

Abstract
Background
Glutamate excitotoxicity may contribute to the pathophysiology of amyotrophic lateral sclerosis (ALS). Studies in ALS animal models show decreased excitatory amino acid transporter 2 (EAAT2) overexpression delays onset and prolongs survival, and that ceftriaxone increases EAAT2 activity in rodent brains. Phase 1, 2, and 3 clinical studies of ceftriaxone for ALS were combined into a three-stage, nonstop study.
Methods
514 participants were randomised to ceftriaxone (n=341) or placebo (n=173); 66 participants were enrolled in stages 1 (pharmacokinetics) and 2 (safety) to determine cerebrospinal fluid and blood pharmacokinetics and safety of two dosages: 2 grams and 4 grams/day of ceftriaxone. All participants continued into stage 3 (efficacy) in blinded fashion with participants who began treatment on the discontinued dose analysed in the same group as those on the dose that that was continued. In stage 3, 44 participants previously assigned to 2 or 4 g ceftriaxone in stage 2 received 4 g ceftriaxone; 21 participants assigned to placebo in stage 2 continued on placebo. 448 new participants were randomized in stage 3 to 4 g ceftriaxone or placebo (2:1). Participants, family members and all site staff were blinded to treatment assignment. Computerized randomisation sequence using permuted blocks of 3 was stratified by riluzole use and blocked by site. Participants received 2g ceftriaxone or placebo BID via a central venous catheter (CVC) administered in the home setting by a trained caregiver. To minimize biliary side effects, participants assigned to ceftriaxone also received 300 mg ursodiol BID in a blinded manner; those assigned to placebo received matched placebo capsules BID. The co-primary efficacy outcomes were survival and functional decline, using the slope of scores on the ALS Functional Rating Scale-Revised (ALSFRS-R). The first participant entered the trial on September 4, 2006 (stage 1); the first stage-3 participant entered on June 4, 2009. The trial was stopped in July 2012.
Findings
During stages 1 and 2, ALSFRS-R functional decline was 0.5076±0.2440 units per month slower in participants taking 4 g ceftriaxone versus those taking placebo (95% CI 0.0196, 0.9956, p=0.0416), yet in stage 3, functional decline differed only by 0.08975±0.07581 units per month (95% CI −0.05919, 0.2387; p=0.2370). No significant differences were seen in stage 3 survival (hazard ratio, 0.904 [95% CI 0.710, 1.152]; p=0.4146). Adverse events rates were higher in the ceftriaxone versus placebo group for gastrointestinal (72% [245/340] vs 56% [97/173]; p=0.0004) and hepatobiliary events (62% [211/340] vs 11% [19/173]; p<0.0001). Add-on ursodiol reduced these events in participants taking ceftriaxone. A significantly larger percentage of ceftriaxone versus placebo participants experienced hepatobiliary serious adverse events (12% [41/340] vs 0% [0/173]).
Interpretation
Despite promising stage-2 efficacy data, the stage-3 ceftriaxone in ALS study failed to show clinical efficacy. The adaptive design approach allowed for seamless movement from one phase to another obviating the need for multiple grant submissions. CVC use in the home setting was shown to be not only possible, but also safe.
Introduction
Amyotrophic lateral sclerosis (ALS) is a neurodegenerative disorder affecting motor neurons of the brain and spinal cord, leading to progressive muscle atrophy and weakness, respiratory failure and death.1,2 Median survival is 2–4 years; 5–10% of patients survive beyond 10 years.3,4 One approved therapy, riluzole, provides a modest survival benefit,5,6 and there is an urgent need for more efficacious treatments. While the etiology of neurodegeneration in ALS is not fully understood, evidence suggests that glutamate excitotoxicity may be a factor in disease progression.7,8 Studies in ALS animal models and human tissue have shown decreased excitatory amino acid transporter 2 (EAAT2; mouse analog GLT-1), which clears synaptic glutamate.9–11 Riluzole inhibited glutamate release in preclinical studies.12,13 Conversely, overexpression of EAAT2/GLT-1 delays onset and prolongs survival in ALS mice.14 Thus, targets that upregulate EAAT2 may be neuroprotective in ALS.
Ceftriaxone, an FDA–approved beta-lactam antibiotic, significantly increases EAAT2 activity and GLT-1 expression in rodent brains.15,16 Ceftriaxone also increases EAAT2 promoter activity and protects motor neurons from excitotoxicity in human astrocyte cultures.15,17,18 Ceftriaxone reduces glutamate excitotoxicity in animal models of spinal muscular atrophy,19 Huntington’s disease,20 ischemia21,22 and multiple sclerosis.23 Ceftriaxone slowed disease progression and prolonged survival in ALS mice.15 When the National Institute of Neurological Disorders and Stroke (NINDS) Neurodegeneration Drug Screening Consortium screened 1040 compounds to identify candidate therapies for neurodegenerative diseases, cephalosporin antibiotics were the only class of compounds active in the majority of ALS-related assays.15,18 Ceftriaxone has the longest half-life of available cephalosporins and is thought to effectively penetrate cerebral spinal fluid (CSF).24
Phase 1, 2 and 3 studies of ceftriaxone for ALS were combined into a three-stage, nonstop study to expedite testing. Stage 1 pharmacokinetic and stage 2 safety results were previously published.25 The objective of the current study was to evaluate the efficacy of ceftriaxone versus placebo during stage 3 of testing, with the per-protocol hypothesis that intravenous (IV) ceftriaxone would slow disease course in patients with ALS.
Methods
Study Design and Participants
This was a randomized, double-blind, placebo-controlled, multicentre, Phase III trial of the efficacy and safety of ceftriaxone versus placebo for treating ALS. Eligible adults (>18 years) had a diagnosis of possible, laboratory-supported probable, probable or definite ALS, vital capacity (VC) >60% of the normal predicted for age and height,26 and symptom duration <3 years before enrolment. Participants were permitted to use riluzole if they were taking a stable dose for ≥30 days. Participants were required to be medically able to undergo central venous catheter (CVC) placement. Screening assessments included medical history, physical examination, concomitant medication review and laboratory tests. Key exclusion criteria included known sensitivity to ceftriaxone, cephalosporins, penicillin, beta-lactams, ursodiol or biliary salts; active biliary or gastrointestinal disease; known immune-compromising illness, history of antibiotic-induced colitis and dependence on mechanical ventilation. A complete listing of the study inclusion and exclusion criteria can be found in webappendix A. Institutional review board (IRB) approval was obtained at each centre and participants provided written informed consent before screening. The study adhered to the International Conference on Harmonisation and Good Clinical Practice guidelines and was performed in accordance with the Declaration of Helsinki. Per protocol, double-blind treatment was planned to continue for all subjects until the last enrolled subject reached 52 weeks of participation. An independent Data Safety Monitoring Board (DSMB) worked with the principal investigator and steering committee to evaluate safety and to establish and revise safety stopping rules as necessary. The DSMB met before study start and after stages 1 and 2. Thereafter, the board met approximately every 6 months and at preplanned interim analyses to make recommendations for modification or termination of the trial. The DSMB was advisory to the NINDS and made recommendations independently from the funding source. The NINDS policies on DSMBs for clinical trials were followed (http://www.ninds.nih.gov/research/clinical_research/policies/data_safety_monitoring.htm). No voting members on the DSMB were from the NINDS.
Randomisation and masking
Biostatisticians at Massachusetts General Hospital (MGH) developed the randomisation plan and documents for participating pharmacies. For stage 1, the computerized randomisation sequence used permuted blocks of 3, where each block contained: placebo, ceftriaxone 2g and ceftriaxone 4g. For stage 3, participants were randomised to 4 g ceftriaxone and placebo in a 2:1 ratio. The permuted blocks of 3 contained: placebo, ceftriaxone 4g and ceftriaxone 4g. Randomisation was stratified by riluzole use and blocked by site to allow for a balanced distribution of participants to treatment groups. Pharmacies received their randomisation schedules and ID numbers used to identify participants’ source documents. Coordination centre clinical trial staff, with the exception of the pharmacy monitor, remained blinded to treatment assignments. Participants, investigators, study monitors, site coordinators and site clinical evaluators also were blinded to treatment group assignment.
Study drug was masked to maintain the study blind. Pediatric multivitamin for infusion (MVI) was used as the placebo to color-match the ceftriaxone syringes. Study drug syringes were tinted semi-transparent orange to further protect the color blind. Frozen and liquid syringes were tested for color, consistency, appearance and odor, and participants were not able to distinguish between active and placebo. The ursodiol and placebo manufacturers created capsules that appeared identical for both ursodiol and ursodiol placebo. Participants were asked at time of study withdrawal what treatment assignment they thought they received.
Procedures
In stage 1, 66 participants from ten US-based sites were randomised to ceftriaxone 2 g, ceftriaxone 4 g or placebo daily for seven days to determine whether one or both dosages achieved a CSF trough concentration ≥1 μM in ≥80% of participants. In stage 2, all stage-1 participants continued their dosages for 20 weeks to determine the safety and tolerability of ceftriaxone versus placebo. The DSMB and two members of the steering committee used this information as well as efficacy data at 20 weeks to select the ceftriaxone 4-g dosage for stage 3.25
The Phase 3 study (stage 3) was conducted at 58 sites in the United States and Canada. Participants who completed stage 2 continued (blinded) to stage 3; new participants were randomised to ceftriaxone 4 g or placebo (2:1). The DSMB approved the use of ursodeoxycholic acid (ursodiol) to manage hepatobiliary adverse events associated with ceftriaxone. Thus, participants randomised to ceftriaxone also received 300 mg ursodiol twice daily. For all stages, placebo was a pediatric MVI matched to ceftriaxone. For stage 3, placebo participants also received a matched ursodiol placebo capsule. Participants and caregivers administered study drug via CVC twice daily. Participants and caregivers were educated in catheter care and safety and underwent on-site evaluation of the proper catheter care technique every 16 weeks and on-site written examinations annually. Typically, there was one caregiver per study participant.
The co-primary endpoints were survival (time to death, tracheostomy or initiation of permanent assisted ventilation [PAV]), and change in function after 1 year of treatment, as assessed using the slope of scores on the ALS Functional Rating Scale-Revised (ALSFRS-R).27 Secondary endpoints included changes from baseline in VC and changes in upper- and lower-limb muscle strength using hand-held dynamometry (HHD). Muscle groups included in the upper and lower limb megascores included shoulder flexion, elbow flexion, elbow extension, hip flexion, knee flexion, knee extension, wrist extension, first dorsal interosseous contraction and ankle dorsiflexion. The ALS-Specific Quality of Life (ALSSQOL) questionnaire28 was used to assess symptom severity, mood, intimacy and social issues. The Caregiver Burden Inventory (CBI)29 was used to assess the quality of life of caregivers in terms of social, emotional, physical, time and developmental aspects of their relationships with the participant.
Safety assessments included physical examination, laboratory testing, abdominal ultrasounds, CVC safety checks, vital signs and adverse event monitoring. Adverse events and serious adverse events were categorized using the Common Terminology Criteria for Adverse Events (CTCAE) and rated for severity and relationship to study drug.
Statistical analyses
With DSMB and sponsor approval (July 2011) the original sample size of 600 participants was recomputed to 500 based on a revision to the estimate for expected on-study mortality based on the mortality data at that time and the fact that length of individual subject participation was longer than initially planned. Enrolment of 500 provided 80% power to detect a 50% increase in median time to death, tracheostomy or PAV. The primary analysis was based on the intention-to-treat principle, and all participants were included regardless if they stopped treatment before study end. Given that participants from all stages are included in the stage-3 analysis, no alpha correction was deemed necessary. The only possible bias was to underestimate any potential effect of treatment, as 22 participants on active treatment initially received a lower dose (2g daily) than did participants ultimately enrolled in stage 3.
The effect of a 2:1 randomisation is to increase the sample size by a factor of 9/8. The investigators felt that a 2:1 randomisation would aid in recruitment rate to more than compensate for the additional participant accrual required.
One-year progression of ALSFRS-R, VC and other longitudinal variables, were analysed using random slopes regression models, and survival was analysed using a log-rank test stratified by riluzole use. For participants who discontinued active treatment, survival was assessed on a bimonthly basis. Survival was assessed for all subjects, regardless of whether study drug was discontinued. Participants who stopped treatment were nonetheless encouraged to continue bimonthly study visits, and at that time, the site staff recorded the date that the subject was seen. Participants who were unable to attend study visits at the site were contacted via telephone to assess vital status.
The trial had a group sequential design and was monitored every six months for efficacy and futility using pre-specified stopping rules (webappendix B). Statistical tests were two sided and p-values were not corrected for multiple comparisons or sequential stopping rules. A second analysis accounted for deaths using a shared parameter model.30 This model reproduced the primary results (data not shown).
Alpha levels were not corrected for co-primary outcome measures in the analysis. A bonferroni correction for the two endpoints was considered, but was felt overly conservative as the two endpoints are related. As this study was not performed for the purpose of regulatory approval, the independent assessment of primary measures was felt to be the most informative way to evaluate them. ALSFRS-R could have been employed as a secondary endpoint; however, it would then not have been appropriate to include it in the futility stopping rule.
Selected post hoc analyses were performed to explore the possibility that ceftriaxone versus placebo may have conferred some beneficial effects not apparent from the results of the primary analysis. To analyse the co-primary endpoints jointly, the Combined Analysis of Function and Survival (CAFS) was used. CAFS ranks clinical outcomes based on survival time and change in the ALSFRS-R score.31
In July 2012, the study was stopped for futility by the DSMB before all participants had 1 year of treatment, as the last participant enrolled on December 14, 2011. Data evaluated at that time are referred to as the interim analyses throughout. The final analyses represent data combined from all three stages, and the results reported herein represent the final analyses unless otherwise specified. The study is registered with ClinicalTrials.gov #NCT00349622.
Role of the funding source
The study was conducted under an Investigational New Drug (68,892) and approved by the MGH coordination centre IRB and all participating centre IRBs. NINDS provided a cooperative agreement (5 U01-NS-049640) and participated in the Steering Committee (Robin Conwit). The NINDS selected the members of the DSMB for the study. The NINDS did not have any role in the data collection or analysis.
Results
Participant disposition
The first participant entered the trial on September 4, 2006 (stage 1); the first stage-3 participant entered on June 4, 2009. The trial was stopped in July 2012. A total of 732 people were screened; 219 were excluded for not meeting the inclusion/exclusion criteria, withdrawing consent or for other reasons (figure 1). Active biliary disease, including gallstones, was the most frequently observed exclusion criterion (n=53); the most frequently unmet inclusion criterion was VC >60% of the predicted normal value (n=45). The final dataset was composed of 513 participants, 340 in the ceftriaxone group and 173 in the placebo group. Follow-up for survival was >95% complete; 50% of participants survived during the study. Participants in both groups were similar in age, gender distribution and baseline characteristics (table 1). Of participants on taking ceftriaxone 46% (156/340) continued on treatment until study end or an endpoint was reached. The 156 participants who discontinued study medication early spent 51% of their time on study taking the medication. The corresponding proportions in the placebo group were similar: 41% (71/173) and 46%. In total, participants on ceftriaxone remained on drug for a mean (SD) of 14.2 (11.7) months while participants on placebo remained on drug for a mean (SD) of 12.3 (9.2) months. About 46% (84/184) of participants who stopped active treatment did so because of an adverse event as did 31% (32/102) of participants who stopped placebo. In the active arm, only 15 of these reported GI or hepatobiliary/pancreatic events as the reason. This accounts for <10% of participants who stopped treatment with ceftriaxone.
Figure 1.
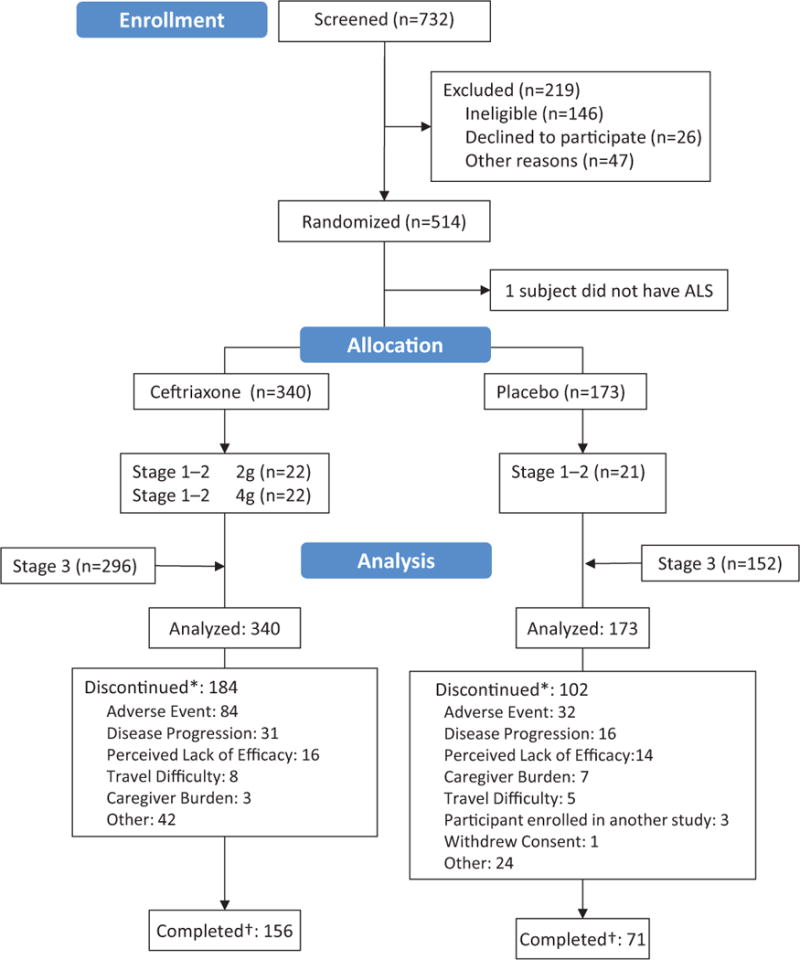
*Participant prematurely discontinued study medication
†Participant completed study treatment assignment
Table 1.
Baseline demographic and clinical characteristics
| Characteristic | Ceftriaxone (n=340) | Placebo (n=173) | All (N=513) |
|---|---|---|---|
| Mean ± SD age at screening, years | 56 ± 10 | 55 ± 10 | 55 ± 10 |
| Mean ± SD years from symptom onset to screening | 1.49 ± 0.7 | 1.50 ± 0.7 | 1.49 ± 0.7 |
| Mean ± SD years from diagnosis to screening | 0.56 ± 0.5 | 0.58 ± 0.5 | 0.57 ± 0.5 |
| Mean ± SD years from symptom onset to diagnosis | 0.93 ± 0.6 | 0.92 ± 0.6 | 0.92 ± 0.6 |
| Male, n (%) | 209 (61) | 101 (58) | 310 (60) |
| Race, n (%) | (n=339) | (n=171) | (n=510) |
| White | 320 (94) | 163 (94) | 483 (94) |
| Black/African American | 8 (2) | 3 (2) | 11 (2) |
| Asian | 6 (2) | 5 (3) | 11 (2) |
| Other | 5 (1) | 0 (0) | 5 (1) |
| Ethnicity, n (%) | (n=337) | (n=171) | (n=508) |
| Hispanic or Latino | 17 (5) | 6 (3) | 23 (5) |
| Family history of ALS, n (%) | (n=333) | (n=169) | (n=502) |
| Yes | 26 (8) | 8 (5) | 34 (7) |
| Site of onset, n (%) | |||
| Limb | 257 (76) | 137 (79) | 394 (77) |
| Bulbar | 75 (22) | 35 (20) | 110 (21) |
| Both | 8 (2) | 1 (0.6) | 9 (2) |
| Riluzole use (yes), n (%) | 249 (73) | 128 (74) | 377 (73) |
| Mean ± SD VC % predicted maximum | 88 ± 17 | 91 ± 18 | 89 ± 17 |
| Mean ± SD ALSFRS-R score | 36.5 ± 6.0 | 36.9 ± 5.4 | 36.7 ± 5.8 |
| Mean ± SD total ALSSQOL* | 398 ± 64 | 397 ± 64 | 398 ± 64 |
| Mean ± SD total CBI | 14.7 ± 11.4 | 14.5 ± 11.8 | 14.6± 11.6 |
ALS=amyotrophic lateral sclerosis; ALSFRS-R=Amyotrophic Lateral Sclerosis Functional Rating Scale-Revised; ALSSQOL=Amyotrophic Lateral Sclerosis-Specific Quality of Life Questionnaire; CBI=Caregiver Burden Inventory; VC=vital capacity.
Maximum possible score is 550.
Efficacy
During stages 1 and 2, functional decline was 0.51±0.24 (95% CI: 0.0196, 0.9956) units per month slower in participants taking 4 g ceftriaxone versus those taking placebo (p=0.0416); there were no other significant between-group differences. In stage 3, there was no significant effect on functional decline after interim (p=0.196) or final analyses (p=0.237; table 2). For survival, log-rank tests showed no significant differences after interim (p=0.3680) or final analyses (p=0.4146) (figure 2).
Table 2.
Summary of results* for primary and secondary endpoints
| Ceftriaxone | Placebo | Difference (95% CI) | p value | |
|---|---|---|---|---|
| ALSFRS-R | −1.13 ± 0.04 (−1.22, −1.05) |
−1.22 ± 0.06 (−1.34, −1.10) |
0.09 ± 0.08 (−0.06, 0.24) |
0.2370 |
| VC | −2.77 ± 0.14 (−3.05, −2.49) |
−3.08 ± 0.20 (−3.47, −2.70) |
0.31 ± 0.24 (−0.17, 0.79) |
0.2016 |
| HHD | ||||
| Upper limb, % baseline | −5.27 ± 0.22 (−5.70, −4.85) |
−5.55 ± 0.30 (−6.15, −4.96) |
0.28 ± 0.37 (−0.46, 1.01) |
0.4560 |
| Lower limb, % baseline | −4.15 ± 0.26 (−4.66, −3.64) |
−4.48 ± 0.36 (−5.19, −3.77) |
0.33 ± 0.45 (−0.55, 1.20) |
0.4623 |
| Upper limb, z score | −0.080 ± 0.004 (−0.088, −0.073) |
−0.077 ± 0.005 (−0.088, −0.067) |
−0.003 ± 0.007 (−0.016, 0.010) |
0.6765 |
| Lower limb, z score | −0.065 ± 0.003 (−0.072, −0.059) |
−0.063 ± 0.004 (−0.072, −0.055) |
−0.002 ± 0.005 (−0.013, 0.009) |
0.7104 |
| ALSQOL | −3.51± 0.33 (−4.16, −2.86) |
−3.44 ± 0.46 (−4.35, −2.53) |
−0.068 ± 0.568 (−1.185, 1.048) |
0.9045 |
| CBI total score | 1.39 ± 0.08 (1.23, 1.54) |
1.31 ± 0.11 (1.09, 1.53) |
0.08 ± 0.14 (−0.19, 0.35) |
0.5538 |
Mean change from baseline through one year of follow up in units per month ± SE (95% CI)
ALS=amyotrophic lateral sclerosis; ALSFRS-R=Amyotrophic Lateral Sclerosis Functional Rating Scale-Revised; ALSSQOL=Amyotrophic Lateral Sclerosis-Specific Quality of Life Questionnaire; CBI=Caregiver Burden Inventory; CI, confidence interval; HHD, hand-held dynamometry; SE, standard error; VC=vital capacity.
Figure 2.
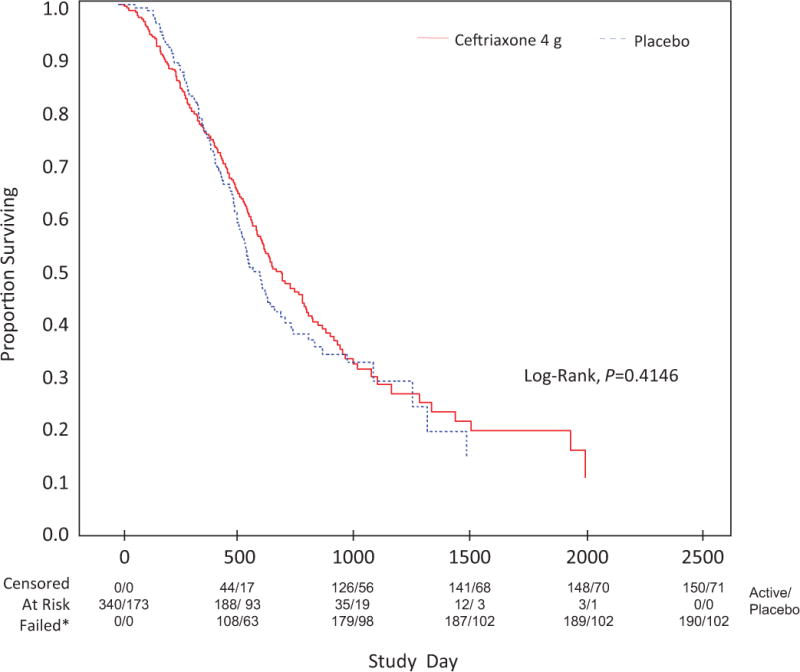
*Death, permanent assisted ventilation, tracheostomy
For VC, no significant between-group differences were noted during stages 1 and 2 or after interim or final analyses. For the interim analysis, the between-group difference in slopes was 0.33±0.24 (95% CI −0.15, 0.81) units per month (p=0.1764). The final analysis showed no significant treatment effects on VC (table 2). HHD results for stages 1 and 2 showed that leg strength but not arm strength declined at a slower rate in the ceftriaxone 4-g group versus the 2-g group. The between-group difference was 0.310±0.243 (95% CI −0.167, 0.787) units per month (p=0.400). The between-group difference between the ceftriaxone 4-g group and placebo was 0.038±0.0192 (95% CI −0.001, 0.076) units per month (p=0.0550). Final analysis of HHD results showed no treatment effect on the percentage of baseline for upper or lower extremities (table 2).
For the subjective assessments of participant and caregiver QoL, there were no significant effects of treatment on the ALSSQOL and CBI, respectively. Results from the final analysis are summarized in table 2.
Safety and tolerability
Overall, 99% (339/340) of ceftriaxone and 97% (167/173) of placebo participants experienced ≥1 adverse event. The most frequently reported adverse events are summarized in webappendix C). Adverse event rates were significantly higher in the ceftriaxone versus placebo group for gastrointestinal (72% [245/340] vs 56% [97/173]), hepatobiliary (62% [211/340] vs 11% [19/173]) and blood/bone marrow (18% [61/340] vs 9% [15/163]) CTCAE categories. Between-group differences in adverse event rates for other categories were minimal. The majority of hepatobiliary events was cholelithiasis, affecting 53% (181/340) and 3% (5/173), of ceftriaxone-and placebo-treated participants, respectively (p<0.0001).
Fifty-two percent (177/340) of ceftriaxone and 47% (80/173) of placebo participants experienced ≥1 serious adverse event. The most frequent serious adverse events were pulmonary. There was no significant difference in rates between the ceftriaxone and placebo groups 26% (88/340) vs 22% (38/173); dyspnea was the most frequently reported by ceftriaxone 22% (74/340) and placebo participants 20% (34/173). A significantly larger percentage of ceftriaxone versus placebo participants experienced hepatobiliary 12% (41/340) vs 0% (0/173) serious adverse events; significantly fewer ceftriaxone versus placebo participants had infection-related serious adverse events 9% (30/340) vs 18% (32/173). Catheter-related serious adverse event rates were low in both groups, with the exception of infection, which was lower among ceftriaxone versus placebo participants 2% (7/340) vs 8% (14/173) (webappendix C).
In stages 1 and 2, 22 participants taking 4 g ceftriaxone but not ursodiol experienced 143 adverse events during a follow-up of period of 71 person-months, corresponding to an adverse event rate of 2.01/month. In stage 3, 296 participants taking 4 g ceftriaxone and prophylactic ursodiol experienced 3316 adverse events during a follow-up period of 3858 person-months, corresponding to an adverse event rate of 0.86/month. Several CTCAE categories, including gastrointestinal and hepatobiliary events, showed significant reductions in frequency between stages 1 and 2 and stage 3 after the addition of ursodiol (webappendix C).
Pharmacokinetics
A repeated measures analysis of variance, controlling for mean ceftriaxone dose over the previous 5 days, showed no effect of riluzole use on plasma levels of ceftriaxone. The least squares geometric mean ceftriaxone level among all stage-3 participants taking riluzole was 29.7 (95% CI 23.8, 37.1) μg/mL and was 28.8 (95% CI 19.9, 41.7) μg/mL among those not taking riluzole (p=0.8911).
Post-hoc analyses
Post hoc analysis of ALSFRS-R, VC and HHD endpoints among completers showed no significant differences between the treatment groups (ALSFRS-R, p=0.4622; VC, p=0.3925; HHD upper extremities percentage of baseline, p=0.5025; HHD lower extremities percentage of baseline, p=0.5259). Post hoc CAFS result showed no significant difference between the ceftriaxone and placebo groups (p=0.5972).
Although ursodiol was associated with reductions in plasma levels of ceftriaxone of ~26% overall, levels varied among participants. Among those participants taking ursodiol, mean plasma levels were similar in stages 1 and 3.
When participants discontinued treatment, study investigators asked them what treatment group they thought they were assigned to. Among those taking ceftriaxone and willing to take a guess (66% [193/292]), 92% (177/193) guessed correctly. Among participants taking placebo and willing to guess (58% [90/154]), 70% (63/90) guessed correctly. This group, however, may not be representative of all study participants.
Discussion
Like many compounds that preceded it, the beta-lactam antibiotic ceftriaxone showed promising neuroprotective effects against glutaminergic excitotoxicity in preclinical studies but failed to show clinical efficacy in this study of individuals with ALS. Further, the promising dose-dependent effects of ceftriaxone versus placebo on the ALSFRS-R and HHD observed at the end of stage 2 was not present in stage 3. During Stages 1 and 2, functional decline slower in participants taking 4 g ceftriaxone versus those taking placebo; there were no other significant between-group differences.
In general, phase 2 trials in ALS are not expected to provide definitive efficacy data, as the primary goal is to monitor safety, enrolment is typically <200, and trial duration is usually <12 months. In multi-stage, adaptive study designs, however, data from earlier stages are used to design subsequent stages, and data collected during early stages are used in the analyses at the final stage.32 The ceftriaxone study was conducted in this way. PK and safety data from stages 1 and 2, respectively, were used to determine whether to proceed with stage 3 and safety and interim efficacy data from stage 2 were used to determine the ceftriaxone dosage for stage 3.
In stage 1, ceftriaxone demonstrated linear PK and there was no effect of riluzole on PK parameters.25 This persisted through stage 3. As expected, ceftriaxone versus placebo treatment was associated with higher rates of cholelithiasis, which was well managed with optional use of ursodiol.25 In stage 3, when all ceftriaxone-treated participants also received blinded add-on ursodiol, rates of hepatobiliary events and other adverse events showed further reductions.
The CVC was well tolerated; thirty serious adverse events were definitely or probably related to the catheter. Although the rate of line infections was higher in placebo versus ceftriaxone participants (0.366 vs 0.174/1000 catheter-days), the overall rate was lower than the CDC– reported rate for catheter care in the home setting (2.9–11.3/1000 catheter-days).33 To our knowledge, this is the first report of non-healthcare specialists providing intravenous treatment in the home setting for this duration of time. The results reflect the level of excellence in training and monitoring procedures, the high calibre of the nursing staff, and the commitment of family members and caregivers.
Despite the failure of ceftriaxone to show clinical efficacy, much was learned from the study’s innovative, adaptive design. The protocol contained clear, detailed stopping and futility rules and provided for pre-specified interim analyses. Some individuals participated in the study up to 4.5 years, demonstrating that CVC use in the home setting is not only possible, but also safe. Finally, validated but simple outcome measures were selected for ease of use.
Some issues in the trial could not be fully resolved. The blinding was imperfect probably due to the adverse event profile of ceftriaxone. Our attempt to prevent gallstones in stage 3 by giving everyone assigned to ceftriaxone ursodiol or those assigned to placebo (MVI), ursodiol placebo did decrease the rate of symptomatic gallstones, but was not entirely effective in preventing possible unblinding. It also may have reduced the effective dose of ceftriaxone somewhat by decreasing blood levels of ceftriaxone. However, in stage 1, CSF ceftriaxone concentrations at 2 grams daily were greater than the pre-specified levels determined from preclinical studies. The 26% reduction in plasma levels at 4 grams daily is clearly above this threshold as well. The potential reduction in symptomatic gallstones with the use of ursodiol is likely to have more than compensated for this reduction in plasma levels. Thus, the co-administration of ursodiol was likely beneficial at the 4-g per day dose.
Another issue affecting trial performance was the unanticipated long duration of the study. This may have increased the proportion of time that participants were off study drug, reducing its intent-to-treat effect. All participants were asked to stay in the study until the last participant completed 12 months of treatment. As ALS progresses, it is harder for participants to continue on study medication, even if follow up visits are conducted by phone or in the home. We were sensitive to this possibility and monitored this during the trial. An alternative design would have been a shorter trial with more participants; however, this approach would be harder to generalise to the long-term treatment that ALS will require. Another alternative would have been a high-dose trial with cholecystectomy to remove the gallstone risk with an observation control group treated without placebo.
As there were no pharmacodynamic markers of ceftriaxone effect on CNS EAAT2 mRNA or protein at the time of this study, it was not possible to determine if the drug activated its target. The lack of a pharmacodynamic marker for target engagement by ceftriaxone renders us unable to conclude that upregulation of glutamate transport is a failed therapeutic target. Further study of drugs that impact glutamate transport and uptake may still be warranted. To this end, EAAT2 positron emission tomography ligands are in development to potentially assess the efficacy of drugs alerting astroglia EAAT2 (Dr. Rothstein, unpublished observations). An effective pharmacodynamic reporting tool will be critical for future ALS clinical trials for the astroglia transporter, and more generally for any new ALS therapeutic.
Research in context
Systematic review
We searched PubMed for articles reporting the results of phase 3 clinical trials for “amyotrophic lateral sclerosis” published in English during the past 10 years. This time period starts approximately when the preclinical data supporting ceftriaxone were known. Ten articles were identified, including the report of the stage 1 and stage 2 findings from the ceftriaxone study.25 Two papers evaluated clinical trial methodology, including the feasibility of sniff nasal inspiratory pressure as an outcome measure34 and the reliability of database controls rather placebo patients in some ALS trials.35 The remaining seven articles evaluated the efficacy of investigational compounds versus placebo in patients with ALS, including dexpramipexole,36 lithium,37 ursodeoxycholic acid,38 insulin-like growth factor type I [IGF-1],39 minocycline,40 TCH346,41 and xaliproden.42 Most reports cited neuroprotective effects in preclinical studies (minocylcine, lithium)37,40 and/or some survival benefit in a pilot (lithium)37 or phase 2 study (dexpramipexole) of ALS patients.36 For IGF-1, preclinical and clinical results have been inconsistent. In all seven studies, the agents were generally well tolerated, but none showed clinical efficacy versus placebo on pre-specified primary or secondary endpoints.
Interpretation
It has been 20 years since the phase 3 studies of riluzole reported a modest survival benefit for patients with ALS. In the interim, many phase 3 trials reporting negative efficacy results were published. Ideally, phase 3 trials are expected to yield positive results, as they are performed only after evidence of potential efficacy is gathered from preclinical research and phase 2 studies. For neurological disorders including ALS however, there is a high rate of failure in phase 3, most likely because of the challenges in phase 2 where there often is a lack of pharmacodynamic and efficacy surrogate biomarker. To date, phase 2 studies are deemed positive often based on non-significant trends in the same endpoints ultimately employed in Phase 3. The predictive value of such studies is clearly less than what would be expected if more sensitive pharmacodynamic markers were available and showed clear effects of treatment. The current study reinforces the need to develop such markers and use them to design more predictive Phase 2 studies.
Despite the lack of benefit of ceftriaxone demonstrated in this study, valuable lessons were learned. Study rationale was based on a novel approach led by the NIH to screen all marketed drugs for preclinical signals of efficacy and to select one that worked in the most assays for human testing. Our negative results do not invalidate this approach. This was the first ALS study to incorporate a Phase I-to-III adaptive design. There is growing use such types of adaptive designs in neurology clinical trials, and knowledge and experience in their value and challenges are important.
The ability to successfully complete our study demonstrates that drugs available for other uses can be successfully studied in ALS. As was the case in previous studies of minocycline, creatine, and lithium, our study successfully enrolled, though the recruitment phase was likely longer than for drugs only available within the context of a study. Such studies are particularly important as patients risk being exposed to harm without benefit by taking off-label medications. Our study further demonstrated that daily home IV infusion can be performed safely in the ALS population, which may inform design of future studies. The knowledge gained regarding management of adverse events of ceftriaxone may also translate to future research. The information from this trial will be helpful for clinicians and scientists in ALS, other neurological disorders and in non-neurological fields.
Supplementary Material
Acknowledgments
The authors thank the study participants and their families. We gratefully acknowledge Carl Leventhal, MD, for serving as the medical monitor and all members of the Data Safety Monitoring Board. We also acknowledge Mary Lou Watson and her colleagues Meghan Hall, Cory Dauphin, Timothy Patrick, Sean Gray, Joseph Thomas, Nazanin Khajouee Nejad, and Dafna Rebibo from the Outcomes Centre at the State University of New York for providing the study monitoring and outcomes training. We thank the Neurological Clinical Research Institute project managers: Julie Berkley, MPH, Kathryn Delaney, MBA, Lauren Mazzapica, RN, Jane McKinley, RN (Canadian ALS Research Network [CALS] manager), Natasha Soodoo, MPH, and Amy Swartz, PT; data managers: Mabel Chan, Jing Deng, Haining Li, and Eric Tustison; systems managers: Igor Katsovskiy, Roger Selsov, Ervin Sinani, Jason Walker, and Karen Wallace; grants manager Bryan Sweet; administrative staff: Nicole Day and Francine Murphy; John Vetrano and Cheryl Reilly-Tremblay from the central pharmacy at Massachusetts General Hospital and Scott Walker from the central pharmacy at Sunnybrook Medical Centre.
This trial was funded by the NINDS (5 U01-NS-049640). Editorial support for the development of this paper was funded by an anonymous philanthropic gift to the Massachusetts General Hospital ALS Program. Linda Goldstein from Excel Scientific Solutions wrote the first draft of the manuscript based on input from the authors, and Joanne King from Excel Scientific Solutions copyedited and styled the manuscript per journal requirements.
Funding National Institute of Neurological Disorders and Stroke 5 U01-NS-049640
Contributors
M E Cudkowicz designed and led the study and analysed and interpreted the study data; S Titus managed the study operations and assisted with organising the data; M Kearney provided oversight for management of study operations and regulatory compliance; H Yu managed the study data and assisted with organizing the data for statistical analysis; A Sherman assisted in protocol development, designed the data capture system for all phases of the study and developed logic check algorithms and procedures for data cleaning; D Schoenfeld was involved in the design of the study and analysis and of the data; D Hayden assisted in the statistical methods and data analysis; A Shui assisted with the statistical analysis; B Brooks referred suitable participants to the local site sub-investigator, served on the advisory committee and reviewed interim and final datasets; R Conwit participated in the design, implementation, and funding of the study and was a member of the executive, steering, ancillary studies, and publications committees; D Felsenstein was a member of the steering committee; as the Infectious Disease Consult to the study, she reviewed issues related to the use and complications from intravenous ceftriaxone and the use of the central venous catheters; DJ Greenblatt participated in the study design for stage 1 and analysed plasma ceftriaxone levels for the PK analysis; M Keroack served as the GI consultant for adverse events related to GI problems from ceftriaxone use; JT Kissel participated in the study design; R Miller participated in trial design, safety review, and analysis of results; J Rosenfeld was a member of the steering committee and collected and interpreted study data; J Rothstein provided preclinical data on the use of ceftriaxone for the study and reviewed the study data; E Simpson served on the steering committee and helped determine outcome measures, study modifications and review serious adverse events; N Tolkoff-Rubin monitored renal treatments for all of the participants as well as their risk for kidney stones and infection as the nephrology consultant; L Zinman served on the steering committee and assisted with management of the Canadian sites; J M Shefner was co-principal investigator for this study, participated in study design, served on the executive and steering committees, and assisted in the interpretation of results.
The principal investigator and steering committee designed the study, held the data collected by investigators, contributed to data interpretation and provided assistance in manuscript preparation. All authors had access to the study data and were members of the writing group. The writing group reviewed and interpreted the study findings, agreed on the content of the paper, contributed to preparation of the manuscript, reviewed drafts and approved the final version for submission. The corresponding author had the final responsibility for the decision to submit the paper for publication.
Ceftriaxone Study Investigators
Canada, Alberta: S Kalra, L Korngut, H Omar-Crawford, R Sekhon, C White; Nova Scotia: T Benstead, I Grant, S Reidy; Ontario: C McIntosh, J McKinley, C Shoesmith, L Zinman; Quebec: S Botez, J-P Bouchard, M D’Amour, A Genge
United States, Arizona: N Hank, T Levine, D Saperstein; California: R Alvarez, C Banda, R Garcia, M Graves, H Gruendler, J Katz, F Lin, C Lomen-Hoerth, V Martin, R Miller, D Moses, T Mozaffar, L Nist, B Oskarsson, J Rosenfeld, E Tsimerinov, P Tully, C Villierme, K Voelz, M Wiedau-Pazos; Colorado: Y Rollins; Connecticut: K Felice; District of Columbia: E Bayat, A N Kelly; Florida: K B Boylan, P DeSaro, D Koggan, A Verma; Georgia: J Bordeau, J Glass, M Polak, B Quarles, M H Rivner; Illinois: P Casey, R Sufit; Indiana: C Bodkin, S Guingrich, J Kincaid, A Micheels, R Pascuzzi, R Snook; Kansas: R J Barohn, A Dick, L Herbelin, M M Dimachkie, A L McVey, M Walsh, Y Wang; Kentucky: J Hutchison, E Kasarskis, J King, T Tandy, S Thomas, K Vanderpool; Maryland: J Rothstein; Massachusetts: P Andres, M Bellanich, M E Cudkowicz, W David, A Goldenberg, DJ Greenblatt, L Krivickas, L Loci, M Majkut, O O’Connor, M Parkinson, D Pulley, J Russell, L Sullivan; Michigan: H Foley, D Gelinas, D S Newman; Minnesota: S Bundlie, T Leviton, S Patel, C Rohde, S Swanson, E Tiryaki; Missouri: A Fann, G Hayat, A Pestronk; Nebraska: G L Pattee, B Weber; New Hampshire: M Keroack; New Jersey: B Belsh, J Belsh, M A Mertz; New York: A M DeNero, T Imperato, D J Lange, N Kassebaum, D MacGowan, H Mitsumoto, S N Scelsa, M Shahbazi, JM Shefner, L Simionescu, M L Watson, J Wymer; North Carolina: E Bravver, B R Brooks, J Caress, M S Cartwright, T Johnston-Crews, C Lary, J Shuh; Ohio: A Bartlett, N Berry, J T Kissel, R Kuenzler, E P Pioro, A Quick; Oregon: B Ash, K Goslin, Pennsylvania: A Deboo, A Giampole, T Heiman-Patterson, D Lacomis, L McCluskey, M Powell, S Rana, L Rojas, D Rowlands, Z Simmons, H E Stephens; South Carolina: M DeCandio, D Stickler; Tennessee: D L Davis, P D Donofrio; Texas: S Halton, S Hand, M Hastings, D Heitzman, L Lay, J McCloskey, E Simpson; Utah: M Bromberg; Virginia: A Joshi, L H Phillips
Data Safety Monitoring Board
United States: L E Chambless (chair) (Department of Biostatistics, Collaborative Studies Coordinating Centre, University of North Carolina at Chapel Hill, Chapel Hill, NC); L I Bruijn (Department of Research, The ALS Association, Washington, DC); G J del Zoppo (Department of Hematology, University of Washington at Harborview Medical Centre, Seattle, WA); B Tilley (Department of Biostatistics, Bioinformatics and Epidemiology, University of Texas, Houston, TX); R Bedlack (Department of Neurology, Duke University Medical Centre, Durham, NC)
Conflicts of Interest
M E Cudkowicz served as consultant to GlaxoSmithKline, Shire, and Teva and was a member of a DSMB for synapse and trophos outside the submitted work. D Schoenfeld received grants from the National Institutes of Health (NIH), Isis Pharmaceuticals, Prize for Life, Immunosuppression (Research Sundry Fund), ALS TDI, DPS (SUNY Subaward), ALS Therapy Alliance -Selection Design, and Mexilitine (Research Sundry Fund) during the conduct of the study; he received personal fees from Neuronova outside the submitted work. B Brooks received grants from the ALS Association, ALS Therapy Alliance, Carolinas ALS Research Fund, Heineman Medical Research Fund, Johns Hopkins University School of Medicine, Muscular Dystrophy Association (MDA), and the National Institute of Neurological Disorders and Stroke (NINDS) during the study; he received educational grants from the Carolinas Healthcare Foundation and Knopp Biosciences for the North American ALS Research Group (ALSRG) Conferences at the 2012 and 2013 ALS-MND symposia, respectively; served as scientific advisor/consultant for Asubio, Biogen Idec, Bristol-Myers-Squibb, Cytokinetic, Countervail Corp, Knopp Biosciences, Nova Biomedical, and NeuroDyn; served on the board of directors for ALSRG, a co-chairman/member of the American Academy of Neurology (AAN) Task Force on ALS Quality Measures, Development of AAN Registry, and a Member of the Centres for Disease Control and Prevention’s Agency for Toxic Substances and Disease Registry/National ALS Registry Oversight Committee; invited speaker at Knopp Biosciences’ Symposium, “Where are we with biomarkers in ALS?” in 2013, and Biogen Idec/Knopp Biosciences’ Symposium, “ALS Treatment and Trial Design–Past, Present, and Future” in 2012 outside the submitted work. D Felsenstein received grants from the NINDS during the study. D J Greenblatt received grants from the NIH during the study. J Rosenfeld received research support from the Hill Rom Corporation, Cytokinetics, Biogen and the Central California Faculty Medical Group and served as a scientific advisor for the Hill Rom Corporation during the study and development of the manuscript; none, overlapped with the scope of the ceftriaxone project. J D Rothstein received grants from the ALS Association, MDA, NIH, and P2ALS and served as a consultant for Ruxton/Psyadon Pharmaceutical during the study; he received consultancy fees from Cytokenetics and has unlicensed patents on the use of drugs (not ceftriaxone) to modulate EAAT2 as a therapy outside the submitted work. J Shefner received grants from the NIH during the study; grants and personal fees from Biogen Idec, Cytokinetics, GlaxoSmithKline, and Isis Pharmaceuticals; grants from Sanofi Aventis; and personal fees from Synaerion, Trophos, and UpToDate outside the submitted work. R Conwit, D Hayden, M Kearney, M Keroack, J T Kissel, R Miller, A Sherman, A Shui, E Simpson, S Titus, N Tolkoff-Rubin, H Yu, and L Zinman have nothing to disclose. Dr Robin Conwit as a NINDS member of the study steering committee took part in discussions on study interpretation and is a co-author of the report.
All co-authors had full access to all the data in the study. The decision to submit the paper for publication was proposed by the study PI (Dr. Cudkowicz) and approved by the entire Steering Committee and all co-authors.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Rowland LP, Shneider NA. Amyotrophic lateral sclerosis. N Engl J Med. 2001;344:1688–700. doi: 10.1056/NEJM200105313442207. [DOI] [PubMed] [Google Scholar]
- 2.Sejvar JJ, Holman RC, Bresee JS, Kochanek KD, Schonberger LB. Amyotrophic lateral sclerosis mortality in the United States, 1979–2001. Neuroepidemiology. 2005;25:144–52. doi: 10.1159/000086679. [DOI] [PubMed] [Google Scholar]
- 3.Chio A, Logroscino G, Hardiman O, et al. Prognostic factors in ALS: A critical review. Amyotroph Lateral Scler. 2009;10:310–23. doi: 10.3109/17482960802566824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.del Aguila MA, Longstreth WT, Jr, McGuire V, Koepsell TD, van Belle G. Prognosis in amyotrophic lateral sclerosis: a population-based study. Neurology. 2003;60:813–9. doi: 10.1212/01.wnl.0000049472.47709.3b. [DOI] [PubMed] [Google Scholar]
- 5.Bensimon G, Lacomblez L, Meininger V. A controlled trial of riluzole in amyotrophic lateral sclerosis. ALS/Riluzole Study Group. N Engl J Med. 1994;330:585–91. doi: 10.1056/NEJM199403033300901. [DOI] [PubMed] [Google Scholar]
- 6.Lacomblez L, Bensimon G, Leigh PN, Guillet P, Meininger V. Dose-ranging study of riluzole in amyotrophic lateral sclerosis. Amyotrophic Lateral Sclerosis/Riluzole Study Group II. Lancet. 1996;347:1425–31. doi: 10.1016/s0140-6736(96)91680-3. [DOI] [PubMed] [Google Scholar]
- 7.Choi DW. Glutamate neurotoxicity and diseases of the nervous system. Neuron. 1988;1:623–34. doi: 10.1016/0896-6273(88)90162-6. [DOI] [PubMed] [Google Scholar]
- 8.Cleveland DW, Rothstein JD. From Charcot to Lou Gehrig: deciphering selective motor neuron death in ALS. Nat Rev Neurosci. 2001;2:806–19. doi: 10.1038/35097565. [DOI] [PubMed] [Google Scholar]
- 9.Rothstein JD. Excitotoxic mechanisms in the pathogenesis of amyotrophic lateral sclerosis. Adv Neurol. 1995;68:7–20. discussion 1–7. [PubMed] [Google Scholar]
- 10.Rothstein JD, Van Kammen M, Levey AI, Martin LJ, Kuncl RW. Selective loss of glial glutamate transporter GLT-1 in amyotrophic lateral sclerosis. Ann Neurol. 1995;38:73–84. doi: 10.1002/ana.410380114. [DOI] [PubMed] [Google Scholar]
- 11.Trotti D, Rolfs A, Danbolt NC, Brown RH, Jr, Hediger MA. SOD1 mutants linked to amyotrophic lateral sclerosis selectively inactivate a glial glutamate transporter. Nat Neurosci. 1999;2:848. doi: 10.1038/12227. [DOI] [PubMed] [Google Scholar]
- 12.Martin D, Thompson MA, Nadler JV. The neuroprotective agent riluzole inhibits release of glutamate and aspartate from slices of hippocampal area CA1. Eur J Pharmacol. 1993;250:473–6. doi: 10.1016/0014-2999(93)90037-i. [DOI] [PubMed] [Google Scholar]
- 13.Mizoule J, Meldrum B, Mazadier M, et al. 2-Amino-6-trifluoromethoxy benzothiazole, a possible antagonist of excitatory amino acid neurotransmission–I. Anticonvulsant properties. Neuropharmacology. 1985;24:767–73. doi: 10.1016/0028-3908(85)90011-5. [DOI] [PubMed] [Google Scholar]
- 14.Guo H, Lai L, Butchbach ME, et al. Increased expression of the glial glutamate transporter EAAT2 modulates excitotoxicity and delays the onset but not the outcome of ALS in mice. Hum Mol Genet. 2003;12:2519–32. doi: 10.1093/hmg/ddg267. [DOI] [PubMed] [Google Scholar]
- 15.Melzer N, Meuth SG, Torres-Salazar D, et al. A beta-lactam antibiotic dampens excitotoxic inflammatory CNS damage in a mouse model of multiple sclerosis. PLoS One. 2008;3:e3149. doi: 10.1371/journal.pone.0003149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rothstein JD, Patel S, Regan MR, et al. Beta-lactam antibiotics offer neuroprotection by increasing glutamate transporter expression. Nature. 2005;433:73–7. doi: 10.1038/nature03180. [DOI] [PubMed] [Google Scholar]
- 17.Lipski J, Wan CK, Bai JZ, Pi R, Li D, Donnelly D. Neuroprotective potential of ceftriaxone in in vitro models of stroke. Neuroscience. 2007;146:617–29. doi: 10.1016/j.neuroscience.2007.02.003. [DOI] [PubMed] [Google Scholar]
- 18.Thone-Reineke C, Neumann C, Namsolleck P, et al. The beta-lactam antibiotic, ceftriaxone, dramatically improves survival, increases glutamate uptake and induces neurotrophins in stroke. J Hypertens. 2008;26:2426–35. doi: 10.1097/HJH.0b013e328313e403. [DOI] [PubMed] [Google Scholar]
- 19.Tikka T, Usenius T, Tenhunen M, Keinanen R, Koistinaho J. Tetracycline derivatives and ceftriaxone, a cephalosporin antibiotic, protect neurons against apoptosis induced by ionizing radiation. J Neurochem. 2001;78:1409–14. doi: 10.1046/j.1471-4159.2001.00543.x. [DOI] [PubMed] [Google Scholar]
- 20.Carreer R, Deby-Dupont G, Deby C, Jadoul L, Mathy M. Oxidant-scavenging activities of beta-lactam agents. Eur J Clin Microbiol Infect Dis. 1998;17:43–6. doi: 10.1007/BF01584363. [DOI] [PubMed] [Google Scholar]
- 21.Lutsar I, McCracken GH, Jr, Friedland IR. Antibiotic pharmacodynamics in cerebrospinal fluid. Clin Infect Dis. 1998;27:1117–27. doi: 10.1086/515003. quiz 28–9. [DOI] [PubMed] [Google Scholar]
- 22.Nau R, Prange HW, Muth P, et al. Passage of cefotaxime and ceftriaxone into cerebrospinal fluid of patients with uninflamed meninges. Antimicrob Agents Chemother. 1993;37:1518–24. doi: 10.1128/aac.37.7.1518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Oksi J, Marjamaki M, Nikoskelainen J, Viljanen MK. Borrelia burgdorferi detected by culture and PCR in clinical relapse of disseminated Lyme borreliosis. Ann Med. 1999;31:225–32. doi: 10.3109/07853899909115982. [DOI] [PubMed] [Google Scholar]
- 24.Yogev R, Shulman ST, Chadwick EG, Davis AT, Glogowski W. Once daily ceftriaxone for central nervous system infections and other serious pediatric infections. Pediatr Infect Dis. 1986;5:298–303. doi: 10.1097/00006454-198605000-00005. [DOI] [PubMed] [Google Scholar]
- 25.Berry JD, Shefner JM, Conwit R, et al. Design and initial results of a multi-phase randomized trial of ceftriaxone in amyotrophic lateral sclerosis. PLoS One. 2013;8:e61177. doi: 10.1371/journal.pone.0061177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Brooks BR, Miller RG, Swash M, Munsat TL. El Escorial revisited: revised criteria for the diagnosis of amyotrophic lateral sclerosis. Amyotrophic lateral sclerosis and other motor neuron disorders : official publication of the World Federation of Neurology, Research Group on Motor Neuron Diseases. 2000;1:293–9. doi: 10.1080/146608200300079536. [DOI] [PubMed] [Google Scholar]
- 27.Cedarbaum JM, Stambler N, Malta E, et al. The ALSFRS-R: a revised ALS functional rating scale that incorporates assessments of respiratory function. BDNF ALS Study Group (Phase III) J Neurol Sci. 1999;169:13–21. doi: 10.1016/s0022-510x(99)00210-5. [DOI] [PubMed] [Google Scholar]
- 28.Simmons Z, Felgoise SH, Bremer BA, et al. The ALSSQOL: balancing physical and nonphysical factors in assessing quality of life in ALS. Neurology. 2006;67:1659–64. doi: 10.1212/01.wnl.0000242887.79115.19. [DOI] [PubMed] [Google Scholar]
- 29.Novak M, Guest C. Application of a multidimensional caregiver burden inventory. Gerontologist. 1989;29:798–803. doi: 10.1093/geront/29.6.798. [DOI] [PubMed] [Google Scholar]
- 30.Vonesh EF, Greene T, Schluchter MD. Shared parameter models for the joint analysis of longitudinal data and event times. Stat Med. 2006;25:143–63. doi: 10.1002/sim.2249. [DOI] [PubMed] [Google Scholar]
- 31.Berry JD, Miller R, Moore DH, et al. The Combined Assessment of Function and Survival (CAFS): a new endpoint for ALS clinical trials. Amyotroph Lateral Scler Frontotemporal Degener. 2013;14:162–8. doi: 10.3109/21678421.2012.762930. [DOI] [PubMed] [Google Scholar]
- 32.Cudkowicz ME, Katz J, Moore DH, et al. Toward more efficient clinical trials for amyotrophic lateral sclerosis. Amyotroph Lateral Scler. 2010;11:259–65. doi: 10.3109/17482960903358865. [DOI] [PubMed] [Google Scholar]
- 33.O’Grady NP, Alexander M, Dellinger EP, et al. Guidelines for the prevention of intravascular catheter-related infections. Am J Infect Control. 2002;30:476–89. doi: 10.1067/mic.2002.129427. [DOI] [PubMed] [Google Scholar]
- 34.Bauer M, Czell D, Hartmann S, Goldman B, Muller D, Weber M. Limitations of sniff nasal pressure as an outcome measurement in amyotrophic lateral sclerosis patients in a clinical trial. Respiration; international review of thoracic diseases. 2012;84:306–11. doi: 10.1159/000339415. [DOI] [PubMed] [Google Scholar]
- 35.Czaplinski A, Haverkamp LJ, Yen AA, Simpson EP, Lai EC, Appel SH. The value of database controls in pilot or futility studies in ALS. Neurology. 2006;67:1827–32. doi: 10.1212/01.wnl.0000244415.48221.81. [DOI] [PubMed] [Google Scholar]
- 36.Cudkowicz ME, van den Berg LH, Shefner JM, et al. Dexpramipexole versus placebo for patients with amyotrophic lateral sclerosis (EMPOWER): a randomised, double-blind, phase 3 trial. The Lancet Neurology. 2013;12:1059–67. doi: 10.1016/S1474-4422(13)70221-7. [DOI] [PubMed] [Google Scholar]
- 37.Morrison KE, Dhariwal S, Hornabrook R, et al. Lithium in patients with amyotrophic lateral sclerosis (LiCALS): a phase 3 multicentre, randomised, double-blind, placebo-controlled trial. The Lancet Neurology. 2013;12:339–45. doi: 10.1016/S1474-4422(13)70037-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Min JH, Hong YH, Sung JJ, Kim SM, Lee JB, Lee KW. Oral solubilized ursodeoxycholic acid therapy in amyotrophic lateral sclerosis: a randomized cross-over trial. Journal of Korean medical science. 2012;27:200–6. doi: 10.3346/jkms.2012.27.2.200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sorenson EJ, Windbank AJ, Mandrekar JN, et al. Subcutaneous IGF-1 is not beneficial in 2-year ALS trial. Neurology. 2008;71:1770–5. doi: 10.1212/01.wnl.0000335970.78664.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gordon PH, Moore DH, Miller RG, et al. Efficacy of minocycline in patients with amyotrophic lateral sclerosis: a phase III randomised trial. The Lancet Neurology. 2007;6:1045–53. doi: 10.1016/S1474-4422(07)70270-3. [DOI] [PubMed] [Google Scholar]
- 41.Miller R, Bradley W, Cudkowicz M, et al. Phase II/III randomized trial of TCH346 in patients with ALS. Neurology. 2007;69:776–84. doi: 10.1212/01.wnl.0000269676.07319.09. [DOI] [PubMed] [Google Scholar]
- 42.Meininger V, Bensimon G, Bradley WR, et al. Efficacy and safety of xaliproden in amyotrophic lateral sclerosis: results of two phase III trials. Amyotrophic lateral sclerosis and other motor neuron disorders : official publication of the World Federation of Neurology, Research Group on Motor Neuron Diseases. 2004;5:107–17. doi: 10.1080/14660820410019602. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.