

Abstract
Background
Computational gene prediction continues to be an important problem, especially for genomes with little experimental data.
Results
I introduce the SNAP gene finder which has been designed to be easily adaptable to a variety of genomes. In novel genomes without an appropriate gene finder, I demonstrate that employing a foreign gene finder can produce highly inaccurate results, and that the most compatible parameters may not come from the nearest phylogenetic neighbor. I find that foreign gene finders are more usefully employed to bootstrap parameter estimation and that the resulting parameters can be highly accurate.
Conclusion
Since gene prediction is sensitive to species-specific parameters, every genome needs a dedicated gene finder.
Background
Complete genomic sequences are becoming more and more abundant. Given a new genome, one of the first and most important tasks is determining the structure of its protein-coding genes. Ab initio gene prediction plays a critical role because it produces gene structures quickly, inexpensively, and in some cases reliably. The accuracy of a gene finder depends on many factors. Chief among these is proper training. Training a gene finder can be a laborious task. In the past, genome projects moved rather slowly, and the trickle of data meant that parameters could be estimated before the genome was complete. Today, sequencing and assembly are so rapid that genomes can appear well in advance of proper training material. This leaves some new genome annotation projects without an appropriate gene finder.
When new genomic sequence emerges, it must be annotated with something, and frequently this is a gene finder for a completely different genome. A particularly good example of this is Genscan [1], which was trained primarily for the human genome, but has been used to annotate genes in worms, flies, fish, fungi, amphioxus, and others [2-6]. Since sequence features such as codon bias [7] and splicing signals [8] vary from organism to organism, it is expected that gene finders may not perform optimally in a foreign genome. While the practice of annotating a genome with a foreign gene finder is commonplace, its consequences are not widely understood.
In this paper, I introduce a new ab initio gene finding program called SNAP. SNAP is similar to Genscan and other generalized hidden Markov model (HMM) gene finders [9-13], but unlike many, it is easily adaptable to a number of organisms and its source code is freely available. I train and evaluate SNAP in the Arabidopsis thaliana (thale cress), Caenorhabditis elegans (nematode), Drosophila melanogaster (fruit fly), and Oryza sativa (rice) genomes, and demonstrate that SNAP is an accurate ab initio gene finder. For newly sequenced genomes without a specific gene finder, I show that annotating with a foreign gene finder may be highly inaccurate and I introduce a bootstrapping procedure that allows one to estimate species-specific parameters de novo.
Results and discussion
SNAP is a high-performance ab initio gene finder
I trained and evaluated SNAP in four genomes (see Methods) and compared its performance to Genscan in all genomes, to HMMGene [12] and Genefinder [14] in C. elegans, and to Augustus [15] in D. melanogaster. Genscan performs as well as recent gene finders designed specifically for Arabidopsis [16], was considered one of the standards for the Drosophila GASP experiments [3], and is one of the gene finders used by the International Rice Genome Sequencing Project [17]. HMMGene and Genefinder are well-established gene prediction programs for C. elegans. Augustus is one of the latest gene prediction programs and has been shown to outperform Genscan, GENIE, and GENEID in Drosophila.
As shown in table 2, SNAP is more accurate than Genscan in every genome. In C. elegans, SNAP performs better than HMMGene and almost as well as Genefinder. In D. melanogaster, SNAP is similar to Augustus. The HMMs employed by SNAP in this study have a minimal genome model without a promoter, poly-A signal, or UTRs, and the reason why SNAP outperforms Genscan is simply that it is trained for each genome. When compared to gene finders tuned for a particular genome, SNAP performs about equally. It may be possible to increase the accuracy of SNAP by including more states in the HMM to model additional genomic features or by using more sophisticated statistical techniques such as interpolated Markov models, maximum dependence decomposition trees or isochore segmentation. Fine-tuning SNAP to particular genomes is not the subject of this study however.
Table 2.
Gene prediction performance Performance figures for SNAP are derived from 5-fold cross-validation. At Arabidopsis thaliana, Ce Caenorhabditis elegans, Dm Drosophila melanogaster, Os Oryza sativa. SN and SP correspond to sensitivity and specificity.
| Nucleotide | Exon | Gene | |||||
| Genome | Gene finder | SN | SP | SN | SP | SN | SP |
| At | SNAP | 97.1 | 95.2 | 82.9 | 81.2 | 54.3 | 46.8 |
| Genscan | 79.9 | 92.9 | 65.3 | 71.2 | 19.5 | 21.3 | |
| Ce | SNAP | 97.6 | 94.2 | 85.5 | 79.3 | 46.0 | 32.5 |
| Genefinder | 98.1 | 95.3 | 89.2 | 86.1 | 51.6 | 48.0 | |
| Genscan | 81.3 | 91.6 | 48.6 | 66.4 | 10.2 | 9.6 | |
| HMMGene | 84.1 | 97.0 | 58.9 | 71.7 | 20.9 | 19.6 | |
| Dm | SNAP | 94.3 | 86.5 | 78.6 | 67.2 | 50.8 | 37.5 |
| Augustus | 92.4 | 88.6 | 77.2 | 68.2 | 50.7 | 31.9 | |
| Genscan | 84.5 | 81.1 | 68.7 | 62.9 | 22.1 | 20.0 | |
| Os | SNAP | 86.2 | 94.0 | 70.2 | 72.4 | 51.2 | 37.0 |
| Genscan | 70.3 | 89.8 | 58.2 | 74.8 | 25.9 | 32.0 | |
Gene prediction in novel genomes can be highly inaccurate
A newly sequenced genome may not have much training material and little experimental data to anchor gene predictions. How can one find genes in such uncharted territory? The common procedure is to use a gene finder from some other genome, perhaps the one that is most phylogenetically similar. To determine the consequences of this practice I evaluated the difference between the intra- and inter-species performance of SNAP. The results are displayed in table 3.
Table 3.
Intra- and inter-species gene prediction accuracy Intra-species performance figures derived from 5-fold cross-validation are along the major diagonal in bold. At Arabidopsis thaliana, Ce Caenorhabditis elegans, Dm Drosophila melanogaster, Os Oryza sativa. SN and SP correspond to sensitivity and specificity.
| Genomic DNA | |||||||||
| At | Ce | Dm | Os | ||||||
| Parameters | Measure | SN | SP | SN | SP | SN | SP | SN | SP |
| At | Nuc | 97.1 | 95.2 | 78.7 | 91.3 | 77.7 | 68.0 | 90.7 | 71.8 |
| Exon | 82.9 | 81.2 | 44.3 | 52.8 | 38.6 | 24.0 | 57.1 | 42.3 | |
| Gene | 54.3 | 46.8 | 20.9 | 11.3 | 18.8 | 5.7 | 20.5 | 9.7 | |
| Ce | Nuc | 83.5 | 91.5 | 97.6 | 94.2 | 81.3 | 73.6 | 79.7 | 74.5 |
| Exon | 40.5 | 49.9 | 85.5 | 79.3 | 42.2 | 29.8 | 27.5 | 26.0 | |
| Gene | 25.7 | 18.1 | 46.0 | 32.5 | 21.9 | 8.8 | 13.9 | 7.3 | |
| Dm | Nuc | 30.0 | 95.3 | 45.9 | 95.0 | 94.3 | 86.5 | 78.4 | 89.8 |
| Exon | 16.5 | 41.3 | 29.9 | 47.2 | 78.6 | 67.2 | 50.0 | 58.4 | |
| Gene | 3.2 | 4.3 | 7.8 | 6.9 | 50.8 | 37.5 | 36.3 | 28.9 | |
| Os | Nuc | 39.3 | 96.3 | 24.9 | 95.5 | 79.8 | 88.7 | 86.2 | 94.0 |
| Exon | 30.7 | 47.6 | 11.1 | 36.6 | 47.4 | 44.4 | 70.2 | 72.4 | |
| Gene | 5.1 | 6.1 | 5.3 | 7.8 | 27.2 | 17.2 | 51.2 | 37.0 | |
Gene prediction accuracy with foreign parameters appears to follow GC content more than phylogenetic relationships. For example, Oryza parameters perform reasonably well in Drosophila sequence (>25% genes correct) but very poorly in A. thaliana (5% genes correct). Similarly, for finding C. elegans genes, one is better off with parameters from A. thaliana than D. melanogaster. Choosing the best foreign gene finder is therefore not simply a matter of using parameters from the closest relative.
Genomes have significant compositional differences
To look more closely at the reasons behind the inaccuracy of foreign parameters, I have examined compositional differences in coding sequence, splice sites, and the translation start. Figure 2 displays the codon frequencies of degenerate codons for each of the four genomes. In general, codon preference is reflected by GC composition. That is, the GC-rich genomes prefer G and C in the 3rd position and the AT-rich genomes prefer A or T. This helps to explain the results of the previous experiment. But even between genomes with similar GC-content there are significant differences among equivalent codons. For example, Oryza prefers CTC for Leucine while Drosophila prefers CTG.
Figure 2.
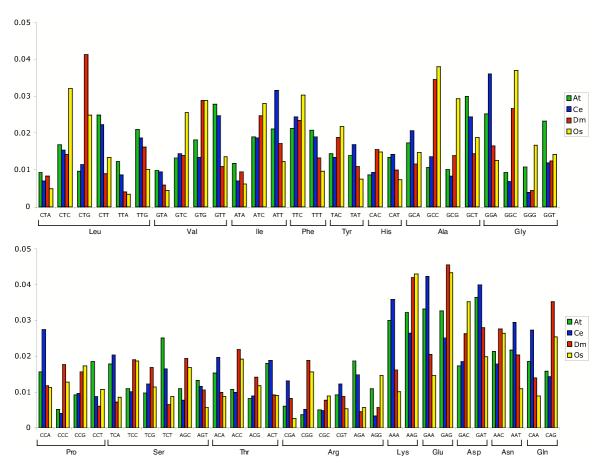
Codon frequency The frequency of each degenerate codon is indicated in a species-specific color (At Arabidopsis thaliana, Ce Caenorhabditis elegans, Dm Drosophila melanogaster, Os Oryza sativa). Codons are grouped by their parent amino acid.
Pictograms [18] for splice sites and the translation start are shown in figure 3. The two plant genomes (Arabidopsis and Oryza) have very similar acceptor sites, but the two animal genomes (Caenorhabditis and Drosophila) each have unique features. The upstream region from -7 to -27, frequently known as the poly-pyrimidine tract is T-rich in plants, AT-rich in Caenorhabditis, and pyrimidine-rich (proximally) in Drosophila. T is the most common nucleotide at positions at -5 and -6 in all genomes, but in Caenorhabditis these are almost invariant. At -4, the plants prefer G while Caenorhabditis prefers T and Drosophila is unbiased. The splice donor sites appear to be similar among all the genomes. However, there may be species-specific higher-order contexts that contribute to some specificity since the tuned architectures (see Methods) are slightly different for each genome. The translation start site appears to have some genome specificity. Given the compositional differences in the various signals, it is not surprising that gene prediction with foreign gene finders can be highly inaccurate.
Figure 3.

Pictograms of splice sites and translation start The height of each letter is proportional to its frequency. At Arabidopsis thaliana, Ce Caenorhabditis elegans, Dm Drosophila melanogaster, Os Oryza sativa. (a) splice acceptor site – canonical AG is at positions -2 and -1, (b) splice donor site – canonical GT is at +1 and +2, (c) translation start site – canonical ATG is at +1 to +3, (d) splice acceptor site consensus derived from gene predictions in A. thaliana with C. elegans parameters.
Parameter estimation in novel genomes
Even though foreign gene finders may perform sub-optimally, their predictions may display compositional properties of the novel genome. For example, when annotating the A. thaliana genome with a C. elegans gene finder, the predictions appeared very much like real A. thaliana genes. Figure 3d shows a splice acceptor pictogram derived from these predictions. Note that the sequence composition broadly resembles true A. thaliana splice acceptors, including a preference for G at -3 and a T-rich upstream sequence. It also retains some C. elegans qualities such as a greater proportion of Ts at -5 and -6.
If predicted genes have compositional properties similar to real genes, it should be possible to train a gene finder for a novel genome in the absence of any data. One simply runs a foreign gene finder (or more than one) to create a virtual training set and estimates parameters for the novel genome from the virtual data. So rather than use foreign gene finders to identify genes, one uses them to bootstrap parameter estimation. To determine if this procedure works, I assumed one of the genomes was novel and evaluated the gene prediction accuracy of bootstrapped parameters derived from one, two, or three foreign gene finders. The results are displayed in table 4.
Table 4.
Performance of foreign and bootstrapped parameters The foreign parameter data (top part of the table) is similar to table 4 but use the default rather than tuned architectures (see Methods). The bold face values are determined by 5-fold cross-validation within the same species. At Arabidopsis thaliana, Ce Caenorhabditis elegans, Dm Drosophila melanogaster, Os Oryza sativa. Sensitivity (NSN) and specificity (NSP) are reported at the nucleotide level. The bootstrapped values (bottom part of the table) are derived from parameter estimates based on gene predictions and no actual data. In these experiments, only inter-species gene parameters were used; dashes represent cells that would contain intra-species predictions.
| Genomic DNA | |||||||||
| At | Ce | Dm | Os | ||||||
| Parameters | NSN | NSP | NSN | NSP | NSN | NSP | NSN | NSP | |
| Actual | At | 97.6 | 94.3 | 81.0 | 90.1 | 75.3 | 63.6 | 90.7 | 68.5 |
| Ce | 86.3 | 91.0 | 98.1 | 92.5 | 85.1 | 72.4 | 79.8 | 73.0 | |
| Dm | 26.0 | 96.0 | 38.6 | 96.0 | 93.8 | 87.0 | 76.1 | 89.8 | |
| Os | 36.0 | 96.9 | 21.7 | 96.1 | 78.5 | 88.7 | 85.1 | 94.2 | |
| Bootstrapped | At | - | - | 95.8 | 88.2 | 94.7 | 76.0 | 92.1 | 76.0 |
| Ce | 96.6 | 93.2 | - | - | 95.7 | 80.3 | 91.0 | 79.2 | |
| Dm | 75.2 | 95.5 | 90.0 | 94.9 | - | - | 74.1 | 88.7 | |
| Os | 85.6 | 95.8 | 76.5 | 94.3 | 92.5 | 86.6 | - | - | |
| At Ce | - | - | - | - | 95.7 | 78.3 | 92.8 | 77.8 | |
| At Dm | - | - | 96.7 | 91.1 | - | - | 85.4 | 80.9 | |
| At Os | - | - | 95.5 | 90.3 | 94.0 | 81.2 | - | - | |
| Ce Dm | 94.3 | 94.4 | - | - | - | - | 84.0 | 83.0 | |
| Ce Os | 94.5 | 94.6 | - | - | 94.7 | 83.3 | - | - | |
| Dm Os | 84.9 | 95.8 | 88.4 | 94.9 | - | - | - | - | |
| At Ce Dm | - | - | - | - | - | - | 88.1 | 80.2 | |
| At Ce Os | - | - | - | - | 95.2 | 80.9 | - | - | |
| At Dm Os | - | - | 95.8 | 91.9 | - | - | - | - | |
| Ce Dm Os | 93.4 | 95.1 | - | - | - | - | - | - | |
Bootstrapped parameters work very well in many cases. In A. thaliana, the best foreign parameters come from C. elegans. But if the gene predictions are used for training rather than the final annotation, the prediction accuracy rises from 86.3/91.0 (nucleotide sensitivity/nucleotide specificity) to 96.6/93.2. The worst foreign performance (D. melanogaster parameters) changes from an abysmal 26.0/96.0 to a respectable 75.2/95.5. Bootstrapped parameters also appear to work very well in C. elegans and D. melanogaster. In C. elegans, the highest performing sets, 96.7/91.1 and 95.8/91.9, come from mixing two or three gene finders. In Drosophila, the best parameters are derived from O. sativa predictions. In general, bootstrapped parameters in these genomes can rival actual data, and even the worst bootstrapped parameters are reasonably accurate.
In O. sativa, bootstrapped parameters are only somewhat helpful. The Arabidopsis and Caenorhabditis parameters improve in both sensitivity and specificity, but the Drosophila foreign parameters are actually better than any of the bootstrapped ones. The reason why O. sativa behaves differently from the others is unclear at this time. In general, however, estimating parameters from gene predictions appears to be a simple and convenient way to train a gene finder for a novel genome. It is important to note that the genomes studied in this paper are all relatively compact. The techniques here may not work well in mammalian genomes or other genomes where exons account for a small fraction of the total sequence. I am currently investigating methods to improve gene finding accuracy and to quickly estimate gene prediction parameters in large genomes.
Conclusions
In this paper, I demonstrated that SNAP is a high performance gene finding program in several genomes. For new genomes without an appropriate gene finder, I showed that one can bootstrap parameter estimation from foreign gene finders.
Methods
Data sets
Genomic data and annotation were retrieved from two sources. The A. thaliana (Release 4.0) and O. sativa (OSA1) data were downloaded from TIGR [19]. Data for C. elegans (version 14.98.1) and D. melanogaster (version 14.3.1) were downloaded from Ensembl [20] via the Ensmart interface. Full-length mRNAs were retrieved using SRS from the Sanger Institute [21] by using the keyword 'complete' in the Description field, selecting 'mrna' for Molecule, and using the specific taxon in the Organism field.
The genomic sequence and annotation were subjected to a battery of tests to select for complete genes with no obvious errors such as overlapping exons, exons out of bounds, exons on the wrong strand, in-frame stop codons, or introns <30 bp. In addition to these "sanity checks", every gene was confirmed by an end-to-end, gap-free alignment between the in silico predicted transcript and a full-length cDNA. This requirement ensures that there are no extra/missed exons and that predicted splice sites are consistent with the actual transcript. Genes with unusual features such as non-canonical splice sites, internal stops, or split start/stop codons were removed. To simplify evaluation, genes with known alternate forms or genes overlapping other genes have been removed. To limit redundancy, all encoded proteins within a genome were aligned to each other, and in cases where there was a BLASTP [22,23] alignment of >90% identity, one of the sequences was removed. The version of BLAST used was 2.0MP-WashU [10-Sep-2003] and the parameters were the defaults plus -wordmask = seg + xnu. Bioperl [24] libraries were used for processing BLAST reports. For the A. thaliana set there were a large number of genes that passed all criteria, so the final set was trimmed by random selection to make it similar to the others.
The amount of sequence upstream and downstream of each gene is variable; the distance is half way to the nearest annotated gene up to a maximum of 1 kb. The average intergenic length in the four genomes is 2.9 kb, 2.7 kb, 6.6 kb, and 4.1 kb for Arabidopsis, Ceanorhabditis, Drosophila, and Oryza. 1 kb was chosen because at longer lengths some unannotated genes were clearly present (data not shown). The GC content, proportion of single-exon genes, and average exon and intron lengths are all similar to the downloaded data for each genome (data not shown).
The gene sets used in this study are summarized in table 1. The Arabidopsis and Caenorhabditis genomes are very AT-rich while the Drosophila and Oryza genomes have a more balanced nucleotide composition. Some genomes have unique features when compared to the others: Caenorhabditis has very few single-exon genes, Drosophila has much longer exons and introns, and Arabidopsis has the shortest introns on average.
Table 1.
Data set characteristics At Arabidopsis thaliana, Ce Caenorhabditis elegans, Dm Drosophila melanogaster, Os Oryza sativa.
| Genome | Sequence | Genes | GC | Single-exon Genes | Mean Exon | Mean Intron |
| At | 1.89 Mb | 631 | 37.3% | 19.8% | 230 bp | 157 bp |
| Ce | 3.02 Mb | 626 | 36.1% | 2.2% | 220 bp | 334 bp |
| Dm | 3.66 Mb | 602 | 43.6% | 24.9% | 394 bp | 948 bp |
| Os | 1.55 Mb | 424 | 44.5% | 22.9% | 237 bp | 350 bp |
Gene model
SNAP models protein coding sequences in genomic DNA via a specialized hidden Markov model similar to the one used in Genscan [1]. There are a few key differences:
1. Genscan is described as having three intron states but SNAP uses six to prevent stop codons at splice junctions (Genscan does not predict genes with stop codons, but its method to prevent stop codons is not described).
2. Genscan models both strands simultaneously while SNAP treats each strand independently. Decoding the strands independently allows genes on opposite strands to overlap. The advantage this is that it allows genes within introns of other genes, which is relatively common in some genomes. The disadvantage is that it also allows overlapping exons, which is not common.
3. The state diagram is fixed in Genscan but is read from a parameter file in SNAP. This allows one to change the HMM to describe a variety of genomic features. The simplest state diagram for predicting single- and multi-exon genes is shown in figure 1.
Figure 1.
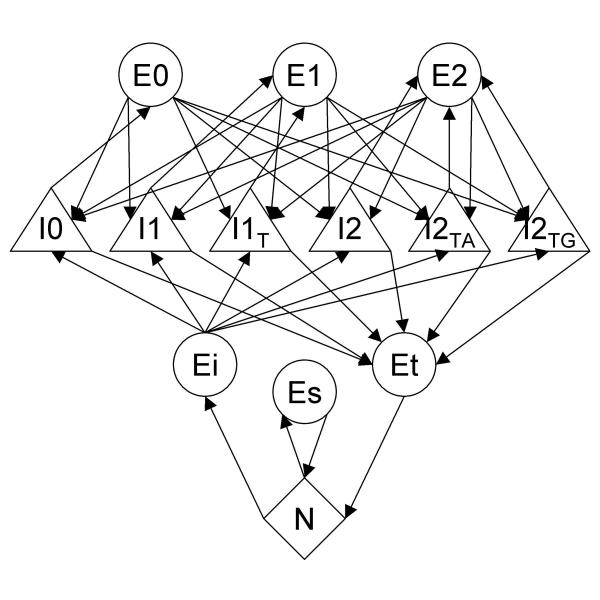
SNAP HMM state diagram Each state of the HMM is represented by a shape and transitions between the states are represented by arrows. States include N: intergenic, Es: single-exon gene, Ei: initial exon, Et terminal exon, E0–E2: exons in phase 0–2, I0–I2: introns in phases 0–2 (subscript of T, TA, or TG denotes the last bp or two bp of the intron – this is used to prevent in-frame stop codons across splice junctions).
4. The sequence feature model architectures in Genscan are fixed but SNAP allows one to employ any length weight matrix and any order Markov model and to embed these models within an array, decision tree, or 3-periodic (coding sequence) framework.
5. Introns may have explicit length distributions over a fixed distance followed by a geometric tail. Using explicit lengths increases the computational load by an amount that depends on the length of the explicit distribution and on exon density. The total runtime is approximately 1.5 times as long for these genomes with a fixed distance of 250 bp.
Parameter estimation
In gene prediction algorithms it is common to employ weight matrices (WM), weight array matrices (WAM), and Markov models (MM) to model compositional features such as the translation start site, splice sites and codon bias [1]. The choice of architectures is partly limited by the amount of data in the training set, but is ultimately determined by what works best in practice. To find a useful combination of models I began with simple weight matrices for the splice acceptor, splice donor, translation start, and translation stop sites, and 4th order Markov models for coding, intron, and intergenic sequence. I did not include models for promoter, poly-A site, UTRs or trans-splicing. These features are often not annotated and I therefore used only the features which could be unambiguously defined in the training data.
The "default" models are as follows: the acceptor WM is 30 bp long with 3 exonic nucleotides, the donor WM is 9 bp with 3 exonic nucleotides, the start WM is 12 bp with 6 coding nucleotides, and the stop WM is 9 bp with 3 bp on either side of the stop codons. The WMs (except the stop which is not shown) are therefore accurately represented by the pictograms in figure 3. These default architectures were used in the gene prediction experiments corresponding to table 4. For the experiments corresponding to tables 2 and 3, the architectures were tuned to find those that work best in each organism. The number of possible combinations of architectures is very large and was not fully explored. Tuned architectures differ from the default by changing the length of the WMs (exonic portions are kept constant) or by adding context to each position (which changes a WM to a WAM). The changes from the aforementioned default architectures are as follows: In A. thaliana, the acceptor is a 20 bp 1st order WAM, the donor is a 9 bp 1st order WAM, and the start is a 12 bp 1st order WAM. In C. elegans, the acceptor is a 15 bp WM, the donor is a 9 bp 2nd order WAM, and the start is a 18 bp 2nd order WAM. In D. melanogaster, the acceptor is a 40 bp 2nd order WAM, the donor is a 15 bp 2nd order WAM, and the start is a 18 bp 1st order WAM. In O. sativa, the acceptor is a 40 bp WM. In addition, the intron models in A. thaliana and C. elegans used explicit durations for a length of 250 bp.
Performance evaluation
All intra-species performance figures were calculated using a 5-fold cross-validation strategy (non-overlapping sets were trained on 4/5 of the data, gene prediction was evaluated on 1/5 of the data, and the performance figures were averaged over the 5 sets). Inter-species performance did not employ 5-fold cross-validation since the training and testing sets are from different genomes. Prior to gene prediction, all sequences were masked with RepeatMasker [25] with appropriate libraries using the MaskerAid [26] enhancement. The repeat library for O. sativa was obtained from TIGR [19]. Other repeat libraries were part of the RepeatMasker distribution.
Genscan was run with the A. thaliana parameter file for A. thaliana sequences and the human parameter file for the others. Genefinder and HMMGene were run with C. elegans parameters. Augustus was run with the "fly_singlestrand_partial.cfg" parameter file. Sensitivity [true positives/(true positives + false negatives)] and specificity [true positives/(true positives + false positives)] were calculated at the nucleotide, exon, and gene levels. Gene sensitivity (fraction of genes predicted exactly correct) is also reported as gene accuracy.
SNAP implementation
SNAP is written in ANSI C as a command line Unix program. The source code is covered by the GNU General Public License and is freely available from the author. SNAP is a relatively efficient program in both memory and time; it requires about 100 megabytes of memory and 30 cpu seconds to decode a 1 Mbp sequence on a 1 Ghz machine. CPU time and memory scale linearly with sequence length.
Authors' contributions
The work in this paper is completely my own.
Supplementary Material
1. The sequences and annotation are in a compressed archive named datasets.tar.gz 2. The SNAP source code is in a compressed archive named snap.tar.gz
Acknowledgments
Acknowledgements
This work was funded by NIH grant K22-00064-01 and by the Wellcome Trust Sanger Institute. Thanks to Richard Durbin and David Carter for comments on the manuscript and especially to the referees for making many useful suggestions.
References
- Burge C, Karlin S. Prediction of complete gene structures in human genomic DNA. J Mol Biol. 1997;268:78–94. doi: 10.1006/jmbi.1997.0951. [DOI] [PubMed] [Google Scholar]
- Webb CT, Shabalina SA, Ogurtsov AY, Kondrashov AS. Analysis of similarity within 142 pairs of orthologous intergenic regions of Caenorhabditis elegans and Caenorhabditis briggsae. Nucleic Acids Res. 2002;30:1233–1239. doi: 10.1093/nar/30.5.1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reese MG, Hartzell G, Harris NL, Ohler U, Abril JF, Lewis SE. Genome annotation assessment in Drosophila melanogaster. Genome Res. 2000;10:483–501. doi: 10.1101/gr.10.4.483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riboldi Tunnicliffe G, Gloeckner G, Elgar GS, Brenner S, Rosenthal A. Comparative analysis of the PCOLCE region in Fugu rubripes using a new automated annotation tool. Mamm Genome. 2000;11:213–219. doi: 10.1007/s003350010039. [DOI] [PubMed] [Google Scholar]
- Kraemer E, Wang J, Guo J, Hopkins S, Arnold J. An analysis of gene-finding programs for Neurospora crassa. Bioinformatics. 2001;17:901–912. doi: 10.1093/bioinformatics/17.10.901. [DOI] [PubMed] [Google Scholar]
- Boeddrich A, Burgtorf C, Francis F, Hennig S, Panopoulou G, Steffens C, Borzym K, Lehrach H. Sequence analysis of an amphioxus cosmid containing a gene homologous to members of the aldo-keto reductase gene superfamily. Gene. 1999;16:207–214. doi: 10.1016/S0378-1119(99)00079-7. [DOI] [PubMed] [Google Scholar]
- Akashi H. Gene expression and molecular evolution. Curr Opin Genet Dev. 2001;11:660–666. doi: 10.1016/S0959-437X(00)00250-1. [DOI] [PubMed] [Google Scholar]
- Lim LP, Burge CB. A computational analysis of sequence features involved in recognition of short introns. Proc Natl Acad Sci U S A. 2001;98:11193–11198. doi: 10.1073/pnas.201407298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solovyev V, Salamov A. The Gene-Finder computer tools for analysis of human and model organisms genome sequences. Proc Int Conf Intell Syst Mol Biol. 1997;5:294–302. [PubMed] [Google Scholar]
- Kulp D, Haussler D, Reese MG, Eeckman FH. A generalized hidden Markov model for the recognition of human genes in DNA. Proc Int Conf Intell Syst Mol Biol. 1996;4:134–142. [PubMed] [Google Scholar]
- Parra G, Blanco E, Guigo R. GeneID in Drosophila. Genome Res. 2000;10:511–515. doi: 10.1101/gr.10.4.511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krogh A. Two methods for improving performance of an HMM and their application for gene finding. Proc Int Conf Intell Syst Mol Biol. 1997;5:179–186. [PubMed] [Google Scholar]
- Cawley SE, Wirth AI, Speed TP. Phat – a gene finding program for Plasmodium falciparum. Mol Biochem Parasitol. 2001;118:167–174. doi: 10.1016/S0166-6851(01)00363-2. [DOI] [PubMed] [Google Scholar]
- Genefinder (Green P.) http://ftp.genome.washington.edu/cgi-bin/genefinder_req.pl
- Stanke M, Waack S. Gene prediction with a hidden Markov model and a new intron submodel. Bioinformatics. 2003;19:II215–II225. doi: 10.1093/bioinformatics/btg1080. [DOI] [PubMed] [Google Scholar]
- Majoros WH, Pertea M, Antonescu C, Salzberg SL. GlimmerM, Exonomy and Unveil: three ab initio eukaryotic genefinders. Nucleic Acids Res. 2003;31:3601–3604. doi: 10.1093/nar/gkg527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakata K, Nagamura Y, Numa H, Antonio BA, Nagasaki H, Idonuma A, Watanabe W, Shimizu Y, Horiuchi I, Matsumoto T, Sasaki T, Higo K. RiceGAAS: an automated annotation system and database for rice genome sequence. Nucleic Acids Res. 2002;30:98–102. doi: 10.1093/nar/30.1.98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pictogram (Burge C) http://genes.mit.edu/pictogram.html
- The Institute for Genomic Research http://www.tigr.org
- Ensembl Genome Browser http://www.ensembl.org
- SRS7 at the Sanger Institute http://srs.sanger.ac.uk
- Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215:403–410. doi: 10.1006/jmbi.1990.9999. [DOI] [PubMed] [Google Scholar]
- WU-BLAST (Gish W) http://blast.wustl.edu
- Bioperl. Stajich JE, Block D, Boulez K, Brenner SE, Chervitz SA, Dagdigian C, Fuellen G, Gilbert JG, Korf I, Lapp H, Lehvaslaiho H, Matsalla C, Mungall CJ, Osborne BI, Pocock MR, Schattner P, Senger M, Stein LD, Stupka E, Wilkinson MD, Birney E. The Bioperl toolkit: Perl modules for the life sciences. Genome Res. 2002;12:1611–1618. doi: 10.1101/gr.361602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- RepeatMasker (Smit, AFA, Green P.) http://repeatmasker.genome.washington.edu
- Bedell JA, Korf I, Gish W. MaskerAid: a performance enhancement to RepeatMasker. Bioinformatics. 2000;16:1040–1041. doi: 10.1093/bioinformatics/16.11.1040. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
1. The sequences and annotation are in a compressed archive named datasets.tar.gz 2. The SNAP source code is in a compressed archive named snap.tar.gz