

Abstract
Activins and Nodal are members of the transforming growth factor beta (TGF-β) family of growth factors. Their Smad2/3-dependent signalling pathway is well known for its implication in the patterning of the embryo after implantation. Although this pathway is active early on at preimplantation stages, embryonic phenotypes for loss-of-function mutations of prominent components of the pathway are not detected before implantation. It is only fairly recently that an understanding of the role of the Activin/Nodal signalling pathway at these stages has started to emerge, notably from studies detailing how it controls the expression of target genes in embryonic stem cells. We review here what is currently known of the TGF-β-related ligands that determine the activity of Activin/Nodal signalling at preimplantation stages, and recent advances in the elucidation of the Smad2/3-dependent mechanisms underlying developmental progression.
Keywords: Activin, Nodal, blastocyst, Smads, pluripotency, epiblast maturation
1. Introduction
The Smad2/3-dependent Activin/Nodal signalling pathway is known to play critical roles in the specification of cell identities in embryonic and extra-embryonic lineages of the postimplantation embryo, notably during processes such as the establishment of the anterior–posterior and left–right axes [1–5]. It has been known for a while that Activin/Nodal signalling is active well before implantation, but the absence of preimplantation defects when components of the pathway are mutated has delayed our understanding of the actual functions of Activin/Nodal signalling at these early stages. This dearth of notable phenotypes may reflect (i) the pathway's robustness, derived from partial functional redundancies between some of its components, (ii) the possible rescue of zygotic deficiencies by molecules of maternal origin, and (iii) the possibility that molecular changes brought about by the inactivation of the pathway before implantation only become detectable at later stages. A review of recent and not so recent studies, conducted both in the embryo and in cultured pluripotent stem cells, allows the relative merits of these alternatives to be assessed, and provides valuable insights into how the Activin/Nodal signalling pathway is operating at preimplantation stages.
2. The Activin/Nodal signalling pathway
Activins and Nodal are secreted as dimerized precursors, which are then cleaved to generate an active ligand [6]. Activins are homo or heterodimers of βA or βB subunits and therefore come in three versions known as Activin A, Activin B and Activin AB, collectively designated as Activin thereafter. Activin and Nodal have in common that they signal via receptor complexes containing the same type I (ALK4 or 7) and type II (ActRIIA or B) serine/threonine kinase receptors, the activation of which leads to the phosphorylation of the cytoplasmic transducers Smad2 or Smad3 (figure 1) [7]. Upon phosphorylation, dimers of Smad2/3 form a ternary complex with Smad4 that translocates to the nucleus where it associates with tissue-specific transcription factors [8,9] to activate the expression of target genes. How this is achieved has been the focus of intense research activity in recent years. These studies all emphasize the role in this process of chromatin modifiers recruited by Smad complexes and their cofactors. These advances have been the subject of several reviews [10–12].
Figure 1.

The TGF-β signalling pathway in the early mouse embryo. See the text for details. TF, transcription factor.
Nodal differs from Activin in that it requires the presence of an EGF–CFC family co-receptor (Cripto or Cryptic) to be able to activate the receptor complex. This critical difference results in Activin and Nodal signals being subjected to distinct regulatory interactions. Among the targets of Smad2/3 signalling are Nodal itself, which can therefore amplify its own expression, and the Lefty1 and Lefty2 genes, which encode Nodal antagonists thus placing Nodal expression under the control of a powerful negative feedback mechanism that will limit how long and how far it can signal [13]. Lefty1,2 are also transforming growth factor beta (TGF-β) family members. They inhibit Nodal signalling by interacting with Nodal or with Cripto. Because Activin signals in a Cripto-independent fashion, it is not sensitive to inhibition by Lefty [14]. Activin is however antagonized by another secreted molecule, called Follistatin. There is also evidence that Cripto can inhibit signalling from Activin, TGF-β and Myostatin, which have in common to signal via Smad2/3 without requiring an EGF–CFC co-receptor to interact with their receptor complex [15–18].
Other TGF-β-related ligands are known to modulate Activin/Nodal signalling. Bone morphogenetic protein (BMP) signals are transduced via the Smad1/5/8 branch of TGF-β signalling. There are now two well-characterized instances where BMP/Smad1,5,8 signalling ensures the proper developmental outcome by countering Activin/Nodal/Smad2/3 signalling via competition for a limited pool of Smad4 or for a shared receptor, ActRIIB [19,20]. Gdf1 and Gdf3, two other TGF-β family members, were found to bind the same receptor complex as Nodal, with the same requirement for an EGF–CFC co-receptor [21]. They are, however, unable to activate the Smad2/3 pathway on their own at physiological concentrations [22–25]. Instead, they seem to act as heterodimers, either to increase the range or strength of Nodal signalling when combined with Nodal, or to inhibit BMP signalling when combined with Bmp4 [22,24–26]. Further interactions between the two signalling pathways involve the inhibitory Smads, Smad6 and Smad7, which are themselves targets of Smad signalling and act either as general inhibitors of Smad-mediated signalling (Smad7), or inhibit more specifically the BMP pathway (Smad6) [27]. Smads can also integrate the input of FGF/RTK signalling, via MAPK-mediated phosphorylation, which can, for example, promote Smad1 degradation, and reduce BMP signal transduction [12,28]. Further information on how Smads integrate input from other signalling pathways can be found in other reviews [7,10,29].
3. Phenotypes of Activin/Nodal signalling pathway mutants
At first glance, genetics seems to offer little support to the notion that Activin/Nodal signalling plays an important role at preimplantation stages. Loss-of-function mutations for components of the Activin/Nodal signalling pathway (reviewed in [4,13] and summarized in table 1) result in a range of phenotypes that broadly fall in three groups: in the first, homozygotes get born, and display either craniofacial defects that lead them to an early death, or milder defects or an absence of defects that let them reach adulthood (Activin A, Activin B, Alk7, Smad3); in the second, homozygotes have primitive streak and/or left–right axis defects, which are sometimes mild enough to allow them to develop to term (Gdf1, ActRIIA, ActRIIB, Cryptic); in the third, homozygotes have early patterning defects and fail to gastrulate normally (Nodal, Gdf3, Alk4, Cripto, Smad2, Smad4). The Nodal−/− phenotype is the one that has been most extensively characterized and therefore provides a convenient standard for comparison with the other mutant phenotypes in that last group. In Nodal−/− embryos, the earliest defects appear shortly after implantation. They are smaller than littermates [30,31], and their epiblast, the pluripotent tissue that gives rise to the embryo proper, differentiates prematurely towards an anterior neural identity [32,33]. In addition, their visceral endoderm (VE), the extraembryonic layer that surrounds both the epiblast and the extraembryonic ectoderm (ExE), is improperly regionalized and fails notably to differentiate distal visceral endoderm (DVE) cells [34], which are essential for the establishment of anterior–posterior polarity. These embryos do not gastrulate [1,31]. Cripto, Cryptic double mutant embryos have a phenotype similar to that of Nodal−/− [46]. Consequently, although there have been suggestions that Cripto and Cryptic may have Nodal-independent functions, their compound mutant phenotype is consistent with the notion that Nodal absolutely requires them to signal. By contrast, the fact that Smad2,3 double mutant embryos [56], as well as Smad4−/− embryos [57], are even smaller than Nodal−/− embryos suggests that zygotic Nodal may not be the only ligand capable of activating the Smad2/3 pathway early on. Consistent with this view, the expression of a reporter transgene for the autoregulatory Smad2/3-dependent Nodal enhancer ASE, called ASE–YFP, was found to be maintained up to embryonic day (E)4.5 in Nodal−/− embryos [23]. In other animal models, there is broad evidence of another TGF-β family member acting upstream of early Nodal expression [58–62]. Vg1 in Xenopus is the prototype of a maternally deposited TGF-β-related ligand that is required to form the organizer and the mesoderm [58], and Vg1-related molecules of maternal origin identified in zebrafish and in sea-urchin appear to have similar properties [59–61]. Gdf1 and Gdf3 in the mouse are the two factors identified as Vg-1-related, however, as we saw, indications are that it is as Nodal partner and BMP antagonist that they are playing a role in the regulation of Activin/Nodal signalling and Nodal expression, not as inducers. Better candidate ligands for the early activation of the Activin/Nodal signalling pathway may thus be activins, which are present in the oviduct, uterine epithelia and blastocysts prior to implantation [63–65]. In any case, what the analysis of the earliest mutant phenotypes indicates is that the Activin/Nodal signalling pathway is not required for the formation of the epiblast and the VE, but that it is important to ensure their proper growth and patterning.
Table 1.
Mutant phenotypes of genes encoding components of the Activin/Nodal signalling pathway. AVE, anterior visceral endoderm; DVE, distal visceral endoderm; Epi, epiblast; ExE, extraembryonic ectoderm; LR, left–right axis; ME, mesendoderm; PS, primitive streak; VE, visceral endoderm.
| gene | mutant phenotype | references |
|---|---|---|
| Nodal | small size; premature Epi differentiation (neuralization); no DVE; defective ExE; no PS; no ME | Conlon et al. [30], with Iannaccone et al. [31], Camus et al. [32], Mesnard et al. [33], Brennan et al. [34] and Lowe et al. [35] |
| Activin A | get born, die within 24 h; craniofacial defects | Matzuk et al. [36] |
| Activin B | get born; maternal Activin B not essential; defective eyelid and female reproduction | Vassalli et al. [37] |
| Activin A, B | additive phenotype; no new defect; no functional redundancy | Matzuk et al. [36] |
| Alk4 | resembles Nodal−/− small; defective from E5.5; reduced ME | Gu et al. [38] |
| Alk7 | no phenotype even when combined with Alk4 or Nodal mutation | Jornvall et al. [39] |
| ActRIIA | get born; craniofacial and reproductive defects; PS defects | Matzuk et al. [36] and Song et al. [40] |
| ActRIIB | get born; LR defects (right isomerism) | Oh et al. [41] |
| ActRIIA, B | resembles Nodal−/−; small; no PS and ME; demonstrates partial redundancy | Song et al. [40] |
| Cripto | form DVE but no AVE; reduced ME | Ding et al. [42] and Xu et al. [43] |
| Cryptic | LR defects (right isomerism and heterotaxia) | Gaio et al. [44] and Yan et al. [45] |
| Cripto, Cryptic | resembles Nodal−/−; defects in Epi, ExE and AVE | Chu et al. [46] |
| Gdf1 | LR defects (resembles Cryptic−/−) | Rankin et al. [47] and Andersson et al. [48] |
| Gdf3 | AVE defects; ME defects | Chen et al. [49] |
| FoxH1 | no anterior PS and its derivatives | Hoodless et al. [50] |
| Smad2 | Epi converted into ExE; no AVE; early patterning defects | Waldrip et al. [51], Heyer et al. [52] and Brennan et al. [34] |
| Smad3 | get born; subtle developmental anomalies | Zhu et al. [53], Datto et al. [54] and Yang et al. [55] |
| Smad2/3 | resembles Smad4−/−; very small; fail to gastrulate and lack ME | Dunn et al. [56] |
| Smad4 | very small; fail to gastrulate | Sirard et al. [57] |
4. Expression dynamics of transforming growth factor beta-related ligands and associated developmental events
After fertilization, the first three rounds of divisions lead the mouse embryo to the 8-cell stage. Symmetric and asymmetric divisions then generate inner and outer cells within the 16- and 32-cell stage embryos [66]. At E3.5, during blastocyst formation, outer cells differentiate into trophoblasts to form the trophectoderm (TE), an extraembryonic tissue that encloses inner cell mass (ICM) cells and the blastocoele cavity. At E4.0, shortly before implantation, the ICM gives rise to the epiblast and to the primitive endoderm (PrE), another extraembryonic layer from which the VE will later derive. Around E4.5, the TE mediates the implantation of the blastocyst in the uterine wall.
Phosphorylated forms of Smad2/3 are detected in cell nuclei of mouse embryos from the 4-cell stage onward [67]. Studies of Activin expression in the mouse embryo showed that there is a stock of maternal Activin present in the egg that gets depleted during early cleavage stages. Zygotic expression is detected in the compacted morula (figure 2). By E3.5, this expression is confined to the ICM of the blastocyst, but at E4.5 it is not detected in the ICM-derived epiblast and PrE and is instead present in the TE [63,68]. Activin is also produced by the oviduct and the uterus at these stages [65], which may explain the lack of an early phenotype in embryos mutant for the βA and βB subunits [36,37]. An indication that this Activin of maternal origin does contribute to embryo development comes from studies showing that cleavage stage embryos developed better in vitro when cultured in the presence of Activin [69,70].
Figure 2.
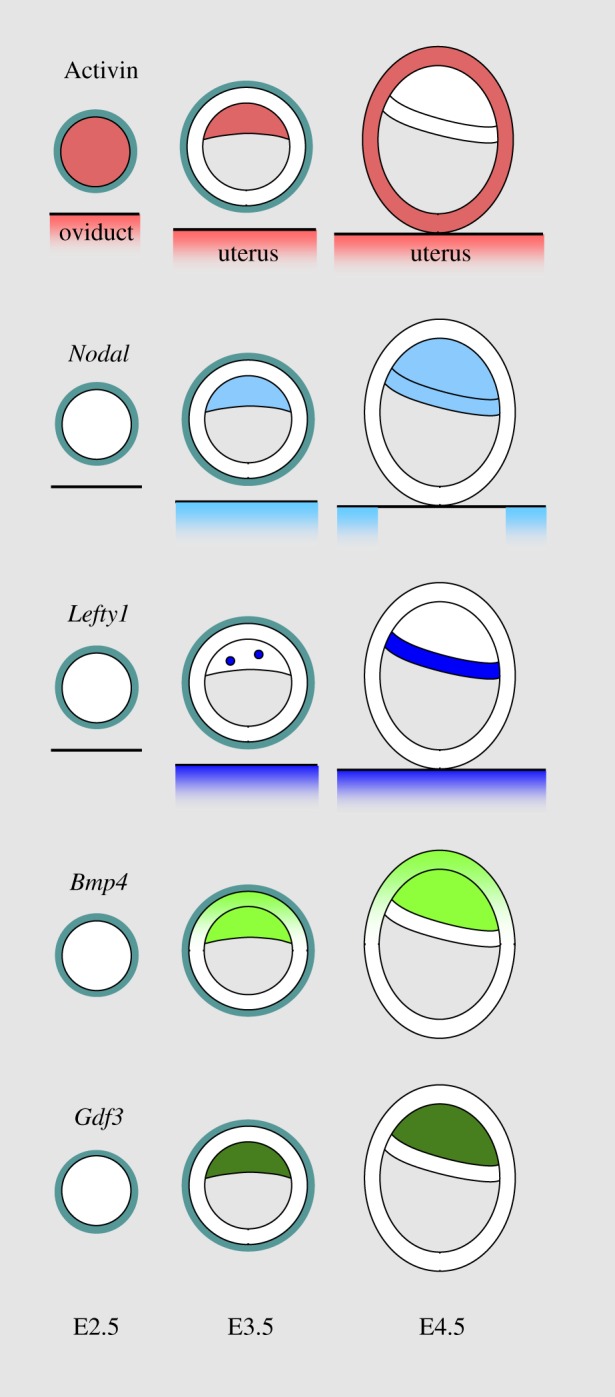
Expression of TGF-β ligands in the early mouse embryo. Expression of Activin, Nodal, Lefty1, Bmp4 and Gdf3 in the morula (E2.5), the early preimplantation blastocyst (E3.5) and the hatched blastocyst (E4.5). See the text for details.
In contrast to this presence of Activin in the embryo from a very early stage, other TGF-β family members of interest all see their embryonic expression begin in the ICM of the E3.5 blastocyst, but from there they follow different dynamics. Nodal expression persists in the epiblast and the PrE when these tissues segregate [23,71]. It is also detected in the endometrium of pregnant females at E3.5 [72], a potential source of Nodal of maternal origin to the blastocyst. Lefty1 expression is detected in a cluster of cells of the PrE when this layer forms [71,73]. The expression of Bmp4 persists in the epiblast but not in the PrE when they appear, and is induced in the polar trophectoderm [74]. Gdf3 expression strengthens in the epiblast, but is not detected in the PrE [23,24,49,71].
The onset of the expression of these genes and their subsequent diverging dynamics mark key transitions in the development of the epiblast and the PrE, events in which Activin/Nodal signalling appears to be implicated. Analysis of Nodal−/− embryos had revealed their failure to properly differentiate the part of the VE overlying the epiblast just after implantation and their subsequent failure to form DVE cells [33,34], thus establishing a requirement for Nodal in the patterning of this extra-embryonic endodermal layer. Use of a conditional gene inactivation strategy showed that it is Nodal expressed in the epiblast that drives the regionalization of this layer [75]. However, the realization that two Nodal-dependent DVE markers, Lefty1 and Cerl, had an earlier phase of expression in a small cluster of PrE cells [71,76], and that Nodal was expressed in the ICM and in the preimplantation epiblast [23,71], suggested that the regionalization of the VE had its origin in earlier events. Lineage-tracing studies indeed showed that descendants from Lefty1 and Cerl-expressing PrE cells give rise to the DVE at E5.5, thus possibly placing the specification of this particular cell identity shortly before implantation [5,76,77]. For Lefty1, the enhancer driving this early PrE expression was shown to be dependent on FoxH1, a well-known effector of Activin/Nodal signalling. Likewise, ASE–YFP, the Smad2/3-dependent reporter transgene, was found to be transiently expressed in a subpopulation of PrE cells before implantation [23]. However, as previously mentioned, this expression is maintained in Nodal−/− embryos. So, although evidence shows that the Activin/Nodal signalling pathway is implicated in the regionalization of the extraembryonic endodermal layer as soon as the PrE is established, these data indicate that, at this early stage, it involves a ligand other than zygotic Nodal, possibly Activin.
There are indications that something similar is taking place in the epiblast. The epiblast that first emerges from the ICM, the preimplantation epiblast, is quite different from that of the egg cylinder, the postimplantation epiblast. Preimplantation epiblast is composed of apolar cells, postimplantation epiblast becomes organized in an epithelium when the proamniotic cavity forms at E5.0. This correlates with marked changes in the properties of the tissue. Single preimplantation epiblast cells injected in a host blastocyst can contribute to all fetal lineages of the resulting chimaera [78,79], whereas epiblast cells isolated at postimplantation stages cannot [80]. These differences extend to the distinct pluripotent stem cell lines derived from the epiblast at these stages: blastocyst-derived embryonic stem cells (ESCs) can contribute to all fetal lineages [81], whereas E5.5 to E6.5 embryo-derived epiblast stem cells (EpiSCs) can differentiate in multiple lineages in vitro but are unable to participate in the formation of a chimaera when injected in a blastocyst [82]. Unsurprisingly, the physiology of these cells is also different. Although ESCs express Nodal and have an active Activin/Nodal signalling pathway, this is not essential to their maintenance [67,83]. In contrast, EpiSCs' capacity to self-renew depends critically on Activin/Nodal signalling [82,84]. Inhibition of the pathway in EpiSCs triggers a drastic downregulation of the pluripotency factor Nanog, which results in the cells differentiating towards a neural identity [82,85]. ESCs can be converted into EpiSCs in vitro when cultured in the presence of Activin and FGF, suggesting that the maturation of the epiblast is dependent on the same signals in vivo [86]. This differentiation has been described as a transition from a ground state of pluripotency to a primed state of pluripotency [81]. The dynamics of certain molecular markers allow us to visualize the beginning of this process in the embryo. Nascent epiblast cells maintain Nanog expression as they emerge from the ICM, but they start to downregulate it, in a salt and paper fashion, as they approach implantation. At the same time, the expression of the ASE–YFP transgene gets activated in epiblast cells with low or no Nanog, so that the two markers briefly display a somewhat complementary pattern in the epiblast of the implanting blastocyst [23]. Nanog then disappears while ASE–YFP is found in all epiblast cells. The observation that the expression of the Smad2/3-dependent ASE–YFP transgene is still present in the epiblast of implanting Nodal−/− embryos suggests that the transition to a primed state of pluripotency is correctly initiated in these mutants, and that it involves a TGF-β-related ligand other than zygotic Nodal. This interpretation is supported by the fact that Nodal−/− epiblast cells prematurely differentiate along the neural pathway, just as EpiSCs do when deprived of Activin/Nodal signalling [32,82,85].
5. Embryonic stem cells as a model to study early function and regulation of Activin/Nodal signalling
Studying what happens when ESCs differentiate into EpiSCs has recently led to important advances in our understanding of epiblast maturation and of the part Activin/Nodal signalling is playing in it. Transcriptomic and epigenomic comparisons between the two cell types has revealed that a global rearrangement of enhancer chromatin landscape is taking place, that leads to a shift in enhancer usage not just for the few genes found to be differentially expressed, but also for those that do not see a change in their expression levels [87,88]. For the latter, enhancers specifically active in ESCs, which tend to be enriched for DNA binding motifs of Smad2/3 and Smad4, are decommissioned once their EpiSC-specific enhancers are activated in differentiating ESCs [88]. A genome-wide characterization of Smad3 binding in different cell types led to the surprising finding that a small set of cell-type-specific master transcription factors direct Smad3 to cell-type-specific binding sites and determines cell-type-specific responses to TGF-β signalling [8]. Thus, in ESCs, it is with the pluripotency factor Oct4 that Smad3 co-occupies the genome. Oct4 is known to act as a pioneer factor at enhancers, opening up the chromatin to allow other factors to access their binding sites. Recent studies have shown that the capacity of ESCs to differentiate is critically dependent on the level of Oct4 not being too low [89,90], indicating the function of Oct4 involves more than allowing the establishment of pluripotency, an assessment supported by its extensive relocalization on the genome during the ESC to EpiSC transition [87].
Other evidence for the implication of Activin/Nodal signalling during this transition has emerged from particular examples of the two sets of genes described above, the ones that are differentially expressed and the ones that are similarly expressed. Gsc, which encodes the transcription factor and mesendoderm regulator Goosecoid, is a poised gene whose expression is induced once differentiation is underway [91]. Work in the Massagué laboratory showed that the stimulation of ESCs with Activin leads to the formation of companion Smad4–Smad2/3 and TRIM33–Smad2/3 complexes, which are both required to activate Gsc expression. The repressive histone mark H3K9me3 at the poised promoter of the gene is bound by TRIM33–Smad2/3, which in turn displaces the chromatin-compacting factor HP1γ to make neighbouring Smad binding elements (SBEs) accessible to the Smad4–Smad2/3 complex, presumably associated with FoxH1, and allows PolII recruitment [91]. Smad4 then promotes further binding of the TRIM33 complex via chromatin modification, ensuring robust gene expression.
Nodal is expressed in both ESCs and EpiSCs. As both a target and an inducer of Activin/Nodal signalling, it has been extensively studied and much is known about its regulation. Recent work led to the identification of a novel Nodal enhancer called HBE that is a hotspot for the binding of pluripotency factors, and to the characterization of its implication in the regulatory shift taking place at the Nodal locus. HBE is the only active Nodal enhancer in ESCs, while it is ASE that is the most active one in EpiSCs [23,92]. Both enhancers are dependent on Activin/Nodal signalling, but their activation relies on pSmad2/3 interacting with distinct transcription factors: the pluripotency factor Oct4 in the case of HBE (figure 3) [8,93], and FoxH1 in that of ASE [94]. Deletion of HBE in ESCs eliminates Nodal expression, confirming that HBE is essential to the expression of the gene in these cells. Deletion of HBE in EpiSCs does not affect Nodal expression, which is consistent with Nodal expression being dependent on ASE in these cells. However, ESCs carrying an HBE-deleted allele of Nodal fail to express this allele when induced to differentiate into EpiSCs, revealing that the activation of ASE is dependent on HBE being present when the cells differentiate. Furthermore, HBE deletion results in the repressive histone mark H3K27me3 accumulating in the vicinity of the ASE [92]. This suggests that multi-transcription factors binding loci (MTLs, as the hotspots for the binding of pluripotency factors are called) may mediate the influence of the pluripotency gene regulatory network by determining the status of adjacent regulatory elements and the timing of their activation. The transition from ESC to EpiSC, from an HBE-driven phase to an ASE-driven phase, correlates with a decrease in the expression of master pluripotency factors known to bind HBE (such as Nanog and Klf4), and an upregulation of Nodal downstream targets [23,82,84]. The exposure of ESCs to FGF and Activin, and the resulting surge in Activin/Nodal signalling, triggers a cascade of events leading them to reach a new equilibrium, which defines their new identity. The molecular mechanisms underlying this transition, notably the role played by physical interactions between components of the Activin/Nodal signalling pathway, Smad2/3-associated transcription factors and chromatin modifiers and specific regulatory sequences, remain to be elucidated.
Figure 3.

Different Smad2/3 complexes cooperate to control the expression of Activin/Nodal target genes in differentiating ESCs. (a) The expression of the poised gene Gsc is initiated by the binding of a TRIM33–Smad2/3 complex which in turn facilitates the recruitment of a FoxH1–Smad4–Smad2/3 complex to a neighbouring Activin response element (ARE). (b) The expression of Nodal in ESCs is dependent on the interaction of its enhancer HBE with an Oct4–Smad2/3 complex. HBE is itself required to activate the FoxH1–Smad4–Smad2/3-dependent ASE enhancer, which becomes the predominant Nodal enhancer in EpiSCs.
6. Conclusions
Current data therefore suggest that Activin/Nodal signalling, although not required for the formation of the epiblast and the PrE, plays an essential role in their development and regionalization as soon as these lineages emerge. Modulations in the activity of Activin/Nodal signalling trigger changes in gene expression via the recruitment of cell-type specific Smad2/3 complexes at cognate regulatory sequences, and the remodelling of adjacent chromatin. Studying the dynamics and the molecular bases of these events in pluripotent stem cells is bound to further our understanding of the role of Activin/Nodal signalling in the early mouse embryo.
Funding statement
Work in J.C.'s laboratory is supported by the CNRS, the ANR and the Ile-de-France Committees of Ligue Nationale contre le Cancer. C.P. was supported by a fellowship from the FRM and by the ANR.
Competing interests
The authors declare no competing interests.
References
- 1.Robertson EJ. 2014. Dose-dependent Nodal/Smad signals pattern the early mouse embryo. Semin. Cell Dev. Biol. 32, 1717–1728. ( 10.1016/j.semcdb.2014.03.028) [DOI] [PubMed] [Google Scholar]
- 2.Rossant J, Tam PP. 2009. Blastocyst lineage formation, early embryonic asymmetries and axis patterning in the mouse. Development 136, 701–713. ( 10.1242/dev.017178) [DOI] [PubMed] [Google Scholar]
- 3.Schier AF. 2009. Nodal morphogens. Cold Spring Harb. Perspect. Biol. 1, a003459 ( 10.1101/cshperspect.a003459) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shen MM. 2007. Nodal signaling: developmental roles and regulation. Development 134, 1023–1034. ( 10.1242/dev.000166) [DOI] [PubMed] [Google Scholar]
- 5.Takaoka K, Yamamoto M, Hamada H. 2011. Origin and role of distal visceral endoderm, a group of cells that determines anterior–posterior polarity of the mouse embryo. Nat. Cell Biol. 13, 743–752. ( 10.1038/ncb2251) [DOI] [PubMed] [Google Scholar]
- 6.Constam DB. 2014. Regulation of TGF-β and related signals by precursor processing. Semin. Cell Dev. Biol. 32, 85–97. ( 10.1016/j.semcdb.2014.01.008) [DOI] [PubMed] [Google Scholar]
- 7.Massague J, Seoane J, Wotton D. 2005. Smad transcription factors. Genes Dev. 19, 2783–2810. ( 10.1101/gad.1350705) [DOI] [PubMed] [Google Scholar]
- 8.Mullen AC, et al. 2011. Master transcription factors determine cell-type-specific responses to TGF-β signaling. Cell 147, 565–576. ( 10.1016/j.cell.2011.08.050) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Oshimori N, Fuchs E. 2012. The harmonies played by TGF-β in stem cell biology. Cell Stem Cell 11, 751–764. ( 10.1016/j.stem.2012.11.001) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gaarenstroom T, Hill CS. 2014. TGF-β signaling to chromatin: how Smads regulate transcription during self-renewal and differentiation. Semin. Cell Dev. Biol. 32, 107–118. ( 10.1016/j.semcdb.2014.01.009) [DOI] [PubMed] [Google Scholar]
- 11.Massague J. 2012. TGF-β signaling in development and disease. FEBS Lett. 586, 1833 ( 10.1016/j.febslet.2012.05.030) [DOI] [PubMed] [Google Scholar]
- 12.Wu MY, Hill CS. 2009. Tgf-β superfamily signaling in embryonic development and homeostasis. Dev. Cell 16, 329–343. ( 10.1016/j.devcel.2009.02.012) [DOI] [PubMed] [Google Scholar]
- 13.Schier AF. 2003. Nodal signaling in vertebrate development. Annu. Rev. Cell Dev. Biol. 19, 589–621. ( 10.1146/annurev.cellbio.19.041603.094522) [DOI] [PubMed] [Google Scholar]
- 14.Cheng SK, Olale F, Brivanlou AH, Schier AF. 2004. Lefty blocks a subset of TGF-β signals by antagonizing EGF–CFC coreceptors. PLoS Biol. 2, E30 ( 10.1371/journal.pbio.0020030) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Adkins HB, et al. 2003. Antibody blockade of the Cripto CFC domain suppresses tumor cell growth in vivo. J. Clin. Invest. 112, 575–587. ( 10.1172/JCI17788) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gray PC, Harrison CA, Vale W. 2003. Cripto forms a complex with activin and type II activin receptors and can block activin signaling. Proc. Natl Acad. Sci. USA 100, 5193–5198. ( 10.1073/pnas.0531290100) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gray PC, Shani G, Aung K, Kelber J, Vale W. 2006. Cripto binds transforming growth factor beta (TGF-β) and inhibits TGF-β signaling. Mol. Cell. Biol. 26, 9268–9278. ( 10.1128/MCB.01168-06) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Guardiola O, et al. 2012. Cripto regulates skeletal muscle regeneration and modulates satellite cell determination by antagonizing myostatin. Proc. Natl Acad. Sci. USA 109, E3231–E3240. ( 10.1073/pnas.1204017109) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Furtado MB, et al. 2008. BMP/SMAD1 signaling sets a threshold for the left/right pathway in lateral plate mesoderm and limits availability of SMAD4. Genes Dev. 22, 3037–3049. ( 10.1101/gad.1682108) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yamamoto M, Beppu H, Takaoka K, Meno C, Li E, Miyazono K, Hamada H. 2009. Antagonism between Smad1 and Smad2 signaling determines the site of distal visceral endoderm formation in the mouse embryo. J. Cell Biol. 184, 323–334. ( 10.1083/jcb.200808044) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Massague J. 2003. Integration of Smad and MAPK pathways: a link and a linker revisited. Genes Dev. 17, 2993–2997. ( 10.1101/gad.1167003) [DOI] [PubMed] [Google Scholar]
- 22.Andersson O, Bertolino P, Ibanez CF. 2007. Distinct and cooperative roles of mammalian Vg1 homologs GDF1 and GDF3 during early embryonic development. Dev. Biol. 311, 500–511. ( 10.1016/j.ydbio.2007.08.060) [DOI] [PubMed] [Google Scholar]
- 23.Granier C, et al. 2011. Nodal cis-regulatory elements reveal epiblast and primitive endoderm heterogeneity in the peri-implantation mouse embryo. Dev. Biol. 349, 350–362. ( 10.1016/j.ydbio.2010.10.036) [DOI] [PubMed] [Google Scholar]
- 24.Levine AJ, Brivanlou AH. 2006. GDF3, a BMP inhibitor, regulates cell fate in stem cells and early embryos. Development 133, 209–216. ( 10.1242/dev.02192) [DOI] [PubMed] [Google Scholar]
- 25.Tanaka C, Sakuma R, Nakamura T, Hamada H, Saijoh Y. 2007. Long-range action of Nodal requires interaction with GDF1. Genes Dev. 21, 3272–3282. ( 10.1101/gad.1623907) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fuerer C, Nostro MC, Constam DB. 2014. Nodal–Gdf1 hetrodimers with bound prodomains enable serum-independent Nodal signaling and endoderm differentiation. J. Biol. Chem. 289, 17 854–17 871. ( 10.1074/jbc.M114.550301) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.ten Dijke P, Hill CS. 2004. New insights into TGF-β–Smad signalling. Trends Biochem. Sci. 29, 265–273. ( 10.1016/j.tibs.2004.03.008) [DOI] [PubMed] [Google Scholar]
- 28.Sapkota G, Alarcon C, Spagnoli FM, Brivanlou AH, Massague J. 2007. Balancing BMP signaling through integrated inputs into the Smad1 linker. Mol. Cell 25, 441–454. ( 10.1016/j.molcel.2007.01.006) [DOI] [PubMed] [Google Scholar]
- 29.Feng XH, Derynck R. 2005. Specificity and versatility in TGF-β signaling through Smads. Annu. Rev. Cell Dev. Biol. 21, 659–693. ( 10.1146/annurev.cellbio.21.022404.142018) [DOI] [PubMed] [Google Scholar]
- 30.Conlon FL, et al. 1994. A primary requirement for nodal in the formation and maintenance of the primitive streak in the mouse. Development 120, 1919–1928. [DOI] [PubMed] [Google Scholar]
- 31.Iannaccone PM, Zhou X, Khokha M, Boucher D, Kuehn MR. 1992. Insertional mutation of a gene involved in growth regulation of the early mouse embryo. Dev. Dyn. 194, 198–208. ( 10.1002/aja.1001940305) [DOI] [PubMed] [Google Scholar]
- 32.Camus A, Perea-Gomez A, Moreau A, Collignon J. 2006. Absence of Nodal signaling promotes precocious neural differentiation in the mouse embryo. Dev. Biol. 295, 743–755. ( 10.1016/j.ydbio.2006.03.047) [DOI] [PubMed] [Google Scholar]
- 33.Mesnard D, Guzman-Ayala M, Constam DB. 2006. Nodal specifies embryonic visceral endoderm and sustains pluripotent cells in the epiblast before overt axial patterning. Development 133, 2497–2505. [DOI] [PubMed] [Google Scholar]
- 34.Brennan J, Lu CC, Norris DP, Rodriguez TA, Beddington RS, Robertson EJ. 2001. Nodal signalling in the epiblast patterns the early mouse embryo. Nature 411, 965–969. ( 10.1038/35082103) [DOI] [PubMed] [Google Scholar]
- 35.Lowe LA, Yamada S, Kuehn MR. 2001. Genetic dissection of nodal function in patterning the mouse embryo. Development 128, 1831–1843. [DOI] [PubMed] [Google Scholar]
- 36.Matzuk MM, Kumar TR, Bradley A. 1995. Different phenotypes for mice deficient in either activins or activin receptor type II. Nature 374, 356–360. ( 10.1038/374356a0) [DOI] [PubMed] [Google Scholar]
- 37.Vassalli A, Matzuk MM, Gardner HA, Lee KF, Jaenisch R. 1994. Activin/inhibin beta B subunit gene disruption leads to defects in eyelid development and female reproduction. Genes Dev. 8, 414–427. ( 10.1101/gad.8.4.414) [DOI] [PubMed] [Google Scholar]
- 38.Gu Z, et al. 1998. The type I activin receptor ActRIB is required for egg cylinder organization and gastrulation in the mouse. Genes Dev. 12, 844–857. ( 10.1101/gad.12.6.844) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jörnvall H, Reissmann E, Andersson O, Mehrkash M, Ibáñez CF. 2004. ALK7, a receptor for nodal, is dispensable for embryogenesis and left–right patterning in the mouse. Mol. Cell. Biol. 24, 9383–9389. ( 10.1128/MCB.24.21.9383-9389.2004) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Song J, Oh SP, Schrewe H, Nomura M, Lei H, Okano M, Gridley T, Li E. 1999. The type II activin receptors are essential for egg cylinder growth, gastrulation, and rostral head development in mice. Dev. Biol. 213, 157–169. ( 10.1006/dbio.1999.9370) [DOI] [PubMed] [Google Scholar]
- 41.Oh SP, Li E. 1997. The signaling pathway mediated by the type IIB activin receptor controls axial patterning and lateral asymmetry in the mouse. Genes Dev. 11, 1812–1826. ( 10.1101/gad.11.14.1812) [DOI] [PubMed] [Google Scholar]
- 42.Ding J, Yang L, Yan YT, Chen A, Desai N, Wynshaw-Boris A. 1998. Cripto is required for correct orientation of the anterior–posterior axis in the mouse embryo. Nature 395, 702–707. ( 10.1038/27215) [DOI] [PubMed] [Google Scholar]
- 43.Xu C, Liguori G, Persico MG, Adamson ED. 1999. Abrogation of the Cripto gene in mouse leads to failure of postgastrulation morphogenesis and lack of differentiation of cardiomyocytes. Development 126, 483–494. [DOI] [PubMed] [Google Scholar]
- 44.Gaio U, et al. 1999. A role of the cryptic gene in the correct establishment of the left–right axis. Curr. Biol. 9, 1339–1342. ( 10.1016/S0960-9822(00)80059-7) [DOI] [PubMed] [Google Scholar]
- 45.Yan YT, et al. 1999. Conserved requirement for EGF–CFC genes in vertebrate left–right axis formation. Genes Dev. 13, 2527–2537. ( 10.1101/gad.13.19.2527) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chu J, Shen MM. 2010. Functional redundancy of EGF–CFC genes in epiblast and extraembryonic patterning during early mouse embryogenesis. Dev. Biol. 342, 63–73. ( 10.1016/j.ydbio.2010.03.009) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rankin CT, Bunton T, Lawler AM, Lee SJ. 2000. Regulation of left–right patterning in mice by growth/differentiation factor-1. Nat. Genet. 24, 262–265. ( 10.1038/73472) [DOI] [PubMed] [Google Scholar]
- 48.Andersson O, Reissmann E, Jörnvall H, Ibáñez CF. 2006. Synergistic interaction between Gdf1 and Nodal during anterior axis development. Dev. Biol. 293, 370–381. ( 10.1016/j.ydbio.2006.02.002) [DOI] [PubMed] [Google Scholar]
- 49.Chen C, et al. 2006. The Vg1-related protein Gdf3 acts in a Nodal signaling pathway in the pre-gastrulation mouse embryo. Development 133, 319–329. ( 10.1242/dev.02210) [DOI] [PubMed] [Google Scholar]
- 50.Hoodless PA, Pye M, Chazaud C, Labbé E, Attisano L, Rossant J, Wrana JL. 2001. FoxH1 (Fast) functions to specify the anterior primitive streak in the mouse. Genes Dev. 15, 1257–1271. ( 10.1101/gad.881501) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Waldrip WR, Bikoff EK, Hoodless PA, Wrana JL, Robertson EJ. 1998. Smad2 signaling in extraembryonic tissues determines anterior–posterior polarity of the early mouse embryo. Cell 92, 797–808. ( 10.1016/S0092-8674(00)81407-5) [DOI] [PubMed] [Google Scholar]
- 52.Heyer J, Escalante-Alcalde D, Lia M, Boettinger E, Edelmann W, Stewart CL, Kucherlapati R. 1999. Postgastrulation Smad2-deficient embryos show defects in embryo turning and anterior morphogenesis. Proc. Natl Acad. Sci. USA 96, 12 595–12 600. ( 10.1073/pnas.96.22.12595) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zhu Y, Richardson JA, Parada LF, Graff JM. 1998. Smad3 mutant mice develop metastatic colorectal cancer. Cell 94, 703–714. ( 10.1016/S0092-8674(00)81730-4) [DOI] [PubMed] [Google Scholar]
- 54.Datto MB, Frederick JP, Pan L, Borton AJ, Zhuang Y, Wang XF. 1999. Targeted disruption of Smad3 reveals an essential role in transforming growth factor beta-mediated signal transduction. Mol. Cell. Biol. 19, 2495–2504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yang X, et al. 1999. Targeted disruption of SMAD3 results in impaired mucosal immunity and diminished T cell responsiveness to TGF-β. EMBO J. 18, 1280–1291. ( 10.1093/emboj/18.5.1280) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Dunn NR, Vincent SD, Oxburgh L, Robertson EJ, Bikoff EK. 2004. Combinatorial activities of Smad2 and Smad3 regulate mesoderm formation and patterning in the mouse embryo. Development 131, 1717–1728. ( 10.1242/dev.01072) [DOI] [PubMed] [Google Scholar]
- 57.Sirard C, et al. 1998. The tumor suppressor gene Smad4/Dpc4 is required for gastrulation and later for anterior development of the mouse embryo. Genes Dev. 12, 107–119. ( 10.1101/gad.12.1.107) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Birsoy B, Kofron M, Schaible K, Wylie C, Heasman J. 2006. Vg 1 is an essential signaling molecule in Xenopus development. Development 133, 15–20. ( 10.1242/dev.02144) [DOI] [PubMed] [Google Scholar]
- 59.Dohrmann CE, Kessler DS, Melton DA. 1996. Induction of axial mesoderm by zDVR-1, the zebrafish orthologue of Xenopus Vg1. Dev. Biol. 175, 108–117. ( 10.1006/dbio.1996.0099) [DOI] [PubMed] [Google Scholar]
- 60.Hagos EG, Fan X, Dougan ST. 2007. The role of maternal Activin-like signals in zebrafish embryos. Dev. Biol. 309, 245–258. ( 10.1016/j.ydbio.2007.07.010) [DOI] [PubMed] [Google Scholar]
- 61.Range R, Lapraz F, Quirin M, Marro S, Besnardeau L, Lepage T. 2007. Cis-regulatory analysis of nodal and maternal control of dorsal–ventral axis formation by Univin, a TGF-β related to Vg1. Development 134, 3649–3664. ( 10.1242/dev.007799) [DOI] [PubMed] [Google Scholar]
- 62.Skromne I, Stern CD. 2001. Interactions between Wnt and Vg1 signalling pathways initiate primitive streak formation in the chick embryo. Development 128, 2915–2927. [DOI] [PubMed] [Google Scholar]
- 63.Albano RM, Groome N, Smith JC. 1993. Activins are expressed in preimplantation mouse embryos and in ES and EC cells and are regulated on their differentiation. Development 117, 711–723. [DOI] [PubMed] [Google Scholar]
- 64.Albano RM, Smith JC. 1994. Follistatin expression in ES and F9 cells and in preimplantation mouse embryos. Int. J. Dev. Biol. 38, 543–547. [PubMed] [Google Scholar]
- 65.Jones RL, Findlay JK, Farnworth PG, Robertson DM, Wallace E, Salamonsen LA. 2006. Activin A and inhibin A differentially regulate human uterine matrix metalloproteinases: potential interactions during decidualization and trophoblast invasion. Endocrinology 147, 724–732. ( 10.1210/en.2005-1183) [DOI] [PubMed] [Google Scholar]
- 66.Johnson MH, Ziomek CA. 1983. Cell interactions influence the fate of mouse blastomeres undergoing the transition from the 16 to the 32 cell stage. Dev. Biol. 95, 211–218. [DOI] [PubMed] [Google Scholar]
- 67.James D, Levine AJ, Besser D, Hemmati-Brivanlou A. 2005. TGF-β/Activin/Nodal signaling is necessary for the maintenance of pluripotency in human embryonic stem cells. Development 132, 1273–1282. ( 10.1242/dev.01706) [DOI] [PubMed] [Google Scholar]
- 68.Lu RZ, Matsuyama S, Nishihara M, Takahashi M. 1993. Developmental expression of activin/inhibin beta A, beta B, and alpha subunits, and activin receptor-IIB genes in preimplantation mouse embryos. Biol. Reprod. 49, 1163–1169. ( 10.1095/biolreprod49.6.1163) [DOI] [PubMed] [Google Scholar]
- 69.Orimo T, Taga M, Matsui H, Minaguchi H. 1996. The effect of activin-A on the development of mouse preimplantation embryos in vitro. J. Assist. Reprod. Genet. 13, 669–674. ( 10.1007/BF02069647) [DOI] [PubMed] [Google Scholar]
- 70.Yoshioka K, Suzuki C, Iwamura S. 1998. Activin A and follistatin regulate developmental competence of in vitro-produced bovine embryos. Biol. Reprod. 59, 1017–1022. ( 10.1095/biolreprod59.5.1017) [DOI] [PubMed] [Google Scholar]
- 71.Takaoka K, Yamamoto M, Shiratori H, Meno C, Rossant J, Saijoh Y, Hamada H. 2006. The mouse embryo autonomously acquires anterior–posterior polarity at implantation. Dev. Cell 10, 451–459. ( 10.1016/j.devcel.2006.02.017) [DOI] [PubMed] [Google Scholar]
- 72.Park CB, Dufort D. 2011. Nodal expression in the uterus of the mouse is regulated by the embryo and correlates with implantation. Biol. Reprod. 84, 1103–1110. ( 10.1095/biolreprod.110.087239) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Chazaud C, Rossant J. 2006. Disruption of early proximodistal patterning and AVE formation in Apc mutants. Development 133, 3379–3387. ( 10.1242/dev.02523) [DOI] [PubMed] [Google Scholar]
- 74.Coucouvanis E, Martin GR. 1999. BMP signaling plays a role in visceral endoderm differentiation and cavitation in the early mouse embryo. Development 126, 535–546. [DOI] [PubMed] [Google Scholar]
- 75.Lu CC, Robertson EJ. 2004. Multiple roles for Nodal in the epiblast of the mouse embryo in the establishment of anterior–posterior patterning. Dev. Biol. 273, 149–159. ( 10.1016/j.ydbio.2004.06.004) [DOI] [PubMed] [Google Scholar]
- 76.Torres-Padilla ME, Richardson L, Kolasinska P, Meilhac SM, Luetke-Eversloh MV, Zernicka-Goetz M. 2007. The anterior visceral endoderm of the mouse embryo is established from both preimplantation precursor cells and by de novo gene expression after implantation. Dev. Biol. 309, 97–112. ( 10.1016/j.ydbio.2007.06.020) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Morris SA, Grewal S, Barrios F, Patankar SN, Strauss B, Buttery L, Alexander M, Shakesheff KM, Zernicka-Goetz M. 2012. Dynamics of anterior–posterior axis formation in the developing mouse embryo. Nat. Commun. 3, 673 ( 10.1038/ncomms1671) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Gardner RL. 1968. Mouse chimeras obtained by the injection of cells into the blastocyst. Nature 220, 596–597. ( 10.1038/220596a0) [DOI] [PubMed] [Google Scholar]
- 79.Gardner RL. 1998. Contributions of blastocyst micromanipulation to the study of mammalian development. Bioessays 20, 168–180. () [DOI] [PubMed] [Google Scholar]
- 80.Rossant J. 2008. Stem cells and early lineage development. Cell 132, 527–531. ( 10.1016/j.cell.2008.01.039) [DOI] [PubMed] [Google Scholar]
- 81.Nichols J, Smith A. 2009. Naive and primed pluripotent states. Cell Stem Cell 4, 487–492. ( 10.1016/j.stem.2009.05.015) [DOI] [PubMed] [Google Scholar]
- 82.Tesar PJ, et al. 2007. New cell lines from mouse epiblast share defining features with human embryonic stem cells. Nature 448, 196–199. ( 10.1038/nature05972) [DOI] [PubMed] [Google Scholar]
- 83.Conlon FL, Barth KS, Robertson EJ. 1991. A novel retrovirally induced embryonic lethal mutation in the mouse: assessment of the developmental fate of embryonic stem cells homozygous for the 413.d proviral integration. Development 111, 969–981. [DOI] [PubMed] [Google Scholar]
- 84.Brons IG, et al. 2007. Derivation of pluripotent epiblast stem cells from mammalian embryos. Nature 448, 191–195. ( 10.1038/nature05950) [DOI] [PubMed] [Google Scholar]
- 85.Vallier L, et al. 2009. Activin/Nodal signalling maintains pluripotency by controlling Nanog expression. Development 136, 1339–1349. ( 10.1242/dev.033951) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Guo G, Yang J, Nichols J, Hall JS, Eyres I, Mansfield W, Smith A. 2009. Klf4 reverts developmentally programmed restriction of ground state pluripotency. Development 136, 1063–1069. ( 10.1242/dev.030957) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Buecker C, et al. 2014. Reorganization of enhancer patterns in transition from naive to primed pluripotency. Cell Stem Cell 14, 838–853. ( 10.1016/j.stem.2014.04.003) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Factor DC, et al. 2014. Epigenomic comparison reveals activation of ‘seed’ enhancers during transition from naive to primed pluripotency. Cell Stem Cell 14, 854–863. ( 10.1016/j.stem.2014.05.005) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Karwacki-Neisius V, et al. 2013. Reduced Oct4 expression directs a robust pluripotent state with distinct signaling activity and increased enhancer occupancy by Oct4 and Nanog. Cell Stem Cell 12, 531–545. ( 10.1016/j.stem.2013.04.023) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Radzisheuskaya A, et al. 2013. A defined Oct4 level governs cell state transitions of pluripotency entry and differentiation into all embryonic lineages. Nat. Cell Biol. 15, 579–590. ( 10.1038/ncb2742) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Xi Q, et al. 2011. A poised chromatin platform for TGF-β access to master regulators. Cell 147, 1511–1524. ( 10.1016/j.cell.2011.11.032) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Papanayotou C, et al. 2014. A novel Nodal enhancer dependent on pluripotency factors and Smad2/3 signaling conditions a regulatory switch during epiblast maturation. PLoS Biol. 12, e1001890 ( 10.1371/journal.pbio.1001890) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Dahle O, Kumar A, Kuehn MR. 2010. Nodal signaling recruits the histone demethylase Jmjd3 to counteract polycomb-mediated repression at target genes. Sci Signal. 3, ra48 ( 10.1126/scisignal.2000841) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Norris DP, Brennan J, Bikoff EK, Robertson EJ. 2002. The Foxh1-dependent autoregulatory enhancer controls the level of Nodal signals in the mouse embryo. Development 129, 3455–3468. [DOI] [PubMed] [Google Scholar]