

Preface
Induced pluripotency is a powerful tool to derive patient-specific stem cells. In addition, it provides a unique assay to study the interplay between transcription factors and chromatin structure. Here, we review the latest insights into chromatin dynamics inherent to induced pluripotency. Moreover, we compare and contrast these events with other physiological and pathological processes involving changes in chromatin and cell state, including germ cell maturation and tumorigenesis. We propose that an integrated view of these seemingly diverse processes could provide mechanistic insights into cell fate transitions in general and might lead to novel approaches in regenerative medicine and cancer treatment.
Introduction
Reprogramming of somatic cells to pluripotency can be achieved by different approaches, including somatic cell nuclear transfer (SCNT) into oocytes, fusion between somatic and pluripotent cells and ectopic expression of defined transcription factors (TFs)1,2. SCNT demonstrated that epigenetic rather than genetic changes are the basis for most differentiation processes during normal development. Cell fusion experiments documented that the pluripotent state is dominant over the somatic state in the context of hybrids. Together, these observations led to the seminal discovery that a small set of TFs, such as Oct4, Sox2, Klf4 and c-Myc (collectively called OKSM), are sufficient to convert differentiated cells into induced pluripotent stem cells (iPSCs)3. Importantly, induced pluripotency provides a biochemically and genetically tractable system to dissect the mechanisms underlying this remarkable cell fate change.
Recent progress in genome-wide technologies and the analysis of small cell numbers has allowed researchers to capture transcriptional and epigenetic snapshots of rare cell populations undergoing cell fate transitions in different biological contexts. These analyses yielded important insights into the type and sequence of molecular changes inherent to transcription factor-induced pluripotency, germ cell reprogramming and cellular transformation. A common theme emerging from these studies is that nascent iPSCs, developing germ cells and premalignant cells utilize different as well as overlapping mechanisms to alter cell identity. The aim of this review is to define those transcriptional, chromatin and epigenetic changes that endow specialized cells with pluripotency as well as the molecular barriers that resist cell fate change.
Mechanisms of Induced Pluripotency
Acquisition of induced pluripotency is a slow (∼2 weeks) and inefficient (0.1-3%) process1,3, indicating that TFs need to overcome a series of epigenetic barriers that have been gradually imposed on the genome during differentiation to stabilize cell identity and to prevent aberrant cell fate changes. Earlier work has shown that cell populations expressing OKSM pass through a sequence of distinct molecular and cellular events (Figure 1). Fibroblasts initially downregulate markers associated with the somatic state and subsequently activate genes associated with pluripotency, suggesting an ordered process4,5. As soon as nascent iPSCs activate endogenous core pluripotency genes including Oct4, Sox2 and Nanog, they acquire a self-sustaining pluripotent state and no longer require exogenous factor expression. The latter events also coincide with the activation of the silenced X chromosome in female somatic cells, the upregulation of telomerase and the acquisition of immortality, which are hallmarks of cultured pluripotent cells5,6. In the following sections, we will review current knowledge of how overexpressed TFs engage with chromatin, collaborate with epigenetic regulators and integrate extracellular signals in order to reprogram cellular identity (Figure 2).
Figure 1. Dynamics of key epigenetic and transcriptional changes during direct reprogramming.

Colored bars represent different cellular and transcriptional events (top panel) or epigenetic modifications (bottom panel) that change in dynamic patterns during iPSC formation. Examples of candidate enzymes that have been associated with these epigenetic marks in the context of direct reprogramming are given on the right. OKSM: Oct4, Klf4, Sox2, cMyc; 5mC: 5′-methyl-Cytosine; 5hmC: 5′-hydroxy-methyl-Cytosine; MET: Mesenchymal-to-epithelial-transition.
Figure 2. Interplay between reprogramming factors and molecules influencing chromatin state.

Shown are different transcription factors that have been shown to trigger induced pluripotency, with the classical combination (Oct4, Sox2, Klf4, c-Myc) highlighted. Below each category are examples of molecules that have been shown to facilitate (green) or inhibit (red) reprogramming. BMPs (Bone Morphogenetic Factors) and Wnts have stage-dependent enhancing or suppressive roles during iPSC formation (black, see text for details). RBPs: RNA Binding Proteins.
Transcription Factors Drive Cell Fate Change
The most commonly used combination of reprogramming TFs comprises OKSM, and we will therefore primarily focus on this set of factors3. Previous results suggested that c-Myc and OKS play distinct roles during the acquisition and maintenance of pluripotency7. Briefly, OKS are the minimally required set of factors for iPSC generation from many cell types under classic reprogramming conditions (i.e. in the presence of serum and the cytokine LIF). OKS cooperatively suppress lineage-specific genes and activate embryonic stem cell (ESC)-related genes, resulting in the establishment of a self-sustaining pluripotency network7. In contrast, ectopic c-Myc expression significantly enhances and accelerates reprogramming but is dispensable for iPSC formation8,9. c-Myc expression functions early during reprogramming, presumably by stimulating cell proliferation and inducing a metabolic switch from an oxidative to a glycolytic state typical of pluripotent cells10,11. More recent evidence suggests that c-Myc may also contribute to reprogramming by inducing pause release and promoter reloading of RNA polymerase, leading to transcriptional amplification of target genes12,13.
It is worth noting that each of the original four reprogramming factors has been functionally replaced by either related TFs, upstream epigenetic modifiers, miRNAs or small compounds1. Moreover, iPSCs have been derived with molecules that do not contain any of the original TFs14,15, indicating a remarkable flexibility and redundancy among reprogramming factors (Figure 2). For example, a recent report demonstrated that the core pluripotency factors Oct4 and Sox2 can be substituted for by lineage specifiers such as Gata3 and Geminin16. These molecules have been associated with mesendodermal and ectodermal differentiation, respectively, and they mutually repress the respective other lineage program, suggesting that the suppression of these major differentiation pathways is sufficient to trigger iPSC induction. This idea is consistent with the observation that many classic pluripotency factors, including Oct4, Sox2 and Nanog, participate in early lineage decisions and hence may also be considered lineage specifiers17. A prediction that follows from these results is that TFs that stabilize the somatic state should be inhibitory to OKSM-mediated reprogramming. Indeed, depletion of the B cell-specifying TF Pax5 or ectopic expression of its antagonist CEB/P-alpha enables the reprogramming of terminally differentiated B cells18. Conversely, forced expression of lineage-specific TFs, in combination with OKSM, significantly impairs iPSC formation by sustaining a somatic gene expression program and preventing activation of pluripotency genes19. Together, these findings demonstrate that reprogramming TFs have to achieve two key tasks, namely the extinction of the somatic program, which is maintained by counteracting TFs, and the induction of a stable pluripotent state typical of ESCs.
Different Types of Chromatin Targets
Developmental progression from pluripotent stem cells via progenitors to terminally differentiated cells is accompanied by a gradual deposition of repressive histone marks, followed by chromatin compaction20-22. A key question is therefore how reprogramming TFs dismantle somatic chromatin and establish an epigenetic state that is compatible with pluripotency. Recent studies assessing OKSM occupancy and histone marks early during the reprogramming of mouse and human fibroblasts into iPSCs have provided first clues to this question10,23,24. Combining these observations, one may categorize OKSM targets into three classes of loci based on (i) chromatin accessibility, (ii) the requirement for additional remodeling, and (iii) the kinetics of transcriptional activation (Figure 3a). Genes with an “open” chromatin state in somatic cells comprise the first group of targets, characterized by increased DNaseI hypersensivity, active H3K4me2/3 marks and the ability to bind OKSM immediately. Downregulated somatic genes and genes linked to a mesenchymal-to-epithelial transition (MET), which specifies early stages of reprogramming25, fall into this group24.
Figure 3. Levels of epigenetic gene regulation during induced pluripotency.

a. Depicted are four categories of genes with associated histone modifications and their transcriptional response during reprogramming. Examples of genes within each category are shown to the right. b. Gain and loss of DNA methylation occurs late in reprogramming, while the acquisition of hydroxymethylation at pluripotency genes takes place at early-to-mid stages of iPSC formation. c. Oct4 (O), Klf4 (K) and Sox2 (S) function as “pioneer factors” that bind to high-density nucleosome regions, enabling chromatin remodeling and the recruitment of other factors including c-Myc (M). d. Changes in long-range chromatin interactions around the Nanog locus during iPSC formation. iPSC formation re-establishes a 3D chromatin network typical of ESCs, and this process depends on Mediator and Cohesin. Colored loops represent somatic-specific (orange), intermediate-specific (green) and pluripotency-specific (blue) Nanog-interacting loci.
A second class of early-bound OKSM targets includes distal regulatory elements, which appear to require additional chromatin remodeling for transcriptional activation24. A subgroup of these elements carries the H3K4me1 mark and exhibits nucleosome depletion as well as DNAse I hypersensitivity, which are chromatin features characteristic of “permissive enhancers”. Permissive enhancers typically bind TFs prior to their associated promoter regions and before transcriptional activation26. The MyoD locus exemplifies this group of enhancers; ectopically expressed Oct4 initially binds to the MyoD enhancer, triggering crosstalk with its promoter and subsequent acquisition of a poised chromatin state26 Another subset of distal regulatory elements comprises DNase I-resistant loci unable to bind c-Myc alone24. Early pluripotency genes such as Sall4 belong to this group. Interestingly, occupancy of these targets by OKS facilitates binding of c-Myc. This observation thus identifies OKS as “pioneer factors” for c-Myc, which defines the ability of TFs to bind closed somatic chromatin and enable chromatin remodeling as well as recruitment of other TFs and cofactors24.
Broad heterochromatic regions enriched for the repressive H3K9me3 mark constitute a third set of OKSM targets. Genes within this category comprise core pluripotency genes, such as Nanog and Sox224. These regions are refractory to immediate OKSM binding and seem to require the most extensive chromatin remodeling for transcriptional activation. Of interest, refractory domains are reminiscent of broad chromatin regions containing extensive H3K9 dimethylation termed LOCKs (Large Organized Chromatin K9 modification)27. LOCKs are generally underrepresented in pluripotent cells and associated with repression of lineage-specific genes in differentiated cells27.
Bivalent genes constitute a separate group of chromatin targets that are marked by both active H3K4 methylation and repressive H3K27 methylation in iPSCs or ESCs28. Genes within this category are transcriptionally silent in ESCs and iPSCs but poised for rapid activation upon lineage commitment. Although the actual number and relevance of bivalent promoters remain controversial29, induced pluripotency gradually restores these domains to an ESC-like pattern10,23. The transcription factor Utf1 has recently been implicated in the regulation of bivalency by a dual mechanisms that involves deposition of the repressive H3K27me3 mark and degradation of residual transcripts30. Notably, Utf1 expression can substitute for some of the original reprogramming factors, providing indirect evidence that the establishment of bivalent promoters may be an important step during the acquisition of pluripotency14. Together, these results illustrate how the initial chromatin state of somatic and pluripotency-related genes determines if and when they become occupied and transcriptionally regulated by OKSM in the course of reprogramming. They further suggest that certain histone marks (e.g., H3K9 methylation) act as potent barriers that resist acquisition of pluripotency. We will therefore next focus on the enzymes that deposit or remove these marks and their impact on cellular reprogramming.
Role of Histone-modifying Enzymes
Histone modifications and chromatin structure are regulated by histone code “writers” such as histone methyltransferases (HMTs) and acetyltransferases (HATs) and “erasers” such as histone demethylases (HDMs) and deacetylases (HDACs) (Figure 2). These enzymes function as coactivators or corepressors of OKSM at different stages of reprogramming and can profoundly influence iPSC derivation. For example, recruitment of PRC2, which deposits the repressive H3K27me3 mark, and inhibition of Dot1L, which establishes the active H3K79me2/me3 marks, have been associated with the downregulation of somatic genes early in reprogramming31,32. Accordingly, loss of PRC2 abrogates whereas loss of Dot1L enhances iPSC formation. Activation of the H3K36 HDMs Jhdm1a/1b33 and suppression of the H3K27 HDM Jmjd3 promote intermediate-to-late stages of iPSC generation by suppressing the Ink4/Arf locus34, which is essential for the acquisition of immortality. An additional early role for Jhdm1b in epithelial gene activation was recently reported35. In contrast, H3K9 HMTs maintain the abovementioned “refractory” heterochromatic state of somatic cells and thus act as major barriers of reprogramming. Consistent with this notion, knockdown of G9a (H3K9me2 HMT) or Suv39h1/h2 and Setd1 (H3K9me3 HMTs), or overexpression of H3K9 HDMs, increases TF accessibility and results in more efficient iPSC generation from somatic cells24,36,37. Altogether, these results demonstrate that histone code writers and erasers are essential components of iPSC formation by either maintaining the somatic state or assisting in the TF-induced establishment of pluripotency.
Reprogramming TFs have been reported to directly interact with histone-modifying enzymes, providing a mechanistic explanation for how they may alter chromatin and cell state during induced pluripotency. Examples include the H3K27 HDM Utx14 and the H3K4me3 “effector” Wdr538, which bind to Oct4 protein and co-occupy many genomic targets in ESCs, thus keeping them in a transcriptionally active state. Depletion of either molecule blunts iPSC formation due to a failure to activate pluripotency genes14,38. Oct4 may play a very specific role in recruiting epigenetic regulators to target genes compared with Sox2, Klf4 and c-Myc, as it cannot be replaced by related family members during induced pluripotency8. In agreement, deletion of a linker domain on Oct4, which is absent on other POU domain family members, associates with chromatin remodelers implicated in reprogramming and its deletion abrogates iPSC formation39. Intriguingly, some reprogramming-associated cofactors function in a chromatin-independent manner. For example, the H3K27 HDM Jmjd3 blocks reprogramming not only by activating the Ink4a/Arf locus but also by targeting the methyl-lysine effector protein Phf20 for ubiquitination; Phf20 is required to activate Oct4 transcription in collaboration with Wdr534. Overall, these and other7 examples document the physical association of reprogramming TFs with a variety of histone modifiers and exemplify the diverse mechanisms by which they assist in reinstating pluripotency in somatic cells.
DNA Methylation: Safeguarding Cell Identity
DNA methylation is considered to be the most stable epigenetic modification, which confers permanent gene silencing during development and in the adult. Changes in histone modifications typically precede the removal or deposition of DNA methylation marks during differentiation20. Similarly, DNA methylation changes almost exclusively occur at the end of the reprogramming process and after chromatin changes have taken place10, indicating a hierarchy of events that recapitulates normal development (Figure 1). DNA methylation is established by the de novo methyltransferases Dnmt3a and Dnmt3b and preserved by the maintenance methyltransferase Dnmt140. Although DNMT3a knockdown promotes iPSC formation in human cells31, the mouse Dnmt3a/b enzymes are dispensable for cellular reprogramming41. This surprising finding suggests that the silencing of lineage-specific genes is mainly achieved through alternative mechanisms such as deposition of repressive H3K27 methylation, which is consistent with PRC2's essential role in iPSC formation32.
In contrast to the dispensability of de novo methylation for iPSC formation, DNA demethylation of pluripotency genes appears to be critical for faithful reprogramming. Demethylation can occur by either active or passive mechanisms40, both of which have been implicated in iPSC generation. Downregulation of Dnmt1 in reprogramming intermediates facilitates their transition towards authentic iPSCs, consistent with a supportive role of passive, replication-dependent demethylation in iPSC formation42. Enzymes associated with active DNA demethylation have been more directly linked with iPSC formation (Figure 1). Tet proteins catalyze the hydroxylation of 5-methylcytosine (5mC) to 5-hydroxymethylcytosine (5hmC), which serves as a substrate for TDG-mediated base excision repair into unmodified cytosines40. Shortly after overexpression of OKSM, Tet2 induces hydroxymethylation of key pluripotency genes such as Nanog and Esrrb, priming them for subsequent demethylation and transcriptional activation (Figures 1 and 3b)43. Interestingly, proteomic and genomic analyses revealed that Tet1/2 directly interact with Nanog and co-occupy many pluripotency targets in ESCs, implicating Nanog in the targeting of Tets44. In agreement, simultaneous overexpression of Tet1/2 and Nanog significantly enhances, while depletion of either factor abrogates iPSC formation43-45. Moreover, Tet1 overexpression can compensate for exogenous Oct4 expression during cellular reprogramming, providing genetic evidence that Tet1 contributes to the activation of somatically silenced pluripotency genes46. A role for activation-induced deaminase (AID) in DNA demethylation during TF-induced reprogramming has also been reported, although the underlying mechanisms are incompletely understood40.
Inefficient DNA demethylation or remethylation has further been suggested to be the main reason for the “epigenetic memory” observed in many iPSC lines. This term describes cell type of origin-dependent transcriptional and epigenetic patterns that can influence the differentiation potential of iPSCs1. Of note, genomic regions that fail to undergo DNA demethylation towards an ESC-like state in human iPSCs were shown to be decorated by broad H3K9me3 domains in somatic cells47, and some of these areas overlap with the abovementioned “refractory domains” that are inaccessible to OKSM early in reprogramming24. These findings therefore suggest that promiscuous OKSM binding or lack of OKSM binding to targets could explain some of the observed differences between ESCs and iPSCs. It should be interesting to assess whether manipulation of H3K9 HDMs or HMTs is sufficient to erase epigenetic memory in iPSCs.
3D Chromatin Architecture in Reprogramming
Accumulating evidence suggests that local and three-dimensional (3D) chromatin architecture provide additional levels of gene regulation in pluripotent stem cells48. However, their roles in cellular reprogramming are incompletely understood. Local chromatin architecture defines the position and density of nucleosomes as well as the presence of histone variants (Figures 1 and 3c)49. Histone variants usually modify the ability of nucleosomes to undergo remodeling and to accommodate active or repressive histone modifications. The histone variant macroH2A has previously been associated with resistance to efficient chromatin remodeling49. In agreement, the presence of macroH2A potently inhibits TF-induced reprogramming of somatic cells to pluripotency by maintaining pluripotency loci in a repressed state50-52.
Local chromatin architecture is regulated by diverse remodeling complexes, which also impact iPSC formation (Figure 1). Components of the SWI/SNF complex, such as Brg1, BAF155 and Brm, are directly recruited by Oct4 to targets in order to relax chromatin structure and facilitate binding of other TFs53. This finding thus corroborates Oct4's role as a pioneer factor by influencing local chromatin structure at silenced targets. Similarly, the CHD remodeling factor Chd1 has been proposed to actively open chromatin during factor-induced reprogramming, and its knockdown impairs iPSC formation54. In contrast, members of the repressive NURD complex including HDAC1 and Mdb3, which are critical for heterochromatin compaction, inhibit reprogramming and their knockdown strongly increases the efficiency of iPSC generation55. Interestingly, Mbd3 associates with loci enriched for Tet1 and 5hmC in ESCs and its expression is essential for global levels of 5hmC56. The latter observation may explain the discrepancy between the observed early deposition of 5hmC at pluripotency loci43 but its delayed conversion into unmodified cytosines during iPSC generation10; Mbd3 may be recruited to these hydroxymethylated pluripotency promoters and block immediate demethylation until unidentified coactivators relieve Mbd3-mediated gene repression.
In addition to local chromatin structure, 3D chromatin architecture has been implicated in pluripotency, differentiation and reprogramming (Figure 3d)48. Differentiation of ESCs is accompanied by a repositioning of pluripotency genes from the nuclear center to the nuclear periphery57 and a disruption of promoter-enhancer looping at key pluripotency loci such as Oct458,59 and Nanog54,55. This raises the question of whether and how 3D chromatin structure is restored in iPSCs. A recent study identified complex pluripotency-specific long-range interactions of the Nanog locus, which become rearranged during differentiation and are largely restored during reprogramming60. The establishment and maintenance of this network is dependent on Mediator and Cohesin complexes, which are known to orchestrate long-range chromatin interactions59. Interestingly, subunits of these complexes were found to directly interact with the reprogramming factors60,61 and their knockdown inhibited iPSC formation60.
Extending these findings, long-range chromatin interactions around the Oct4 promoter were recently implicated in the reprogramming of murine and human cells61,62. Importantly, these interactions took place specifically in those rare cells that were poised to form iPSCs, and they preceded transcriptional activation, suggesting a causal effect of 3D chromatin structure on transcription. These results, together with previous studies documenting a role for Oct4 and Klf1 (highly similar to Klf4) in mediating long-range chromatin interactions63,64, support the idea that reprogramming factors do not merely activate or silence genes but also function as chromatin organizers, which rearrange chromatin architecture from a somatic to a pluripotent state. This interpretation is further consistent with the recent discovery of “super-enhancers”, which are broad distal regulatory elements characterized by cooperative and excessive binding of Mediator components and cell-type specific transcription factors, such as Esrrb and Klf4, in ESCs65. Given the documented role of super-enhancers in controlling the expression of master regulatory genes in different cell types, it is likely that the resetting of somatic-specific to pluripotency-specific super-enhancers constitutes another roadblock for iPSCs formation. The dynamics of this switch during induced pluripotency and the potential role of super-enhancers in 3D chromatin architecture certainly warrants further investigation.
Cell Signaling and Chromatin
External cues are critical to direct cells expressing OKSM towards a stable pluripotent state and to prevent acquisition of alternative cell fates7. Extracellular signals can either support or inhibit iPSC formation (Figure 2). For example, dual chemical inhibition of the Gsk3b and Erk1/2 pathways (“2i” condition) enhances the transition of partially reprogrammed cells into iPSCs66. Similarly, activation of the Jak/Stat3 pathway by the cytokine LIF is limiting for iPSC formation67. By contrast, the TFG-b pathway negatively affects iPSC generation and its suppression by chemical inhibitors significantly increases the acquisition of pluripotency68,69.
Recent data offer new mechanistic insights into how external signals communicate with chromatin structure in pluripotency and reprogramming. For instance, the downstream effector of LIF signaling, Stat3, requires the chromatin remodeler subunit Brg1 in order to keep targets accessible and prevent their repression by the PRC2 complex70. Moreover, culture of ESCs in 2i endows cells with a so-called “naïve” or “ground” state that is characterized by altered H3K27me3 distribution and a decreased number of bivalent promoters as well as global DNA hypomethylation71,72. Mechanistically, growth of ESCs in 2i induces activation of the TF Prdm14, which directly represses Dnmt3a/b and inhibits differentiation-inducing Fgfr signaling71. It is important to mention here that signaling molecules can also have opposite effects on different stages of reprogramming. BMPs promote an early MET in a miRNA-dependent manner25, while they block the late conversion of partially reprogrammed cells into iPSCs by targeting the repressive H3K9 HMTs Suv39h1 and Setdb136. Conversley, Wnt/Tcf3 signaling is inhibitory early but stimulatory late in reprogramming73.
Nutrients and cofactors present in the extracellular environment represent a final class of molecules that influence the epigenome and cellular reprogramming. A point in case is ascorbic acid (vitamin C), which has been shown to strongly enhance the efficiency and kinetics of reprogramming74 and to increase the quality of mouse iPSCs by preventing aberrant hypermethylation75. Ascorbic acid presumably functions both as an antioxidant and as a cofactor for specific epigenetic modifiers such as the H3K36 HDMs Jmjd1a/1b33. Furthermore, ascorbic acid was suggested to be a cofactor for H3K9 HDMs and Tet enzymes according to recent studies, which reported a global decrease of the repressive H3K9me2/3 marks36 and genome-wide DNA hypomethylation76, respectively, in nascent iPSCs exposed to this compound. Together, these observations provide compelling new evidence for the tight communication between reprogramming-associated signaling molecules and TFs in order to rewire epigenetic regulatory circuits.
Sequence of Molecular Events
An unresolved question is whether the rare cells that successfully give rise to iPSCs undergo a defined sequence of transcriptional and epigenetic events. Different approaches have been used to resolve this issue. A number of reports utilized surface marker combinations to prospectively identify those rare cell populations that are destined to form iPSCs10,77-79. One of these studies identified two major waves of gene expression changes that coincided with the early extinction of somatic genes and with the late activation of core pluripotency genes, respectively10. The lack of major transcriptional changes during the intermediate phase suggested that cells undergo gradual epigenetic alterations in order to prime the genome for transcriptional activation of pluripotency genes. In agreement with this notion is the observation that histone marks associated with pluripotency enhancers are established at early and intermediate stages of reprogramming, whereas DNA methylation changes occur late, coinciding with transcriptional activation of associated genes10. Integrating observations from other studies, this intermediate period is also characterized by the establishment of pluripotency-specific long-range chromatin interactions60,61 and Tet-mediated conversion of 5mC into 5hmC at pluripotency promoters43, further supporting the interpretation that cells undergo a series of chromatin changes in preparation for stable pluripotency.
An independent study analyzed transcriptional changes of 48 genes in single cells undergoing reprogramming15 and concluded that the initial response to reprogramming factor expression is quite heterogeneous and consistent with a stochastic process. However, later events, leading to the activation of pluripotency genes, occur in a hierarchical fashion. This analysis led to the inference that activation of the endogenous Sox2 locus is a critical upstream event in cells that undergo successful reprogramming. A parallel study examined clonal populations of cells expressing OKSM and defined “maturation” and “stabilization” phases of reprogramming that were distinguished by differential expression of pluripotency genes80. Unexpectedly, the authors discovered that the transition to the stabilization phase is dependent on a different set of factors (e.g., GDNF signaling and meiosis genes) from those that control maintenance of pluripotency.
Several groups reported transient upregulation of developmental regulators, such as epidermal, extraembryonic and epiblast-associated genes, at intermediate stages of reprogramming10,16,42,73,77. While the molecular mechanisms underlying this observation remain elusive, it is tempting to speculate that reprogramming intermediates transiently pass through a state with increased developmental plasticity, which could represent stages of normal development7. Alternatively, these genes might be activated as a consequence of aberrant TF binding11 or unspecific effects incurred by small compounds81. Regardless, recent studies showed that depletion of some of these transiently expressed genes impairs reprogramming into iPSCs, suggesting functional relevance78,81.
In conclusion, while the overall gene expression trends are similar among different studies, variability related to the exact sequence of molecular events and the relative contribution of stochastic and deterministic events remain to be resolved. Another fundamental question that needs to be addressed is whether cells expressing alternative reprogramming factors pass through the same sequence of events, described here, and encounter the same barriers as cells expressing OKSM. This question is particularly relevant with respect to reprogramming approaches that involve small compounds81 or TFs that have not been directly implicated in pluripotency16.
Lessons from Other Reprogramming Systems
In this section, we will compare induced pluripotency with other processes involving epigenetic reprogramming such as SCNT, cell fusion and germline specification with the goal of identifying mechanistic similarities and differences (Table 1).
Table 1. Comparison of different types of cellular reprogramming.
Key molecular and cellular events during different reprogramming strategies with examples of implicated molecules. SCNT, somatic cell nuclear transfer; PGC, primordial germ cell; EGC, embryonic germ cell; NPC, neural progenitor cell; ESC, embryonic stem cell; TF, transcription factor. Note that some references refer to review articles covering the discussed molecule.
| Types of Reprogramming |
Extinction of cell-of- origin expression program |
Activation of pluripotency genes |
X chromosome reactivation |
Histone variants (chaperones) |
Histone modifiers |
DNA methylation factors |
Requirement for endogenous TFs |
Relevant signaling pathways |
|---|---|---|---|---|---|---|---|---|
| Direct Reprogramming (OKSM factors) | 1-2 days (e.g., Thy1 and Snai1 during fibroblast reprogramming)(5) | 8-12 days (Oct4-GFP reporter)(4,5) | ∼12 days (X-GFP reporter) (5) | macroH2A (50-52) | PRC1/2 (32) Utx (14) Dot1L (31) HDACs (1) (see also Figure 1) |
Dnmt1 (42) Tet1/2 (43,44) AID (40) |
Nanog (45) Stat3 (67) |
Wnt (1,73) LIF (67) BMP (25,36) ERK/GSK inhibition (“2i”)(66) |
| SCNT | 22-24 hrs (2-cell embryo; global silencing of somatic genes/activation of zygotic genes)(82) | <36h hrs (4-cell embryo; Oct4-GFP reporter)(82) | ∼3 days (morula embryo; X-GFP reporter) | H1/B4 (86) H3.3 (89) H2A.X (90) H2A.Z (90) macroH2A (91) |
HDACs (91) | Dnmt1 Tet3 (83) |
(Oct4 not required)(84) | N/A |
| Cell Fusion | 24-48 hrs (e.g., Nestin, Glur6 in NPC-ESC hybrids)(92) | 24-48 hrs (Oct4-GFP reporter)(92) | ∼9 days (94) | N/A | G9a (96) Jhdm2a (96) PRC1/2 (97) |
Tet1/2 (93) AID (2) |
Nanog (45) Oct4 (2) (Sox2 not required)(2) |
LIF Wnt |
| PGC maturation in vivo | ∼2 days, btw. E6.25-E8.5 (e.g., Hox genes)(100) | 1-2 days, btw. E7.25-E8.5 (e.g., Sox2, Dppa3, Nanog)(100) | ∼3 days, btw. E11.5-E13.5 (X-GFP reporter)(101) | H1 (99) H2A.Z (99) (Nap-1 Hira)(99) |
Glp (100) Utx (14) Prmt5 (103) |
Dnmt3a/b/L (107) Uhrf1 (107) Tet1/2 (105) AID (1,40) |
Blimp1 (100) Prdm14 (100) Tcfap2c (100) Nanog (100) Oct4 (100) Sox2 (100) |
BMP (100) |
| PGC-to-EGC conversion in vitro | 1-2 days (e.g., Blimp1)(103) | 3-4 days (e.g., Klf4, Stat3)(103) | 3-4 days (X-GFP reporter)(101) | N/A | N/A | N/A | N/A | LIF/Stat3+FGF+ SCF or ERK/GSK inhibition (“2i”)(71) |
Somatic Cell Nuclear Transfer
Remarkably, the somatic gene expression program is downregulated within 22 hours of SCNT in mice (Dieter Egli, pers. comm.)82. This observation is reminiscent of iPSC formation and suggests that extinction of somatically expressed genes is rapid and efficient in both approaches. Somatic Oct4 activation after SCNT occurs with a similar kinetics (24-36 hours)82 and was suggested to require Tet383, whereas it takes a minimum of 8 days to detect Oct4 expression during induced pluripotency82 and this process appears to involve Tet243. This marked difference in pluripotency gene activation poses the key question of whether SCNT and induced pluripotency depend on the same TFs for reprogramming; both Oct4 and Sox2 proteins are in fact detectable in oocytes. Schöler and colleagues recently addressed this question by genetically depleting maternal Oct4 protein from mouse oocytes prior to SCNT84. Surprisingly, loss of Oct4 did not abrogate the oocyte's ability to reprogram somatic nuclei, indicating compensation by other TFs. An alternative explanation is that SCNT may depend on an entirely different suite of reprogramming factors. The identification of the oocyte-enriched Glis1 protein85 capable of enhancing iPSC formation in the context of OKS expression indeed supports this hypothesis.
A key molecular event that distinguishes induced pluripotency from SCNT is the rapid histone exchange between somatic nucleus and oocyte, as was demonstrated in Xenopus SCNT experiments. Specifically, the somatic linker histone H1 is replaced within hours of SCNT for the oocyte-specific counterpart B4, and this process is essential for pluripotency gene activation in reconstructed oocytes86. This particular exchange of histone types might contribute to reprogramming by depleting somatic chromatin from epigenetic repressors known to interact with histone H1, such as Dnmt1/3b and H3K9 methyltransferases87,88. Concomitant with the replacement of “repressive” histone variants, incorporation of “active” histone variants including H3.3 and H2A.X into the somatic chromatin facilitates efficient chromatin remodeling of embryonic genes89,90. Despite these effective mechanisms, some epigenetic marks including those of the silenced X chromosome (Xi) in female somatic nuclei appear to be more recalcitrant to remodeling by the oocyte compared with pluripotency genes, indicating differential susceptibility of some genomic loci to the oocyte's reprogramming machinery91. The relative resistance of X reactivation to efficient remodeling during Xenopus SCNT has been functionally linked to the repressive histone variant macroH2A, since its knockdown resulted in a more efficient reactivation of the Xi91. Thus, the eviction of macroH2A represents a rate-limiting step for successful reprogramming in the context of both SCNT and iPSC induction. Given the prominent role of “active” and “repressive” histone variants during SCNT, it should be informative to systematically test their function and that of associated chaperones49 during iPSC generation.
Cell-Cell Fusion
Downregulation of somatic genes in ESC-somatic hybrids also occurs within the first 1-2 days of fusion92. When examining the same Oct4-GFP reporter that was used for SCNT and iPSC formation, Oct4 reactivation in fusion hybrids took place with a similar kinetics as SCNT (24-48 hours) 92 and this process was reported to involve Tet293. This finding suggests that ESCs, like oocytes, contain additional reprogramming factors that are limiting during iPSC formation. Corroborating this point, Nanog overexpression promotes cell fusion reprogramming and drives maturation of iPSCs, whereas its absence blunts both types of reprogramming45. Surprisingly, Oct4 protein is required, whereas Sox2 protein is dispensable for fusion-mediated reprogramming, identifying a notable difference to induced pluripotency94. It is further important to mention that the reprogramming of hybrids is not complete upon activation of somatic pluripotency genes 2 days post fusion. Activation of the silenced X chromosome in female hybrids takes several days, which is reminiscent of the delayed X reactivation observed during Xenopus SCNT95. Consistently, transcriptome-wide analysis of hybrid formation documented that some silenced ESC-associated genes are activated more rapidly than others96. These results are in line with the sequential activation of pluripotency-associated genes during iPSC derivation4,5 and likely reflect different chromatin accessibility of associated genomic loci to ESC-derived reprogramming factors.
At the chromatin level, inhibition of the H3K9 HMT G9a or overexpression of the H3K9 HDM Jhdm2a increases cell fusion-directed reprogramming, which is in accordance with their supportive roles in iPSC generation97. Conversely, depletion of PRC1 or PRC2 subunits decreases cell fusion-mediated reprogramming and induced pluripotency, underscoring the general importance of H3K27me3-mediated gene repression for the acquisition of pluripotency98. Collectively, these findings support the interpretation that iPSC formation and cell fusion-directed reprogramming face similar epigenetic barriers and are stimulated by the same TFs, consistent with the fact that iPSC factors were initially identified in ESCs3. Based on the accelerated kinetics of pluripotency activation in hybrids compared with nascent iPSCs, it should be feasible to devise genetic screens in order to identify other Nanog-like molecules that are limiting for efficient TF-induced reprogramming.
Primordial Germ Cell Reprogramming
PGC maturation represents yet another type of reprogramming, which occurs naturally and encompasses major epigenetic remodeling events that prepare the developing germ line for totipotency99,100. Remarkably, PGCs that have completed reprogramming exhibit two active X chromosomes in females101, ESC-like transcriptional patterns and bivalent domains102. This includes expression of potent reprogramming factors, such as Oct4, Sox2, Nanog and Prdm14100,103. Each of these factors is essential for PGC formation in vivo, although their precise roles in PGC reprogramming remain elusive. Importantly, PGCs are unipotent in vivo (i.e., they can only produce oocyte or sperm) but they have the unique potential to give rise to pluripotent stem cells, coined “embryonic germ cells” (EGCs), upon explantation in culture1. Expression of these ESC-associated TFs might thus endow PGCs with the latent ability to acquire pluripotency upon isolation from the gonads and exposure to appropriate extracellular cues. Because germ cell reversion into EGCs rarely occurs in vivo, except in cases of spontaneous teratocarcinomas (i.e., pluripotent tumors), potent mechanisms must be in place to preserve the PGC state. Blimp1 is a possible candidate molecule owing to its role as a repressor of c-Myc and Klf4 expression in PGCs103. It should be possible to test this hypothesis by assessing whether acute loss of Blimp1 is sufficient to convert PGCs into EGCs in vitro and to cause teratocarcinomas in vivo. Notably, Blimp1's putative role in suppressing the acquisition of a pluripotent state in PGCs might be taken over by Dmrt1 at subsequent stages of male germ line development. Male mice lacking this TF in the germline aberrantly express pluripotency factors and develop testicular teratomas with almost full penetrance104. The notion that Blimp1 and Drmt1 might actively resist acquisition of pluripotency is analogous to the inhibitory effect that somatic TFs have during iPSC formation19.
With respect to chromatin dynamics, global loss of H3K9 methylation is among the most striking changes in developing PGCs99. Notably, downregulation of H3K9 HMTs appears to be critical for efficient reprogramming in the context of PGC specification100, cell fusion97 and iPSC formation24,36,37. This particular chromatin alteration may therefore represent a general requirement for cellular reprogramming in these very different cellular contexts. Similarly, loss of inhibitory H3K27 methylation at pluripotency loci through catalysis by Utx seems to be another required step during both PGC reprogramming and iPSC formation14. Lastly, Tet1 and Tet2 enzymes have been functionally associated with DNA demethylation in in vitro-derived PGCs105, revealing similarities with SCNT83 and cell fusion93. However, it is noteworthy that genetic loss of both Tet1 and Tet2 is not essential for viability and fertility in vivo106, suggesting compensation by other mechanisms. Indeed, passive demethylation was recently suggested to contribute to PGC reprogramming through downregulation of the de novo methyltransferases Dnmt3a/b and the Dnmt1 cofactor Uhfr1107. Combined with the enhancing effects of Tet1/2 overexpression44 and Dnmt1/Dnmt3a depletion31,42 on iPSC formation, these data show that the erasure of somatic DNA methylation patterns is a general roadblock for successful epigenetic reprogramming in different cellular settings. Cells utilize a combination of “passive” and “active” DNA demethylation strategies to overcome this barrier, although their relative contribution varies depending on the reprogramming context.
Induced Pluripotency and Tumorigenesis
Several lines of evidence support the idea that induced pluripotency and transformation are related processes at a cellular level (see Box). Reprogramming, like cancer, is a rare, multi-step process that ultimately leads to the formation of a small population of immortal cells with tumorigenic potential; iPSCs, like ESCs, have the ability to give rise to teratomas (benign tumors containing derivatives of the three germ layers) upon transplantation under the skin of mice1. Another similarity between reprogramming and some types of cancer is the observation that somatic stem and progenitor cells are more susceptible to both iPSC formation108,109 and tumorigenesis110,111 compared to mature cells. This observation may indicate that the epigenetic state of the starting cell provides a permissive environment for both oncogenic and reprogramming factors. Further, TF-mediated reprogramming induces a metabolic switch from an oxidative to a glycolytic state typical of most cancer cells112. Lastly, teratocarcinomas represent a special type of cancer that originates from transformed PGCs and contains pluripotent cells, documenting a rare example of spontaneous reprogramming of committed (germ) cells into pluripotent malignant cells1. An important distinction, however, between these examples of cancer and induced pluripotency is the fact that iPSCs are normal diploid cells that support development when re-introduced into embryos, whereas most cancer cells are aneuploid and characterized by aberrant differentiation patterns. It should thus be informative to study those chromatin and epigenetic events that transiently endow iPSCs with immortality and tumorigenic properties in addition to increased differentiation potential, as this might lead to new strategies to reverse malignancy.
Molecular data support the cellular commonalities between reprogramming and malignancy. First, each of the four classic reprogramming factors has been shown to be oncogenic in mice, and some of these genes are amplified or mutated in human cancer113, suggesting that they might destabilize cell state in tumors akin to their role in reprogramming. Chromatin regulators that cooperate with OKSM during reprogramming, such as Jhdm1b and macroH2A, have also been associated with tumorigenesis. Jhdm1b expression has been causally linked with myeloid transformation of hematopoietic progenitors through silencing of the ink4b gene114 and with pancreatic adenocarcinoma formation through silencing of developmental genes in collaboration with PRC2115. In contrast to the cancer- and reprogramming-promoting role of Jhdm1b, expression of macroH2A provides a barrier for both iPSC formation and the malignant progression of melanoma cells. MacroH2A's promoting effect on melanoma invasion is in part exerted through upregulation of the cell cycle regulator CDK8, which differs from the pluripotency genes targeted by this histone variant during induced pluripotency116. These and several other examples113 hence document that premalignant cells and nascent iPSCs target some of the same chromatin regulators to manipulate cell identity, although their targets may vary.
Both cellular reprogramming and cancer are also characterized by similar global changes in chromatin structure and DNA methylation. Cancer cells, like ESCs, are devoid of LOCKs compared to normal differentiated cells27. Given the observation that H3K9 methylation is a major barrier for iPSC formation24,36,37, this finding suggests that many cancers have acquired a developmentally more primitive epigenetic state that might be required for the maintenance of malignancy. Another hallmark of most cancer genomes are altered methylation patterns, which can manifest as aberrant hypermethylation or hypomethylation. It is interesting to mention in this regard that reduced methylation levels, induced by hypomorphic expression of Dnmt1, causes T cell lymphomas in mice117 and promotes iPSC formation in vitro42. Likewise, mutations in Dnmt3A have been observed in AML113, and knock-down of this enzyme facilitates human reprogramming into iPSCs31. These similarities between reprogramming and tumorigenesis are thus further consistent with the view that cancer cells need to override some of the same somatic barriers as iPSCs to alter cellular states.
Of note, the correlation between reprogramming and cancer is not absolute. In fact, some epigenetic regulators and histone modifications have been shown to play opposite roles during reprogramming and malignancy. For example, loss of Tet2 causes myeloid transformation in mice118, consistent with a tumor suppressor function, whereas depletion of Tet2 protein abrogates reprogramming43. Similarly, the H3K79 methyltransferase Dot1L promotes leukemia formation induced by MLL-AF9 translocations119 although it prevents iPSC formation31. Components of the SWI/SNF complex, which facilitates reprogramming, act as potent tumor-suppressors and are frequently mutated in cancer120, indicating opposite functions. Lastly, genome-wide methylation analyses of somatic cells and iPSCs identified a set of reprogramming-specific differentially methylated regions (R-DMRs), which showed significant overlap with DMRs that changed during the transformation of normal into malignant cells121. However, R-DMRs that become hypomethylated and bivalent during iPSC generation are typically hypermethylated in cancer, whereas R-DMRs that become hypermethylated in iPSCs lack bivalent marks and are usually hypomethylated in cancer. Given the importance of bivalently marked genes in multilineage differentiation, their methylation silencing in cancer may be a secure way to keep cells in a self-perpetuating undifferentiated state while this change would be detrimental for pluripotency in iPSCs. In summary, the abovementioned discrepancies between tumorigenesis and reprogramming are likely explained by strongly stage and cell-context dependent roles these enzymes and their modifications play during tumorigenesis and reprogramming.
A testable prediction following the abovementioned observations is that some cancer cells should be susceptible to epigenetic reprogramming into a non-malignant state. Indeed, both SCNT and iPSC experiments demonstrated that malignant cells such as melanoma122 and medulloblastoma123 can be reprogrammed into a pluripotent state that supports differentiation into a number of normal cell types. Thus, some cancers are not irreversibly locked in a tumorigenic state but instead amenable to epigenetic reversion into a phenotypically normal state.
Outlook
Extensive functional genomics and screening approaches over the past few years have provided important insights into the epigenetic mechanisms occurring during normal, induced and pathological examples of cell fate change. While the drivers of cell fate change may be quite different in distinct contexts (e.g., OKSM in reprogramming, unidentified factors during SCNT and oncogenes in tumorigenesis), the resultant chromatin and epigenetic changes leading to altered cell identities are often conserved. This observation probably underlies the fact that different reprogramming approaches face some of the same molecular barriers that have been established during development and terminal differentiation to resist aberrant cell fate changes. We therefore conclude that TF-induced reprogramming provides a powerful tool to interrogate those chromatin and epigenetic mechanisms that stabilize cell fates during development and that become corrupted in cancer. These analyses have implications for both regenerative medicine and cancer biology. A better understanding of the molecular steps leading to pluripotency and the roadblocks resisting cell fate change in different contexts have already enabled researchers to interfere in a rationalized way with defined molecules or pathways to promote or prevent desired cell fate changes50-52,91. An interesting future challenge will be to isolate and stabilize intermediate stages of reprogramming, which might represent natural or artificial cellular states with increased differentiation potential. Dissecting the molecular roadblocks of reprogramming has also relevance for studying and treating cancer. Given that premalignant cells utilize some of the same epigenetic mechanisms as nascent iPSCs to change cell identity, their manipulation may lead to novel strategies that reverse malignancy by altering cellular state rather than cell survival. While the concept of reprogramming cancer cells to pluripotency has already been demonstrated, additional work is needed to develop more specific approaches that reverse malignant cells into a non-pluripotent state by targeting defined transcription factors or epigenetic regulators. Recent work on the conversion of leukemia and lymphoma cells into non-tumorigenic, quiescent macrophages by a single TF124 is a promising step in this direction.
Similarities between induced pluripotency and malignant transformation.
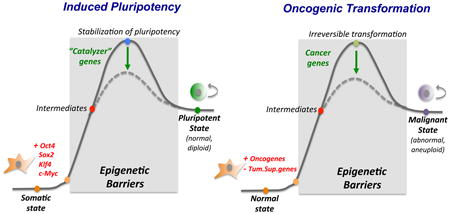
Reprogramming of somatic cells to pluripotency and transformation of normal cells into malignant cells are illustrated as biochemical reactions with defined reactants (somatic cells) and products (iPSCs or cancer cells). Reprogramming is initiated by the overexpression of OKSM, while cellular transformation begins with the activation of oncogenes and/or repression of tumor suppressor genes. Intermediates of each reaction remain largely elusive. Both processes can be enhanced by modulation of additional genes, which can be considered “catalyzers” of the reaction. At a cellular level, both induced pluripotency and tumorigenesis are multi-step processes that require proliferation and result in a change of cell identity or differentiation potential. The “end product” is in both cases an immortal cell with tumorigenic potential. However, cancer cells almost always acquire genetic aberrations and become aneuploid while iPSCs retain a normal diploid genome. At a molecular level, many cancer cells have, like iPSCs, reduced levels of H3K9 methylation and altered DNA methylation patterns compared with differentiated cells. Overall, these findings suggest that nascent iPSCs and premalignant cells face some of the same epigenetic barriers to alter cell identity. This notion may explain why the same epigenetic regulators are involved in both processes, such as Utx, macroH2A, Jhdm1b, Ezh2, Tet2 or Dnmts. This idea is further consistent with the finding that certain somatic progenitor stem and cells are more susceptible to tumorigenesis and reprogramming than differentiated cells, indicating a more permissive epigenetic environment. A prediction that follows is that the malignant state should be reversible upon exposure of cancer cells to reprogramming factors, which is indeed consistent with recent results (see text for details).
Acknowledgments
We thank Alex Bortvin, Dieter Egli, Vincent Pasque, Bernhard Payer, Bing Ren, Xie Wei, Mamta Tahiliani, and members of the laboratory for helpful suggestions, and Bill Lowry, Kathrin Plath and Matthias Stadtfeld for critical reading of the manuscript. We apologize to those colleagues whose work we could not cite due to space contraints. Support to E.A. was by HHMI and to K.H. by HHMI and NIH (R01HD058013).
References
- 1.Stadtfeld M, Hochedlinger K. Induced pluripotency: history, mechanisms, and applications. Genes Dev. 2010;24:2239–2263. doi: 10.1101/gad.1963910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Yamanaka S, Blau HM. Nuclear reprogramming to a pluripotent state by three approaches. Nature. 2010;465:704–712. doi: 10.1038/nature09229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006;126:663–676. doi: 10.1016/j.cell.2006.07.024. [DOI] [PubMed] [Google Scholar]
- 4.Brambrink T, et al. Sequential expression of pluripotency markers during direct reprogramming of mouse somatic cells. Cell stem cell. 2008;2:151–159. doi: 10.1016/j.stem.2008.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Stadtfeld M, Maherali N, Breault D, Hochedlinger K. Defining molecular cornerstones during fibroblast to iPS cell reprogramming in mouse. Cell stem cell. 2008;2:230–240. doi: 10.1016/j.stem.2008.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Maherali N, et al. Directly reprogrammed fibroblasts show global epigenetic remodeling and widespread tissue contribution. Cell stem cell. 2007;1:55–70. doi: 10.1016/j.stem.2007.05.014. [DOI] [PubMed] [Google Scholar]
- 7.Orkin S, Hochedlinger K. Chromatin connections to pluripotency and cellular reprogramming. Cell. 2011;145:835–850. doi: 10.1016/j.cell.2011.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nakagawa M, et al. Generation of induced pluripotent stem cells without Myc from mouse and human fibroblasts. Nat Biotechnol. 2008;26:101–106. doi: 10.1038/nbt1374. [DOI] [PubMed] [Google Scholar]
- 9.Wernig M, Meissner A, Cassady J, Jaenisch R. c-Myc is dispensable for direct reprogramming of mouse fibroblasts. Cell stem cell. 2008;2:10–12. doi: 10.1016/j.stem.2007.12.001. [DOI] [PubMed] [Google Scholar]
- 10.Polo J, et al. A molecular roadmap of reprogramming somatic cells into iPS cells. Cell. 2012;151:1617–1632. doi: 10.1016/j.cell.2012.11.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sridharan R, et al. Role of the murine reprogramming factors in the induction of pluripotency. Cell. 2009;136:364–377. doi: 10.1016/j.cell.2009.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lin CY, et al. Transcriptional amplification in tumor cells with elevated c-Myc. Cell. 2012;151:56–67. doi: 10.1016/j.cell.2012.08.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rahl PB, et al. c-Myc regulates transcriptional pause release. Cell. 2010;141:432–445. doi: 10.1016/j.cell.2010.03.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mansour A, et al. The H3K27 demethylase Utx regulates somatic and germ cell epigenetic reprogramming. Nature. 2012;488:409–413. doi: 10.1038/nature11272. [DOI] [PubMed] [Google Scholar]
- 15.Buganim Y, et al. Single-cell expression analyses during cellular reprogramming reveal an early stochastic and a late hierarchic phase. Cell. 2012;150:1209–1222. doi: 10.1016/j.cell.2012.08.023. This study performed the first single cell transcriptional analysis of nascent iPSCs and identified an early stochastic and a late deterministic phase. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shu J, et al. Induction of Pluripotency in Mouse Somatic Cells with Lineage Specifiers. Cell. 2013;153:963–975. doi: 10.1016/j.cell.2013.05.001. This report demonstrated that iPSCs can be derived through ectopic expression of lineage specifiers rather than “classic” pluripotency factors. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Loh KM, Lim B. A precarious balance: pluripotency factors as lineage specifiers. Cell stem cell. 2011;8:363–369. doi: 10.1016/j.stem.2011.03.013. [DOI] [PubMed] [Google Scholar]
- 18.Hanna J, et al. Direct reprogramming of terminally differentiated mature B lymphocytes to pluripotency. Cell. 2008;133:250–264. doi: 10.1016/j.cell.2008.03.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hikichi T, et al. Transcription factors interfering with dedifferentiation induce cell type-specific transcriptional profiles. Proceedings of the National Academy of Sciences of the United States of America. 2013;110:6412–6417. doi: 10.1073/pnas.1220200110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gifford CA, et al. Transcriptional and Epigenetic Dynamics during Specification of Human Embryonic Stem Cells. Cell. 2013 doi: 10.1016/j.cell.2013.04.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Xie W, et al. Epigenomic analysis of multilineage differentiation of human embryonic stem cells. Cell. 2013;153:1134–1148. doi: 10.1016/j.cell.2013.04.022. These studies (20 and 21) established the first comprehensive chromatin and DNA methylation maps of pluripotent and lineage-restricted cells. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhu J, et al. Genome-wide Chromatin State Transitions Associated with Developmental and Environmental Cues. Cell. 2013;152:642–654. doi: 10.1016/j.cell.2012.12.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Koche R, et al. Reprogramming factor expression initiates widespread targeted chromatin remodeling. Cell stem cell. 2011;8:96–105. doi: 10.1016/j.stem.2010.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Soufi A, Donahue G, Zaret K. Facilitators and impediments of the pluripotency reprogramming factors' initial engagement with the genome. Cell. 2012;151:994–1004. doi: 10.1016/j.cell.2012.09.045. This publication determined how somatic chromatin structure influences access by reprogramming factors and identified important barriers to iPSC formation. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Polo JM, Hochedlinger K. When fibroblasts MET iPSCs. Cell stem cell. 2010;7:5–6. doi: 10.1016/j.stem.2010.05.018. [DOI] [PubMed] [Google Scholar]
- 26.Taberlay P, et al. Polycomb-repressed genes have permissive enhancers that initiate reprogramming. Cell. 2011;147:1283–1294. doi: 10.1016/j.cell.2011.10.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wen B, Wu H, Shinkai Y, Irizarry RA, Feinberg AP. Large histone H3 lysine 9 dimethylated chromatin blocks distinguish differentiated from embryonic stem cells. Nature genetics. 2009;41:246–250. doi: 10.1038/ng.297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bernstein BE, et al. A bivalent chromatin structure marks key developmental genes in embryonic stem cells. Cell. 2006;125:315–326. doi: 10.1016/j.cell.2006.02.041. [DOI] [PubMed] [Google Scholar]
- 29.Marks H, et al. The transcriptional and epigenomic foundations of ground state pluripotency. Cell. 2012;149:590–604. doi: 10.1016/j.cell.2012.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jia J, et al. Regulation of pluripotency and self- renewal of ESCs through epigenetic-threshold modulation and mRNA pruning. Cell. 2012;151:576–589. doi: 10.1016/j.cell.2012.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Onder T, et al. Chromatin-modifying enzymes as modulators of reprogramming. Nature. 2012;483:598–602. doi: 10.1038/nature10953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fragola G, et al. Cell reprogramming requires silencing of a core subset of polycomb targets. PLoS genetics. 2013;9:e1003292. doi: 10.1371/journal.pgen.1003292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang T, et al. The histone demethylases Jhdm1a/1b enhance somatic cell reprogramming in a vitamin-C-dependent manner. Cell stem cell. 2011;9:575–587. doi: 10.1016/j.stem.2011.10.005. [DOI] [PubMed] [Google Scholar]
- 34.Zhao W, et al. Jmjd3 inhibits reprogramming by upregulating expression of INK4a/Arf and targeting PHF20 for ubiquitination. Cell. 2013;152:1037–1050. doi: 10.1016/j.cell.2013.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Liang G, He J, Zhang Y. Kdm2b promotes induced pluripotent stem cell generation by facilitating gene activation early in reprogramming. Nature cell biology. 2012;14:457–466. doi: 10.1038/ncb2483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chen J, et al. H3K9 methylation is a barrier during somatic cell reprogramming into iPSCs. Nature genetics. 2013;45:34–42. doi: 10.1038/ng.2491. [DOI] [PubMed] [Google Scholar]
- 37.Sridharan R, et al. Proteomic and genomic approaches reveal critical functions of H3K9 methylation and heterochromatin protein-1gamma in reprogramming to pluripotency. Nature cell biology. 2013;15:872–882. doi: 10.1038/ncb2768. These reports (36 and 37) identified H3K9 methylation as a major chromatin barrier of cellular reprogramming into iPSCs. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ang YS, et al. Wdr5 mediates self-renewal and reprogramming via the embryonic stem cell core transcriptional network. Cell. 2011;145:183–197. doi: 10.1016/j.cell.2011.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Esch D, et al. A unique Oct4 interface is crucial for reprogramming to pluripotency. Nature cell biology. 2013;15:295–301. doi: 10.1038/ncb2680. [DOI] [PubMed] [Google Scholar]
- 40.Kohli RM, Z Y. Tet, TDG and the Tet, TDG and the Dynamic Cycle of DNA DemethylationDynamic Cycle of DNA Demethylation. Nature. 2013 [Google Scholar]
- 41.Pawlak M, Jaenisch R. De novo DNA methylation by Dnmt3a and Dnmt3b is dispensable for nuclear reprogramming of somatic cells to a pluripotent state. Genes Dev. 2011;25:1035–1040. doi: 10.1101/gad.2039011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mikkelsen T, et al. Dissecting direct reprogramming through integrative genomic analysis. Nature. 2008;454:49–55. doi: 10.1038/nature07056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Doege C, et al. Early-stage epigenetic modification during somatic cell reprogramming by Parp1 and Tet2. Nature. 2012;488:652–655. doi: 10.1038/nature11333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Costa Y, et al. NANOG-dependent function of TET1 and TET2 in establishment of pluripotency. Nature. 2013 doi: 10.1038/nature11925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Silva J, et al. Nanog is the gateway to the pluripotent ground state. Cell. 2009;138:722–737. doi: 10.1016/j.cell.2009.07.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gao Y, et al. Replacement of Oct4 by Tet1 during iPSC Induction Reveals an Important Role of DNA Methylation and Hydroxymethylation in Reprogramming. Cell stem cell. 2013;12:453–469. doi: 10.1016/j.stem.2013.02.005. [DOI] [PubMed] [Google Scholar]
- 47.Lister R, et al. Hotspots of aberrant epigenomic reprogramming in human induced pluripotent stem cells. Nature. 2011;471:68–73. doi: 10.1038/nature09798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Duboule D, dl W. The many faces of developmental enhancers. Nature. 2013 doi: 10.1038/nature12753. [DOI] [PubMed] [Google Scholar]
- 49.Skene PJ, Henikoff S. Histone variants in pluripotency and disease. Development. 2013;140:2513–2524. doi: 10.1242/dev.091439. [DOI] [PubMed] [Google Scholar]
- 50.Gaspar-Maia A, et al. MacroH2A histone variants act as a barrier upon reprogramming towards pluripotency. Nat Commun. 2013;4:1565. doi: 10.1038/ncomms2582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pasque V, et al. Histone variant macroH2A marks embryonic differentiation in vivo and acts as an epigenetic barrier to induced pluripotency. J Cell Sci. 2012;125:6094–6104. doi: 10.1242/jcs.113019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Barrero MJ, et al. Macrohistone Variants Preserve Cell Identity by Preventing the Gain of H3K4me2 during Reprogramming to Pluripotency. Cell reports. 2013;3:1005–1011. doi: 10.1016/j.celrep.2013.02.029. [DOI] [PubMed] [Google Scholar]
- 53.Singhal N, et al. Chromatin-Remodeling Components of the BAF Complex Facilitate Reprogramming. Cell. 2010;141:943–955. doi: 10.1016/j.cell.2010.04.037. [DOI] [PubMed] [Google Scholar]
- 54.Gaspar-Maia A, et al. Chd1 regulates open chromatin and pluripotency of embryonic stem cells. Nature. 2009;460:863–868. doi: 10.1038/nature08212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Luo M, et al. NuRD Blocks Reprogramming of Mouse Somatic Cells into Pluripotent Stem Cells. Stem Cells. 2013;31:1278–1286. doi: 10.1002/stem.1374. [DOI] [PubMed] [Google Scholar]
- 56.Yildirim O, et al. Mbd3/NURD complex regulates expression of 5-hydroxymethylcytosine marked genes in embryonic stem cells. Cell. 2011;147:1498–1510. doi: 10.1016/j.cell.2011.11.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Peric-Hupkes D, et al. Molecular maps of the reorganization of genome-nuclear lamina interactions during differentiation. Molecular cell. 2010;38:603–613. doi: 10.1016/j.molcel.2010.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Phillips-Cremins JE, et al. Architectural Protein Subclasses Shape 3D Organization of Genomes during Lineage Commitment. Cell. 2013 doi: 10.1016/j.cell.2013.04.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kagey MH, et al. Mediator and cohesin connect gene expression and chromatin architecture. Nature. 2010;467:430–435. doi: 10.1038/nature09380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Apostolou E, et al. Genome-wide Chromatin Interactions of the Nanog Locus in Pluripotency, Differentiation, and Reprogramming. Cell stem cell. 2013 doi: 10.1016/j.stem.2013.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wei Z, et al. Klf4 organizes long-range chromosomal interactions with the oct4 locus in reprogramming and pluripotency. Cell stem cell. 2013;13:36–47. doi: 10.1016/j.stem.2013.05.010. [DOI] [PubMed] [Google Scholar]
- 62.Zhang H, et al. Intrachromosomal Looping Is Required for Activation of Endogenous Pluripotency Genes during Reprogramming. Cell stem cell. 2013;13:30–35. doi: 10.1016/j.stem.2013.05.012. The above studies (60-62) identified pluripotency-specific, genome-wide interaction networks around pluripotency loci and implicate Mediator and Cohesin in their regulation. [DOI] [PubMed] [Google Scholar]
- 63.Levasseur D, Wang J, Dorschner M, Stamatoyannopoulos J, Orkin S. Oct4 dependence of chromatin structure within the extended Nanog locus in ES cells. Genes & development. 2008;22:575–580. doi: 10.1101/gad.1606308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Schoenfelder S, et al. Preferential associations between co-regulated genes reveal a transcriptional interactome in erythroid cells. Nature genetics. 2010;42:53–61. doi: 10.1038/ng.496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Whyte WA, et al. Master transcription factors and mediator establish super-enhancers at key cell identity genes. Cell. 2013;153:307–319. doi: 10.1016/j.cell.2013.03.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Silva J, et al. Promotion of reprogramming to ground state pluripotency by signal inhibition. PLoS Biol. 2008;6:e253. doi: 10.1371/journal.pbio.0060253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Yang J, et al. Stat3 activation is limiting for reprogramming to ground state pluripotency. Cell stem cell. 2010;7:319–328. doi: 10.1016/j.stem.2010.06.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ichida J, et al. A small-molecule inhibitor of tgf-Beta signaling replaces sox2 in reprogramming by inducing nanog. Cell stem cell. 2009;5:491–503. doi: 10.1016/j.stem.2009.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Maherali N, Hochedlinger K. Tgfbeta signal inhibition cooperates in the induction of iPSCs and replaces Sox2 and cMyc. Curr Biol. 2009;19:1718–1723. doi: 10.1016/j.cub.2009.08.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ho L, et al. esBAF facilitates pluripotency by conditioning the genome for LIF/STAT3 signalling and by regulating polycomb function. Nature cell biology. 2011;13:903–913. doi: 10.1038/ncb2285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Leitch HG, et al. Naive pluripotency is associated with global DNA hypomethylation. Nat Struct Mol Biol. 2013;20:311–316. doi: 10.1038/nsmb.2510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Yamaji M, et al. PRDM14 ensures naive pluripotency through dual regulation of signaling and epigenetic pathways in mouse embryonic stem cells. Cell stem cell. 2013;12:368–382. doi: 10.1016/j.stem.2012.12.012. The above two references characterize the effect of combined Gsk3/Mapk signal inhibition (“2i”) on the epigenome of ESCs and provide an intriguing link to Prdm14. [DOI] [PubMed] [Google Scholar]
- 73.Ho R, Papp B, Hoffman JA, Merrill BJ, Plath K. Stage-specific regulation of reprogramming to induced pluripotent stem cells by wnt signaling and T cell factor proteins. Cell reports. 2013;3:2113–2126. doi: 10.1016/j.celrep.2013.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Esteban M, et al. Vitamin C enhances the generation of mouse and human induced pluripotent stem cells. Cell stem cell. 2010;6:71–79. doi: 10.1016/j.stem.2009.12.001. [DOI] [PubMed] [Google Scholar]
- 75.Stadtfeld M, et al. Ascorbic acid prevents loss of Dlk1-Dio3 imprinting and facilitates generation of all-iPS cell mice from terminally differentiated B cells. Nature genetics. 2012;44:398–405. S391–392. doi: 10.1038/ng.1110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Blaschke K, et al. Vitamin C induces Tet-dependent DNA demethylation and a blastocyst-like state in ES cells. Nature. 2013 doi: 10.1038/nature12362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.O'Malley J, et al. High-resolution analysis with novel cell-surface markers identifies routes to iPS cells. Nature. 2013;499:88–91. doi: 10.1038/nature12243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Hansson J, et al. Highly coordinated proteome dynamics during reprogramming of somatic cells to pluripotency. Cell reports. 2012;2:1579–1592. doi: 10.1016/j.celrep.2012.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Tanabe K, Nakamura M, Narita M, Takahashi K, Yamanaka S. Maturation, not initiation, is the major roadblock during reprogramming toward pluripotency from human fibroblasts. Proceedings of the National Academy of Sciences of the United States of America. 2013;110:12172–12179. doi: 10.1073/pnas.1310291110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Golipour A, et al. A late transition in somatic cell reprogramming requires regulators distinct from the pluripotency network. Cell stem cell. 2012;11:769–782. doi: 10.1016/j.stem.2012.11.008. [DOI] [PubMed] [Google Scholar]
- 81.Hou P, et al. Pluripotent Stem Cells Induced from Mouse Somatic Cells by Small-Molecule Compounds. Science. 2013 doi: 10.1126/science.1239278. This provocative report suggests that induced pluripotency can be achieved with four small compounds and without the use of transcription factors. [DOI] [PubMed] [Google Scholar]
- 82.Egli D, et al. Reprogramming within hours following nuclear transfer into mouse but not human zygotes. Nat Commun. 2011;2:488. doi: 10.1038/ncomms1503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Gu TP, et al. The role of Tet3 DNA dioxygenase in epigenetic reprogramming by oocytes. Nature. 2011;477:606–610. doi: 10.1038/nature10443. [DOI] [PubMed] [Google Scholar]
- 84.Wu G, et al. Establishment of totipotency does not depend on Oct4A. Nature cell biology. 2013 doi: 10.1038/ncb2816. This study discovered that Oct4 is dispensable for reprogramming somatic nuclei by SCNT, pointing to important mechanistic differences with induced pluripotency. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Maekawa M, et al. Direct reprogramming of somatic cells is promoted by maternal transcription factor Glis1. Nature. 2011;474:225–229. doi: 10.1038/nature10106. [DOI] [PubMed] [Google Scholar]
- 86.Jullien J, Astrand C, Halley-Stott RP, Garrett N, Gurdon JB. Characterization of somatic cell nuclear reprogramming by oocytes in which a linker histone is required for pluripotency gene reactivation. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:5483–5488. doi: 10.1073/pnas.1000599107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Lu X, et al. Drosophila H1 regulates the genetic activity of heterochromatin by recruitment of Su(var)3-9. Science. 2013;340:78–81. doi: 10.1126/science.1234654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Yang SM, Kim BJ, Norwood Toro L, Skoultchi AI. H1 linker histone promotes epigenetic silencing by regulating both DNA methylation and histone H3 methylation. Proceedings of the National Academy of Sciences of the United States of America. 2013;110:1708–1713. doi: 10.1073/pnas.1213266110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Nashun B, Akiyama T, Suzuki MG, Aoki F. Dramatic replacement of histone variants during genome remodeling in nuclear-transferred embryos. Epigenetics. 2011;6:1489–1497. doi: 10.4161/epi.6.12.18206. [DOI] [PubMed] [Google Scholar]
- 90.Jullien J, et al. HIRA dependent H3.3 deposition is required for transcriptional reprogramming following nuclear transfer to Xenopus oocytes. Epigenetics Chromatin. 2012;5:17. doi: 10.1186/1756-8935-5-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Pasque V, Gillich A, Garrett N, Gurdon JB. Histone variant macroH2A confers resistance to nuclear reprogramming. EMBO J. 2011;30:2373–2387. doi: 10.1038/emboj.2011.144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Han DW, et al. Pluripotential reprogramming of the somatic genome in hybrid cells occurs with the first cell cycle. Stem Cells. 2008;26:445–454. doi: 10.1634/stemcells.2007-0553. [DOI] [PubMed] [Google Scholar]
- 93.Piccolo FM, et al. Different Roles for Tet1 and Tet2 Proteins in Reprogramming-Mediated Erasure of Imprints Induced by EGC Fusion. Mol Cell. 2013;49:1023–1033. doi: 10.1016/j.molcel.2013.01.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Pereira CF, et al. Heterokaryon-based reprogramming of human B lymphocytes for pluripotency requires Oct4 but not Sox2. PLoS genetics. 2008;4:e1000170. doi: 10.1371/journal.pgen.1000170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Do JT, et al. Enhanced reprogramming of Xist by induced upregulation of Tsix and Dnmt3a. Stem Cells. 2008;26:2821–2831. doi: 10.1634/stemcells.2008-0482. [DOI] [PubMed] [Google Scholar]
- 96.Foshay K, et al. Embryonic stem cells induce pluripotency in somatic cell fusion through biphasic reprogramming. Molecular cell. 2012;46:159–170. doi: 10.1016/j.molcel.2012.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Ma DK, Chiang CH, Ponnusamy K, Ming GL, Song H. G9a and Jhdm2a regulate embryonic stem cell fusion-induced reprogramming of adult neural stem cells. Stem Cells. 2008;26:2131–2141. doi: 10.1634/stemcells.2008-0388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Pereira CF, et al. ESCs require PRC2 to direct the successful reprogramming of differentiated cells toward pluripotency. Cell stem cell. 2010;6:547–556. doi: 10.1016/j.stem.2010.04.013. [DOI] [PubMed] [Google Scholar]
- 99.Hajkova P, et al. Chromatin dynamics during epigenetic reprogramming in the mouse germ line. Nature. 2008;452:877–881. doi: 10.1038/nature06714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Magnusdottir E, Gillich A, Grabole N, Surani MA. Combinatorial control of cell fate and reprogramming in the mammalian germline. Current opinion in genetics & development. 2012;22:466–474. doi: 10.1016/j.gde.2012.06.002. [DOI] [PubMed] [Google Scholar]
- 101.Chuva de Sousa Lopes SM, et al. X chromosome activity in mouse XX primordial germ cells. PLoS genetics. 2008;4:e30. doi: 10.1371/journal.pgen.0040030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Sachs M, et al. Bivalent chromatin marks developmental regulatory genes in the mouse embryonic germline in vivo. Cell reports. 2013;3:1777–1784. doi: 10.1016/j.celrep.2013.04.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Durcova-Hills G, Tang F, Doody G, Tooze R, Surani MA. Reprogramming primordial germ cells into pluripotent stem cells. PLoS One. 2008;3:e3531. doi: 10.1371/journal.pone.0003531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Krentz AD, et al. The DM domain protein DMRT1 is a dose-sensitive regulator of fetal germ cell proliferation and pluripotency. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:22323–22328. doi: 10.1073/pnas.0905431106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Hackett JA, et al. Germline DNA demethylation dynamics and imprint erasure through 5-hydroxymethylcytosine. Science. 2013;339:448–452. doi: 10.1126/science.1229277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Dawlaty MM, et al. Combined deficiency of Tet1 and Tet2 causes epigenetic abnormalities but is compatible with postnatal development. Dev Cell. 2013;24:310–323. doi: 10.1016/j.devcel.2012.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Kagiwada S, Kurimoto K, Hirota T, Yamaji M, Saitou M. Replication-coupled passive DNA demethylation for the erasure of genome imprints in mice. EMBO J. 2013;32:340–353. doi: 10.1038/emboj.2012.331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Eminli S, et al. Differentiation stage determines potential of hematopoietic cells for reprogramming into induced pluripotent stem cells. Nature genetics. 2009;41:968–976. doi: 10.1038/ng.428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Tan KY, Eminli S, Hettmer S, Hochedlinger K, Wagers AJ. Efficient generation of iPS cells from skeletal muscle stem cells. PLoS One. 2011;6:e26406. doi: 10.1371/journal.pone.0026406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.White AC, et al. Defining the origins of Ras/p53-mediated squamous cell carcinoma. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:7425–7430. doi: 10.1073/pnas.1012670108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Barker N, et al. Crypt stem cells as the cells-of-origin of intestinal cancer. Nature. 2009;457:608–611. doi: 10.1038/nature07602. [DOI] [PubMed] [Google Scholar]
- 112.Folmes CD, et al. Somatic oxidative bioenergetics transitions into pluripotency-dependent glycolysis to facilitate nuclear reprogramming. Cell Metab. 2011;14:264–271. doi: 10.1016/j.cmet.2011.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Suva ML, Riggi N, Bernstein BE. Epigenetic reprogramming in cancer. Science. 2013;339:1567–1570. doi: 10.1126/science.1230184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.He J, Nguyen AT, Zhang Y. KDM2b/JHDM1b, an H3K36me2-specific demethylase, is required for initiation and maintenance of acute myeloid leukemia. Blood. 2011;117:3869–3880. doi: 10.1182/blood-2010-10-312736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Tzatsos A, et al. KDM2B promotes pancreatic cancer via Polycomb-dependent and -independent transcriptional programs. J Clin Invest. 2013;123:727–739. doi: 10.1172/JCI64535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Kapoor A, et al. The histone variant macroH2A suppresses melanoma progression through regulation of CDK8. Nature. 2010;468:1105–1109. doi: 10.1038/nature09590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Gaudet F, et al. Induction of tumors in mice by genomic hypomethylation. Science. 2003;300:489–492. doi: 10.1126/science.1083558. [DOI] [PubMed] [Google Scholar]
- 118.Moran-Crusio K, et al. Tet2 loss leads to increased hematopoietic stem cell self-renewal and myeloid transformation. Cancer Cell. 2011;20:11–24. doi: 10.1016/j.ccr.2011.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Krivtsov AV, et al. H3K79 methylation profiles define murine and human MLL-AF4 leukemias. Cancer Cell. 2008;14:355–368. doi: 10.1016/j.ccr.2008.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Kadoch C, et al. Proteomic and bioinformatic analysis of mammalian SWI/SNF complexes identifies extensive roles in human malignancy. Nature genetics. 2013;45:592–601. doi: 10.1038/ng.2628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Doi A, et al. Differential methylation of tissue- and cancer-specific CpG island shores distinguishes human induced pluripotent stem cells, embryonic stem cells and fibroblasts. Nature genetics. 2009;41:1350–1353. doi: 10.1038/ng.471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Hochedlinger K, et al. Reprogramming of a melanoma genome by nuclear transplantation. Genes Dev. 2004;18:1875–1885. doi: 10.1101/gad.1213504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Stricker SH, et al. Widespread resetting of DNA methylation in glioblastoma-initiating cells suppresses malignant cellular behavior in a lineage-dependent manner. Genes Dev. 2013;27:654–669. doi: 10.1101/gad.212662.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Rapino F, et al. C/EBPalpha induces highly efficient macrophage transdifferentiation of B lymphoma and leukemia cell lines and impairs their tumorigenicity. Cell reports. 2013;3:1153–1163. doi: 10.1016/j.celrep.2013.03.003. [DOI] [PubMed] [Google Scholar]