufficient hydrophobicity of the Sali–DMHA coencapsulation system, we tried using a second hydrophobic chemical to supplement more hydrophobicity. Our choice was α-tocopherol, a neutrally charged hydrophobic chemical.31 We observed improvements in both the loading efficiency and the release profile of Sali when DMHA and α-tocopherol were used in combination. We believe that this innovative, dual-additive approach may find its application in many encapsulation-based delivery systems, given that there could be many cases where the drugs to be encapsulated do not have an ideal charge.
Sali, when delivered by iTEP–Sali NPs, has a higher accumulation in the tumor and a longer system circulation than those of free Sali. Consistent with these advantages, iTEP–Sali NPs deplete CSCs and decrease mammosphere-forming capacity of the 4T1 tumors more efficiently than free Sali. However, the NPs do not inhibit tumor growth more effectively, a result consistent with a previous report that was obtained by using a xenograph tumor model.23 In fact, neither the NPs nor free Sali stabilized or abolished tumors. There are at least two possible reasons for the discrepancy between the CSC eradication effect and the overall tumor inhibition effect of the NPs. First, iTEP–Sali NPs did not eradicate CSCs completely, a deficiency that may contribute to tumor re-regrowth. The regrowth, in turn, may have canceled out some tumor inhibition effect of the NPs. Given this possibility, it is important to develop new carriers to expand the advantage of the Sali carrier in accumulating Sali in tumors. The carriers that could meet this need may be either a carrier that offers more stable encapsulation and slower release or a carrier that recognizes tumor cells through its targeting moieties. The second reason stems from the Sali’s toxicity characteristics, its higher toxicity to CSCs compared to differentiated cells in tumors. So, it is reasonable to find that CSCs, rather than bulk tumors, respond well to the increased Sali accumulation caused by the iTEP–Sali NPs. The toxicity characteristics advocate for a combination therapy that, concurrently or sequentially, uses the NPs together with conventional chemotherapeutics such as PTX. The combinational therapy promises to target all cell types in tumors, and hence is able to either stabilize or abolish the tumors. Another application niche of the iTEP–Sali NPs is their application in preventing tumor recurrence or MDR, given that the NPs have been shown to strengthen the toxicity of Sali to CSCs and that CSCs have been shown as one factor driving the tumor MDR.
Acknowledgments
We thank Dr. Ashutosh Chilkoti’s for generous support and mentoring to M.C. in the early phase of this study. We appreciate Dr. Mark Dewhirst’s suggestions for this project and comments on the manuscript. This research is supported by NIH Grant 4R00CA153929 and a University of Utah startup fund.
Supporting Information Available
Additional experimental details. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Al-Hajj M.; Wicha M. S.; Benito-Hernandez A.; Morrison S. J.; Clarke M. F. Prospective identification of tumorigenic breast cancer cells. Proc. Natl. Acad. Sci. U.S.A. 2003, 10073983–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Todaro M.; Alea M. P.; Di Stefano A. B.; Cammareri P.; Vermeulen L.; Iovino F.; Tripodo C.; Russo A.; Gulotta G.; Medema J. P.; Stassi G. Colon cancer stem cells dictate tumor growth and resist cell death by production of interleukin-4. Cell Stem cell 2007, 14389–402. [DOI] [PubMed] [Google Scholar]
- Dylla S. J.; Beviglia L.; Park I.-K.; Chartier C.; Raval J.; Ngan L.; Pickell K.; Aguilar J.; Lazetic S.; Smith-Berdan S.; Clarke M. F.; Hoey T.; Lewicki J.; Gurney A. L. Colorectal Cancer Stem Cells Are Enriched in Xenogeneic Tumors Following Chemotherapy. PLoS One 2008, 36e2428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eramo A.; Lotti F.; Sette G.; Pilozzi E.; Biffoni M.; Di Virgilio A.; Conticello C.; Ruco L.; Peschle C.; De Maria R. Identification and expansion of the tumorigenic lung cancer stem cell population. Cell Death Differ. 2008, 153504–14. [DOI] [PubMed] [Google Scholar]
- Bao S.; Wu Q.; McLendon R. E.; Hao Y.; Shi Q.; Hjelmeland A. B.; Dewhirst M. W.; Bigner D. D.; Rich J. N. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature 2006, 4447120756–60. [DOI] [PubMed] [Google Scholar]
- Li C.; Heidt D. G.; Dalerba P.; Burant C. F.; Zhang L.; Adsay V.; Wicha M.; Clarke M. F.; Simeone D. M. Identification of pancreatic cancer stem cells. Cancer Res. 2007, 6731030–7. [DOI] [PubMed] [Google Scholar]
- Yu F.; Yao H.; Zhu P.; Zhang X.; Pan Q.; Gong C.; Huang Y.; Hu X.; Su F.; Lieberman J.; Song E. let-7 Regulates Self Renewal and Tumorigenicity of Breast Cancer Cells. Cell 2007, 13161109–1123. [DOI] [PubMed] [Google Scholar]
- Hermann P. C.; Huber S. L.; Herrler T.; Aicher A.; Ellwart J. W.; Guba M.; Bruns C. J.; Heeschen C. Distinct Populations of Cancer Stem Cells Determine Tumor Growth and Metastatic Activity in Human Pancreatic Cancer. Cell Stem Cell 2007, 13313–323. [DOI] [PubMed] [Google Scholar]
- Gupta P. B.; Chaffer C. L.; Weinberg R. A. Cancer stem cells: mirage or reality?. Nat. Med. 2009, 1591010–2. [DOI] [PubMed] [Google Scholar]
- Li X.; Lewis M. T.; Huang J.; Gutierrez C.; Osborne C. K.; Wu M.-F.; Hilsenbeck S. G.; Pavlick A.; Zhang X.; Chamness G. C.; Wong H.; Rosen J.; Chang J. C. Intrinsic Resistance of Tumorigenic Breast Cancer Cells to Chemotherapy. J. Natl. Cancer Inst. 2008, 1009672–679. [DOI] [PubMed] [Google Scholar]
- Guan Y.; Hogge D. E. Proliferative status of primitive hematopoietic progenitors from patients with acute myelogenous leukemia (AML). Leukemia 2000, 14122135–41. [DOI] [PubMed] [Google Scholar]
- Lou H.; Dean M. Targeted therapy for cancer stem cells: the patched pathway and ABC transporters. Oncogene 2007, 2691357–60. [DOI] [PubMed] [Google Scholar]
- Zhou S.; Schuetz J. D.; Bunting K. D.; Colapietro A. M.; Sampath J.; Morris J. J.; Lagutina I.; Grosveld G. C.; Osawa M.; Nakauchi H.; Sorrentino B. P. The ABC transporter Bcrp1/ABCG2 is expressed in a wide variety of stem cells and is a molecular determinant of the side-population phenotype. Nat. Med. 2001, 791028–34. [DOI] [PubMed] [Google Scholar]
- Magni M.; Shammah S.; Schiro R.; Mellado W.; Dalla-Favera R.; Gianni A. M. Induction of cyclophosphamide-resistance by aldehyde-dehydrogenase gene transfer. Blood 1996, 8731097–103. [PubMed] [Google Scholar]
- Vermeulen L.; Todaro M.; de Sousa Mello F.; Sprick M. R.; Kemper K.; Perez Alea M.; Richel D. J.; Stassi G.; Medema J. P. Single-cell cloning of colon cancer stem cells reveals a multi-lineage differentiation capacity. Proc. Natl. Acad. Sci. U.S.A. 2008, 1053613427–13432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dean M.; Fojo T.; Bates S. Tumour stem cells and drug resistance. Nat. Rev. Cancer 2005, 54275–84. [DOI] [PubMed] [Google Scholar]
- Shackleton M.; Quintana E.; Fearon E. R.; Morrison S. J. Heterogeneity in Cancer: Cancer Stem Cells versus Clonal Evolution. Cell 2009, 1385822–829. [DOI] [PubMed] [Google Scholar]
- Gupta P. B.; Onder T. T.; Jiang G.; Tao K.; Kuperwasser C.; Weinberg R. A.; Lander E. S. Identification of Selective Inhibitors of Cancer Stem Cells by High-Throughput Screening. Cell 2009, 1384645–659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang B.; Wang X.; Cai F.; Chen W.; Loesch U.; Zhong X. Y. Antitumor properties of salinomycin on cisplatin-resistant human ovarian cancer cells in vitro and in vivo: involvement of p38 MAPK activation. Oncol. Rep. 2013, 2941371–8. [DOI] [PubMed] [Google Scholar]
- Zhou J.; Li P.; Xue X.; He S.; Kuang Y.; Zhao H.; Chen S.; Zhi Q.; Guo X. Salinomycin induces apoptosis in cisplatin-resistant colorectal cancer cells by accumulation of reactive oxygen species. Toxicol. Lett. 2013, 2222139–145. [DOI] [PubMed] [Google Scholar]
- Lu D.; Choi M. Y.; Yu J.; Castro J. E.; Kipps T. J.; Carson D. A. Salinomycin inhibits Wnt signaling and selectively induces apoptosis in chronic lymphocytic leukemia cells. Proc. Natl. Acad. Sci. U.S.A. 2011, 1083213253–13257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riccioni R.; Dupuis M. L.; Bernabei M.; Petrucci E.; Pasquini L.; Mariani G.; Cianfriglia M.; Testa U. The cancer stem cell selective inhibitor salinomycin is a p-glycoprotein inhibitor. Blood Cells, Mol., Dis. 2010, 45186–92. [DOI] [PubMed] [Google Scholar]
- Zhang Y.; Zhang H.; Wang X.; Wang J.; Zhang X.; Zhang Q. The eradication of breast cancer and cancer stem cells using octreotide modified paclitaxel active targeting micelles and salinomycin passive targeting micelles. Biomaterials 2012, 332679–91. [DOI] [PubMed] [Google Scholar]
- Boehmerle W.; Endres M. Salinomycin induces calpain and cytochrome c-mediated neuronal cell death. Cell Death Dis. 2011, 2, e168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Story P.; Doube A. A case of human poisoning by salinomycin, an agricultural antibiotic. N. Z. Med. J. 2004, 1171190U799. [PubMed] [Google Scholar]
- Ojo O. O.; Bhadauria S.; Rath S. K. Dose-dependent adverse effects of salinomycin on male reproductive organs and fertility in mice. PLoS One 2013, 87e69086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacKay J. A.; Chen M.; McDaniel J. R.; Liu W.; Simnick A. J.; Chilkoti A. Self-assembling chimeric polypeptide-doxorubicin conjugate nanoparticles that abolish tumours after a single injection. Nat. Mater. 2009, 812993–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrari M. Cancer nanotechnology: opportunities and challenges. Nat. Rev. Cancer 2005, 53161–71. [DOI] [PubMed] [Google Scholar]
- Wei X.; Senanayake T. H.; Warren G.; Vinogradov S. V. Hyaluronic acid-based nanogel-drug conjugates with enhanced anticancer activity designed for the targeting of CD44-positive and drug-resistant tumors. Bioconjugate Chem. 2013, 244658–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roche. Lab FAQs: Find a Quick Solution, 4th ed.; 2013. [Google Scholar]
- Forrest M. L.; Won C. Y.; Malick A. W.; Kwon G. S. In vitro release of the mTOR inhibitor rapamycin from poly(ethylene glycol)-b-poly(epsilon-caprolactone) micelles. J. Controlled Release 2006, 1102370–7. [DOI] [PubMed] [Google Scholar]
- Satoh T.; Higuchi Y.; Kawakami S.; Hashida M.; Kagechika H.; Shudo K.; Yokoyama M. Encapsulation of the synthetic retinoids Am80 and LE540 into polymeric micelles and the retinoids’ release control. J. Controlled Release 2009, 1363187–95. [DOI] [PubMed] [Google Scholar]
- Dontu G.; Abdallah W. M.; Foley J. M.; Jackson K. W.; Clarke M. F.; Kawamura M. J.; Wicha M. S. In vitro propagation and transcriptional profiling of human mammary stem/progenitor cells. Genes Dev. 2003, 17101253–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsumura Y.; Maeda H. A New Concept for Macromolecular Therapeutics in Cancer Chemotherapy: Mechanism of Tumoritropic Accumulation of Proteins and the Antitumor Agent Smancs. Cancer Res. 1986, 4612 Part 16387–6392. [PubMed] [Google Scholar]
- Fang J.; Nakamura H.; Maeda H. The EPR effect: Unique features of tumor blood vessels for drug delivery, factors involved, and limitations and augmentation of the effect. Adv. Drug Delivery Rev. 2011, 633136–51. [DOI] [PubMed] [Google Scholar]
- Chen M.; McDaniel J. R.; Mackay J. A.; Chilkoti A. Nanoscale Self-Assembly for Delivery of Therapeutics and Imaging Agents. Technol. Innovation 2011, 1315–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sokolić M.; Pokorny M. Comparative determination of salinomycin by high-performance liquid chromatography, microbiological and calorimetric methods in testing production processes and animal feed preparations. J. Pharm. Biomed. Anal. 1991, 910–121047–1053. [DOI] [PubMed] [Google Scholar]
- Ponti D.; Costa A.; Zaffaroni N.; Pratesi G.; Petrangolini G.; Coradini D.; Pilotti S.; Pierotti M. A.; Daidone M. G. Isolation and In vitro Propagation of Tumorigenic Breast Cancer Cells with Stem/Progenitor Cell Properties. Cancer Res. 2005, 65135506–5511. [DOI] [PubMed] [Google Scholar]
- Hirsch H. A.; Iliopoulos D.; Tsichlis P. N.; Struhl K. Metformin Selectively Targets Cancer Stem Cells, and Acts Together with Chemotherapy to Block Tumor Growth and Prolong Remission. Cancer Res. 2009, 69197507–7511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang D.; Huang J.; Wang X.; Yu Y.; Zhang H.; Chen Y.; Liu J.; Sun Z.; Zou H.; Sun D.; Zhou G.; Zhang G.; Lu Y.; Zhong Y. The eradication of breast cancer cells and stem cells by 8-hydroxyquinoline-loaded hyaluronan modified mesoporous silica nanoparticle-supported lipid bilayers containing docetaxel. Biomaterials 2013, 34317662–7673. [DOI] [PubMed] [Google Scholar]
- Kontos S.; Hubbell J. A. Drug development: longer-lived proteins. Chem. Soc. Rev. 2012, 4172686–95. [DOI] [PubMed] [Google Scholar]
- De Groot A. S.; Scott D. W. Immunogenicity of protein therapeutics. Trends Immunol. 2007, 2811482–90. [DOI] [PubMed] [Google Scholar]
- Yokoyama M.; Fukushima S.; Uehara R.; Okamoto K.; Kataoka K.; Sakurai Y.; Okano T. Characterization of physical entrapment and chemical conjugation of adriamycin in polymeric micelles and their design for in vivo delivery to a solid tumor. J. Controlled Release 1998, 501–379–92. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.