

Abstract
Objective
Glucagon-like peptide-1 (GLP-1) plays a major role in pancreatic β-cell function and survival by increasing cytoplasmic cAMP levels, which are thought to affect transcription through activation of the basic leucine zipper (bZIP) transcription factor CREB. Here, we test CREB function in the adult β-cell through inducible gene deletion.
Methods
We employed cell type-specific and inducible gene ablation to determine CREB function in pancreatic β-cells in mice.
Results
By ablating CREB acutely in mature β-cells in tamoxifen-treated CrebloxP/loxP;Pdx1-CreERT2 mice, we show that CREB has little impact on β-cell turnover, in contrast to what had been postulated previously. Rather, CREB is required for GLP-1 to elicit its full effects on stimulating glucose-induced insulin secretion and protection from cytokine-induced apoptosis. Mechanistically, we find that CREB regulates expression of the pro-apoptotic gene p21 (Cdkn1a) in β-cells, thus demonstrating that CREB is essential to mediating this critical aspect of GLP-1 receptor signaling.
Conclusions
In sum, our studies using conditional gene deletion put into question current notions about the importance of CREB in regulating β-cell function and mass. However, we reveal an important role for CREB in the β-cell response to GLP-1 receptor signaling, further validating CREB as a therapeutic target for diabetes.
Keywords: CREB, Leucine zipper transcription factor, cAMP signaling, Conditional gene ablation, Apoptosis, p21
1. Introduction
The cyclic AMP response element (CRE) binding protein (CREB) is a basic leucine zipper transcription factor that belongs to the CREB/ATF1 gene family [1]. Early in vitro studies suggested a role of CREB in transcription of insulin [2], glucagon [3,4], somatostatin [5], and islet amyloid polypeptide [6], implicating CREB as an indispensable component of the transcriptional network of the endocrine pancreas. This is perhaps best exemplified by the suggestion that CREB is among the primary targets responsible for the therapeutic benefit of GLP-1 analogs, receptor agonists, and DPPIV inhibitors in the β-cell [7–12]. These compounds have proven to be useful treatments for type 2 diabetes based on their ability to promote β-cell function and survival [13–16]. However, the mechanisms underlying the role that CREB plays in these pathways remain unclear.
Despite the preponderance of reports implicating CREB as a key determinant of gene expression in islets, surprisingly little is known about the in vivo function of CREB in the endocrine pancreas. Since germline deletion of CREB results in perinatal lethality due to defects in multiple tissues [17], our current understanding of CREB function in the pancreas has largely been derived from dominant-negative approaches, which have yielded conflicting results.
Mice transgenically expressing a dominant-negative peptide termed ‘A-CREB’ under the control of the rat insulin promoter, and thus in islet β-cells, developed a diabetic phenotype at about 8 weeks of age due to an increase in apoptosis causing a progressive decrease in β-cell mass [18]. This defect in β-cell survival was attributed to downregulation of insulin receptor substrate-2 (IRS-2), which was proposed as a direct target of CREB activation [18]. A second model employing β-cell-restricted, transgenic overexpression of the inducible cAMP early repressor (ICER), a splice variant of the cAMP response element modulator (CREM) gene that can inhibit CREB and related proteins, also elicited a diabetic phenotype, but by an entirely different mechanism [19]. Strikingly, these mice were already severely hyperglycemic on postnatal day 7, much earlier than the phenotype onset observed in the A-CREB model. Furthermore, there was no evidence of increased apoptosis in ICER transgenic animals despite a loss in β-cell mass. Rather, reduced islet mass in this mouse model was attributed to impaired β-cell proliferation due to reduced levels of the cell cycle regulator cyclin A2 [19]. In sum, studies using dominant-negative forms of CREB came to contradictory conclusions regarding the function of CREB in the β-cell [16,17]. A limitation of both dominant negative-based studies is that they target multiple related leucine zipper proteins, and thus do not directly test the function of CREB itself [20].
Therefore, to assess the specific role of CREB in the β-cell in vivo, we employed a conditional gene ablation approach using CrebloxP/loxP mice. By deleting CREB in adult islet β-cells using the Pdx1-CreERT2 transgene, we demonstrate that while CREB plays only a limited role in regulating basal β-cell function or turnover, it is essential for mediating the effects of GLP-1 in stimulating glucose-induced insulin secretion and protection from cytokine-induced β-cell apoptosis.
2. Materials and methods
2.1. Animals
A 12 kb DNA fragment containing exon 11 of the Creb gene was retrieved from C57BL/6J mouse BAC clone RP23-31C24 via bacterial recombination [21]. One loxP site was inserted upstream of exon 11 while a pair of loxP sites flanking a neomycin selection cassette was placed downstream of exon 11. The linearized targeting vector was electroporated into B6 ES cells (Chemicon) and surviving selection was screened for homologous recombination by Southern blot analysis. Targeted clones were injected into C57BL/6J-derived blastocyts that were then transferred to pseudopregnant females. Male offspring were mated to C57BL/6J females and ES cell-derived offspring were identified by PCR-based genotyping. Mice harboring the targeted insertion of the three loxP sites in the Creb locus were crossed to the EIIa-Cre line to achieve mosaic germline deletion of loxP-flanked sequences [22]. Offspring determined to contain the intended loxP allele (CrebloxP/loxP) were identified and crossed to the Pdx1-CreERT2 line [23,24]. CrebL/+ offspring also heterozygous for the Pdx1CreERT2 transgene were then intercrossed with CrebL/+mice to yield mice homozygous for loxP allele and heterozygous for the Pdx1-CreERT2 transgene (CrebloxP/loxP;Pdx1-CreERT2). Creb+/+, Creb+/+;Pdx1-CreERT2, CrebL/+ and CrebloxP/loxP littermates were phenotypically indistinguishable and were used as controls. 8–10 weeks old male and female mice were used for glucose tolerance test. Male and female mice were fed either normal chow (10 kcal% fat) (Research Diets) or high-fat (60 kcal% fat) (Research Diets) diets for 10–11 weeks starting when the mice were 4–5 weeks old. Mice were treated with 0.1 mg/g body weight tamoxifen daily for 3 days via intraperitoneal injection, and glucose tolerance tests and tissue harvesting were performed one week after tamoxifen administration. We did not observe any worsening of the phenotypes in the mice analyzed 30 days after tamoxifen administration (data not shown). All protocols were approved by the Institutional Animal Care and Use Committee of the University of Pennsylvania.
2.2. Islet isolation
Adult islets for all experiments were prepared based on standard collagenase procedures, as described previously in Ref. [25].
2.3. RNA isolation and quantitative RT-PCR analysis
Total cellular RNA was extracted from isolated islets using the RNeasy Kit (Qiagen) and TRIzol (Invitrogen), and then assayed for quantity and quality with the Agilent 2100 Bioanalyzer (Agilent Technologies). RNA was reverse transcribed using oligo (dT) and Superscript II reverse transcriptase (Invitrogen). Quantitative polymerase chain reaction (PCR) reactions were performed using SyBr Green QPCR Master Mix (Agilent Technologies) on an Mx3000 PCR cycler (Agilent Technologies). Reactions were performed in triplicate and normalized relative to the ROX reference dye. Median cycle threshold values were determined and used for analyses. Expression levels were normalized to those of Glyceraldehyde 3-phosphate dehydrogenase (Gapdh) and TATA box binding protein (Tbp) as the internal controls. Primer information is available at http://www.med.upenn.edu/kaestnerlab/index.shtml.
2.4. Immunostaining
Tissues were fixed in 4% paraformaldehyde overnight at 4 °C before embedding in paraffin. Slides (8–10 μm sections) were subjected to microwave antigen retrieval in 10 mM citric acid buffer (pH 6.0) and blocked in PBT (1× PBS, 2% BSA, 0.1% Triton X-100) at room temperature. Slides were incubated with primary antibodies diluted in Zymed Antibody Diluent overnight at 4 °C, and appropriate secondary antibodies diluted in Zymed Antibody Diluent were added for 2 h at room temperature. For immunofluoresecence staining of CREB, frozen sections from OCT compound (Sakura Finetek)-embbeded tissues were blocked with CAS-Block (Invitrogen). Primary and secondary antibodies were diluted in CAS-Block (Invitrogen). TUNEL staining was performed with the Chemicon Apoptag Peroxidase In Situ Apoptosis Detection Kit.
For endocrine cell mass determinations, serial sections were stained for chromogranin A, insulin, or glucagon. IPVision software was then used to morphometrically quantify the stained area relative to the size of the tissue section. Ratios were then multiplied by pancreas weight to obtain cell mass.
The following antibodies were used: guinea pig anti-insulin (Linco Research Inc.), rabbit anti-glucagon (prediluted; Zymed Laboratories Inc.), rabbit anti-CREB (Cell Signaling 48H2), biotinylated goat anti-guinea pig IgG (Vector Laboratories), biotinylated goat anti-rabbit IgG (Vector Laboratories), Cy2-conjugated donkey anti-guinea pig IgG (Jackson), Cy3-conjugated donkey anti-guinea pig IgG (Jackson), and Cy5-conjugated donkey anti-rabbit IgG (Jackson).
2.5. Determination of blood glucose levels and glucose tolerance tests
Blood was sampled from the tail vein of CrebloxP/loxP;Pdx1-CreERT2 mice and control mice allowed to feed ad libitum or fasted overnight for 16 h. Glucose levels were measured with a OneTouch Ultra Glucometer (Lifescan Inc.). For glucose tolerance tests, animals were fasted overnight before being injected intraperitoneally (IPGTT) or gavaged (OGTT) with 2 g of glucose (Sigma–Aldrich) per kilogram of body weight for. Glucose levels were measured just before injection (0 min), then at the indicated times post-injection. For determination of serum insulin concentrations during glucose tolerance tests, blood was collected from the tail vein at 0, 2, 5, 15, and 30 min after injection. Plasma insulin measurements were performed by ELISA (Mercodia). Breeze 2 Glucometer (Bayer) was used to determine glucose levels of CrebloxP/loxP mice and CrebloxP/loxP;Pdx1-CreERT2 mice.
2.6. Perifusion
Perifusion experiments were performed by the University of Pennsylvania Diabetes Research Center (P30-DK19525). Briefly, islets were hand-picked and cultured for 3–4 days. Islets were perifused with Krebs bicarbonate buffer (2.2 mM Ca2+, 0.25% bovine serum albumin, 10 mM HEPES [acid], and 95% O2 and 5% CO2 equilibration, pH 7.4) to reach baseline hormone secretion values before the addition of the appropriate secretagogues. Samples were collected at regular intervals with a fraction collector (Waters Corporation) and insulin content was determined using a radioimmunoassay (University of Pennsylvania Diabetes Research Center).
2.7. Glucose-induced insulin secretion
Islets were handpicked then cultured overnight in mouse islet medium (1× RPMI, 10% FBS, 1× penicillin/streptomycin, 50 μg/mL Gentamycin, 2 mM l-glutamine, 5 mM Hepes, 5 mM glucose), then transferred to Krebs bicarbonate buffer for 2 h in the presence or absence of 100 nM exendin-4. Islets were then transferred to fresh Krebs bicarbonate buffer with 2 mM glucose in the presence or absence of 100 nM exendin-4. After 30 min, the supernatant was sampled and then supplemented with glucose to 15 mM. After 30 min, the supernatant was sampled again. Insulin measurements were performed by ELISA (Mercodia).
2.8. Induction of apoptosis
Islets were isolated from four CrebloxP/loxP mice and three CrebloxP/loxP;Pdx1-CreERT2 mice and cultured overnight in islet medium. Islets from each mouse were divided evenly and transferred to islet medium with or without 100 nM exendin-4 for 2.5 h. The islets were then cultured in islet medium with or without cytokines (10 ng/mL interleukin-1 beta, 25 ng/mL tumor necrosis factor alpha, and 10 ng/mL interferon gamma) for 24 h, in the continued presence or absence of exendin-4 [26]. Terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) assay and staining for insulin were performed as described previously in Ref. [27]. Immunofluoresence was detected using a Nikon Eclipse Ti-U confocal microscope (University of Pennsylvania Molecular Pathology and Imaging Core).
2.9. Microarray analyses
Hybridization of samples and data analyses were performed by University of Pennsylvania Functional Genomics Core (P30-DK19525). Briefly, total RNAs from extracted from islets of CrebloxP/loxP and CrebloxP/loxP;Pdx1-CreERT2 high-fat diet-fed female mice treated with 0.1 mg/g body weight tamoxifen daily for 3 days (8 mice per each group) were amplified and labeled using Low Input Quick Amp Labeling Kits (Agilent Technologies). Labeled samples were hybridized overnight to the Agilent 4X44 Whole Mouse Genome Array, and arrays were scanned with the model G2565B Agilent DNA microarray Scanner (Agilent Technologies). The subsequent analysis was performed as previously in Ref. [28]. Gene functional classification was performed on differentially expressed genes with more than 1.5-fold and false discovery rate less than 10, using Ingenuity Pathways Analysis (Ingenuity® Systems, www.ingenuity.com).
2.10. ChIP-Seq analyses
Islet chromatin was prepared from approximately 600 mouse islets each for two biological replicates, as previously described in Ref. [29]. Immunoprecipitations were performed using anti-CREB (Santa Cruz Biotech, sc-186) as previously described in Ref. [30]. Multiplexed libraries were prepared using the NEBNext ChIP-Seq Library Prep Reagent Set (NEB E6200) and sequenced on an Illumina hiSeq2000. Sequence reads were mapped to the mouse genome (mm8) using ELAND. Only those reads with a unique best match containing up to 2 mismatches were used for further analysis. Multiple reads mapping to identical positions were condensed to a single read to remove amplification artifacts. Peak calling was carried out by HOMER (v3.16, 0.1% false discovery rate, default settings) [31].
2.11. RNA-Seq analyses
Following glucose-stimulated insulin secretion assays, islets were collected from four control and for CREB-deficient mice each and total RNA was extracted as described above. Multiplexed RNA-Seq libraries were prepared from 200 ng of total RNA using the NEBNext Ultra RNA Library Prep Kit (NEB). mRNA enrichment was performed using the NEBNext Poly(A) Magnetic Isolation Module (NEB). Libraries were single-end sequenced on an Illumina hiSeq2000. Sequencing reads were analyzed and gene expression profiles were determined as previously described in Ref. [32]. Reads from ribosomal RNA and genomic repeats were identified by aligning the 5′ 50 bp of each read to ribosomal sequences and mouse repeats in RepBase using Bowtie [33], allowing up to three mismatches. The remaining reads were processed with RUM [34] and aligned to the set of known transcripts included in RefSeq, UCSC known genes, and ENSEMBL transcripts, and the mouse genome (mm9). Transcript-, exon-, and intron-level quantification was done using only the uniquely aligning reads. To analyze global gene expression profiles, the number of uniquely aligning read counts to mRNA transcripts in RefSeq were extracted from the RUM output and processed by EDGE. The p-values from EDGE were corrected for multiple testing using the Benjamini & Hochberg mode of the R function p.adjust. We then summarized these data for individual genes by selecting a “representative transcript” with the highest read counts for each gene. The fold-change computed in EDGE expressed as the ratio of mutant versus control is shown in Figure 5C, with those determined to be statistically significantly different indicated by stars.
Figure 5.
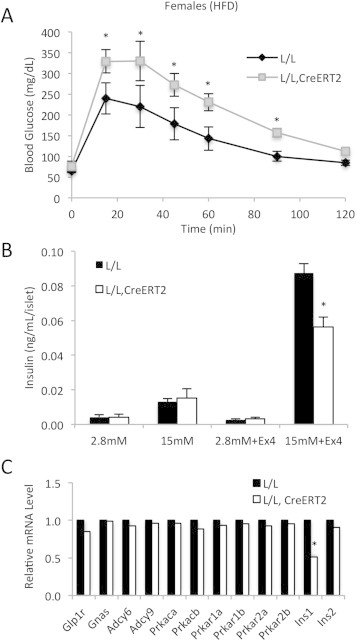
Impaired insulinotropic effect of GLP-1 signaling CrebloxP/loxP;Pdx1CreERT2 mutants. (A) Blood glucose levels during oral glucose tolerance tests of control (L/L) and mutant (L/L, CreERT2) female mice (n = 4) after tamoxifen administration. Female mice were fed a high-fat diet for 10–11 weeks starting when the mice were 4–5 weeks old. *p < 0.05 by ANOVA. (B) Glucose-stimulated insulin secretion was measured in islets from tamoxifen-treated control and mutant mice incubated in the presence or absence of 100 nM exendin-4 (n = 4). *p < 0.05. All values are expressed as mean + SEM. (C) RNA-Seq analyses to measure relative levels of insulin and components of the GLP-1 signaling pathway in control (n = 4) and mutant islets (n = 4) incubated in the presence of 100 nM exendin-4. *False discovery rate <1% by EDGE analysis (see Section 2.11 for details).
2.12. Statistical analysis
Standard ANOVA with Tukey's post-test was used to determine the significance of differences between groups in GTT and perifusion assays. Student t-tests with equal variance and two-tailed distribution were used for all other comparisons. A p-value of 0.05 was considered statistically significant.
3. Results
3.1. Islet-specific deletion of CREB in CrebloxP/loxP;Pdx1-CreERT2 mice
For conditional deletion of CREB, we employed CrebloxP/loxP mice, in which two loxP sites flank exon 11, which encodes the basic leucine zipper domain of the Creb gene [35] (Figure 1A), which is essential for gene function [17]. To assess the role of CREB in mature β-cells, we inducibly deleted CREB in adult mice using the Pdx1-CreERT2 transgenic line [23]. One week following tamoxifen administration, CREB was efficiently deleted in islets isolated from CrebloxP/loxP;Pdx1-CreERT2 mice as assessed by quantitative PCR analysis (Figure 1B), while immunohistochemical staining confirmed the specific loss of CREB protein in most β-cells (Figure 1C). Note that CREB protein is still present in insulin-negative islets cells, because the Pdx1 promoter is not active in α-cells. In accordance with previously reported models of CREB ablation [17,36–38], we also observed an upregulation of CREM message in the islets of CrebloxP/loxP;Pdx1-CreERT2 mice, illustrating that CREB normally functions to attenuate CREM expression (Figure 1B). mRNA levels of activating transcription factor-1 (ATF-1), another CREB family member that can be phosphorylated by protein kinase A, on the other hand, were unchanged in mutants islets (Figure 1B).
Figure 1.
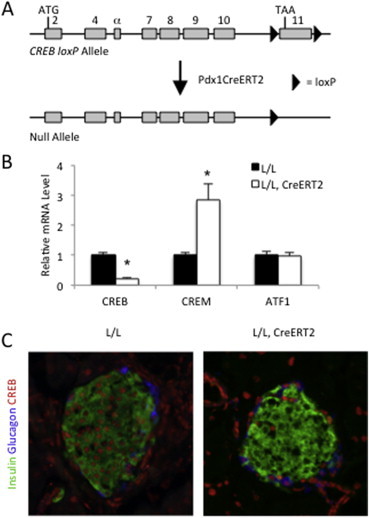
Derivation and validation of the CrebloxP/loxP;Pdx1CreERT2 mouse. (A) loxP sites were positioned to flank exon 11 of the Creb gene, which codes for much of the leucine zipper domain. Mice harboring the CrebloxP allele were crossed to mice expressing tamoxifen-inducible Cre recombinase (CreERT2) under control of the Pdx1 enhancer driving expression in β-cells. Recombination between loxP sites only occurs following tamoxifen administration. (B) Quantitative RT-PCR to measure Creb, Crem and Atf1 mRNA levels in islets isolated from tamoxifen-treated control (L/L) and mutant (L/L, CreERT2) mice. Female mice were fed a high-fat diet (HFD) for 10–11 weeks starting when the mice were 4–5 weeks old. Transcript levels in control mice were assigned an arbitrary value of 1 for comparison (n = 8). Values are expressed as mean + SEM; *p < 0.05. (C) Immunostaining of pancreas sections demonstrating efficient deletion of the Creb protein in islet β-cells. Female mice were fed a high-fat diet for 10–11 weeks starting when the mice were 4–5 weeks old. In control (L/L) animals, Creb-positive nuclei (red) are present in all cell types including insulin-positive β-cells (green) and glucagon-positive α-cells (blue) in endocrine islets, while Creb protein is selectively absent from β-cells in mutant (L/L, CreERT2) pancreata following tamoxifen injection.
3.2. β-Cell mass is not reduced in CrebloxP/loxP;Pdx1-CreERT2 mice
Prior studies using dominant-negative approaches have suggested that CREB plays an important role in maintaining β-cell mass [18,19]. Jhala and colleagues observed massive β-cell apoptosis in their transgenic mouse model and attributed that to downregulation of insulin receptor substrate-2 (IRS-2) [18]. In contrast, Inada and colleagues found no evidence for increased apoptosis in their transgenic model of ICER overexpression, but rather observed reduced β-cell replication, which they ascribed to attenuated cyclin A2 expression [19]. Therefore, we assessed β-cell mass, as well as expression of these two proposed targets of CREB activity in β-cells, in our genetic model of CREB ablation. Surprisingly, β-cell mass was not altered in high-fat diet-fed female mutant mice compared to controls (Figure 2A). Likewise, mRNA levels of insulin and glucagon did not change following deletion of CREB (Figure 2B), indicating that CREB deficiency in mature β-cell does not affect β-cell mass or hormone gene expression. Furthermore, mRNA levels of both the insulin receptor substrate 2 (Irs2) and cyclin A2 (Ccna2) were unaffected in CREB-deficient islets (Figure 2C). In addition, gene expression of Irs1, cyclin D1 (Ccnd1), and B-cell lymphoma 2 (Bcl2), three additional proposed CREB targets that could have an impact on β-cell turnover, were similarly unchanged in the islets of CREB mutants (Figure 2C). Finally, proliferation rates of β-cells detected by continuous 5-bromo-2′-deoxyuridine (BrdU) labeling remained unaltered by CREB deficiency (Figure 2D). These data strongly suggest that the previously reported phenotypes in the dominant-negative transgenic mouse models were the result of off-target effects (see Section 4).
Figure 2.
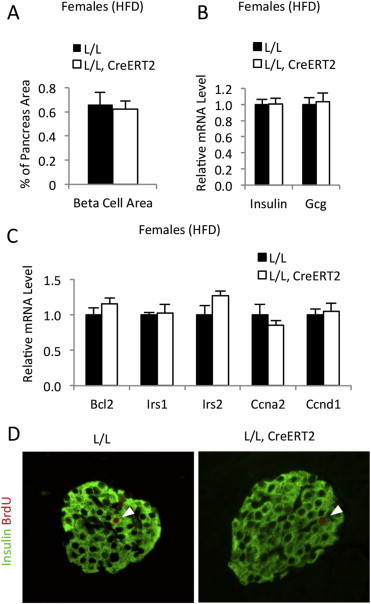
β-cell Mass is not affected in high-fat diet-fed CrebloxP/loxP;Pdx1CreERT2 mice. (A) Quantification of β-cell mass in control (L/L) and mutant (L/L, CreERT2) mice (n = 4–5). Female mice were fed a high-fat diet for 10–11 weeks starting when the mice were 4–5 weeks old. (B) Quantitative RT-PCR to measure relative mRNA levels of insulin (Ins1 and Ins2) and glucagon (Gcg) in islets isolated from control (L/L) and mutant (L/L, CreERT2) female mice fed a high-fat diet (n = 10–14). (C) Quantitative RT-PCR to measure relative mRNA levels of presumed Creb target genes that may be associated with β-cell turnover in islets isolated from tamoxifen-treated control (L/L) and mutant (L/L, CreERT2) female mice fed a high-fat diet. Transcript levels in control mice were assigned an arbitrary value of 1.0 for comparison (n = 6). All values are expressed as mean + SEM. (D) Immunostaining of pancreas sections demonstrating that replication of islet β-cells is unaffected by Creb deficiency. Female mice were fed a high-fat diet for 10–11 weeks starting when the mice were 4–5 weeks old. Insulin-positive β-cells (green) and 5-bromo-2′-deoxyuridine (BrdU)-positive cells (red) are shown. Arrowheads indicate β-cells labeled with BrdU. BrdU were administered in drinking water (1 g/L) for one week.
3.3. Glucose homeostasis is not dramatically affected in CrebloxP/loxP;Pdx1-CreERT2 mice
Next, we assessed the effect of CREB deficiency in the adult β-cell on glucose homeostasis in vivo by measuring fed and fasted blood glucose levels in control and mutant mice. Compared with controls, no differences in fed or fasted blood glucose levels were observed in mice maintained on normal chow (Figure 3A). Furthermore, there was no indication of impairments in glucose clearance in normal chow-fed CrebloxP/loxP;Pdx1-CreERT2 mice during intraperitoneal glucose tolerance tests (IPGTT) (Figure 3B). There were no differences in fed and fasted insulin levels (Figure 3C) as well as in insulin secretion during IPGTT between control and mutant female mice fed normal chow, which is low in triglycerides (Figure 3D).
Figure 3.
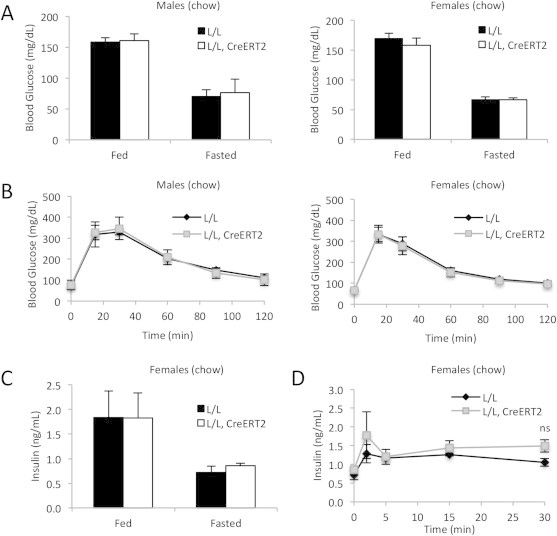
CrebloxP/loxP;Pdx1CreERT2 mice are not glucose intolerant on a normal chow diet. (A) Fed and fasted blood glucose levels of control (L/L) and mutant (L/L, CreERT2) male (n = 6) and female mice (n = 7) after tamoxifen administration. Male and female mice were fed normal chow for 10–11 weeks starting when the mice were 4–5 weeks old. (B) Blood glucose levels during IP glucose tolerance tests of control (L/L) and mutant (L/L, CreERT2) male (n = 6) and female mice (n = 7) one week after tamoxifen administration. (C) Serum insulin levels of control (n = 3) and mutant (n = 3) female mice. (D) Serum insulin levels of control and mutant female mice following glucose injection (n = 3). ns = not significant. All values are expressed as mean + SEM.
After ten weeks on a high-fat diet to metabolically stress β-cells, male mutants displayed glucose clearance comparable to controls (Figure 4A). However, glucose levels were slightly but significantly elevated during the IPGTT in CrebloxP/loxP;Pdx1-CreERT2 females that had been fed a high-fat diet, indicating an impairment in glucose tolerance specifically in that state (Figure 4A). While blood glucose levels and circulating insulin in fed and fasted CREB mutant mice were not significantly different even in mice fed a high-fat diet (Figure 4B and C), insulin secretion during IPGTT experiments tended to be lower in mutants (Figure 4D), and insulin secretion in response to glucose during perifusion assays was impaired in islets isolated from mutant mice compared to controls, especially the first-phase response, which was reduced by more than 60% (Figure 4E and F). Thus, as significant phenotypes were only observed in metabolically challenged female mutant mice, CREB appears to have only a limited involvement in the response of the β-cell to glucose when administered intraperitoneally.
Figure 4.
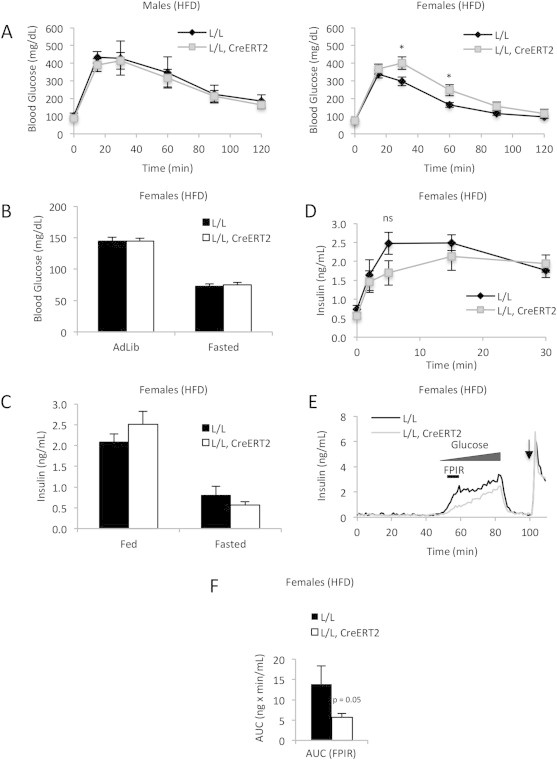
CrebloxP/loxP;Pdx1CreERT2 female mice are mildly glucose intolerant on a high-fat diet. (A) Blood glucose levels during IP glucose tolerance tests of control (L/L) and mutant (L/L, CreERT2) male (n = 4–8) and female mice (n = 12–24) fed a high-fat diet for 10–11 weeks starting when the mice were 4–5 weeks old. *p < 0.05 by ANOVA. (B) Fed and fasted blood glucose levels of control and mutant female mice (n = 12–24). (C) Serum insulin levels of control (n = 8) and mutant (n = 11) female mice. (D) Serum insulin levels of control and mutant female mice following glucose injection (n = 8–11). (E) Insulin secretion in islets isolated from control and mutant female mice in response to a glucose ramp and KCl stimulation (arrow) (n = 3). FPIR indicates the period of first-phase insulin release, which is the time interval used for the area under the curve analysis shown in F. (F) Area under the insulin curve (AUC) during the first-phase insulin response (55–60 min) during the islet perifusion experiment shown in (E) (n = 3). Values are expressed as mean + SEM.
3.4. Exendin-4-potentiated insulin secretion is compromised in CrebloxP/loxP;Pdx1-CreERT2 mice
Intraperitoneal and intravenous glucose tolerance tests bypass exposure of the intestinal epithelium to glucose, and thus do not evaluate the effects of incretins, such as GLP-1 and GIP, on glucose disposal. To test if the effects of incretins on islet β-cells are CREB-dependent, we first performed oral glucose tolerance tests (OGTT). The OGTT revealed a larger impact on glucose excursion in CREB mutants (Figure 5A) than was observed in the IPGTT (Figure 4A), demonstrating that CREB is critical to the ability of incretin signaling to stimulate glucose clearance.
To test the involvement of CREB in the augmentation of insulin secretion by GLP-1, we performed static insulin secretion experiments on isolated islets in the presence or absence of exendin-4, a GLP-1 analog. As expected of islets from mice maintained on a normal chow diet, no differences in glucose-stimulated insulin secretion were observed between controls and mutants not subjected to exendin-4 stimulation (Figure 5B). However, in the presence of exendin-4, insulin secretion in response to 15 mM glucose was markedly blunted in mutant islets compared with controls (Figure 5B). Control and mutant islets incubated in the presence of exendin-4 were used for subsequent RNA-Seq analysis (Figure 5C). Expression levels of the components of the GLP-1 signaling pathway upstream of CREB including GLP-1 receptor (Glp1r), the stimulatory G protein alpha subunit (Gnas), adenylate cyclases (Adcy6 and Adcy9), and subunits of protein kinase A (Prkaca, Prkacb, Prkar1a, Prkar1b, Prkar2a, Prkar2b) remained unchanged. The mRNA levels of Ins1 were lower in mutant islets compared with control islets after exdendin-4 treatment. However, the mRNA levels of Ins2 remained unaffected. Because the mRNA levels of Ins2 were 3.8-fold greater than Ins1 (data not shown), it unlikely that changes in insulin secretion in Figure 5B are primarily attributable to decreased Ins1 steady-state mRNA levels. In addition, the fact that basal insulin mRNA levels of control and mutants mice were same (Figure 2B) suggests that it is unlikely that impaired insulin transcription in response to acute oral glucose administration in mutant mice led to impaired glucose tolerance in Figure 5A. Collectively, these results clearly indicate that CREB is necessary in the adult mouse β-cell for GLP-1 to fully potentiate insulin secretion.
3.5. Increased β-cell apoptosis in CREB-deficient islets
Several studies have documented that chronic inflammation in obese mouse models results in cytokine-mediated β-cell dysfunction, and cytokine-induced β-cell loss is thought to be a major contributor to diabetes [39,40]. Furthermore, it has been postulated that exendin-4 protects β-cells from cytokine-induced apoptosis [15,16,41,42], while previous overexpression and dominant-negative approaches have indicated that CREB might be required for this effect on β-cell survival [7,9]. To test this hypothesis in our model of genetic deletion, we isolated islets from chow-fed tamoxifen-treated CrebloxP/loxP mice and CrebloxP/loxP;Pdx1-CreERT2 mice, and incubated them in medium containing cytokines with or without exendin-4. As expected for mice with no dramatic basal metabolic defect, there was no difference in the percentage of apoptotic β-cells from control and mutant mice when islets were exposed to cytokines at baseline (Figure 6A and B). In control islets, we observed a dramatic decrease in the number of apoptotic β-cells when islets were exposed to both cytokines and exendin-4, confirming the protective effect of GLP-1 signaling reported previously [7,9]. However, the protection conferred by exendin-4 against cytokine-induced cell death was abolished in CREB-deficient islets, indicating that deletion of CREB leads to increased susceptibility of β-cells to inflammatory stress.
Figure 6.
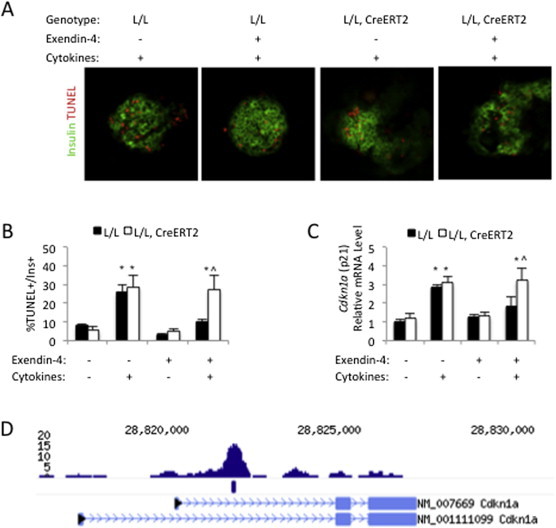
Increased apoptosis in islets isolated from CrebloxP/loxP;Pdx1CreERT2 mice. (A) Islets were isolated from normal chow-fed, control (L/L) and mutant (L/L, CreERT2) female mice, after tamoxifen administration, and then incubated in medium containing cytokines with (+) or without (−) exendin-4. Immunostaining was performed to detect insulin-positive (green) and TUNEL-positive (red) cells (n = 5–6). (B) Quantification of TUNEL-positive cells and insulin-positive cells in isolated islets incubated in medium with (+) or without (−) cytokines and exendin-4 (n = 5–6). (C) Quantitative RT-PCR to measure relative mRNA levels of Cdkn1a in isolated islets incubated in medium with (+) or without (−) cytokines and exendin-4 (n = 4). Transcript levels in untreated islets were assigned an arbitrary value of 1 for comparison. All values are expressed as mean + SEM; *p < 0.05 between treatment groups. ˆp < 0.05 within treatment group. (D) ChIP-Seq analysis of Creb binding in mouse islets identifies a Creb binding site in the first intron of the Cdkn1a gene. A consensus CRE is also found at this locus. The blue mark below the peak indicates that the site was identified as Creb-bound by statistical HOMER analysis.
In order to identify the CREB-regulated target genes that might sensitize CREB-deficient β-cells to cytokines, we performed expression profiling using islets isolated from high-fat diet-fed CrebloxP/loxP and CrebloxP/loxP;Pdx1-CreERT2 female mice, as high-fat diet feeding causes β-cell stress. We identified 27 down-regulated and 38 up-regulated genes in mutant islets (Supplemental Tables 1 and 2). Ingenuity pathway analysis of genes differentially expressed upon acute CREB ablation revealed that one of the top functional categories was ‘cell death’ (Supplemental Table 3). In particular, we observed that expression levels of the cyclin-dependent kinase inhibitor 1A (Cdkn1a or p21), which promotes apoptosis of β-cells in response to various stressors [43–46], was increased by ∼2-fold in mutants islets (Supplemental Table 2). On the other hand, expression of Irs2 and Ccna2, mediators of CREB function in the β-cell proposed by others [18,19], were unchanged following deletion of CREB, consistent with the results shown in Figure 3. To determine if Cdkn1a is a direct target of CREB in the endocrine pancreas, we performed chromatin immunoprecipitation followed by sequencing (ChIP-Seq) analysis using mouse islet chromatin. Our results indicate strong binding of CREB to an evolutionarily conserved region in the first intron of the Cdkn1a gene that contains a consensus CRE (Figure 6C).
Next, to evaluate the involvement of CREB in the regulation of Cdkn1a in response to cytokines and exendin-4, we performed quantitative RT-PCR analysis on islets from normal chow-fed mice. As expected for mice not subject to the additional metabolic stress of a high-fat diet, no differences were observed in baseline levels of Cdkn1a comparing controls to mutants. Cytokines stimulated the expression of Cdkn1a in both groups, while exendin-4 alone had no impact on Cdkn1a levels. However, while exendin-4 prevented the induction of Cdkn1a by cytokines in controls, it was not able to block this increase in CREB-deficient islets (Figure 6D). In summary, our results indicate that augmented β-cell apoptosis in CREB-deficient islets is associated with increased expression of Cdkn1a. Thus, CREB mediates the protective effects of the intestinal hormone GLP-1 on β-cell survival.
4. Discussion
CREB has been proposed as a key determinant of GLP-1 activity in pancreatic islets responsible for the therapeutic benefit of several prominent diabetes drugs. Multiple groups have attempted to define CREB's mechanism of action within the islet β-cell using cell-type specific dominant-negative [18,19] or genetic dominant-active approaches [47]. Both dominant-negative manipulations resulted in severe diabetic phenotypes, albeit through entirely different suggested mechanisms. Importantly, none of these models directly or specifically addressed the importance of CREB, the preeminent factor associated with regulation through CRE sequences, in the endocrine pancreas, due to the well-documented off-target effects of both dominant negative proteins [20].
Here, we used the Cre-loxP system to conditionally delete CREB in the adult endocrine pancreas. In contrast to the dramatic hyperglycemic phenotypes provoked by the action of the dominant-negative peptides, Creb deletion in the adult β-cell had very little impact on glucose homeostasis in mice fed standard rodent chow. Only when challenged by high-fat feeding did mutant female mice display mild glucose intolerance. CREB deficiency in β-cells of male mice was insufficient to elicit a metabolic phenotype regardless of the diet. While our knowledge on the gender effects on CREB expression or signaling in pancreatic β-cells is limited, multiple reports have demonstrated that the levels of CREB expression and/or phospho-CREB are different between males and females in brain [48–50].
While both prior transgenic models described dramatic alterations in β-cell turnover, we detected no changes to β-cell mass in CrebloxP/loxP;Pdx1CreERT2 mutants. Previous studies have also suggested that CREB regulates the expression of the survival genes IRS2 and Bcl-2 as well as proliferation genes such as cyclin A and cyclin D [8,9,18,19]. We found that the expression levels of none of these genes were impacted by CREB deletion, clearly demonstrating that CREB is not necessary for their transcriptional regulation.
This discrepancy, along with the limited impact of CREB deletion on glucose homeostasis and β-cell turnover in vivo, is likely due to the specificity of our model relative to over-expression of dominant-negative peptides that influence the activity of multiple CRE-binding factors. It has been established, for instance, that both A-CREB and ICER can affect the binding of other endogenous leucine zipper-containing proteins [20,51–53]. Furthermore, as A-CREB-containing dimers are incapable of DNA binding, CREs are prone to aberrant occupancy by factors that are not normally able to interact with those sequences when CREB family members are engaged, thus predisposing target genes to abnormal regulation. ICER, on the other hand, is a naturally occurring gene product that consists of only the basic leucine-zipper domain of CREM but lacks an activation domain. Therefore, dimers containing ICER retain the ability to bind CRE sequences, but are unable to activate transcription [54,55]. Thus, ICER represents an extremely potent but non-specific inhibitor of all factors capable of interacting with CREs, not just CREB. In support of this notion, expression levels of many direct CREB targets, including insulin and glucagon, were unaffected by CREB deficiency (Figure 2), suggesting functional overlap with other CRE-associated proteins.
Our findings indicate, however, that CREB has very specific and unique functions within the endocrine pancreas that cannot be fulfilled by related factors. Specifically, we determined that CREB is required in β-cells for the incretin GLP-1 to exert its full effect in potentiating insulin secretion. Thus, in addition to acutely stimulating insulin granule exocytosis through the Epac2/Rap1 pathway [56], elevated cAMP levels in response to GLP-1 receptor activation exert part of their effect through CREB-dependent mechanisms.
In addition, we discovered that CREB is required in order for GLP-1 receptor signaling to assert its protective effects against cytokine-induced cell death, in part through its regulation of targets including Cdkn1a. Other groups have demonstrated that Cdkn1a is regulated by CREB in melanoma cells [57] and myeloid leukemia cells [58], further supporting our results. Notably, our microarray analyses indicated that mRNA levels of Cbp/p300-interacting transactivator 2 (CITED2) were downregulated in CREB mutant mice. CITED2 was shown to be an anti-apoptotic factor that inhibits expression of Cdkn1a in hematopoetic stem cells [59], pointing to a likely CREB-CITED2-p21 axis.
5. Conclusions
Collectively, our studies using conditional gene ablation approaches dispel current opinions about the importance of CREB in regulating β-cell function and mass, but reveal a critical role for CREB in the mechanisms that govern specific aspects of the β-cell response to GLP-1 receptor signaling, further validating CREB as a therapeutic target for diabetes.
Acknowledgments
We thank Sophia Hammani, Beth Helmbrecht, Karrie Brondell and Tia Bernard-Banks for their help in managing the mouse colony. These studies were facilitated by Molecular Pathology and Imaging Core of the Penn Center for Molecular Studies in Digestive and Liver Disease (P30-DK50306) and the University of Pennsylvania Diabetes Center (P30-DK19525) Radioimmunoassay and Biomarker and Functional Genomics Cores. This work was supported by National Institute of Diabetes and Digestive and Kidney Diseases R01-DK055342 and American Diabetes Association mentor-based postdoctoral fellowship award No.: 7-08-MN-28 to K.H.K.
Conflict of interest
The authors have declared that no conflict of interest exists.
Appendix A. Supplementary data
The following is the supplementary data related to this article:
References
- 1.Mayr B., Montminy M. Transcriptional regulation by the phosphorylation-dependent factor CREB. Nature Reviews Molecular Cell Biology. 2001;2(8):599–609. doi: 10.1038/35085068. [DOI] [PubMed] [Google Scholar]
- 2.Oetjen E. Distinct properties of the cAMP-responsive element of the rat insulin I gene. The Journal of Biological Chemistry. 1994;269(43):27036–27044. [PubMed] [Google Scholar]
- 3.Miller C.P., Lin J.C., Habener J.F. Transcription of the rat glucagon gene by the cyclic AMP response element-binding protein CREB is modulated by adjacent CREB-associated proteins. Molecular and Cellular Biology. 1993;13(11):7080–7090. doi: 10.1128/mcb.13.11.7080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schwaninger M. Membrane depolarization and calcium influx induce glucagon gene transcription in pancreatic islet cells through the cyclic AMP-responsive element. The Journal of Biological Chemistry. 1993;268(7):5168–5177. [PubMed] [Google Scholar]
- 5.Montminy M.R., Bilezikjian L.M. Binding of a nuclear protein to the cyclic-AMP response element of the somatostatin gene. Nature. 1987;328(6126):175–178. doi: 10.1038/328175a0. [DOI] [PubMed] [Google Scholar]
- 6.Novials A. Mutation at position -132 in the islet amyloid polypeptide (IAPP) gene promoter enhances basal transcriptional activity through a new CRE-like binding site. Diabetologia. 2004;47(7):1167–1174. doi: 10.1007/s00125-004-1439-y. [DOI] [PubMed] [Google Scholar]
- 7.Sarkar S.A. Dominant negative mutant forms of the cAMP response element binding protein induce apoptosis and decrease the anti-apoptotic action of growth factors in human islets. Diabetologia. 2007;50(8):1649–1659. doi: 10.1007/s00125-007-0707-z. [DOI] [PubMed] [Google Scholar]
- 8.Kim M.J. Exendin-4 induction of cyclin D1 expression in INS-1 beta-cells: involvement of cAMP-responsive element. Journal of Endocrinology. 2006;188(3):623–633. doi: 10.1677/joe.1.06480. [DOI] [PubMed] [Google Scholar]
- 9.Velmurugan K. Antiapoptotic actions of exendin-4 against hypoxia and cytokines are augmented by CREB. Endocrinology. 2012;153(3):1116–1128. doi: 10.1210/en.2011-1895. [DOI] [PubMed] [Google Scholar]
- 10.Klinger S. Increasing GLP-1-induced beta-cell proliferation by silencing the negative regulators of signaling cAMP response element modulator-alpha and DUSP14. Diabetes. 2008;57(3):584–593. doi: 10.2337/db07-1414. [DOI] [PubMed] [Google Scholar]
- 11.Skoglund G., Hussain M.A., Holz G.G. Glucagon-like peptide 1 stimulates insulin gene promoter activity by protein kinase A-independent activation of the rat insulin I gene cAMP response element. Diabetes. 2000;49(7):1156–1164. doi: 10.2337/diabetes.49.7.1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sonoda N. Beta-Arrestin-1 mediates glucagon-like peptide-1 signaling to insulin secretion in cultured pancreatic beta cells. Proceedings of the National Academy of Sciences of the United States of America. 2008;105(18):6614–6619. doi: 10.1073/pnas.0710402105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yabe D., Seino Y. Two incretin hormones GLP-1 and GIP: comparison of their actions in insulin secretion and beta cell preservation. Progress in Biophysics & Molecular Biology. 2011;107(2):248–256. doi: 10.1016/j.pbiomolbio.2011.07.010. [DOI] [PubMed] [Google Scholar]
- 14.Garber A.J., Spann S.J. An overview of incretin clinical trials. The Journal of Family Practice. 2008;57(9 Suppl.):S10–S18. [PubMed] [Google Scholar]
- 15.Li Y. Glucagon-like peptide-1 receptor signaling modulates beta cell apoptosis. The Journal of Biological Chemistry. 2003;278(1):471–478. doi: 10.1074/jbc.M209423200. [DOI] [PubMed] [Google Scholar]
- 16.Wang Q., Brubaker P.L. Glucagon-like peptide-1 treatment delays the onset of diabetes in 8 week-old db/db mice. Diabetologia. 2002;45(9):1263–1273. doi: 10.1007/s00125-002-0828-3. [DOI] [PubMed] [Google Scholar]
- 17.Rudolph D. Impaired fetal T cell development and perinatal lethality in mice lacking the cAMP response element binding protein. Proceedings of the National Academy of Sciences of the United States of America. 1998;95(8):4481–4486. doi: 10.1073/pnas.95.8.4481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jhala U.S. cAMP promotes pancreatic beta-cell survival via CREB-mediated induction of IRS2. Genes & Development. 2003;17(13):1575–1580. doi: 10.1101/gad.1097103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Inada A. Overexpression of inducible cyclic AMP early repressor inhibits transactivation of genes and cell proliferation in pancreatic beta cells. Molecular and Cellular Biology. 2004;24(7):2831–2841. doi: 10.1128/MCB.24.7.2831-2841.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ahn S. A dominant-negative inhibitor of CREB reveals that it is a general mediator of stimulus-dependent transcription of c-fos. Molecular and Cellular Biology. 1998;18(2):967–977. doi: 10.1128/mcb.18.2.967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Copeland N.G., Jenkins N.A., Court D.L. Recombineering: a powerful new tool for mouse functional genomics. Nature Reviews Genetics. 2001;2(10):769–779. doi: 10.1038/35093556. [DOI] [PubMed] [Google Scholar]
- 22.Holzenberger M. Cre-mediated germline mosaicism: a method allowing rapid generation of several alleles of a target gene. Nucleic Acids Research. 2000;28(21):E92. doi: 10.1093/nar/28.21.e92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gu G., Dubauskaite J., Melton D.A. Direct evidence for the pancreatic lineage: NGN3+ cells are islet progenitors and are distinct from duct progenitors. Development. 2002;129(10):2447–2457. doi: 10.1242/dev.129.10.2447. [DOI] [PubMed] [Google Scholar]
- 24.Heiser P.W. Stabilization of beta-catenin impacts pancreas growth. Development. 2006;133(10):2023–2032. doi: 10.1242/dev.02366. [DOI] [PubMed] [Google Scholar]
- 25.Scharp D.W. The use of ficoll in the preparation of viable islets of langerhans from the rat pancreas. Transplantation. 1973;16(6):686–689. doi: 10.1097/00007890-197312000-00028. [DOI] [PubMed] [Google Scholar]
- 26.Cornu M. Glucagon-like peptide-1 protects beta-cells against apoptosis by increasing the activity of an IGF-2/IGF-1 receptor autocrine loop. Diabetes. 2009;58(8):1816–1825. doi: 10.2337/db09-0063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mohanty S. Overexpression of IRS2 in isolated pancreatic islets causes proliferation and protects human beta-cells from hyperglycemia-induced apoptosis. Experimental Cell Research. 2005;303(1):68–78. doi: 10.1016/j.yexcr.2004.09.011. [DOI] [PubMed] [Google Scholar]
- 28.Rieck S. The transcriptional response of the islet to pregnancy in mice. Molecular Endocrinology. 2009;23(10):1702–1712. doi: 10.1210/me.2009-0144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tuteja G. Cis-regulatory modules in the mammalian liver: composition depends on strength of Foxa2 consensus site. Nucleic Acids Research. 2008;36(12):4149–4157. doi: 10.1093/nar/gkn366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Le Lay J. CRTC2 (TORC2) contributes to the transcriptional response to fasting in the liver but is not required for the maintenance of glucose homeostasis. Cell Metabolism. 2009;10(1):55–62. doi: 10.1016/j.cmet.2009.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Heinz S. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Molecular Cell. 2010;38(4):576–589. doi: 10.1016/j.molcel.2010.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bramswig N.C. Epigenomic plasticity enables human pancreatic alpha to beta cell reprogramming. The Journal of Clinical Investigation. 2013;123(3):1275–1284. doi: 10.1172/JCI66514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Langmead B. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biology. 2009;10(3):R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Grant G.R. Comparative analysis of RNA-Seq alignment algorithms and the RNA-Seq unified mapper (RUM) Bioinformatics. 2011;27(18):2518–2528. doi: 10.1093/bioinformatics/btr427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lee D., Le Lay J., Kaestner K.H. The transcription factor CREB has no non-redundant functions in hepatic glucose metabolism in mice. Diabetologia. 2014;57(6):1242–1248. doi: 10.1007/s00125-014-3203-2. [DOI] [PubMed] [Google Scholar]
- 36.Hummler E. Targeted mutation of the CREB gene: compensation within the CREB/ATF family of transcription factors. Proceedings of the National Academy of Sciences of the United States of America. 1994;91(12):5647–5651. doi: 10.1073/pnas.91.12.5647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Valverde O. Modulation of anxiety-like behavior and morphine dependence in CREB-deficient mice. Neuropsychopharmacology. 2004;29(6):1122–1133. doi: 10.1038/sj.npp.1300416. [DOI] [PubMed] [Google Scholar]
- 38.Blendy J.A. Targeting of the CREB gene leads to up-regulation of a novel CREB mRNA isoform. EMBO Journal. 1996;15(5):1098–1106. [PMC free article] [PubMed] [Google Scholar]
- 39.Dobrian A.D. Dipeptidyl peptidase IV inhibitor sitagliptin reduces local inflammation in adipose tissue and in pancreatic islets of obese mice. American Journal of Physiology. Endocrinology and Metabolism. 2011;300(2):E410–E421. doi: 10.1152/ajpendo.00463.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Donath M.Y. Islet inflammation impairs the pancreatic beta-cell in type 2 diabetes. Physiology. 2009;24:325–331. doi: 10.1152/physiol.00032.2009. [DOI] [PubMed] [Google Scholar]
- 41.Ferdaoussi M. Exendin-4 protects beta-cells from interleukin-1 beta-induced apoptosis by interfering with the c-Jun NH2-terminal kinase pathway. Diabetes. 2008;57(5):1205–1215. doi: 10.2337/db07-1214. [DOI] [PubMed] [Google Scholar]
- 42.Natalicchio A. Exendin-4 prevents c-Jun N-terminal protein kinase activation by tumor necrosis factor-alpha (TNFalpha) and inhibits TNFalpha-induced apoptosis in insulin-secreting cells. Endocrinology. 2010;151(5):2019–2029. doi: 10.1210/en.2009-1166. [DOI] [PubMed] [Google Scholar]
- 43.Huo J.X., Metz S.A., Li G.D. p53-independent induction of p21(waf1/cip1) contributes to the activation of caspases in GTP-depletion-induced apoptosis of insulin-secreting cells. Cell Death & Differentiation. 2004;11(1):99–109. doi: 10.1038/sj.cdd.4401322. [DOI] [PubMed] [Google Scholar]
- 44.Kaneto H. Oxidative stress induces p21 expression in pancreatic islet cells: possible implication in beta-cell dysfunction. Diabetologia. 1999;42(9):1093–1097. doi: 10.1007/s001250051276. [DOI] [PubMed] [Google Scholar]
- 45.Yamada T. WFS1-deficiency increases endoplasmic reticulum stress, impairs cell cycle progression and triggers the apoptotic pathway specifically in pancreatic beta-cells. Human Molecular Genetics. 2006;15(10):1600–1609. doi: 10.1093/hmg/ddl081. [DOI] [PubMed] [Google Scholar]
- 46.Zhang S. Induction of apoptosis by human amylin in RINm5F islet beta-cells is associated with enhanced expression of p53 and p21WAF1/CIP1. FEBS Letters. 1999;455(3):315–320. doi: 10.1016/s0014-5793(99)00894-7. [DOI] [PubMed] [Google Scholar]
- 47.Hussain M.A. Increased pancreatic beta-cell proliferation mediated by CREB binding protein gene activation. Molecular and Cellular Biology. 2006;26(20):7747–7759. doi: 10.1128/MCB.02353-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kudo K. A selective increase in phosphorylation of cyclic AMP response element-binding protein in hippocampal CA1 region of male, but not female, rats following contextual fear and passive avoidance conditioning. Brain Research. 2004;1024(1–2):233–243. doi: 10.1016/j.brainres.2004.08.007. [DOI] [PubMed] [Google Scholar]
- 49.Lin Y. Sex differences in the effects of acute and chronic stress and recovery after long-term stress on stress-related brain regions of rats. Cerebral Cortex. 2009;19(9):1978–1989. doi: 10.1093/cercor/bhn225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mogi K. Sex difference in the response of melanin-concentrating hormone neurons in the lateral hypothalamic area to glucose, as revealed by the expression of phosphorylated cyclic adenosine 3′,5′-monophosphate response element-binding protein. Endocrinology. 2005;146(8):3325–3333. doi: 10.1210/en.2005-0078. [DOI] [PubMed] [Google Scholar]
- 51.Hai T., Curran T. Cross-family dimerization of transcription factors Fos/Jun and ATF/CREB alters DNA binding specificity. Proceedings of the National Academy of Sciences of the United States of America. 1991;88(9):3720–3724. doi: 10.1073/pnas.88.9.3720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ryseck R.P., Bravo R. c-JUN, JUN B, and JUN D differ in their binding affinities to AP-1 and CRE consensus sequences: effect of FOS proteins. Oncogene. 1991;6(4):533–542. [PubMed] [Google Scholar]
- 53.Sassone-Corsi P. Transcription factors responsive to cAMP. Annual Review of Cell and Developmental Biology. 1995;11:355–377. doi: 10.1146/annurev.cb.11.110195.002035. [DOI] [PubMed] [Google Scholar]
- 54.Molina C.A. Inducibility and negative autoregulation of CREM: an alternative promoter directs the expression of ICER, an early response repressor. Cell. 1993;75(5):875–886. doi: 10.1016/0092-8674(93)90532-u. [DOI] [PubMed] [Google Scholar]
- 55.Stehle J.H. Adrenergic signals direct rhythmic expression of transcriptional repressor CREM in the pineal gland. Nature. 1993;365(6444):314–320. doi: 10.1038/365314a0. [DOI] [PubMed] [Google Scholar]
- 56.Seino S. Roles of cAMP signalling in insulin granule exocytosis. Diabetes Obesity and Metabolism. 2009;11(Suppl. 4):180–188. doi: 10.1111/j.1463-1326.2009.01108.x. [DOI] [PubMed] [Google Scholar]
- 57.Melnikova V.O. CREB inhibits AP-2alpha expression to regulate the malignant phenotype of melanoma. PLoS One. 2010;5(8):e12452. doi: 10.1371/journal.pone.0012452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Pellegrini M. Expression profile of CREB knockdown in myeloid leukemia cells. BMC Cancer. 2008;8:264. doi: 10.1186/1471-2407-8-264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Du J. HIF-1alpha deletion partially rescues defects of hematopoietic stem cell quiescence caused by Cited2 deficiency. Blood. 2012;119(12):2789–2798. doi: 10.1182/blood-2011-10-387902. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.