

Abstract
Detection of adenovirus DNA in human tonsillar T cells in the absence of active virus replication suggests that T cells may be a site of latency or of attenuated virus replication in persistently infected individuals. The lytic replication cycle of Ad5 in permissive epithelial cells (A549) was compared to the behavior of Ad5 in four human T-cell lines, Jurkat, HuT78, CEM, and KE37. All four T-cell lines expressed the integrin coreceptors for Ad2 and Ad5, but only Jurkat and HuT78 express detectable surface levels of the coxsackie adenovirus receptor (CAR). Jurkat and HuT78 cells supported full lytic replication of Ad5, albeit at a level ∼10% of that of A549, while CAR-transduced CEM and KE37 cells (CEM-CARhi and KE37-CARhi, respectively) produced no detectable virus following infection. All four T-cell lines bind and internalize fluorescently labeled virus. In A549, Jurkat, and HuT78 cells, viral proteins were detected in 95% of cells. In contrast, only a small subpopulation of CEM-CARhi and KE37-CARhi cells contained detectable viral proteins. Interestingly, Jurkat and HuT78 cells synthesize four to six times more copies of viral DNA per cell than did A549 cells, indicating that these cells produce infectious virions with much lower efficiency than A549. Similarly, CEM-CARhi and KE37-CARhi cells, which produce no detectable infectious virus, synthesize three times more viral genomes per cell than A549. The observed blocks to adenovirus gene expression and replication in all four human T-cell lines may contribute to the maintenance of naturally occurring persistent adenovirus infections in human T cells.
Subgroup C adenoviruses are ubiquitous in the human population and cause an acute infection in the upper respiratory tract that resolves within 7 to 10 days. In addition to acute disease, the subgroup C adenoviruses also establish persistent infections characterized by intermittent excretion from immunocompetent hosts (22). Roughly half of primary infections are followed by prolonged fecal shedding of virus months and even years after virus is no longer detected in nasopharyngeal washings (21, 22). Restriction analysis of viruses isolated up to 4 years after initial infection suggested chronic persistent infection rather than reinfection with the same serotype (1). Early studies suggested that the site of adenovirus persistence was mucosal lymphoid tissues (17, 34). Group C adenovirus DNA was detected in human tonsillar lymphocytes in the apparent absence of virus production (57). Recent studies using cell separation and sorting techniques to isolate lymphocyte populations from tonsil and adenoid tissues have confirmed this observation and revealed that adenovirus DNA was enriched in T lymphocytes (23). While some replicating group C adenovirus could be detected in most tonsil and adenoid lymphocyte preparations, the vast majority of viral DNA present in mucosal lymphocytes is not actively replicating at the time of surgical removal (C. T. Garnett, J. A. Mahr, and L. R. Gooding, unpublished data).
These studies suggest that the group C adenoviruses produce chronic or persistent infection of mucosal lymphocytes. In support of this notion are several reports of established human lymphocyte cell lines that sustain prolonged, noncytopathic adenovirus infections. Infection of an Epstein-Barr virus (EBV)-transformed culture of human umbilical cord blood lymphocytes with group C adenoviruses results in virus production for months in the absence of noticeable cytopathic effect (3). Interestingly, subculture of these infected cells in neutralizing antibody for extended periods of time failed to cure the infection, hinting at an ability of adenovirus to establish a nonlytic persistent infection in human lymphocytes (3). A lymphoblastoid cell line derived from a bone marrow transplant recipient with adenovirus pneumonia was also reported to support prolonged nonlytic virus replication (20). Both B- and T-cell lines have been shown to grow while yielding infectious virus for a period of several months (52 and our unpublished observations). Additionally, a human monocyte line was observed to support a persistent infection with low levels of viral genome replication and virus production for up to a year (14).
The ability of adenoviruses to establish a persistent infection likely involves an atypical infection cycle in a cell type(s) that can maintain the viral genome for a long period of time without succumbing to virus-induced apoptosis of the infected cell. The finding that human tonsillar T lymphocytes harbor adenovirus DNA, apparently in the absence of virus replication (23), suggests that T cells may be a reservoir of persistent adenovirus infection. Therefore, this study was undertaken to test the hypothesis that some T cells are capable of controlling gene expression and/or replication of adenovirus. To this end, infection was characterized in four human T-cell lines by evaluating virus receptor expression, infectious virus production, binding and internalization of virus, kinetics of viral protein expression, and viral genome replication.
MATERIALS AND METHODS
Cell lines.
A549, Jurkat, HuT78, and CEM cell lines were purchased from the American Type Culture Collection (ATCC). The KE37 cell line was obtained from Periasamy Selveraj (Emory University). The packaging cell line BOSC23 (48) for retrovirus production was obtained from Joshy Jacob (Emory University). Jurkat, HuT78, CEM, and KE37 cells were grown in RPMI medium with 10% fetal calf serum (FCS) (HyClone, Logan, Utah) and 10 mM glutamine (RPMI complete). A549 and BOSC23 cells were grown in Dulbecco's modified Eagle medium (DMEM) with 4.5 μg of glucose/ml, 10% FCS, and 10 mM glutamine.
Human CAR was introduced into cells by transduction with the LXSN-hCAR retrovirus obtained from James DeGregori (University of Colorado Health Sciences) (39). Retroviruses were produced as described previously (41). Briefly, the vector control LXSN or the LXSN-CAR plasmids were transfected into BOSC23 cells using FuGene6 transfection reagent (Roche) following the manufacturer's protocol. Two days posttransfection the retroviral-containing supernatants were harvested and filtered with a 0.45-μm-pore-size syringe filter, and then polybrene (Sigma) was added to a final concentration of 8 μg/ml. CEM and KE37 cells were transduced with either retrovirus by adding 1 ml of the virus stock to 2 × 106 cells in 2 ml of RPMI complete medium, and the mixture was centrifuged for 45 min at 25°C and 500 × g. Transduced cells were selected by using G418 sulfate (CellGro) 48 h posttransduction, and these cultures were maintained in G418 (1 mg/ml).
Adenoviruses.
The Ad-CMV-GFP, Ad-RSV-GFP, and Ad-E1A-GFP viruses (39) were obtained from James DeGregori. These replication-defective viruses lack E1A, and expression of green fluorescent protein (GFP) is driven by the cytomegalovirus (CMV), respiratory syncytial virus (RSV), or E1A promoters, respectively. The wild-type rec700 adenovirus was obtained from William Wold (St. Louis University). This type 5/2/5 recombinant adenovirus contains the Ad2 EcoRI-D fragment (62). In some experiments, the phenotypically wild-type virus dl309 (36) was used instead of rec700. Virus was grown and purified as described by Goodrum and Ornelles (24).
Fluorescently labeled adenovirus (Ad-568) was made by coupling Alexa Fluor 568 (Molecular Probes, Eugene, Oreg.) to rec700, using a method adapted from a previously described protocol (26). A 0.1-ml aliquot of rec700 virus stock was dialyzed against 100 ml of 0.1 M sodium bicarbonate (pH 8.2) at 4°C for 15 min using Slide-A-Lyzer mini-dialysis units (Pierce), with three exchanges of buffer. The Alexa Flour 568 dye was reconstituted per the manufacturer's instructions, and the dialyzed virus was mixed with 5,000-fold-molar-excess dye and incubated with rocking for 1 h at 25°C. To block excess labeling, 1 M Tris (pH 8.0) was added and the reaction mixture was incubated for 0.5 h at 25°C. Unincorporated dye was removed by dialysis at 4°C for 15 min against 100 ml of virus storage buffer, with four exchanges of buffer. To concentrate the virus stock a final dialysis was done against virus storage buffer with 75% glycerol. Infectivity of the labeled virus was tested on A549 cells and was observed to be unchanged relative to unlabeled virus (data not shown).
Infection of cell lines.
Lymphocyte cell lines were infected by washing cells in serum-free (SF) RPMI and adjusting to a density of 107 cells/ml in SF RPMI. Virus was added to the cell suspension at 30 PFU/cell (10 focus-forming units [ffu]/cell) for recombinant GFP viruses (39). Following a 90-min incubation at 37°C, the cells were washed three times with RPMI complete medium using 1 ml of solution/106 cells. Cells were then resuspended in RPMI complete medium at a density of 106 cells/ml and were incubated for the indicated times before harvesting. Cells were harvested by collecting 1 ml of culture and washing it in 1 ml of phosphate-buffered saline (PBS). The samples were then resuspended in 1 ml of PBS and divided into two aliquots for further analyses with the following method. A 0.5-ml aliqot was centrifuged, and the cell pellet was stored at −20°C and used for quantifying adenovirus DNA by real time PCR. Another 0.5 ml was fixed with 1% formaldehyde, stored at 4°C, and used for intracellular staining for viral proteins. Cells infected with GFP recombinant viruses were washed, resuspended in PBS with 1% FCS, and analyzed by flow cytometry for GFP expression as described below.
Adherent A549 cells (106) were plated onto duplicate 6-well plates and allowed to adhere for 12 to 16 h. Monolayers were washed twice in SF DMEM, and virus was added at 30 PFU/cell in 0.5 ml of SF DMEM and was incubated for 90 min at 37°C. Cells were washed three times with complete DMEM, and 3 ml was added to each well. Infected cells were incubated for the indicated times before harvesting. The cells were harvested by one washing with 2 ml of DMEM complete and once with 2 ml of PBS. Cells on one duplicate well were harvested with 0.25% trypsin in PBS, fixed in 1% formaldehyde, and stored at 4°C prior to staining for intracellular viral proteins. One milliliter of DMEM was added to the other well, and the cells were collected by scraping. The sample was divided into two aliquots: 0.5 ml was centrifuged and the pellet was stored at −20°C and used for quantitating viral DNA by real time PCR, and 0.5 ml was stored at −20°C and used to measure infectious virus production by plaque assay.
Flow cytometry assays.
Sources of antibodies used for flow cytometry assays were as follows. The anti-CAR clone RmCB was obtained from James De Gregori (39). The mouse antiadenovirus, specific for the hexon protein, was purchased from Chemicon International (catalog no. MAB8051). The goat F(ab′)2 anti-mouse-phycoerythrin (PE) was purchased from Southern Biotechnology Associates (catalog no. 1032-09). The anti-E1A hybridoma clones M1 and M73 were obtained from Ed Harlow (29) and were used together at a 1:1 dilution. The isotype control immunoglobulin G1 antibody used in the intracellular staining assay was purchased from Pharmingen (catalog no. 03171D). The anti-integrin antibodies specific for αVβ3 (catalog no. MAB1976F) or αvβ5(catalog no. MAB1961H) were obtained from Chemicon International. The anti-adenovirus penton monocloal antibody was clone 2Pb-1 (NIHC3) (10).
Intracellular staining for adenovirus proteins was performed essentially as described by Weaver and Kadan (60). The primary antibodies used were anti-adenovirus (10 μg/ml), anti-E1A (1:25 dilution), anti-DBP clone B6-8 (1:50 dilution) (25), anti-penton clone 2Pb-1 (1:25 dilution), or the immunoglobulin G1 isotype control (10 μg/ml). Cells were analyzed on a FACS-Calibur fluorescence-activated cell sorter (FACS) flow cytometer (Becton Dickinson) with Cell Quest software.
Surface staining for CAR expression was performed by incubating live cells with an isotype control or the RmCB anti-CAR (1:20 dilution) (39) on ice for 30 min. After being washed, cells were incubated with goat F(ab′)2 anti-mouse-PE (2.5 μg/ml) for 30 min on ice. Cells were washed and analyzed as described above.
CARhi cell lines were selected by staining the CEM and KE37 cells transduced with LXSN-hCAR with RmCB as described above and were sorted into CAR-high and CAR-low populations by using a MoFlo flow cytometer (Cytomation) at the Yerkes Regional Primate Center. For CEM-CAR cells, the 12% of cells expressing the highest level of surface CAR were collected, and post-sort analysis showed them to be 97.6% positive for CAR expression. For the KE37-CAR cells, the 47% of cells expressing the highest level of surface CAR were collected, and post-sort analysis showed these were 98.4% positive for CAR expression.
Quantitation of viral genomes.
Quantitative real-time PCR was performed as described previously with primers specific to the hexon gene (23). DNA was extracted from samples of equal volume collected from cultures of infected cells as described above. Each sample represents 0.5 × 106 or 106 cells for A549 and all T-cell lines, respectively. Analyses were done in triplicate and are reported as the average values with standard errors.
Infection with Ad-568.
Lymphocytes were washed with SF RPMI, adjusted to 107 cells per ml in SF RPMI medium, and infected with 30 PFU of Ad-568 per cell as determined by titer on A549 cells. The concentrated cells and virus were maintained on ice for 60 min with periodic agitation. Samples analyzed at 0 h postinfection (p.i.) (the end of the 60-min incubation on ice) were immediately transferred to ice-cold PBS with 0.1% sodium azide. For all other time points, the infected cells were collected by centrifugation at 200 × g for 5 min at room temperature, suspended in normal growth medium at 106 cells per ml, and maintained at 37°C. At the indicated times, a portion of the infected cells was removed, collected by centrifugation, and suspended in ice-cold PBS with 0.1% sodium azide.
Sample processing and fluorescence microscopy.
Approximately 5 × 104 lymphocytes in PBS with sodium azide were collected by centrifugation, washed with ice-cold PBS, and suspended in 0.02 ml of PBS. The cells were allowed to attach to a polylysine-coated glass slide in a humidified chamber for 10 min at room temperature. Excess liquid was removed, and the cells were fixed in place by adding an excess of freshly prepared 4% formaldehyde in PBS. The cells were allowed to fix for 15 min at room temperature before the fixative was removed. The attached and fixed cells were rinsed first with PBS, next with Tris-buffered saline (pH 7.8) containing 0.05% Tween 20, 5 mg of glycine, and 5 mg of bovine serum albumin per ml, and then again with PBS for 2 to 5 min for each rinse. Excess liquid was drained from the slide, and a coverslip was mounted by using a polyvinyl alcohol-based mounting medium containing the fluorescent DNA dye 4′,6-diamidino-2-phenylindole (DAPI) at 0.1 μg per ml. By comparing the number of cells placed on the slide before fixation with the number that remained after mounting, few, if any, cells that made contact with the glass slide were lost during processing.
Fluorescent samples were analyzed by epifluorescence and differential interference microscopy with a Nikon TE300 inverted microscope fitted with filters appropriate for DAPI and Alexa Fluor 568 excitation. Images were acquired as 12-bit TIF images with a monochrome Retiga EX 1350 digital camera (QImaging Corp., Burnaby, British Columbia, Canada) using a 20×/0.45 numeric aperture CFI Plan Fluor ELWD and a 100×/1.4 numeric aperture CFI Plan APO oil immersion objective. Micrographs were generated from the TIF images by using Photoshop 5.5 (Adobe Systems Inc., San Jose, Calif.) to adjust brightness and contrast in a uniform manner for each cell type. The figures were assembled with the use of Canvas 5.0 (Deneba, Miami, Fla.).
Quantitative fluorescence microscopy.
Low-magnification images of 20 to 300 cells from at least eight randomly selected fields were collected with the 20× objective. This objective provided sufficient depth of focus to ensure that all emitted light was in the plane of focus. The exposure time (0.2 to 4 s) was adjusted for each preparation to avoid saturation, thus insuring linearity between fluorescence intensity and the recorded signal. The mean fluorescence intensity over background was measured for each cell by using ImageJ, version 1.29 (http://rsb.info.nih.gov/ij/; Wayne Rasband, National Institutes of Health), operating on a Macintosh microcomputer. Typical background values ranged from 0.8 to 8.5 (of 4,096 possible) arbitrary units per pixel. The strongest fluorescent signals recorded registered as approximately 2,880 U in a single pixel. The area of each cell varied between 1,000 to 2,200 pixels. The maximum mean fluorescent value over background measured for all cells was approximate 330 U per pixel. In some experiments the phosphatidylinositol 3 (PI3)-kinase inhibitor LY294002 (Cell Signaling Technologies, Beverly, Mass.) was added to a concentration of 50 μM 5 min prior to infection and was maintained at this concentration throughout the time course to inhibit virus internalization (59).
Laser-scanning confocal microscopy.
The three-dimensional distribution of Ad-568 in infected lymphocytes was determined by confocal microscopy with a Zeiss LSM 510 (Carl Zeiss, Inc., Thornwood, N.Y.) confocal laser scanning device fitted to a Zeiss Axioplan 2 microscope with a 60×/1.4 numeric aperture oil immersion objective. The Alexa Fluor 568 dye was excited by helium-neon laser illumination. Successive serial images approximately 0.5 μm in depth with approximately 0.1 μm of overlap spanning the depth of representative cells were collected as 8-bit TIF files. The brightness and contrast of each stack of images was separately adjusted by using the LSM 510 software. Fifteen sequential sections (to encompass the entire cell) were presented in gallery fashion.
Southern blot analysis of viral DNA.
Lymphocyte cell lines were infected with dlI309 as described above. One milliliter of infected cells, representing 106 cells at the time of infection, was harvested immediately after the infection (day 0) and every 24 h thereafter for days 1, 2, and 3. Total DNA was isolated from the harvested cells by washing the cells twice with PBS before treating them with 0.2 mg of proteinase K per ml in 0.2 M Tris (pH 8.0), 50 mM EDTA, 0.5% sodium dodecyl sulfate in a volume of 0.2 ml. After 3 to 8 h at 60°C, the nucleic acid was recovered by phenol extraction and ethanol precipitation.
DNA obtained from 4 × 105 cells at the time of infection was cleaved to completion with the restriction enzyme HindIII and separated by electrophoresis through 0.7% agarose in Tris-borate buffer at 1.5 V per cm. The DNA was transferred to a nylon support (Nytran N; Schleicher and Schuell, Keene, N.H.) by alkaline capillary transfer. Adenovirus DNA was detected by hybridization to a radioactive probe (15) generated by random primer extension of genomic dl309 DNA in the presence of [α-32P]dATP (19). The viral DNA was visualized and quantified by phosphorescence imaging (ImageQuant; Molecular Dynamics, Sunnyvale, Calif.). The exposure for the blot was adjusted to represent the full range of the signal, requiring approximately five times the sensitivity for the CEM-CARhi and KE37-CARhi cell lines as for the HuT78 and Jurkat cell lines.
RESULTS
Adenovirus receptor and coreceptor expression on human T-cell lines.
Susceptibility to infection of human T cells was analyzed by using four T-cell lines: Jurkat, HuT78, CEM, and KE37. Jurkat, CEM, and KE37 were isolated from the peripheral blood of patients with acute lymphoblastic leukemia (50, 58). HuT78 was derived from a patient with Sezary Syndrome (9). Infection with human group C adenoviruses is dependent upon cellular expression of the virus receptor and coreceptors (reviewed in reference 47). Thus, T-cell lines were first analyzed for surface expression of these proteins. CAR mediates the high-affinity interaction between the fiber capsid protein and the host cell (7, 56). CAR expression on these lymphocyte lines was measured by staining cells with anti-CAR antibody, followed by flow cytometric analysis (Fig. 1). The fully permissive A549 cell line expressed relatively high levels of surface CAR, as did the Jurkat and HuT78 T cells. CAR expression on CEM and KE37 cells was very low or absent.
FIG. 1.
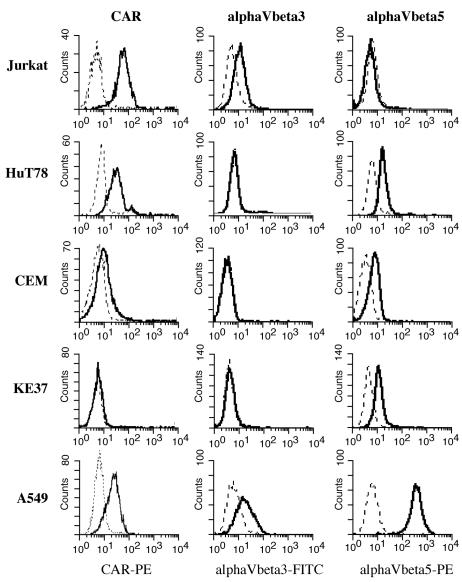
Surface expression of adenovirus CAR receptor and integrin coreceptors. Cells were stained with antibody specific for CAR (solid lines) or with an isotype control antibody (dashed lines), incubated with goat anti-mouse F(ab′)2-PE, and analyzed by using a FACS Calibur flow cytometer and CellQuest software. Integrin staining was done by using directly conjugated antibodies as described in Materials and Methods. FITC, fluorescein isothiocyanate.
The αV integrins, specifically αVβ3 and αVβ5, are coreceptors for adenoviruses and function to mediate virus internalization in part by binding to the penton base on the virus capsid (47, 61). Thus, the surface expression level of these integrins may also contribute to infection permissivity. The levels of these proteins on the T-cell lines were also measured by surface staining and flow cytometry (Fig. 1). αVβ3 was detected at low levels on Jurkat and A549 cells and was absent on the other cell lines. A549 cells expressed high levels of αVβ5, and low but detectable levels of αVβ5 were found on HuT78, CEM, and KE37.
Preliminary experiments revealed that CEM and KE37 cells were not permissive to adenovirus infection (data not shown), and the low levels of CAR on these cell lines likely contribute to this state. In order to evaluate postinternalization events in the virus life cycle among the T-cell lines, CAR levels were increased on CEM and KE37 cells by transducing the cells with a retrovirus expressing human CAR (LXSN-hCAR) or the empty vector control (LXSN) (39). Transduced CEM and KE37 cells were selected in G418 and analyzed for surface CAR expression (Fig. 2). Retroviral transduction and G418 selection produced cells that were heterogeneous for CAR expression, with 28% of CEM (Fig. 2A) and 59% of KE37 (Fig. 2C) cells expressing CAR. Transduction with the control vector, LXSN, had no effect on CAR expression. CAR-expressing cells were further selected by staining cells with anti-CAR and sorting them into CAR-high and CAR-low subsets. The cells expressing high levels of CAR were retained, and the postsort CAR expression of the CEM-CARhi and KE37-CARhi cells is shown in Fig. 2B and D, respectively. The high levels of CAR expression were stable for months in culture in the presence of G418 (data not shown). These CARhi cells were used for most subsequent analyses.
FIG. 2.
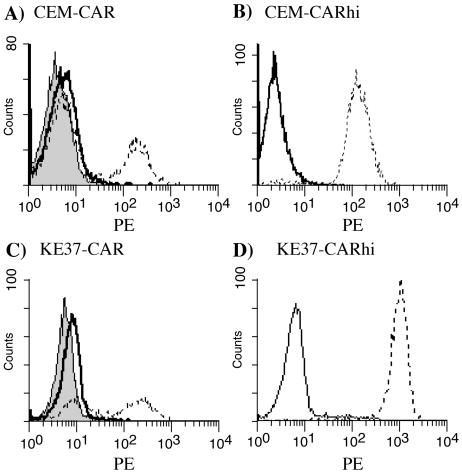
CAR expression on transduced cells. CEM (A) and KE37 (C) cells were infected with either the control LXSN retrovirus (solid lines) or the LXSN-hCAR retrovirus (dashed lines), grown in G418 selection, stained with anti-CAR, and analyzed by flow cytometry as described in the legend to Fig. 1. Shaded peaks represent staining with an isotype control antibody. CEM-CAR and KE37-CAR were sorted to isolate cells with the highest levels of CAR. (B and D) Surface CAR expression (dashed lines) 1 week post-sort on the CEM-CARhi and KE37-CARhi cells, respectively. Solid lines represent staining with an isotype control.
Quantitation of virus production.
In a fully permissive cell, thousands of infectious progeny adenovirus virions are released when the infected cell dies. Thus, the definition of complete permissivity is the ability to produce high levels of infectious virus as quantified by plaque assay. To measure permissivity of human T-cell lines to adenovirus infection, cells were infected with a wild-type adenovirus (rec700) at 30 PFU/cell. Cultures were harvested at 90 min and 4 days p.i. At 90 min, the end of incubation with virus, cells were washed to remove unbound virus and were returned to culture. An aliquot of washed cells was taken at the 90-min time point to determine the amount of infectious input virus remaining associated with cells prior to virus replication. Freeze/thaw lysates were prepared, and infectious virus was determined by plaque assay. The 4-day time point was chosen on the basis of preliminary experiments indicating that in Jurkat and HUT78 this was the earliest point at which maximal virus production could be detected and at which most cells were dead. This is in contrast to permissive A549 cells, in which all cells are dead by 2 days p.i. At day 4, both supernatant and cell pellets were collected and subjected to two rounds of freeze/thaw to release any cell-associated virus. Infectious virus was measured by plaque assay, and the results are reported as PFU per cell in Table 1. In A549 cells virtually no infectious virus was detected immediately following infection (90 min), which increased to approximately 7,300 PFU/cell by 2 days. Similarly, little or no infectious virus was detected in the Jurkat and HuT78 cultures at 90 min p.i., indicating that any virus associated with these cells is no longer infectious. At 4 days p.i., several hundred PFU of virus per cell were detected in HuT78 and Jurkat samples, indicating that these cells supported the full replication cycle of the virus, albeit quantitatively less effectively than A549 cells. CEM and KE37 cells that lack the CAR receptor contained no infectious virus at either time point, indicating, as expected, that little or no virus bound to or entered the cells. Surprisingly, however, even introduction of high levels of CAR into these cells was insufficient to permit virus production from either CEM-CARhi or KE37-CARhi. Infectious virus remained associated with both cell lines at 90 min p.i., at levels indistinguishable from that of input virus (30 PFU/cell). This suggests that in these cells, virus binding to the surface is not immediately followed by virus internalization, which would cause loss of infectivity (27). At 4 days p.i., infectious virus associated with CEM-CARhi and KE37-CARhi remained at roughly the same level, suggesting that either input virus remained cell associated and infectious or that the level of virus replication in these cells was only sufficient to reproduce the quantities of input virus.
TABLE 1.
Virus production by cell lines infected with wild-type adenovirusa
| Cell line | Virus yield (PFU per cell)
|
||
|---|---|---|---|
| 90 min p.i. | 2 days p.i. | 4 days p.i. | |
| Jurkat | 1.0 ± 0.4 | 628 ± 458 | |
| HuT78 | 1.2 ± 0.4 | 176 ± 82.8 | |
| CEM-CARhi | 15.3 ± 2.3 | 36.5 ± 3.7 | |
| KE37-CARhi | 40.4 ± 14.0 | 18.6 ± 1.0 | |
| CEM-LXSN | 0.06 ± 0.03 | 0.4 ± 0.07 | |
| KE37-LXSN | 0.1 ± 0.04 | 0.06 ± 0.02 | |
| A549 | 1.1 ± 0.5 | 7,300 ± 5,602 | |
At the indicated times p.i., aliquots from cell cultures infected as described in Materials and Methods were collected and analyzed by plaque assay on A549 cells. Values reported are the averages and standard errors of four measurements.
The virus production data suggest that Jurkat and HuT78 are capable of replicating live virus while CEM-CARhi and KE37-CARhi are not. To rule out the possibility that this difference is due to defects in the retrovirally expressed CAR, its function was evaluated in the EL4 mouse T-lymphoma cell line. EL4 cells cannot be infected with human adenoviruses as measured by GFP expression from recombinant vectors; however, retroviral expression of human CAR allows these cells to be infected (39). We reproduced this observation with an Ad-CMV-GFP adenovirus to show that retrovirally expressed CAR confers sensitivity to infection to the EL4 cell line (data not shown). This assay confirmed the biological activity of the retroviral CAR construct used in these studies. Thus, the CAR expressed by the LXSN retrovirus functioned to convert a cell that cannot be infected to a cell that can be. Therefore, the block to infection in the CEM and KE37 cell lines probably occurs at a postattachment step in the lytic cycle.
Another possibility is that CEM and KE37 cells lack sufficient levels of the integrin coreceptor to permit virus internalization. However, direct staining revealed similar levels of αVβ5 integrin on KE37, CEM, and permissive HuT78 cells (Fig. 1). Furthermore, levels of integrins too low to be detected by FACS are sufficient to facilitate virus entry into mouse EL4 cells (39).
Virion binding and uptake by human T cells.
To determine what fraction of cells in the T-cell populations actually bind adenovirus, virus localization on these cells was examined by using purified adenovirus labeled with the fluorescent dye Alexa 568 (Ad-568) and fluorescence microscopy. For these experiments, virus was bound to the cells on ice and then unbound virus was removed by washing, and membrane movement was stopped with sodium azide and fixation with formaldehyde prior to microscopy. By visual analysis at low magnification following incubation with virus on ice, 95 to 100% of cells from each of the T-cell lines bound detectable virus (data not shown). Higher magnification revealed that immediately following infection individual virions were detected in a diffuse pattern on the cell surface in each of the four cell lines. At 24 h p.i., clusters of virus were apparent on the cell lines rather than the uniform punctate pattern that was observed at the earlier time points. Also, at later time points the intensity of fluorescence appeared to decrease as individual virions are dismantled and their component proteins are degraded (27).
Fluorescence intensity was quantified on infected cells at various times p.i., and the results are shown in Fig. 3. The higher initial levels of fluorescence on CEM-CARhi and KE37-CARhi probably reflect increased virus binding due to the higher levels of CAR on these cells relative to Jurkat and HuT78 (Fig. 1 and 2). However, over the 40-h time course, the fluorescence signal is seen to decrease on all four cell lines. This decrease in fluorescence could reflect either loss of virus by internalization and degradation of the virion or loss of virus from the surface by shedding. Binding of the virus to the integrin coreceptors activates PI3 kinase signaling, which is necessary for virus internalization (40). To determine whether the time-dependent loss of fluorescence was due to virus internalization, KE37-CARhi and CEM-CARhi cells were infected with Ad-568 in the presence or absence of a PI3 kinase inhibitor, LY294002 (59), which specifically inhibits kinase-dependent virus internalization. In both cell lines loss of fluorescence was inhibited by LY294002, indicating that virus internalization is the primary cause of the decreased fluorescence seen in these cell lines (Fig. 4). Further, the decrease in fluorescence signal is similar among both permissive and nonpermissive cell lines, suggesting that a block in virus binding and internalization does not account for the differences in permissivity observed among the T-cell lines.
FIG. 3.
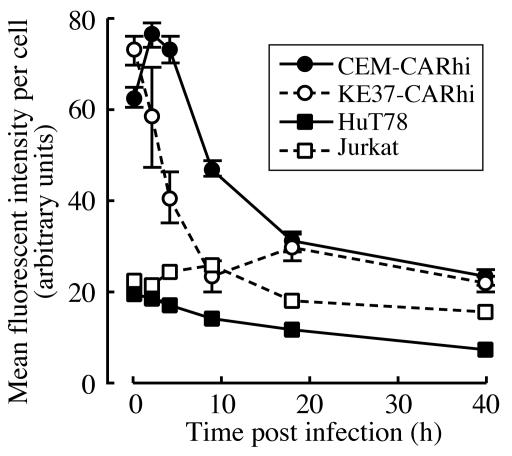
Time course of Ad-568 fluorescence following infection. The indicated cells were infected with 30 PFU of Ad-568 per cell for 1 h on ice and then were returned to normal growth conditions. At each time point indicated a portion of the infected cells was processed for quantitative fluorescent microscopy as described in Materials and Methods. The mean fluorescence intensity, representing the amount of Ad-568 per cell, was determined for each of 20 to 300 cells. The data are expressed as average values ± standard errors of the means.
FIG. 4.
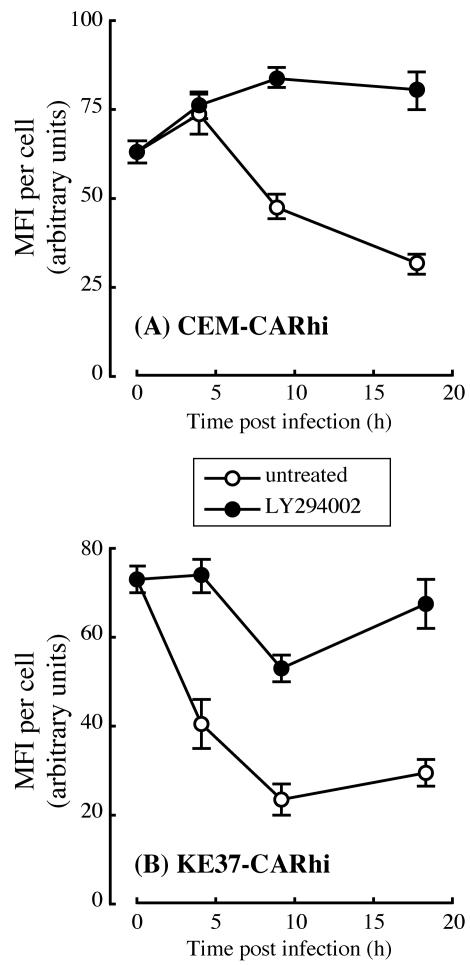
Inhibition of internalization blocks time-dependent loss of Ad-568 fluorescence. CEM-CARhi (A) or KE37-CARhi (B) cells were treated with a 50 μM concentration of the PI3 kinase inhibitor LY294002 or vehicle (dimethyl sulfoxide) as a control for 5 min. Drug-treated cells (filled circles) were kept in the presence of the inhibitor throughout the duration of the experiment. The cells were infected with 30 PFU of Ad-568 per cell for 1 h on ice and then were returned to normal growth conditions. At each time point indicated a portion of the infected cells was processed for quantitative fluorescent microscopy as described in Materials and Methods, and the mean fluorescence intensity (MFI) per cell was determined for each of 40 to 300 cells. The data are expressed as average values ± standard errors of the means.
To confirm that virus internalization follows binding to the surface of infected T cells, individual cells were analyzed by confocal microscopy immediately after incubation with virus and at 18 h p.i. (Fig. 5). Initially, Ad-568 was distributed uniformly about the surface of the infected T cell, as seen by the rim-staining pattern and absence of fluorescence within the cell. However, by 18 h p.i. the number of fluorescent particles on the cell surface had diminished and intracellular particles (marked by an asterisk in the figure panels) could readily be detected, indicating that virus is being internalized.
FIG. 5.
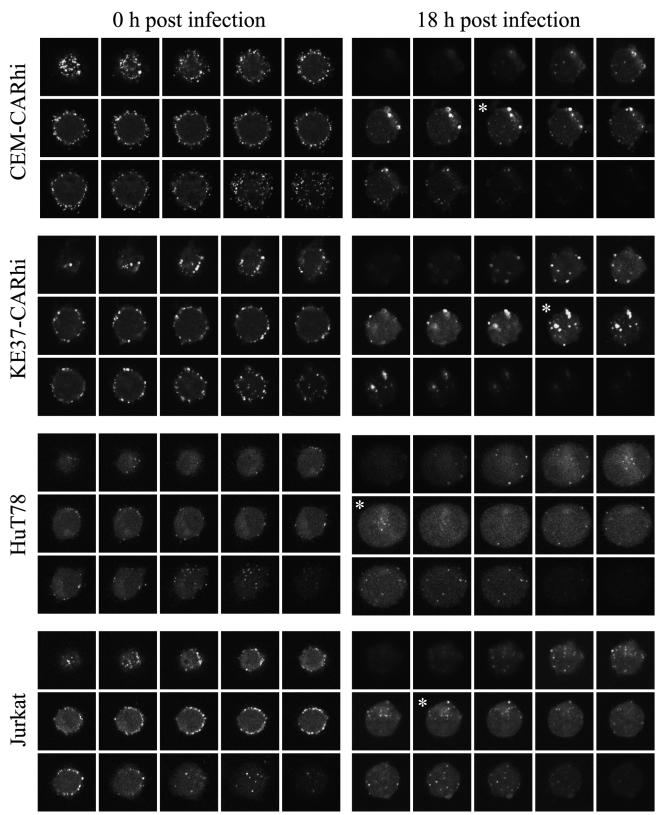
Internalized Ad-568 particles are detected in each lymphocytic cell line at 18 h p.i. The cell lines indicated on the left of each pair of panels were infected with 30 PFU of Ad-568 and either were washed extensively and fixed (0 h p.i.) or were returned to normal growth conditions for 18 h before being fixed and processed for confocal laser scanning microscopy. Fifteen successive serial images through the extent of a representative cell is shown in each gallery. Sections marked with an asterisk contain fluorescent Ad-568 particles in the interior of the cell. Each micrograph represents a field that is 18 to 20 μm wide.
Kinetics of adenovirus protein expression.
The results described above indicate that while all four human T-cell lines are capable of binding and internalizing virus, only two of the lines, Jurkat and HuT78, support a productive infection. To explore further where the block in the infection cycle occurs, intracellular staining for viral proteins and flow cytometric analysis were used to detect early and late viral proteins in infected cells as a function of time p.i. The immediate-early E1A and the early DNA binding protein (DBP) as well as two virion proteins produced late in infection, hexon and penton, were measured at various times p.i. (Fig. 6). For comparison, viral protein expression was measured in permissive A549 human epithelial cells. As shown in Fig. 6G, DBP and E1A were detected at 6 h p.i. in A549 cells, while both late proteins were first detected at 12 h p.i. The percentage of cells expressing these proteins increased during the time course, with approximately 90% of A549 cells expressing viral proteins at 24 h p.i.
FIG. 6.
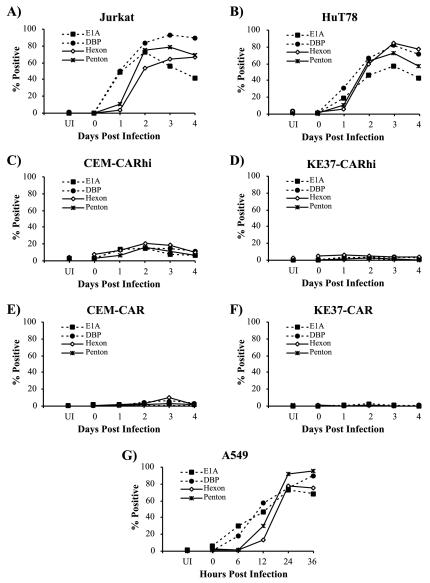
Time course of viral protein expression. Jurkat (A), HuT78 (B), CEM-CARhi (C), KE37-CARhi (D), CEM-CAR (E), KE37-CAR (F), and A549 (G) cell lines were infected with rec700 at 30 PFU/cell as described in Materials and Methods. Cells were harvested and stained at the indicated times p.i. for the intracellular expression of E1A, DBP, hexon, and penton as indicated. Data are representative of at least three independent experiments.
The expression of viral proteins in the T-cell lines is shown in Fig. 6A to F. Both early and late proteins were readily detected in Jurkat and HuT78 cells, although the time course was significantly delayed compared to that of A549. Early protein synthesis again preceded detection of late proteins, and viral protein expression peaked at 72 h p.i., with 80 to 90% of cells positive for viral protein expression (Fig. 6A and B). In contrast, CEM-CARhi displayed a small percentage of cells (<20%) expressing viral proteins, and viral protein expression was detected in only 1 to 5% of KE37-CARhi cell lines (Fig. 6C and D). These values were not greatly increased over those measured in the CAR heterogeneous cell lines (Fig. 6E and F). No viral protein expression was detected in the LXSN control cell lines (data not shown). Thus, the increased expression of CAR on the CARhi cell lines did not significantly overcome the block to viral gene expression in these cells.
The failure of most CEM-CARhi and KE37-CARhi cells to express detectable levels of either early or late viral proteins could be due to selective inhibition of adenoviral promoters in these cells. To test this possibility, T-cell lines were infected with adenoviral vectors expressing GFP from heterologous promoters, and GFP expression was monitored by flow cytometry (Fig. 7). The majority of A549 and Jurkat cells express GFP following infection with viruses that control GFP expression from CMV, RSV, or E1A promoters. In contrast, the majority of CEM-CARhi and KE37-CARhi cells fail to express detectable levels of GFP under the same conditions. Thus, the failure of adenoviral gene expression in these cells is not unique to adenoviral promoters. Interestingly, comparing the intensity of fluorescence in the GFP-positive cells, particularly for the CMV promoter vector, all of the T-cell lines are comparably bright (Fig. 7B). This finding suggests that there is no inherent difference in transcription levels among the three T-cell lines tested in the subset of cells in which transcription is initiated.
FIG. 7.
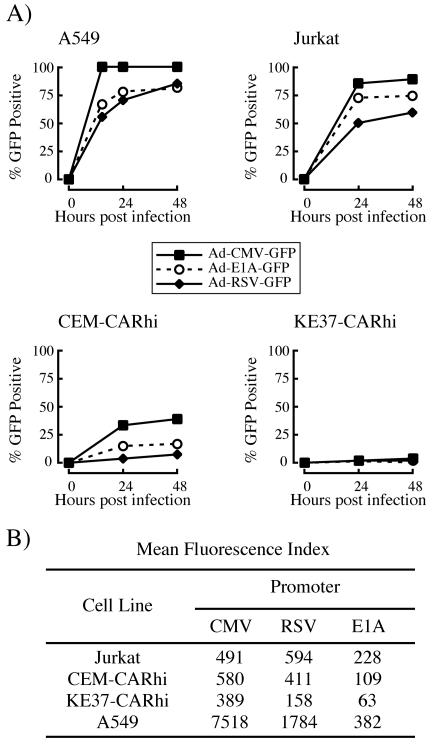
Expression of GFP from isogenic adenoviral vectors in human T-cell lines. Cells were infected with recombinant adenoviruses expressing GFP directed by either the E1A, CMV immediate-early, or RSV promoters, and expression was measured by flow cytometry as detailed in Materials and Methods. (A) Percentage of cells expressing GFP. (B) Mean fluorescence intensity of the GFP-positive population at 24 h p.i.
Maintenance and replication of adenovirus DNA.
The reduction or absence of viral protein production in CEM-CARhi and KE37-CARhi cells indicated that expression of the viral genome is regulated differently in these cells. To determine whether or not the viral genome remained associated with these cells during infection, the amount of viral DNA was measured in all T-cell lines during infection by using real-time PCR. Samples were collected from both the infected T cell and A549 cultures used for the analyses shown in Fig. 6, and the viral genomes were quantified (Fig. 8). A549 cultures of 0.5 × 106 cells contained 1.2 × 107 copies of viral DNA per sample at 90 min p.i. This number increased nearly 600-fold by 48 h p.i. (Fig. 8A). This level of replication was consistent with the virus production data shown in Table 1. Analysis of the T-cell lines showed that 0.5 × 109 to 4 × 109 viral DNA copies were associated with 0.5 × 106 cells at 90 min p.i., with the CARhi cell lines having the highest amounts. The viral genome levels in Jurkat and HuT78 cells increased by approximately 50-fold over the 4-day time course, consistent with the observed expression of both early and late proteins (Fig. 6) and production of infectious virus (Table 1). Interestingly, although the fold replication of viral genomes in Jurkat and HuT78 was only 10% of that seen in A549 cells, overall the T-cell lines produced 4 to 6 times more DNA per cell than did A549. In contrast, viral genomes in the CEM-CARhi and KE37-CARhi cell lines replicated only 13- and 5.5-fold, respectively. In CEM-CARhi cells, this low level of viral DNA replication may occur in the small percentage of cells that expres viral proteins (Fig. 6C). The amount of viral genome produced was similar in CEM-CARhi and KE37-CARhi cells, although relatively few KE37-CARhi cells express viral proteins compared to CEM-CARhi cells (Fig. 6D). However, the high levels of bound genomes at the beginning of the virus infectious cycle in CEM and KE37 cells (Fig. 8C) masks the fact that, at the end of the incubation period, both cell lines have produced roughly 3 times more viral DNA per cell than A549.
FIG. 8.
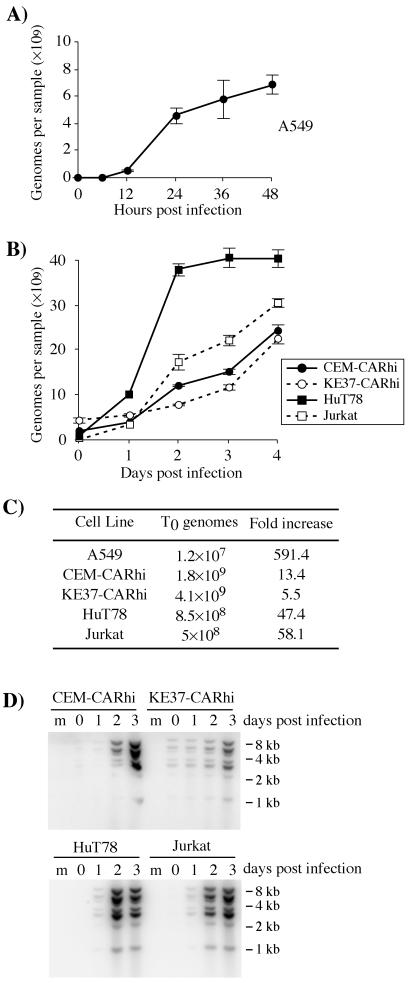
Adenoviral genome replication in A549 and human T-cell lines. All cells were infected with 30 PFU/cell of rec700 (A, B, and C) or dl309 (D) as described in Materials and Methods. The genome/PFU ratio for the CsCl-banded rec700 virus used in these experiments was 350. Thus, in each sample there were 5 × 109 input genomes as measured by real-time PCR for hexon. Real-time PCR was used to quantitate genome copy numbers at the noted times p.i. (A, B, and C). (D) In separate experiments, Southern blotting was used to confirm replication of viral DNA in infected lymphocytes. Lane m, mock-infected cells. The approximate migration of molecular size standards in kilobase pairs is indicated to the right of each blot.
Similar conclusions were reached when viral DNA in the infected T-cell lines was analyzed by Southern blotting. Total DNA isolated from infected cells was digested to completion with HindIII, separated by electrophoresis, and analyzed by Southern blotting using a radioactive probe derived from the entire viral genome (Fig. 8D). Within each lane the intensity of hybridization signal is proportional to the mass of the DNA fragment, indicating that the restriction fragments of the viral genome are represented in equimolar amounts. Note that the signals at 5.5 and 2.9 kbp appear twice as intense, because the signal is due to two restriction fragments that were not resolved under these conditions. Furthermore, the increase in viral DNA over the 3-day time course for each cell line is in good agreement with the measurements obtained by real-time PCR. To the resolution afforded by Southern blotting, the entire viral genome appeared to replicate over the course of the infection in each of the T-cell lines.
DISCUSSION
Early investigations established that group C adenoviruses form a long-lasting, asymptomatic infection in young children (21, 22). However, the cell type that harbors the virus and the nature of this interaction, whether persistent or latent, are not known. This laboratory recently reported that tonsillar T lymphocytes are a site of adenovirus persistence in naturally infected people (23). Given previous research showing atypical adenovirus infection patterns in lymphocyte cell lines, the present study was undertaken to analyze more thoroughly adenovirus infection of human T lymphocytes in vitro. Two patterns of behavior were observed among the four T-cell lines characterized. Both patterns diverge significantly from the lytic replication cycle seen in epithelial cells. Two of the cell lines (Jurkat and HuT78) were fully permissive for viral replication, synthesizing readily detectable early and late viral proteins, although on a per-cell basis they produce less than 10% of the level of infectious virus seen in A549 cells. Nonetheless, Jurkat and HuT78 synthesize four- to sixfold more adenoviral DNA per cell than A549 cells do. The other two cell lines investigated in this study (CEM-CARhi and KE37-CARhi) failed to produce any detectable live virus despite the presence of viral DNA replication. Thus, although all four human T-cell lines replicate viral DNA, they produce infectious virus either inefficiently or not at all. While both CEM-CARhi and KE37-CARhi cells internalize and uncoat incoming virus, unlike Jurkat and HuT78 they produce little viral early or late proteins, which would account for the failure to produce virion. In addition, robust viral genome replication was detected in the CEM-CARhi and KE37-CARhi cell lines despite the fact that the majority of these cells express no detectable E1A, DBP, penton, or hexon. Assuming that 100% of the cells contribute to viral genome replication, these cells produce almost three times more viral DNA per cell at day 4 than do A549 at the end of lytic replication. Alternatively, if viral DNA is replicating only in the subset of cells in which viral proteins are detected, then infected KE37-CARhi cells would be making 50- to 130-fold more viral DNA per cell than A549. In either case, these high levels of genome replication in the comparative absence of viral protein expression is quite unexpected, because products of the E2 transcription unit, including DBP, are required for adenoviral DNA replication in epithelial cells (51). We are aware of only one exception to the requirement for DBP function: four out of five EBV genome-positive lymphocyte cell lines were shown to replicate the genome of a temperature-sensitive DBP mutant adenovirus at the restrictive temperature (31). The authors suggested that some EBV function was complementing the mutation in DBP, but they did not test EBV-negative lymphocyte cell lines. Thus, the complementation could have been due to a cellular factor. It is possible that CEM-CARhi and KE37-CARhi also contain this factor and are able to complement the lack of DBP expression.
It is striking that while nearly all CEM-CARhi and KE37-CARhi cells were able to internalize virus, the majority of these cells expressed no detectable viral proteins. One simple explanation for this observation would be that the internalized virus is being degraded rapidly in these two cell lines. This possibility seems unlikely given the steady increase in viral DNA levels associated with these cells during the 4-day time course of infection. However, further experiments are needed to determine the possible extent of viral DNA degradation in these cells. As mentioned previously, it is possible that a small percentage of cells are replicating the viral genome at such a huge rate as to mask the degradation of the viral genome in the majority of cells.
Adenovirus replicates in the nucleus. Therefore, the question of whether the viral genome successfully enters the nuclei of most CEM-CARhi and KE37-CARhi cells is a very important issue. There are some interesting reports of conditions under which DNA viruses are internalized but do not transit to the nucleus. Naranatt et al. showed that chemical inhibitors of protein kinase C, MEK, or ERK kinases during human herpesvirus 8 infection of human foreskin fibroblasts inhibits transit of the viral genome to the nucleus but not internalization into the cell (46). Adenovirus transit to the nucleus, but not internalization, has been shown to require signaling through protein kinase A and p38-mitogen-activated protein kinase (54), and thus a deficiency in these pathways might result in inefficient nuclear localization and could explain the failure of the majority of CEM and KE37 to express adenoviral genes. We are unaware of any studies which assess the integrity of either of these two kinase signaling pathways in either of these cell lines.
Once inside the nucleus, it remains possible for viral genomes to remain inactive. Replication of several different DNA viruses, including adenovirus, is known to start at specific intranuclear domains called ND10 domains (or PML nuclear bodies) (18, 33, 49). ND10s are interchromatinic regions defined by the presence of several resident proteins. While the functions of these proteins have yet to be elucidated, it is interesting that several of them are upregulated by interferons, and several groups have suggested that these domains function in an antiviral manner (8, 12, 13). ND10s are dispersed during lytic infection with herpes simplex virus, EBV, HCMV, and adenovirus (2, 5, 33). Very recently it has been shown that latent EBV genomes are not associated with ND10s but are instead found associated with cellular chromatin. Upon reactivation of the EBV lytic cycle, these genomes become associated with ND10 (5). Thus, it could be that the adenovirus genome does enter the nucleus of CEM and KE37 cells but is not targeted to the correct location for the complete program of viral gene expression.
In addition to the failure to localize at ND10s, other mechanisms that restrict virus gene expression and replication have been described for DNA viruses that establish latent infections. At the simplest level, host transcriptional repressors have been shown to bind to and repress transcription from key lytic phase promoters of EBV (28, 37, 38, 44) and HCMV (4, 32, 53). Transcriptional repressors act by recruiting histone deacetylases, which remove acetyl groups from specific lysine residues in histones, thus generating higher order chromatin structures which occlude positive-acting transcription factors. In the case of both EBV and CMV, the viral genome is organized into chromatin by cellular histones, and the transition from latent to lytic replication is associated with the acetylation of histones covering key lytic phase promoters (35, 42, 45). Although one early report suggested that the adenovirus genome in infected cells might be organized into nucleosomes by cellular histones (16), others have shown that instead the adenovirus core pVII protein organizes the genome into nucleosome-like structures (11). The effect of this organization on viral transcription, and whether it is modulated similarly to cellular chromatin, is unknown. Initial experiments with inhibitors of histone deacetylases using trichostatin A and/or cellular DNA methyltransferases using 5-azacytidine have not resulted in increased viral gene expression in CEM-CARhi or KE37-CARhi cells (data not shown).
The concept of active intracellular defense mechanisms that restrict the replication of viruses has recently emerged. Human immunodeficiency virus replication in certain T-cell lines is prevented by cytosine deaminases, which cause C-to-U transitions in the single-stranded cDNA intermediates during replication, leading to destruction of the viral genome or the accumulation of fatal mutations (reviewed in reference 30). As previously mentioned, some researchers have proposed that one function of nuclear ND10 domains is antiviral in nature. Lymphotoxin can reversibly restrict replication of HCMV in human foreskin fibroblasts (6); interestingly, these researchers showed that the restriction was caused by an induction of α/β interferon that required both lymphotoxin and HCMV infection. This implies that viruses are able to cooperate with host cell signaling pathways in order to facilitate a latent state.
The recent observation that human mucosal T cells are a site of adenovirus persistence (23), combined with the results reported here, suggests that this cell type plays a critical role in adenoviral persistence in young children. For a virus to successfully maintain a latent infection cycle it must be quiescent in a cell from which it can later reactivate. Lymphocytes are ideal for hosting these cycles, because they are long lived and undergo several phases of activation and differentiation punctuated by periods of quiescence. In one of the most well-studied examples of viral latency in lymphocytes, EBV infects naïve B cells and forces them to differentiate into long-lived memory-type cells (43, 55). In these quiescent memory cells, the virus expresses very few, if any, genes. Lytic EBV replication is later triggered in a subset of these cells by poorly understood signals, resulting in viral recrudescence and spread between individuals. Further characterization of adenovirus infection in T lymphocytes may reveal a similar lifestyle for this virus. Adenovirus acute and latent infection phases may by regulated by tissue tropism. During an acute infection, the virus may initially enter a T cell that maintains viral DNA and suppresses gene expression. Activation of the cell may then lead to initiation of viral transcription and DNA replication, accounting for the episodic nature of virus shedding (21, 22).
Acknowledgments
This work was supported by Public Health Service grants CA-58736 (L.R.G.) and CA-77342 (D.O.) from the National Cancer Institute and AI-52280 (L.R.G.) from the National Institute of Allergy and Infectious Diseases and by a grant from the Emory University Research Committee (L.R.G.). Confocal microscopy was performed through the Micromed facility of the Comprehensive Cancer Center of Wake Forest University, which is supported in part by the grant CA-12197 from the National Cancer Institute.
We thank Ken Grant for his expert technical assistance with confocal microscopy and Emily Phillips for assistance with quantitative fluorescence microscopy.
REFERENCES
- 1.Adrian, T., G. Schafer, M. Cooney, J. Fox, and R. Wigand. 1988. Persistent enteral infections with adenovirus types 1 and 2 in infants; no evidence of reinfection. Epidemol. Infect. 101:503-509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ahn, J., and G. Hayward. 1997. The major immediate-early proteins IE1 and IE2 of human cytomegalovirus colocalize with and disrupt PML-associated nuclear bodies at very early times in infected permissive cells. J. Virol. 71:4599-4613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Andiman, W. A., and G. Miller. 1982. Persistent infection with adenovirus types 5 and 6 in lymphoid cells from humans and woolly monkeys. J. Infect. Dis. 145:83-88. [DOI] [PubMed] [Google Scholar]
- 4.Bain, M., M. Mendelson, and J. Sinclair. 2003. Ets-2 repressor factor (ERF) mediates repression of the human cytomegalovirus major immediate-early promoter in undifferentiated non-permissive cells. J. Gen. Virol. 84:41-49. [DOI] [PubMed] [Google Scholar]
- 5.Bell, P., P. Lieberman, and G. Maul. 2000. Lytic but not latent replication of Epstein-Barr virus is associated with PML and induces sequential release of nuclear domain 10 proteins. J. Virol. 74:11800-11810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Benedict, C. A., T. A. Banks, L. Senderowicz, M. Ko, W. J. Britt, A. Angulo, P. Ghazal, and C. F. Ware. 2001. Lymphotoxins and cytomegalovirus cooperatively induce interferon-beta, establishing host-virus detente. Immunity 15:617-626. [DOI] [PubMed] [Google Scholar]
- 7.Bergelson, J. M., J. A. Cunningham, G. Droguett, E. A. Kurt-Jones, A. Krithivas, J. S. Hong, M. S. Horwitz, R. L. Crowell, and R. W. Finberg. 1997. Isolation of a common receptor for coxsackie B viruses and adenoviruses 2 and 5. Science 275:1320-1323. [DOI] [PubMed] [Google Scholar]
- 8.Bonilla, W., D. Pinschewer, P. Klenerman, V. Rousson, M. Gaboli, P. Pandolfi, R. Zinkernagel, M. Salvato, and H. Hengartner. 2002. Effects of promyelocytic leukemia protein on virus-host balance. J. Virol. 76:3810-3818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bunn, P. A., Jr., and F. M. Foss. 1996. T-cell lymphoma cell lines (HUT102 and HUT78) established at the National Cancer Institute: history and importance to understanding the biology, clinical features, and therapy of cutaneous T-cell lymphomas (CTCL) and adult T-cell leukemia-lymphomas (ATLL). J. Cell Biochem. Suppl. 24:12-23. [DOI] [PubMed] [Google Scholar]
- 10.Cepko, C. L., P. S. Changelian, and P. A. Sharp. 1981. Immunoprecipitation with two-dimensional pools as a hybridoma screening technique: production and characterization of monoclonal antibodies against adenovirus 2 proteins. Virology 110:385-401. [DOI] [PubMed] [Google Scholar]
- 11.Chatterjee, P. K., M. E. Vayda, and S. J. Flint. 1986. Adenoviral protein VII packages intracellular viral DNA throughout the early phase of infection. EMBO J. 5:1633-1644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chee, A., P. Lopez, A. Pandolfi, and B. Roizman. 2003. Promyelocytic leukemia protein mediates interferon-based anti-herpes simplex virus 1 effects. J. Virol. 77:7101-7105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chelbi-Alix, M., F. Quignon, L. Pelicano, M. Koken, and H. Dethe. 1998. Resistance to virus infection conferred by the interferon-induced promyelocytic leukemia protein. J. Virol. 72:1043-1051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chu, Y., K. Sperber, L. Mayer, and M. T. Hsu. 1992. Persistent infection of human adenovirus type 5 in human monocyte cell lines. Virology 188:793-800. [DOI] [PubMed] [Google Scholar]
- 15.Church, G. M., and W. Gilbert. 1984. Genomic sequencing. Proc. Natl. Acad. Sci. USA 81:1991-1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dery, C. V., M. Toth, M. Brown, J. Horvath, S. Allaire, and J. M. Weber. 1985. The structure of adenovirus chromatin in infected cells. J. Gen. Virol. 66:2671-2684. [DOI] [PubMed] [Google Scholar]
- 17.Evans, A. 1958. Latent adenovirus infections of the human respiratory tract. Am. J. Hyg. 67:256-266. [DOI] [PubMed] [Google Scholar]
- 18.Everett, R. 2001. DNA viruses and viral proteins that interact with PML nuclear bodies. Oncogene 20:7266-7273. [DOI] [PubMed] [Google Scholar]
- 19.Feinberg, A. P., and B. Vogelstein. 1983. A technique for radiolabeling DNA restriction endonuclease fragments to high specific activity. Anal. Biochem. 132:6-13. [DOI] [PubMed] [Google Scholar]
- 20.Flomenberg, P., V. Piaskowski, J. Harb, A. Segura, and J. T. Casper. 1996. Spontaneous, persistent infection of a B-cell lymphoma with adenovirus. J. Med. Virol. 48:267-272. [DOI] [PubMed] [Google Scholar]
- 21.Fox, J. P., C. D. Brandt, F. E. Wassermann, C. E. Hall, I. Spigland, A. Kogon, and L. R. Elveback. 1969. The virus watch program: a continuing surveillance of viral infections in metropolitan New York families. VI. Observations of adenovirus infections: virus excretion patterns, antibody response, efficiency of surveillance, patterns of infections, and relation to illness. Am. J. Epidemiol. 89:25-50. [DOI] [PubMed] [Google Scholar]
- 22.Fox, J. P., C. E. Hall, and M. K. Cooney. 1977. The Seattle virus watch. VII. Observations of adenovirus infections. Am. J. Epidemiol. 105:362-386. [DOI] [PubMed] [Google Scholar]
- 23.Garnett, C. T., D. Erdman, W. Xu, and L. R. Gooding. 2002. Prevalence and quantitation of species C adenovirus DNA in human mucosal lymphocytes. J. Virol. 76:10608-10616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Goodrum, F. D., and D. A. Ornelles. 1997. The early region 1B 55-kilodalton oncoprotein of adenovirus relieves growth restrictions imposed on viral replication by the cell cycle. J. Virol. 71:548-561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Goodrum, F. D., and D. A. Ornelles. 1998. p53 status does not determine outcome of E1B 55-kilodalton mutant adenovirus lytic infection. J. Virol. 72:9479-9490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Greber, U. F., M. Nakano, M. Suomalainen. 1999. Adenovirus entry into cells: a quantitative fluorescence microscopy approach, p. 217-230. In W. S. Wold (ed.), Adenovirus methods and protocols, vol. 21. Humana Press, Inc., Totowa, N.J. [Google Scholar]
- 27.Greber, U. F., M. Willetts, P. Webster, and A. Helenius. 1993. Stepwise dismantling of adenovirus 2 during entry into cells. Cell 75:477-486. [DOI] [PubMed] [Google Scholar]
- 28.Gruffat, H., E. Manet, and A. Sergeant. 2002. MEF2-mediated recruitment of class II HDAC at the EBV immediate early gene BZLF1 links latency and chromatin remodeling. EMBO Rep. 3:141-146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Harlow, E., B. R. Franza, Jr., and C. Schley. 1985. Monoclonal antibodies specific for adenovirus early region 1A proteins: extensive heterogeneity in early region 1A products. J. Virol. 55:533-546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Harris, R. S., A. M. Sheehy, H. M. Craig, M. H. Malim, and M. S. Neuberger. 2003. DNA deamination: not just a trigger for antibody diversification but also a mechanism for defense against retroviruses. Nat. Immunol. 4:641-643. [DOI] [PubMed] [Google Scholar]
- 31.Horvath, J., C. Faxing, and J. M. Weber. 1991. Complementation of adenovirus early region 1a and 2a mutants by Epstein-Barr virus immortalized lymphoblastoid cell lines. Virology 184:141-148. [DOI] [PubMed] [Google Scholar]
- 32.Huang, T. H., T. Oka, T. Asai, T. Okada, B. W. Merrills, P. N. Gertson, R. H. Whitson, and K. Itakura. 1996. Repression by a differentiation-specific factor of the human cytomegalovirus enhancer. Nucleic Acids Res. 24:1695-1701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ishov, A., and G. Maul. 1996. The periphery of nuclear domain 10 (ND10) as site of DNA virus deposition. J. Cell Biol. 134:815-825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Israel, M. 1962. The viral flora of enlarged tonsils and adenoids. J. Pathol. Bacteriol. 84:169-176. [Google Scholar]
- 35.Jenkins, P. J., U. K. Binne, and P. J. Farrell. 2000. Histone acetylation and reactivation of Epstein-Barr virus from latency. J. Virol. 74:710-720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jones, N., and T. Shenk. 1979. Isolation of adenovirus type 5 host range deletion mutants defective for transformation of rat embryo cells. Cell 17:683-689. [DOI] [PubMed] [Google Scholar]
- 37.Kraus, R. J., S. J. Mirocha, H. M. Stephany, J. R. Puchalski, and J. E. Mertz. 2001. Identification of a novel element involved in regulation of the lytic switch BZLF1 gene promoter of Epstein-Barr virus. J. Virol. 75:867-877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kraus, R. J., J. G. Perrigoue, and J. E. Mertz. 2003. ZEB negatively regulates the lytic-switch BZLF1 gene promoter of Epstein-Barr virus. J. Virol. 77:199-207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Leon, R. P., T. Hedlund, S. J. Meech, S. Li, J. Schaack, S. P. Hunger, R. C. Duke, and J. DeGregori. 1998. Adenoviral-mediated gene transfer in lymphocytes. Proc. Natl. Acad. Sci. USA 95:13159-13164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Li, E., D. Stupack, R. Klemke, D. A. Cheresh, and G. R. Nemerow. 1998. Adenovirus endocytosis via alpha(v) integrins requires phosphoinositide-3-OH kinase. J. Virol. 72:2055-2061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.McNees, A. L., C. T. Garnett, and L. R. Gooding. 2002. The adenovirus E3 RID complex protects some cultured human T and B lymphocytes from Fas-induced apoptosis. J. Virol. 76:9716-9723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Meier, J. L. 2001. Reactivation of the human cytomegalovirus major immediate-early regulatory region and viral replication in embryonal NTera2 cells: role of trichostatin A, retinoic acid, and deletion of the 21-base-pair repeats and modulator. J. Virol. 75:1581-1593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Miyashita, E., B. Yang, G. Babcock, and D. Thorley-Lawson. 1997. Identification of the site of Epstein-Barr virus persistence in vivo as a resting B cell. J. Virol. 71:4882-4891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Montalvo, E. A., M. Cottam, S. Hill, and Y. J. Wang. 1995. YY1 binds to and regulates cis-acting negative elements in the Epstein-Barr virus BZLF1 promoter. J. Virol. 69:4158-4165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Murphy, J. C., W. Fischle, E. Verdin, and J. H. Sinclair. 2002. Control of cytomegalovirus lytic gene expression by histone acetylation. EMBO J. 21:1112-1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Naranatt, P., S. Akula, C. Zien, H. Krishnan, and B. Chandran. 2003. Kaposi's sarcoma-associated herpesvirus induces the phosphatidylinositol 3-kinase-PKC-ζ-MEK-ERK signaling pathway in target cells early during infectioni: implications for infectivity. J. Virol. 77:1524-1539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Nemerow, G. R., and P. L. Stewart. 1999. Role of alpha(v) integrins in adenovirus cell entry and gene delivery. Microbiol. Mol. Biol. Rev. 63:725-734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pear, W. S., G. P. Nolan, M. L. Scott, and D. Baltimore. 1993. Production of high-titer helper-free retroviruses by transient transfection. Proc. Natl. Acad. Sci. USA 90:8392-8396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Regad, T., and M. Chelbi-Alix. 2001. Role and fate of PML nuclear bodies in response to interferon and viral infections. Oncogene 20:7274-7286. [DOI] [PubMed] [Google Scholar]
- 50.Schneider, U., H. U. Schwenk, and G. Bornkamm. 1977. Characterization of EBV-genome negative “null” and “T” cell lines derived from children with acute lymphoblastic leukemia and leukemic transformed non-Hodgkin lymphoma. Int. J. Cancer 19:621-626. [DOI] [PubMed] [Google Scholar]
- 51.Shenk, T. 1996. Adenoviridae: the viruses and their replication, p. 2111-2148. In B. N. Fields, D. M. Knipe, and P. M. Howley (ed.), Fields virology, 3rd ed. Lippincott-Raven, Philadelphia, Pa.
- 52.Silver, L., and C. W. Anderson. 1988. Interaction of human adenovirus serotype 2 with human lymphoid cells. Virology 165:377-387. [DOI] [PubMed] [Google Scholar]
- 53.Sissons, J. G., M. Bain, and M. R. Wills. 2002. Latency and reactivation of human cytomegalovirus. J. Infect. 44:73-77. [DOI] [PubMed] [Google Scholar]
- 54.Suomalainen, M., M. Y. Nakano, K. Boucke, S. Keller, and U. F. Greber. 2001. Adenovirus-activated PKA and p38/MAPK pathways boost microtubule-mediated nuclear targeting of virus. EMBO J. 20:1310-1319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Thorley-Lawson, D. A. 2001. Epstein-Barr virus: exploiting the immune system. Nat. Rev. Immunology 1:75-82. [DOI] [PubMed] [Google Scholar]
- 56.Tomko, R. P., R. Xu, and L. Philipson. 1997. HCAR and MCAR: the human and mouse cellular receptors for subgroup C adenoviruses and group B coxsackieviruses. Proc. Natl. Acad. Sci. USA 94:3352-3356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.van der Veen, J., and M. Lambriex. 1973. Relationship of adenovirus to lymphocytes in naturally infected human tonsils and adenoids. Infect. Immun. 7:604-609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Venuat, A. M., C. Soria, J. Soria, P. Krief, M. Mirshahi, L. He, A. Thomaidis, A. Houllier, M. Billard, and C. Boucheix. 1988. High frequency of plasminogen activator secretion by malignant human lymphoid cell lines of T-cell type origin. Cancer 62:1952-1957. [DOI] [PubMed] [Google Scholar]
- 59.Vlahos, C. J., W. F. Matter, K. Y. Hui, and R. F. Brown. 1994. A specific inhibitor of phosphatidylinositol 3-kinase, 2-(4-morpholinyl)-8-phenyl-4H-1-benzopyran-4-one (LY294002). J. Biol. Chem. 269:5241-5248. [PubMed] [Google Scholar]
- 60.Weaver, L. S., and M. J. Kadan. 2000. Evaluation of adenoviral vectors by flow cytometry. Methods 21:297-312. [DOI] [PubMed] [Google Scholar]
- 61.Wickham, T. J., P. Mathias, D. A. Cheresh, and G. R. Nemerow. 1993. Integrins alpha v beta 3 and alpha v beta 5 promote adenovirus internalization but not virus attachment. Cell 73:309-319. [DOI] [PubMed] [Google Scholar]
- 62.Wold, W. S., S. L. Deutscher, N. Takemori, B. M. Bhat, and S. C. Magie. 1986. Evidence that AGUAUAUGA and CCAAGAUGA initiate translation in the same mRNA region E3 of adenovirus. Virology 148:168-180. [DOI] [PubMed] [Google Scholar]