

Abstract
An Epstein-Barr virus (EBV) genome in Burkitt's lymphoma-derived cell line Akata was cloned into a bacterial artificial chromosome (BAC) vector. The BAC clone, designated AK-BAC, was rapidly and precisely modified by means of efficient homologous recombination in Escherichia coli. This system was used to produce recombinant EBVs with transgenes. An expression cassette of green fluorescent protein (GFP) was inserted into AK-BAC, and the resultant BAC clone, AK-BAC-GFP, was transfected into Akata cells. We found that transfected BAC plasmids efficiently formed episomes in EBV-positive Akata cells. Mixtures of wild-type and AK-BAC-GFP viruses were then produced and used to infect EBV-negative Akata cells. We obtained cell clones that harbored only AK-BAC-GFP but no wild-type episome. These cell clones produced infectious viruses after stimulating virus production, and the recombinant viruses of AK-BAC-GFP efficiently immortalized primary B lymphocytes. We further revised the method so that any kind of cDNA could be rapidly inserted into the unique I-PpoI site that had been artificially introduced into AK-BAC. The AK-BAC system will have a broad range of applications, such as genetic analyses of various viral gene products and development of viral vectors for human gene therapy.
Epstein-Barr virus (EBV) is a ubiquitous human herpesvirus that establishes latent infection in B lymphocytes (23). EBV is associated with various lymphoid and epithelial malignancies, such as Burkitt's lymphoma, nasopharyngeal carcinoma, lymphoproliferative diseases in immunosuppressed patients, and gastric carcinoma (23). In vitro, EBV transforms peripheral human B lymphocytes into indefinitely proliferating lymphoblastoid cell lines (LCLs).
Latently infected B cells maintain EBV genomes as 170-kb circular plasmids, referred to as episomes, and express only limited numbers of viral gene products (23). In LCLs, EBV establishes type III latency, in which six nuclear antigens (EBNA-1, EBNA-2, EBNA-3A, EBNA-3B, EBNA-3C, and EBNA-LP), three latent membrane proteins (LMP-1, LMP-2A, and LMP-2B), two small nonpolyadenylated RNAs (EBER-1 and EBER-2), and transcripts from the BamHI-A region (BARTs) are expressed (23). Understanding how these latently expressed viral gene products contribute to B-cell transformation has been attracting intense interest, as this process represents, at least partly, the oncogenic ability of EBV (11).
On the other hand, EBV is a potential vector for human gene therapy (35), although it is identified as a tumor virus. For example, recombinant EBVs with transgenes combine B-cell immortalization and gene transfer into a single step and will be used to express transgenes in LCLs. Alternatively, replication-incompetent vectors with minimal viral elements can be produced as amplicon vectors (2, 34). Amplicon vectors can accommodate transgenes up to 120 kb (36), and such large transgene capacity makes the EBV vector a promising one to deliver genomic transgenes for gene therapy.
Like other herpesviruses, generating recombinant EBVs are essential not only for delineating the molecular functions of viral gene products but also for developing EBV-based vectors. However, mutagenesis of the EBV genome is actually very inefficient, mainly because there is no host cell that fully supports lytic replication immediately after EBV infection. Until recently, site-directed mutagenesis of EBV could be performed only by homologous recombination in EBV-positive cells after transfection of DNA fragments carrying mutations (7, 11). However, the frequency of homologous recombination is usually very low due to poor replication efficiency. Identifying cells with a homologously recombined genome is time-consuming and tedious work. As a result, in contrast to the plentiful selection of herpes simplex virus mutants available (24), limited numbers of EBV mutants are available. Therefore, there is a strong demand for a novel experimental strategy that allows more efficient generation of recombinant EBVs.
Recently, introducing the bacterial artificial chromosome (BAC) system into the genetics of herpesviruses has started to change the situation (4, 33). In the BAC system, the entire viral genome can be propagated in Escherichia coli, and mutations can be rapidly and precisely introduced into any kind of viral genes. Following the successful cloning of other herpesviruses, the B95-8 strain of EBV was cloned in a BAC vector (8). The system employed epithelial 293 cells as virus-producing cells, and virus production was induced by transfecting an expression vector encoding a viral immediate-early protein BZLF1 (8). To date, this system is the only available BAC-based system that can produce recombinant EBVs.
In this report, we describe establishment of the second BAC-based system for generating EBV recombinants. We chose Burkitt's lymphoma-derived cell line Akata as a virus-producing cell line (31). In Akata cells, production of progeny viruses can be induced by cross-linking surface immunoglobulin G (IgG) molecules using anti-IgG antibodies (30, 32). Therefore, large quantities of pure recombinant viruses can readily be produced by anti-IgG antibody treatment (28). In contrast, it is unpractical to obtain equivalent amounts of viruses using the 293 cell system, as virus production always requires BZLF1 transfection.
We show that the Akata-derived BAC clone, designated AK-BAC, can be modified by applying a homologous recombination system in E. coli, called GET recombination (19). Combining the AK-BAC and GET recombination systems enabled rapid and precise modification of the EBV genome. We applied the system for generating replication-competent vectors with transgenes and found that BAC-derived recombinant viruses retained the ability to transform B cells. The system was further revised so that any kind of cDNA could be rapidly inserted into the EBV genome by simple in vitro ligation.
MATERIALS AND METHODS
Cells and virus production.
Akata, a human Burkitt's lymphoma-derived cell line carrying EBV episomes (31), and an EBV-negative Akata cell line (27) were grown in RPMI 1640 medium (Sigma-Aldrich Fine Chemicals, St. Louis, Mo.) supplemented with 10% fetal bovine serum. Virus production from Akata cells was induced by cross-linking surface IgG using rabbit anti-human IgG (DakoCytomation, Carpinteria, Calif.) as described previously (30, 32). EBV-negative Akata cells were infected with diluted (1:5 and 1:10) culture supernatant, and infected cells were isolated by selection with G418 (Sigma) (700 μg/ml) as previously described (28).
BAC cloning of EBV genome derived from Akata cells.
The targeting construct, TXneoV-BAC, used for BAC cloning of the EBV genome derived from Akata cells is essentially the same as the one used to generate a recombinant EBV expressing enhanced green fluorescent protein (EGFP-EBV) (18) except for the presence of the BAC vector sequence instead of the EGFP expression cassette. Briefly, TXneoV-BAC had a 2,010-bp EcoRV-SalI fragment (containing the neomycin resistance gene [Neor gene]) of pcDNA3 (Invitrogen) and a 6,877-bp NotI fragment (containing the BAC vector sequence and chloramphenicol resistance gene [Cmr gene]) of pBeloBAC11 (Genome Systems) inserted into the BamHI X fragment. This insertion site corresponds to nucleotide 131290 of the wild-type EBV sequence (GenBank accession number AJ507799) (6), where the SmaI site is located.
The HindIII F fragment (15.2 kb) of TXneoV-BAC (see Fig. 1A), which was 9.2 kb longer than the wild-type fragment due to the insertion of a neomycin resistance gene and a BAC vector sequence, was excised and introduced to EBV-positive Akata cells by electroporation (Bio-Rad Gene Pulser II; 190 V, 950 μF). Transfected cells were then plated at 104 cells per well in 96-well tissue culture plates in medium containing 375 μg of G418 (Sigma) per ml. Half of the culture medium was replaced with fresh G418-containing medium every 5 days. Drug-resistant clones were screened by Southern blotting for the presence of homologously recombined viral DNAs.
FIG. 1.
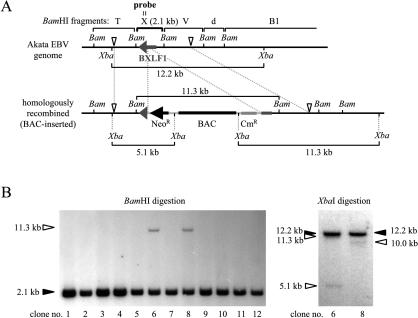
Insertion of a BAC vector sequence into the EBV genome derived from Akata cells. (A) Map of the genome of Akata strain EBV surrounding the targeted region before and after the insertion of a BAC vector sequence. The positions of BamHI (Bam) and XbaI (Xba) sites are indicated. White arrowheads indicate the positions of HindIII sites located at the ends of the linear targeting construct (HindIII F fragment). The positions of the BAC vector sequence (BAC), chloramphenicol resistance gene (CmR) (works in E. coli), and neomycin resistance gene (NeoR) (works in mammalian cells) are shown. The BamHI X fragment was used as a probe for Southern blot analyses. (B) Southern blot analyses using BamHI- or XbaI-digested genomic DNAs of G418-resistant cell clones. The bands derived from BAC-inserted EBV (white arrowheads) and the bands derived from wild-type EBV (black arrowheads) are indicated. Note that only clone 6, but not clone 8, exhibits an expected band pattern.
Southern blot analysis.
Genomic DNAs were extracted by the standard proteinase K-sodium dodecyl sulfate method, followed by phenol-chloroform extraction and ethanol precipitation. Southern blotting was performed as previously described (18).
Plasmids.
A BamHI fragment of pUC4K (Pharmacia) containing a kanamycin resistance marker gene was cloned into the BamHI site of pBS246 (Life Technologies) to make pBS246-km. A SalI (blunted)-BamHI fragment of pSG5 (Stratagene) was cloned between the AseI (blunted) and BamHI sites of pEGFPN1 (Clontech) to produce p(SG5)-EGFP. The EcoRI-NotI fragment of p(SG5)-EGFP containing the EGFP gene was replaced with a synthetic oligonucleotide (5′-AATTCTATGACTCTCTTAAGGTAGCCAAAAGC-3′) containing a recognition sequence of I-PpoI to make p(SG5)-IPpoI. pBS246-km, p(SG5)-EGFP, and p(SG5)-IPpoI were used as PCR templates to produce linear targeting constructs for GET recombination.
pGEM9Zf(−)-IPpoI has a unique I-PpoI site replacing the T7 promoter of pGEM9Zf(−) and is supplied as a control vector of I-PpoI enzyme (Promega). The synthetic I-PpoI site (described above) was cloned into EcoRI-NotI-digested pGEM9Zf(−)-IPpoI to construct a double I-PpoI vector with two I-PpoI sites. A BamHI fragment (blunted) of APR-Muc1 (3) (kindly provided by Keiichi Kontani, Shiga University of Medical Science, Otsu, Japan, and Toshio Kudo, Tohoku University, Sendai, Japan) containing Muc-1 cDNA was cloned into the HincII site of a double I-PpoI vector to construct pIPpoI-Muc1.
Electrotransformation of E. coli.
Electrocompetent E. coli DH10B bacterial cells were prepared as described previously (26). Electrotransformation was performed using a Bio-Rad Gene Pulser II electroporation system (0.1-cm cuvette, 1.25 kV, 25 μF, 100 Ω, 40 μl of cells).
BACmid preparation and DNA analysis.
For minipreparations, each BAC plasmid (BACmid) DNA was isolated from a 1.5-ml portion of a bacterial culture using an alkaline lysis procedure according to a protocol [Current protocols in human genetics. Unit 5.15. Construction of bacterial artificial chromosome (BAC/PAC) libraries] found at the Murdoch Children's Research Institute website (http://murdoch.rch.unimelb.edu.au/). Each miniprep DNA sample prepared from a 1.5-ml portion of a bacterial culture was used for one reaction of restriction enzyme digestion in a total volume of 40 μl. For maxipreparations, BACmid DNA was isolated from each 500-ml bacterial culture using a Nucleobond BAC100 kit (Macherey-Nagel, Duren, Germany) according to the manufacturer's instructions. BACmid DNAs digested with restriction enzymes were resolved by 0.8% agarose gel electrophoresis for 15 h at 40 V and visualized by ethidium bromide staining.
Pulsed-field gel electrophoresis was performed using Genofield AE-8900 (ATTO Corporation, Tokyo, Japan) according to the manufacturer's instructions.
Modifications of AK-BAC in E. coli.
Modifications of AK-BAC were performed in E. coli using GET recombination as previously described (19, 20). Briefly, the plasmid pGETrec (kindly provided by Panayiotis Ioannou, Murdoch Institute, Melbourne, Victoria, Australia) was transformed into E. coli DH10B harboring a BACmid to be modified. DH10B cells containing both BACmid and pGETrec were selected on Luria-Bertani (LB) plates containing 12.5 μg of chloramphenicol per ml and 50 μg of ampicillin per ml. The resulting resistant DH10B cells were made electrocompetent after induction of Redγ, RecE, and RecT by 0.2% arabinose addition.
Linear targeting constructs for GET recombination were prepared by PCR as described previously (19). To construct AK-BACΔneo, pBS246-km was used as a PCR template. A pair of 77-mer oligonucleotides was designed to carry a 57-mer oligonucleotide homologous to the target region of AK-BAC at their 5′ ends and a 20-mer oligonucleotide homologous to the PCR template at their 3′ ends. The resultant PCR product (1.6 kb long) was gel purified, digested with DpnI to remove methylated PCR template DNA, and ethanol precipitated. Approximately 200 ng of the linear PCR product was electroporated into recombinase-induced E. coli DH10B electrocompetent cells (harboring AK-BAC and pGETrec). Colonies of recombinant BACmids were identified by plating cells on LB plates containing chloramphenicol (12.5 μg/ml) and kanamycin (50 μg/ml). Modified BAC clones were purified away from pGETrec by miniprep DNA isolation and electroporation into DH10B cells.
Removal of the kanamycin resistance marker gene from AK-BACΔneo was performed by treating miniprep DNA of AK-BACΔneo with purified cre recombinase (Novagen) in vitro according to the manufacturer's instructions. One microliter of the reaction mixture was then used to transform electrocompetent DH10B cells, followed by selection on LB plates containing chloramphenicol. Bacterial clones with AK-BACΔneoΔkm were chosen by checking their sensitivity to kanamycin.
To construct AK-BAC-GFP and AK-BAC-IPpoI, p(SG5)-EGFP and p(SG5)-IPpoI were used as PCR templates to prepare linear targeting constructs (4.2 and 3.6 kb long), respectively. This insertion site corresponds to nucleotide 145342 of the wild-type EBV sequence (GenBank accession number AJ507799) (6). AK-BAC-GFP2 has a transgene (identical to that of AK-BAC-GFP except for the presence of an additional loxP site) inserted downstream of the BAC vector sequence within the BamHI X region. GET recombination was performed using recombinase-induced DH10B electrocompetent cells harboring AK-BACΔneoΔkm and pGETrec.
The sequences of 77-mer oligonucleotides and the PCR conditions used for GET recombination are available upon request.
In vitro construction of AK-BAC-Muc1.
AK-BAC-IPpoI was linearized by I-PpoI (Promega). A 170-kb linear vector of AK-BAC-IPpoI was then treated with shrimp alkaline phosphatase (Promega), followed by heat inactivation according to the manufacturer's instructions. A 4.2-kb I-PpoI fragment of pIPpoI-Muc1 was ligated to the linear and dephosphorylated AK-BAC-IPpoI vector in a total volume of 10 μl at 16°C for 2 h using the LigaFast rapid ligation system (Promega). The ligation mixture was then diluted fivefold, and 1 μl of the diluted reaction mixture was used for transformation of electrocompetent DH10B cells.
Transfection of BACmids into Akata cells.
EBV-positive Akata cells (5 × 106) were transfected with 5 to 10 μg of BACmid DNA (prepared using a Nucleobond BAC100 kit) via electroporation (Bio-Rad Gene Pulser II; 190 V, 950 μF). Transfected cells were resuspended in 5 ml of culture medium and plated into six-well dishes. At 2 days posttransfection, cells were plated at 104 cells per well in 96-well tissue culture plates in medium containing 400 μg of G418 (Sigma) per ml. Half of the culture medium was replaced with fresh G418-containing medium every 5 days. G418-resistant clones were screened for the presence of episomally maintained BACmids. EBV-negative Akata cells or EBV-negative, EBNA1-positive Akata cells (with constitutive EBNA1 expression, hygromycin resistant) were also used as recipient cells.
Episomal DNA preparation and BAC rescue.
EBV episomes were isolated from Akata cells by an alkaline lysis procedure as described previously (29) with minor modifications. Briefly, 2 × 106 cells were washed once in phosphate-buffered saline (without calcium and magnesium), pelleted by centrifugation at 1,500 × g, and resuspended in 200 μl of lysis buffer (50 mM NaCl, 2 mM EDTA, 1% sodium dodecyl sulfate; buffer adjusted to pH 12.45 with NaOH). The cells were lysed by vortexing them (model G560; Scientific Industries, Bohemia, N.Y.) at the highest speed for 1 min, followed by incubation at 30°C for 30 min. The lysis buffer was neutralized by the addition of 40 μl of 1 M Tris-HCl (pH 7.0), 22 μl of 5 M NaCl, and 1 μl of 20-mg/ml proteinase K. The cellular proteins were partially degraded by incubation at 37°C for 30 min. The cell lysates were extracted with 88 μl of phenol (saturated with 0.2 M NaCl-0.2 M Tris-HCl [pH 8.0]) by gently inverting the tubes. The tubes were then chilled slowly to 4°C, and the phases were separated by centrifugation at 15,000 × g for 15 min at 4°C. The aqueous phase (240 μl) was recovered using wide-bore tips, and 80 μl of chloroform-isoamyl alcohol mixture (24:1) was added. The phases were separated as described above, and 200 μl of the aqueous phase was mixed with 400 μl of ethanol. The precipitated episomal DNAs were recovered by centrifugation at 15,000 × g at 4°C for 15 min, washed once with 70% ethanol, and redissolved in 5 μl of H2O. One microliter of each episomal preparation was used to transform electrocompetent DH10B cells.
Infection of cord blood lymphocytes.
Serially diluted (10−1 to 10−5) culture supernatants containing recombinant EBVs of EGFP-EBV (18) or AK-BAC-GFP were used to infect purified cord blood mononuclear cells (106 cells for each infection). Infected cells were then plated at 2 × 105 cells per well in 96-well tissue culture plates (4 wells for each infection). Half of the culture medium was replaced with fresh medium every 5 days. The number of wells with growing cells was counted at 5 weeks postinfection, and 50% transforming doses (TD50/ml) were calculated by the Reed-Muench method (5).
Immunofluorescence.
Expression of EBV capsid antigen was tested on acetone-fixed cells by indirect immunofluorescence with a monoclonal antibody (MAb) Cl.50-1 specific for gp110 (encoded by BALF4) and a Cy3-conjugated anti-mouse IgG (Jackson ImmunoResearch). Muc-1 expression was tested on cells (that had been fixed by 3.7% formaldehyde) by indirect immunofluorescence with MAb CD227 specific for tandem repeats of Muc-1 protein (PharMingen).
Immunoblotting and RT-PCR.
Immunoblotting was performed using whole-cell extracts essentially as previously described (37). Expression of EBNA proteins was detected with human serum reactive to six EBNA proteins as a primary antibody and a horseradish peroxidase-conjugated anti-human IgG as a secondary antibody, while expression of LMP1 was detected using MAb S12 (specific for LMP1) and peroxidase-conjugated anti-mouse IgG. Expression of BARTs was examined by reverse transcription-PCR (RT-PCR) analysis as previously described (14).
RESULTS
Cloning the Akata cell-derived EBV genome into a BAC vector.
Several transgenes that include a GFP gene and a neomycin resistance gene have been successfully inserted into the BamHI X region of the EBV genome derived from Akata cells (Akata strain EBV) without affecting viral replication and production of progeny viruses (18, 28). As insertion of transgenes into this site disrupts the BXLF1 open reading frame encoding viral thymidine kinase (Fig. 1A), viral thymidine kinase is apparently nonessential for viral replication and infection. Therefore, we decided to put a BAC vector sequence into exactly the same location. Akata cells carrying EBV episomes were transfected with a targeting construct containing a BAC vector sequence, chloramphenicol resistance gene (Cmr gene, a bacterial selection marker), and neomycin resistance gene (Neor gene, a mammalian selection marker) (Fig. 1A), and transfected cells were selected by G418. G418-resistant cell clones were screened by Southern blotting for the presence of cell clones with homologously recombined EBV episome (Fig. 1A). The results of Southern blotting revealed that one of the cell clones (clone 6 in Fig. 1B) exhibited the bands derived from homologously recombined EBV episome in addition to the bands derived from wild-type EBV episome.
Episomal DNA was prepared from clone 6 and used to transform electrocompetent DH10B bacterial cells. As a BAC vector sequence and a chloramphenicol resistance gene were inserted into EBV genomes (Fig. 1A), the transformed bacterial cells were selected by chloramphenicol. Several chloramphenicol-resistant colonies were then grown, and DNA was extracted from bacterial cells. Restriction enzyme analyses revealed that, in all the clones examined, BamHI- and EcoRI-digested band patterns fit very well with the available restriction enzyme mapping data of the genome of Akata strain EBV (Fig. 2A). These results also fit well with the previous data on restriction enzyme-digested Akata virion DNA (31). We also tested several other restriction enzymes and found that all the tested enzymes generated DNA fragments of the expected sizes (data not shown). Therefore, we conclude that the genome of Akata strain EBV has been successfully subcloned into a BAC vector.
FIG. 2.
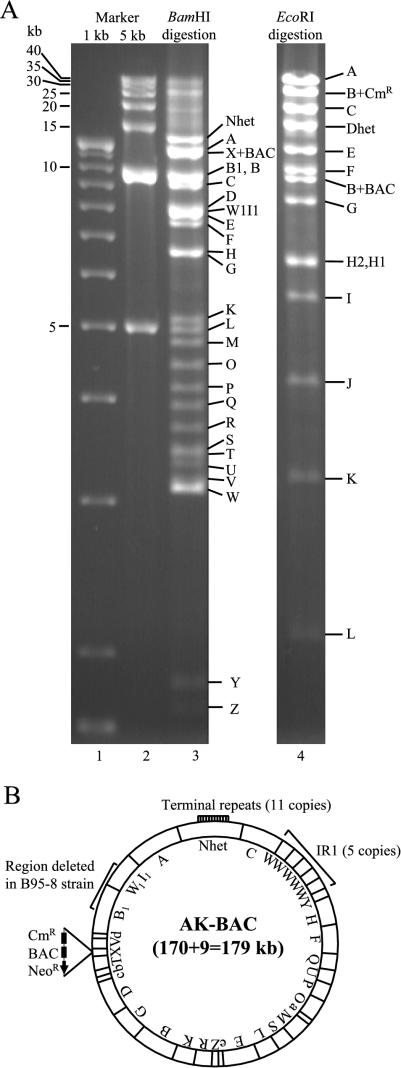
Characterization of AK-BAC. (A) Restriction digestion analysis of a BAC clone harboring the genome of Akata strain EBV. BAC DNAs digested with BamHI or EcoRI, together with DNA size markers, were resolved by 0.8% agarose gel electrophoresis. The positions of the different DNA fragments (capital letters) are indicated to the right of the gels. Note that the BamHI X fragment (originally 2.1 kb) had increased in size to 11.3 kb due to the insertion of a BAC vector sequence. (B) Schematic representation of AK-BAC. The positions of BamHI fragments (capital letters) are indicated. The numbers of terminal repeats and W repeats (IR1) are shown. The region of EBV genome that is deleted in B95-8 strain of EBV is also indicated.
This BAC clone was designated AK-BAC (for Akata-derived BAC clone of EBV), and its schematic structure is shown in Fig. 2B. AK-BAC contains a BAC vector sequence and Cmr and Neor genes inserted into the BamHI X region. Detailed mapping of AK-BAC revealed that it has 5 copies of W repeats and 11 copies of terminal repeats. The genome of Akata strain EBV retains the 12-kb region that is missing in B95-8 strain (6, 21) and is thought to be a prototype EBV. The overall size of AK-BAC is calculated to be 179 kb.
Rapid modification of the EBV genome via homologous recombination in E. coli.
Our initial attempt to reintroduce AK-BAC into Akata cells via transfection was unsuccessful, presumably because the neomycin resistance gene of AK-BAC was not efficient enough to confer drug resistance to transfected cells (data not shown). Therefore, we decided to remove this marker gene and instead put another neomycin resistance gene with a different origin into another location within AK-BAC.
We took advantage of the very efficient homologous recombination in E. coli to modify AK-BAC. The recombination strategy we employed in this study is called GET recombination (19, 20), which utilizes Redγ of λ phage and RecE and RecT of Rac phage. The methods used to remove the neomycin resistance gene are summarized in Fig. 3A. First, the neomycin resistance cassette was replaced with a kanamycin resistance gene (Kmr gene) (Fig. 3A) that was flanked with two loxP sites (AK-BACΔneo). Subsequently, the kanamycin resistance gene was removed by treating the BAC DNA with purified cre recombinase in vitro. The resultant BAC clone, designated AK-BACΔneoΔkm, had only a BAC vector sequence and a chloramphenicol marker gene inserted into the BamHI X region of the genome of Akata strain EBV. These modifications were verified by agarose gel analyses of parental and modified BAC clones. We observed expected changes of band sizes of BamHI-digested and EcoRI-digested DNAs (Fig. 3B). Other bands remained unchanged throughout, demonstrating that unwanted and illegitimate mutations were unlikely. As repeated sequences within the EBV genome may become unstable during recombinogenic engineering, we routinely checked copy numbers of terminal repeats (TR), internal repeat 1 (IR1), and IR4. The length of TR can easily be checked by BamHI or EcoRI digestion, while the length of IR1 and IR4 can be checked by NcoI digestion. The results revealed that their copy numbers were maintained stably in modified BAC clones (Fig. 3B).
FIG. 3.
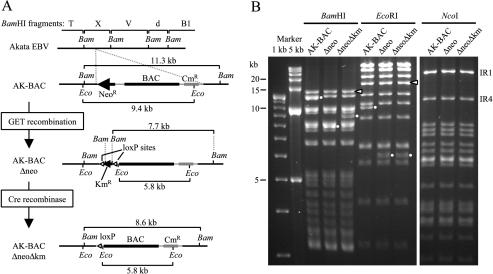
Deleting a neomycin resistance gene from AK-BAC. (A) Map of AK-BAC surrounding the neomycin resistance gene (NeoR) to be deleted. GET recombination was used to replace the Neor gene of AK-BAC with the kanamycin resistance gene (KmR), and the Kmr gene was subsequently removed by in vitro cre recombinase treatment. The positions of loxP sites used to excise the Kmr gene are shown. The sizes of the BamHI (Bam) and EcoRI (Eco) fragments demonstrating the successful modifications are indicated. (B) Restriction digestion analysis of BAC clones shown in panel A. The bands demonstrating successful modification are indicated by small white circles. Note that the bands containing terminal repeats (white arrowheads) and IR1 and IR4 are identical in these clones.
Putting a GFP/neomycin resistance transgene into AK-BAC.
A cassette consisting of a GFP gene and neomycin and kanamycin resistance genes (GFP/Neor/Kmr cassette) was then inserted into AK-BACΔneoΔkm. The GFP gene is driven by the simian virus 40 (SV40) enhancer and promoter, while the Neor and Kmr genes are under the control of both bacterial and mammalian promoters (SV40 enhancer and promoter). Therefore, kanamycin selection can be used for GET recombination in E. coli, while G418 selection can be used for human cells. In order to examine the feasibility of inserting transgenes into various loci within the EBV genome, the cassette was inserted into two different loci of AK-BACΔneoΔkm (Fig. 4A). One locus is downstream of the right lytic replication origin (oriLyt) (9) (located in the BamHI B1 fragment), which is in the middle of the region deleted in the B95-8 strain of EBV (6, 21). The other locus is just downstream of the BAC vector sequence within the BamHI X fragment.
FIG. 4.
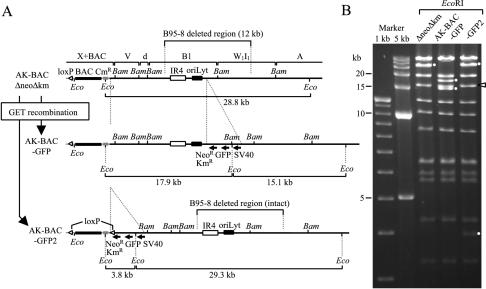
Putting a GFP transgene into two different loci in the EBV genome via GET recombination. (A) Map of the transgene insertion location. The GFP/Neor/Kmr transgene was inserted downstream of the right oriLyt (AK-BAC-GFP) or downstream of the BAC vector sequence (AK-BAC-GFP2) of AK-BACΔneoΔkm. An additional loxP site is included in the transgene of AK-BAC-GFP2 so that the BAC vector sequence can be removed in case it is required. The sizes of EcoRI (Eco) fragments demonstrating successful transgene insertion are indicated. The locations of IR4 and right oriLyt are also shown. The positions of BamHI (Bam) sites are also shown. (B) EcoRI digestion of BAC clones before and after transgene insertion. The bands demonstrating successful modification are indicated by small white circles. The position of the band containing terminal repeats is indicated by the white arrowhead.
Successful insertion of the GFP/Neor/Kmr cassette was verified by agarose gel analyses of parental (AK-BACΔneoΔkm) and transgene-inserted BAC clones (Fig. 4B). We observed the expected changes in band sizes of EcoRI-digested DNAs (Fig. 4B) and BamHI-digested DNAs (data not shown). The resultant BAC clones with the GFP/Neor/Kmr cassette were designated AK-BAC-GFP (transgene inserted into the region deleted in strain B95-8) and AK-BAC-GFP2 (transgene inserted into the BamHI X region). Again, the copy numbers of TR, IR1, and IR4 were identical in AK-BAC, AK-BAC-GFP, and AK-BAC-GFP2 (data not shown). Successful insertions of transgenes into two different loci within the EBV genome demonstrate that GET recombination is a highly reliable method to modify the EBV genome.
Successful transfer of BACmids into Akata cells.
BACmids with transgenes were then reintroduced into Akata cells (EBV positive or negative) via electroporation. Transfected cells were selected by G418, and G418-resistant cell clones with varied levels of GFP expression appeared. Episomal DNA fractions were prepared from G418-resistant cell clones, and they were used for transforming competent E. coli DH10B cells. This strategy enabled us to rescue episomally maintained BACmids as bacterial clones and to determine which cell clones contained episomally maintained BACmids. We found that transfected BACmids efficiently formed episomes in EBV-positive Akata cells. For example, when AK-BAC-GFP was transfected into EBV-positive Akata cells, we examined 27 G418-resistant cell clones and found that 15 of these clones harbored episomal BACmids, while the rest (12 clones) did not (Table 1, experiment 1). The BAC clones rescued from transfected EBV-positive Akata cells were analyzed by restriction enzyme digestion. We found that two cell clones of the 15 episome-positive cell clones contained intact AK-BAC-GFP (Table 1, experiment 1), as the BAC clones rescued from these cells exhibited an EcoRI-digested band pattern identical to that of AK-BAC-GFP (Fig. 5, cell clones 4 and 12). This result was further confirmed by restriction digestion analyses using BamHI and NcoI (data not shown). The rest of the episome-positive cell clones (13 clones) turned out to contain episomes with different degrees of rearrangement (data not shown). We repeated the experiment and obtained cell clones with intact AK-BAC-GFP with similar efficiency (Table 1, experiment 2). AK-BAC-GFP2 (Fig. 4) was also tested to verify the reproducibility of this experiment. We found that intact AK-BAC-GFP2 was successfully introduced into EBV-positive Akata cells with similar efficiency (Table 1, experiments 5 and 6). We conclude that intact BACmids can reproducibly be introduced into EBV-positive Akata cells via transfection.
TABLE 1.
Frequency of episome formation after transfection of various BACmids into EBV-positive or -negative Akata cells
| BAC clone | Recipient cell | Expt no. | No. of cell clones obtained
|
||
|---|---|---|---|---|---|
| G418-resistant | Episome-positive | Intact episome-positive | |||
| AK-BAC-GFP | EBV-positive Akata | 1 | 27 | 15 | 2 |
| 2 | 16 | 14 | 2 | ||
| AK-BAC-GFP | EBV-negative Akata | 3 | 6 | 0 | 0 |
| 4 | 5 | 0 | 0 | ||
| AK-BAC-GFP2 | EBV-positive Akata | 5 | 6 | 5 | 1 |
| 6 | 15 | 14 | 3 | ||
| AK-BAC-Muc1 | EBV-positive Akata | 7 | 8 | 2 | 0 |
| 8 | 22 | 8 | 3 | ||
FIG. 5.
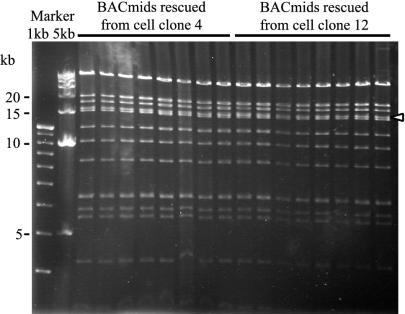
Successful introduction of AK-BAC-GFP back into Akata cells. AK-BAC-GFP was transfected into EBV-positive Akata cells, and the transfected cells were selected by G418. Episomal DNAs were prepared from G418-resistant clones, and the rescued BAC clones were analyzed by EcoRI digestion and agarose gel analysis. The results from 16 independent BAC clones, 8 clones each from cell clones 4 and 8, are shown. Note that the band pattern of these BAC clones is identical to that of AK-BAC-GFP generated in E. coli (shown in Fig. 4B). The position of the band containing terminal repeats is indicated by the white arrowhead.
In contrast, when AK-BAC-GFP was transfected into EBV-negative Akata cells, the number of G418-resistant cell clones that appeared after transfection and G418 selection was much lower than the number for EBV-positive Akata cells, and BACmids were not rescued from those cell clones (Table 1, experiments 3 and 4). As EBNA1 protein is necessary and sufficient for episomal maintenance of transfected oriP plasmids (15), we examined whether stable expression of EBNA1 protein in EBV-negative Akata cells could increase the efficiency of episome formation of transfected BACmids. After transfection of AK-BAC-GFP into EBV-negative, EBNA1-positive Akata cells, 28 cell clones of 55 G418-resistant cell clones (51%) turned out to contain episomes. The result is comparable to the episome formation efficiency of AK-BAC-GFP after transfection into EBV-positive Akata cells (29 cell clones of 43 G418-resistant cell clones [67%]; Table 1, experiments 1 and 2). These data strongly suggest that efficient episome formation occurring in EBV-positive Akata cells is due to EBNA1 protein, which is supplied from wild-type EBV episomes in trans.
Generating cell clones which produce recombinant viruses of AK-BAC-GFP.
Cell clone 12 (Fig. 5), which was obtained by transfecting AK-BAC-GFP into EBV-positive Akata cells, was subjected to further experiments. We next tried to establish cell lines that contained only AK-BAC-GFP and lacked wild-type virus. For this purpose, virus production was induced from clone 12, and the mixture of wild-type and BAC-containing viruses was produced. The mixture was then used to infect EBV-negative Akata cells (28). After G418 selection, all of the G418-resistant cell clones turned out to be GFP positive. G418-resistant cells that appeared in individual wells of culture plates were examined for the induction of gp110 protein (BALF4 gene product) after anti-IgG treatment. A cell clone exhibiting good gp110 induction was further subcloned by limiting dilution protocol. As a result, we obtained cell clones with good gp110 induction level.
One of the cell clones, designated clone 12-15, was chosen because it exhibited the strongest gp110 induction after anti-IgG treatment, and the clone was used for further analyses. We prepared genomic DNAs from EBV-positive Akata cells (harboring wild-type genome), clone 12 (harboring wild-type and AK-BAC-GFP) (Fig. 5), and clone 12-15 before and after stimulating virus production by anti-IgG treatment. Southern blotting using prepared genomic DNAs revealed that clone 12 actually exhibited bands derived from both wild-type genome and AK-BAC-GFP, while clone 12-15 exhibited only the AK-BAC-GFP bands. Southern blotting also revealed that, in clone 12-15, the AK-BAC-GFP band was significantly enhanced after anti-IgG treatment in the absence of wild-type EBV (Fig. 6A). Moreover, the production of gp110 protein observed in clone 12-15 after anti-IgG treatment was comparable to that observed in Akata cells having wild-type virus (Fig. 6B). Culture supernatant of anti-IgG-treated clone 12-15 was readily used to infect EBV-negative Akata cells. We found that more than 50% of recipient cells expressed GFP shortly after infection (data not shown). These results demonstrate that AK-BAC-GFP is competent for making infectious viruses after anti-IgG treatment in the absence of wild-type virus.
FIG. 6.
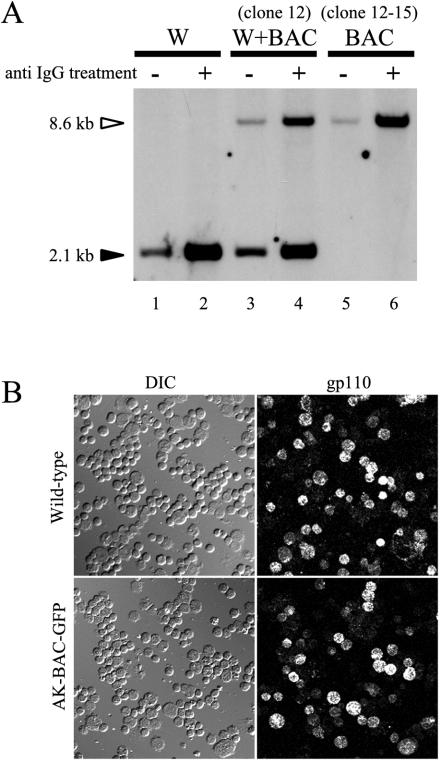
Amplification of AK-BAC-GFP in the absence of wild-type EBV. (A) Genomic DNAs of Akata cells harboring wild-type EBV (W), cell clone 12 harboring wild-type EBV and AK-BAC-GFP (W+BAC), and cell clone 12-15 harboring only AK-BAC-GFP (BAC) were prepared before (−) and after (+) anti-IgG treatment. BamHI-digested genomic DNA was analyzed by Southern blotting using the BamHI X fragment as a probe. The positions and sizes of the band derived from AK-BAC-GFP (white arrowhead) and the band derived from wild-type EBV (black arrowhead) are indicated. Note that cells harboring only AK-BAC-GFP exhibit amplification of viral DNAs in the absence of wild-type virus (lanes 5 and 6). (B) Expression of viral capsid antigen (gp110) after anti-IgG treatment in cells harboring wild-type EBV episome and in cell clone 12-15 harboring AK-BAC-GFP. The corresponding differential interference contrast (DIC) images are also shown.
The same experimental strategy described above was used to obtain cell clones with AK-BAC-GFP2 but without wild-type EBV episomes, and we found that AK-BAC-GFP2 was also competent for making infectious viruses in the absence of wild-type episomes (data not shown).
LCLs with type III latent gene expression were established with BAC virus.
Culture supernatant containing recombinant viruses derived from AK-BAC-GFP was used to infect cord blood lymphocytes in order to determine its infectivity and transforming titer. As a comparison, culture supernatant of EGFP-EBV (18), which had been made by conventional strategy, was subjected to the same assay. EGFP-EBV carries a GFP transgene inserted into the BamHI X region of the EBV genome derived from Akata cells but lacks a BAC sequence. We found that recombinant viruses derived from AK-BAC-GFP efficiently immortalized B lymphocytes and that the resultant LCLs strongly expressed GFP (Fig. 7A). The calculated 50% transforming dose of AK-BAC-GFP virus was 105.2 TD50/ml, which is comparable to the 105.7 value obtained with EGFP-EBV. This result demonstrates that insertion of a BAC vector sequence into the EBV genome does not affect the ability of EBV to transform B lymphocytes. In addition, we found that LCLs established by BAC viruses exhibited type III latency (23), in which all six EBNA proteins and LMP1 protein were expressed (Fig. 7B). Therefore, during the process of recombinogenic engineering and propagation of BAC clones in E. coli, AK-BAC-GFP fully retained the ability to express viral latent gene products. We also examined the expression of BARTs, the transcripts of which are transcribed across the region deleted in strain B95-8. Despite the presence of the GFP/Neor/Kmr transgene, BART mRNA could still be detected in Akata cells and LCLs harboring AK-BAC-GFP (Fig. 7C).
FIG. 7.
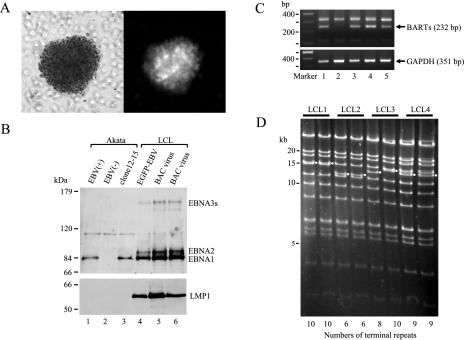
LCLs established by infection with AK-BAC-GFP virus. (A) Phase-contrast (left) and fluorescence (right) images of an established LCL. Note that proliferating cells are clustered and 100% GFP positive. (B) Expression of EBNA proteins and LMP1 protein in various cell lines. Whole-cell extracts of Akata cells with wild-type EBV [EBV(+)], EBV-negative [EBV(−)] Akata cells, Akata cells harboring AK-BAC-GFP (clone 12-15), and LCLs established with EGFP-EBV or with AK-BAC-GFP virus were analyzed for the expression of EBNA and LMP1 proteins. Note that LCLs derived from AK-BAC-GFP virus exhibit type III latency (lanes 5 and 6), while EBV-positive Akata cells (lane 1) and Akata cells harboring AK-BAC-GFP (lane 3) exhibit type I latency. (C) RT-PCR analysis demonstrating the expression of BARTs derived from AK-BAC-GFP. The lanes are numbered identically to the lanes in panel B. Expression of glyceraldehyde-3-phosphate dehydrogenase (GAPDH) is presented as a control. (D) EcoRI digestion of BAC clones rescued from four independent LCLs established by AK-BAC-GFP virus. The bands containing terminal repeats are indicated by small white circles, and the copy numbers of terminal repeats estimated from the sizes of these bands are indicated at the bottom of the gel.
We next examined whether we could rescue intact BACmids from established LCLs. Episomal DNAs were prepared from the established LCLs, and they were transferred back into E. coli. Restriction enzyme digestion revealed that recovered BAC clones exhibited identical DNA fragments, except for the altered copy numbers of terminal repeats compared to those of AK-BAC-GFP, which had been used for transfection of Akata cells (Fig. 7D). These results indicate that the structure of AK-BAC-GFP, including the inserted BAC vector sequence, was stably maintained during the processes of virus production, infection, and lymphocyte transformation.
Rapid insertion of transgenes using an I-PpoI site artificially introduced into AK-BAC.
Experimental data described above clearly demonstrate that AK-BAC can be used as a replication-competent vector for expressing various transgenes in LCLs. As LCLs expressing transgenes can be used as antigen-presenting cells for immunotherapy (13, 22), expediting the process of transgene insertion into the EBV genome is a matter of importance. Therefore, we generated a derivative of AK-BAC, to which any kind of cDNA cassette could be inserted by simple in vitro ligation. For this purpose, we utilized a rare-cutting restriction enzyme, called I-PpoI, which has been used for modifying a large construct (>100 kb) (36). As the recognition sequence of I-PpoI is longer than 15 bp (1), no I-PpoI site exists in the genome of Akata strain EBV as well as in various AK-BAC derivatives. A cassette consisting of an SV40 enhancer and promoter, a synthetic I-PpoI site, a polyadenylation signal, and Neor and Kmr genes was inserted into the same location of AK-BACΔneoΔkm, where the GFP/Neor/Kmr cassette had been inserted to construct AK-BAC-GFP (compare Fig. 4A and 8A). The successful introduction of this cassette was first verified by restriction enzyme digestion (EcoRI, BamHI, and NcoI) and agarose gel analyses (data not shown). The resultant BAC clone, designated AK-BAC-IPpoI, was then digested with I-PpoI and analyzed by pulsed-field gel analyses. The results revealed that I-PpoI digestion of two independent clones of AK-BAC-IPpoI generated a unique 170-kb band (Fig. 8B), indicating the successful introduction of an artificial I-PpoI site.
FIG. 8.
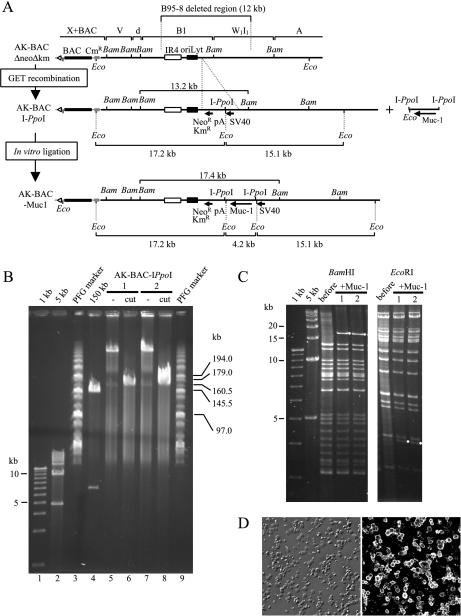
Insertion of the Muc-1 transgene into the genome of Akata strain EBV in vitro. (A) Map of the transgene insertion location. GET recombination was first used to insert a cassette containing an I-PpoI site. AK-BAC-IPpoI has a unique I-PpoI site flanked with an SV40 enhancer and promoter and a polyadenylation signal (pA). An I-PpoI fragment containing a Muc-1 cDNA was inserted into the I-PpoI site of AK-BAC-IPpoI by in vitro ligation to generate AK-BAC-Muc1. The positions of BamHI (Bam) and EcoRI (Eco) sites are indicated. (B) DNAs of two independent clones of AK-BAC-IPpoI before (lanes 5 and 7) and after I-PpoI digestion (lanes 6 and 8) were analyzed by pulsed-field gel electrophoresis. For a control, a BAC clone with a 150-kb NotI insert was digested by NotI and analyzed on the same gel (lane 4). DNA size markers of 1-kb ladder (lane 1), 5-kb ladder (lane 2), together with MidRange I pulsed-field gel (PFG) markers (New England BioLabs) (lanes 3 and 9), were resolved on the same gel. (C) Restriction digestion analysis of BAC clones before and after Muc-1 insertion (two independent clones). The bands that appeared after Muc-1 insertion are indicated by small white circles. (D) A fluorescence image demonstrating Muc1 expression in established LCLs (right) is shown with a corresponding differential interference contrast image (left).
AK-BAC-IPpoI can then be used to subclone transgenes by simple in vitro ligation. As an example, we chose a transgene of Muc-1 mucin (3), one of the tumor-associated antigens frequently used for cancer immunotherapy. AK-BAC-IPpoI was linearized with I-PpoI and then dephosphorylated, and the linear 170-kb vector was ligated with an I-PpoI fragment containing Muc-1 cDNA. We successfully obtained BAC clones that had a correctly orientated Muc-1 cDNA inserted into the unique I-PpoI site of AK-BAC-IPpoI (Fig. 8C).
AK-BAC-Muc1 was then transfected into EBV-positive Akata cells via electroporation, and transfected cells were selected by G418. BACmids were rescued from G418-resistant cell clones and analyzed for the presence of intact AK-BAC-Muc1. We obtained three cell clones in which intact episomes of AK-BAC-Muc1 coexisted with wild-type EBV episomes (Table 1, experiments 7 and 8). Treating the cell lines with anti-IgG readily produced the mixture of wild-type and AK-BAC-Muc1 viruses, and the mixture was then used for infecting cord blood lymphocytes. The established LCLs were initially 30 to 40% Muc1 positive, but they became 100% Muc1 positive after G418 selection (Fig. 8D). Intact AK-BAC-Muc1 was rescued from the resultant LCLs, and a PCR-based assay revealed minimum contamination of wild-type EBV episome. Therefore, we conclude that AK-BAC-Muc1 retains the ability to transform B lymphocytes and that I-PpoI site-mediated cloning can greatly accelerate the establishment of LCLs expressing various transgenes.
DISCUSSION
We cloned the genome of Akata strain EBV as a BAC clone, and the resultant BAC clone was designated AK-BAC. All the restriction enzyme digestion data of this study fit an available restriction map of the genome of Akata strain EBV (K. Takada, unpublished data), strongly arguing that the BAC clone contains the entire EBV genome without any deletion. Although B95-8 strain of EBV genome has been cloned as an infectious BAC clone (8), our AK-BAC is the first to clone the entire EBV genome including the region which is deleted in B95-8 strain of EBV (6, 21). We successfully produced infectious viruses of AK-BAC-GFP (Fig. 4 to 7), AK-BAC-GFP2 (Fig. 4), and AK-BAC-Muc1 (Fig. 8). They were maintained as episomes in infected cells and could be rescued from cells and transferred to E. coli as intact BAC clones, indicating that AK-BAC can shuttle between human cells and E. coli. Importantly, insertion of a BAC vector sequence and transgenes into the genome of Akata strain EBV had little, if any, effect on its transforming ability.
Our system is the first to use B cells (Akata cells) for producing recombinant viruses that have been engineered using the BAC system. We also tested the feasibility of using epithelial cells (293 cells) for producing AK-BAC-GFP viruses according to the previous report (8). We successfully produced infectious viruses after stimulating virus production by BZLF1 transfection, but the amount of recombinant virus was far less than that obtained from anti-IgG-stimulated Akata cells (data not shown). Although there is plenty of room for improving the 293 cell-based system, the Akata cell system has the advantage of producing large quantities of recombinant viruses by simple anti-IgG treatment. AK-BAC-GFP and AK-BAC-GFP2 presented in this study would serve as starting materials for genetic analyses of EBV, and such analyses can be performed using Akata cells under physiological conditions.
We found that EBV-positive Akata cells, but not EBV-negative ones, efficiently supported episome formation of transfected AK-BAC-GFP (Table 1). One interpretation of this result is that EBNA1 expression was insufficient after transfection of naked DNA (AK-BAC-GFP) into EBV-negative Akata cells. EBNA1 protein is essential for episomal replication of oriP plasmids (15) as well as for minimizing mitotic loss of oriP plasmids via its chromosome tethering ability (10, 12, 17). On the basis of our observation that EBV-negative, EBNA1-positive Akata cells are apparently more competent to support episome formation than EBV-negative Akata cells, it is most likely that transfected BACmids efficiently form episomes in EBV-positive Akata cells, as EBNA1 protein is supplied from wild-type EBV episomes in trans. We are currently testing whether EBV-negative, EBNA1-positive Akata cells can substitute for EBV-positive Akata cells in our system. On the other hand, AK-BAC-GFP efficiently formed episomes after transfection into 293 cells and HeLa cells (data not shown), and supplying EBNA1 protein in trans was not necessary. The discrepancy of episome formation efficiency between Akata cells and epithelial cells (293 cells and HeLa cells) may be due to the difference of the EBNA1 expression efficiency from transfected BACmid DNAs.
A recombination strategy called GET recombination (19, 20) was successfully used for mutagenesis of several other herpesviruses, including Marek's disease virus (25), Kaposi's sarcoma virus (38), and bovine herpesvirus 1 (16). We used GET recombination to construct EBV recombinants for the first time. The beauty of this protocol is that homology arms of only around 50 bp are needed to obtain homologous recombinants. Therefore, to generate linear targeting constructs, one can conveniently use PCR amplification with a pair of synthetic primers having approximately 50-bp sequences homologous to EBV genome sequences on their 5′ ends (19). Such flexibility would greatly accelerate the generation of various EBV recombinants. Although BAC clones having EBV genomes were propagated in E. coli for many generations during mutagenesis, all the tested BAC clones were very stably maintained in E. coli.
AK-BACΔneoΔkm, which we used for transgenesis, has a BAC vector sequence and a chloramphenicol resistance gene inserted into the BamHI X region of the Akata strain EBV genome (Fig. 3 and 4). By further inserting a GFP/Neor/Kmr cassette into AK-BACΔneoΔkm, we have shown that at least two loci (the region deleted in strain B95-8 and the BamHI X region) can accommodate additional 4.2-kb sequence (the size of GFP/Neor/Kmr) without impairing the ability of producing progeny viruses. We have also shown that a single-cutting I-PpoI site generated within the region deleted in strain B95-8 can be used to quickly insert transgenes into the EBV genome. AK-BAC-Muc1, which was constructed by I-PpoI site-mediated cloning, was efficiently packaged into infectious virions, and LCLs expressing Muc1 mucin were readily established (Fig. 8D). Such LCLs should be useful for stimulating Muc1-specific cytotoxic T cells ex vivo for cancer immunotherapy (13, 22). The most appropriate locus for transgene insertion and the size limitations of transgenes remain to be further clarified.
Introduction of BAC-based technology has enabled various modifications of EBV genomes that had been practically impossible using conventional methodologies. The ability to modify the EBV genome in E. coli, including GET recombination, cre-mediated excision, and I-PpoI site-mediated cloning, enables unprecedented flexibility of genome engineering. AK-BAC system will find a broad range of applications, such as genetic analyses of various viral gene products and development of viral vectors for human gene therapy.
Acknowledgments
We thank P. Ioannou for generously providing us with pGETrec plasmid. We also thank K. Kontani and T. Kudo for providing APR-Muc1 and R. White for helpful suggestions about I-PpoI enzyme. We thank H. Yoshiyama and S. Maruo for helpful discussions and M. Sato and A. Saito for technical assistance.
This work was supported in part by a Grant-in-Aid for Scientific Research from the Ministry of Education, Culture, Sports, Science and Technology, Japan (T.K. and K.T.) and by a grant from the Akiyama Memorial Foundation (T.K.).
REFERENCES
- 1.Argast, G. M., K. M. Stephens, M. J. Emond, and R. J. Monnat, Jr. 1998. I-PpoI and I-CreI homing site sequence degeneracy determined by random mutagenesis and sequential in vitro enrichment. J. Mol. Biol. 280:345-353. [DOI] [PubMed] [Google Scholar]
- 2.Banerjee, S., E. Livanos, and J. M. Vos. 1995. Therapeutic gene delivery in human B-lymphoblastoid cells by engineered nontransforming infectious Epstein-Barr virus. Nat. Med. 1:1303-1308. [DOI] [PubMed] [Google Scholar]
- 3.Batra, S. K., H. F. Kern, A. J. Worlock, R. S. Metzgar, and M. A. Hollingsworth. 1991. Transfection of the human Muc 1 mucin gene into a poorly differentiated human pancreatic tumor cell line, Panc1: integration, expression and ultrastructural changes. J. Cell Sci. 100:841-849. [DOI] [PubMed] [Google Scholar]
- 4.Brune, W., M. Messerle, and U. H. Koszinowski. 2000. Forward with BACs: new tools for herpesvirus genomics. Trends Genet. 16:254-259. [DOI] [PubMed] [Google Scholar]
- 5.Condit, R. C. 2001. Principles of virology, p. 19-51. In D. M. Knipe, P. M. Howley, D. E. Griffin, R. A. Lamb, M. A. Martin, B. Roizman, and S. E. Straus (ed.), Fields virology, 4th ed. Lippincott Williams & Wilkins, Philadelphia, Pa.
- 6.de Jesus, O., P. R. Smith, L. C. Spender, C. Elgueta Karstegl, H. H. Niller, D. Huang, and P. J. Farrell. 2003. Updated Epstein-Barr virus (EBV) DNA sequence and analysis of a promoter for the BART (CST, BARF0) RNAs of EBV. J. Gen. Virol. 84:1443-1450. [DOI] [PubMed] [Google Scholar]
- 7.Delecluse, H. J., and W. Hammerschmidt. 2000. The genetic approach to the Epstein-Barr virus: from basic virology to gene therapy. Mol. Pathol. 53:270-279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Delecluse, H. J., T. Hilsendegen, D. Pich, R. Zeidler, and W. Hammerschmidt. 1998. Propagation and recovery of intact, infectious Epstein-Barr virus from prokaryotic to human cells. Proc. Natl. Acad. Sci. USA 95:8245-8250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hammerschmidt, W., and B. Sugden. 1988. Identification and characterization of oriLyt, a lytic origin of DNA replication of Epstein-Barr virus. Cell 55:427-433. [DOI] [PubMed] [Google Scholar]
- 10.Hung, S. C., M. S. Kang, and E. Kieff. 2001. Maintenance of Epstein-Barr virus (EBV) oriP-based episomes requires EBV-encoded nuclear antigen-1 chromosome-binding domains, which can be replaced by high-mobility group-I or histone H1. Proc. Natl. Acad. Sci. USA 98:1865-1870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Izumi, K. M. 2001. Identification of EBV transforming genes by recombinant EBV technology. Semin. Cancer Biol. 11:407-414. [DOI] [PubMed] [Google Scholar]
- 12.Kanda, T., M. Otter, and G. M. Wahl. 2001. Coupling of mitotic chromosome tethering and replication competence in Epstein-Barr virus-based plasmids. Mol. Cell. Biol. 21:3576-3588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kilger, E., G. Pecher, A. Schwenk, and W. Hammerschmidt. 1999. Expression of mucin (MUC-1) from a mini-Epstein-Barr virus in immortalized B-cells to generate tumor antigen specific cytotoxic T cells. J. Gene Med. 1:84-92. [DOI] [PubMed] [Google Scholar]
- 14.Kitagawa, N., M. Goto, K. Kurozumi, S. Maruo, M. Fukayama, T. Naoe, M. Yasukawa, K. Hino, T. Suzuki, S. Todo, and K. Takada. 2000. Epstein-Barr virus-encoded poly(A)− RNA supports Burkitt's lymphoma growth through interleukin-10 induction. EMBO J. 19:6742-6750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mackey, D., and B. Sugden. 1999. Applications of oriP plasmids and their mode of replication. Methods Enzymol. 306:308-328. [DOI] [PubMed] [Google Scholar]
- 16.Mahony, T. J., F. M. McCarthy, J. L. Gravel, L. West, and P. L. Young. 2002. Construction and manipulation of an infectious clone of the bovine herpesvirus 1 genome maintained as a bacterial artificial chromosome. J. Virol. 76:6660-6668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Marechal, V., A. Dehee, R. Chikhi-Brachet, T. Piolot, M. Coppey-Moisan, and J. C. Nicolas. 1999. Mapping EBNA-1 domains involved in binding to metaphase chromosomes. J. Virol. 73:4385-4392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Maruo, S., L. Yang, and K. Takada. 2001. Roles of Epstein-Barr virus glycoproteins gp350 and gp25 in the infection of human epithelial cells. J. Gen. Virol. 82:2373-2383. [DOI] [PubMed] [Google Scholar]
- 19.Narayanan, K., R. Williamson, Y. Zhang, A. F. Stewart, and P. A. Ioannou. 1999. Efficient and precise engineering of a 200 kb beta-globin human/bacterial artificial chromosome in E. coli DH10B using an inducible homologous recombination system. Gene Ther. 6:442-447. [DOI] [PubMed] [Google Scholar]
- 20.Orford, M., M. Nefedov, J. Vadolas, F. Zaibak, R. Williamson, and P. A. Ioannou. 2000. Engineering EGFP reporter constructs into a 200 kb human beta-globin BAC clone using GET recombination. Nucleic Acids Res. 28:e84. [DOI] [PMC free article] [PubMed]
- 21.Parker, B. D., A. Bankier, S. Satchwell, B. Barrell, and P. J. Farrell. 1990. Sequence and transcription of Raji Epstein-Barr virus DNA spanning the B95-8 deletion region. Virology 179:339-346. [DOI] [PubMed] [Google Scholar]
- 22.Pecher, G., and O. J. Finn. 1996. Induction of cellular immunity in chimpanzees to human tumor-associated antigen mucin by vaccination with MUC-1 cDNA-transfected Epstein-Barr virus-immortalized autologous B cells. Proc. Natl. Acad. Sci. USA 93:1699-1704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rikinson, A. B., and E. Kieff. 2001. Epstein-Barr virus, p. 2575-2627. In D. M. Knipe, P. M. Howley, D. E. Griffin, R. A. Lamb, M. A. Martin, B. Roizman, and S. E. Straus (ed.), Fields virology, 4th ed. Lippincott Williams & Wilkins, Philadelphia, Pa.
- 24.Roizman, B., and D. M. Knipe. 2001. Herpes simplex viruses and their replication, p. 2399-2459. In D. M. Knipe, P. M. Howley, D. E. Griffin, R. A. Lamb, M. A. Martin, B. Roizman, and S. E. Straus (ed.), Fields virology, 4th ed. Lippincott Williams & Wilkins, Philadelphia, Pa.
- 25.Schumacher, D., B. K. Tischer, W. Fuchs, and N. Osterrieder. 2000. Reconstitution of Marek's disease virus serotype 1 (MDV-1) from DNA cloned as a bacterial artificial chromosome and characterization of a glycoprotein B-negative MDV-1 mutant. J. Virol. 74:11088-11098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sharma, R. C., and R. T. Schimke. 1996. Preparation of electrocompetent E. coli using salt-free growth medium. BioTechniques 20:42-44. [DOI] [PubMed] [Google Scholar]
- 27.Shimizu, N., A. Tanabe-Tochikura, Y. Kuroiwa, and K. Takada. 1994. Isolation of Epstein-Barr virus (EBV)-negative cell clones from the EBV-positive Burkitt's lymphoma (BL) line Akata: malignant phenotypes of BL cells are dependent on EBV. J. Virol. 68:6069-6073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shimizu, N., H. Yoshiyama, and K. Takada. 1996. Clonal propagation of Epstein-Barr virus (EBV) recombinants in EBV-negative Akata cells. J. Virol. 70:7260-7263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Simpson, K., and C. Huxley. 1996. A shuttle system for transfer of YACs between yeast and mammalian cells. Nucleic Acids Res. 24:4693-4699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Takada, K. 1984. Cross-linking of cell surface immunoglobulins induces Epstein-Barr virus in Burkitt lymphoma lines. Int. J. Cancer 33:27-32. [DOI] [PubMed] [Google Scholar]
- 31.Takada, K., K. Horinouchi, Y. Ono, T. Aya, T. Osato, M. Takahashi, and S. Hayasaka. 1991. An Epstein-Barr virus-producer line Akata: establishment of the cell line and analysis of viral DNA. Virus Genes 5:147-156. [DOI] [PubMed] [Google Scholar]
- 32.Takada, K., and Y. Ono. 1989. Synchronous and sequential activation of latently infected Epstein-Barr virus genomes. J. Virol. 63:445-449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wagner, M., Z. Ruzsics, and U. H. Koszinowski. 2002. Herpesvirus genetics has come of age. Trends Microbiol. 10:318-324. [DOI] [PubMed] [Google Scholar]
- 34.Wang, F., D. C. Seldin, B. Annis, A. Trocha, and R. P. Johnson. 1998. Immune modulation of human B lymphocytes by gene transfer with recombinant Epstein-Barr virus amplicons. J. Virol. Methods 72:81-93. [DOI] [PubMed] [Google Scholar]
- 35.Wang, J., and J. M. Vos. 2002. Infectious Epstein-Barr virus vectors for episomal gene therapy. Methods Enzymol. 346:649-660. [DOI] [PubMed] [Google Scholar]
- 36.White, R. E., R. Wade-Martins, and M. R. James. 2002. Infectious delivery of 120-kilobase genomic DNA by an Epstein-Barr virus amplicon vector. Mol. Ther. 5:427-435. [DOI] [PubMed] [Google Scholar]
- 37.Yang, L., S. Maruo, and K. Takada. 2000. CD21-mediated entry and stable infection by Epstein-Barr virus in canine and rat cells. J. Virol. 74:10745-10751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhou, F. C., Y. J. Zhang, J. H. Deng, X. P. Wang, H. Y. Pan, E. Hettler, and S. J. Gao. 2002. Efficient infection by a recombinant Kaposi's sarcoma-associated herpesvirus cloned in a bacterial artificial chromosome: application for genetic analysis. J. Virol. 76:6185-6196. [DOI] [PMC free article] [PubMed] [Google Scholar]