

Abstract
The early response to a homologous rotavirus infection in mice includes a T-cell-independent increase in the number of activated B lymphocytes in the Peyer's patches. The mechanism of this activation has not been previously determined. Since rotavirus has a repetitively arranged triple-layered capsid and repetitively arranged antigens can induce activation of B cells, one or more of the capsid proteins could be responsible for the initial activation of B cells during infection. To address this question, we assessed the ability of rotavirus and virus-like particles to induce B-cell activation in vivo and in vitro. Using infectious rotavirus, inactivated rotavirus, noninfectious but replication-competent virus, and virus-like particles, we determined that neither infectivity nor RNA was necessary for B-cell activation but the presence of the rotavirus outer capsid protein, VP7, was sufficient for murine B-cell activation. Preincubation of the virus with neutralizing VP7 antibodies inhibited B-cell activation. Polymyxin B treatment and boiling of the virus preparation were performed, which ruled out possible lipopolysaccharide contamination as the source of activation and confirmed that the structural conformation of VP7 is important for B-cell activation. These findings indicate that the structure and conformation of the outer capsid protein, VP7, initiate intestinal B-cell activation during rotavirus infection.
Rotavirus is the leading cause of viral gastroenteritis in pediatric patients worldwide (34). Symptoms include severe dehydrating diarrhea, vomiting, and fever. Over 600,000 deaths occur annually worldwide resulting from dehydration secondary to infection mainly in developing countries (47). Infection in developed countries does not cause significant mortality but results in high morbidity and a significant economic impact on the health care industry (26, 29). Unfortunately, the development of a safe and effective vaccine has remained elusive, in part due to conflicting evidence on the nature of protective immune responses against rotavirus. Specifically, deciphering the importance of individual rotavirus proteins to protective immunity has been difficult.
Rotavirus is a member of the family Reoviridae and is composed of 11 segments of double-stranded RNA surrounded by three concentric spherical protein coats (34). The 11 segments of double-stranded RNA code for both structural proteins, which compose three concentric protein layers that surround and protect the RNA, and nonstructural proteins, which are involved in viral replication (34). There are four major structural proteins that comprise the capsids of rotavirus: viral proteins (VPs) 2, 4, 6, and 7. The innermost layer is composed of VP2, the middle layer is composed of VP6, and the outermost layer is composed of glycoprotein VP7 and protease-sensitive VP4 spikes that emanate from VP7 (34). Antibody responses following rotavirus infection are directed toward both structural and nonstructural proteins. VP6, the most prevalent protein in the rotavirus particle, is highly immunogenic (34). Antibodies to VP6 are nonneutralizing but induce protective immunity in some animal models (12). The outer capsid proteins VP4 and VP7 induce neutralizing antibodies (34). Epitope-specific antibodies to VP4 and VP7 correlate with protection from infection (27, 41); however, there is conflicting evidence on the importance of VP4 and VP7 antibodies for protection (34).
Rotavirus infection induces a rapid humoral antibody response, and protective immunity is thought to correlate with intestinal immunoglobulin A (IgA) (34). The correlation of IgA antibody production with protection has suggested a critical role for B cells in the immune response to rotavirus. In fact, mice deficient in B cells are unable to establish complete protective immunity against reinfection with rotavirus (23). We previously demonstrated in mice that large numbers of activated B cells, but not T cells, are present as early as 2 days postinfection in the Peyer's patches (PP) (7), small lymphoid nodules located on the antimesenteric side of the small intestine. The increased numbers of activated PP B cells are also observed in rotavirus-infected T-cell-deficient mice, indicating that the early B-cell activation response to rotavirus infection occurs via T-cell-independent mechanisms (7). These findings of rotavirus induced T-cell-independent B-cell activation are supported by previous reports that rotavirus induces intestinal IgA in mice lacking T cells (24).
Investigations elucidating the molecular mechanisms of B-cell activation by rotavirus are important initial steps in determining the role of specific antibodies in protective immunity to rotavirus. Although rotavirus induces T-cell-independent B-cell activation, the viral proteins required for B-cell activation are unknown. Because of the difficulty in determining the viral moiety responsible for B-cell activation in vivo, we designed an in vitro system using murine PP or splenic B cells to determine the critical rotavirus antigen for the induction of B-cell activation. To do this, we treated murine B cells in vitro with virus or noninfectious virus-like particles (VLPs) consisting of various combinations of the capsid rotavirus proteins and found that neither infectivity nor rotavirus RNA is necessary for the induction of B-cell activation. However, the activation of B cells is observed in the presence of the outer capsid protein, VP7, and that activation is dependent on conformation of the protein.
MATERIALS AND METHODS
Virus and VLPs.
Wild-type murine rotavirus ECwt was obtained from Harry Greenberg (Stanford University Medical School, Palo Alto, Calif.) (22). A stock of ECwt was prepared as a gut homogenate, and the 50% infectious dose (ID50) was determined as described previously (46). Wild-type murine rotavirus EDIM was a gift from Richard Ward (Cincinnati Children's Hospital Medical Center, Cincinnati, Ohio) (59). An original isolate of the rhesus rotavirus strain RRV (56) was also obtained from H. Greenberg and was grown in MA104 cells in the presence of trypsin and purified with a cesium chloride gradient. Infectious virus titers ranged from 108 to 1011 PFU/ml. Rotavirus strains SA11 Cl3 (19), SA11 4F (10), SA11 Cl28 (19), and HAL 1166 (8) were similarly propagated in MA104 cells as described previously, in the presence of trypsin, and purified with cesium chloride gradients. Double-layered RRV particles were isolated from cesium chloride gradients, and the absence of VP7 and VP4 was confirmed by electron microscopy and Western blotting. RRV was inactivated by UV radiation treatment and psoralen, and inactivation was confirmed as described previously (13). 2/6-, 2/6/7-, 2/4/6/7-, and Norwalk virus VLPs were prepared as described previously (17, 32), using a baculovirus expression system in Sf9 insect cells followed by cesium chloride or sucrose gradient purification. Endotoxin levels of all reagents were determined by the Limulus amebocyte lysate assay (Associates of Cape Cod, Inc., Woods Hole, Mass.), and only reagents containing less lipopolysaccharide (LPS) than the level required for the induction of B-cell activation (50 pg) were utilized in all experiments (data not shown).
Rotavirus infection in mice.
Female CD-1 (body weight, 22 to 25 g) outbred mice were obtained from Charles River Breeding Laboratories (Wilmington, Mass.). All mice were housed in microisolation cages and fed ad libitum. Mice were inoculated orally with either 100 μl of PBS and cell lysate containing 104 ID50 of EDIM (59) or 108 PFU of RRV (56) or 100 μl of PBS and gut homogenate containing 105 ID50 of ECwt (22). Control mice received 100 μl of PBS containing an equivalent volume of normal gut homogenate and uninfected cell lysate. Infection was confirmed by collection of fecal samples that were analyzed by enzyme-linked immunosorbent assay (ELISA) for rotavirus-specific antigen (46).
Isolation of lymphocytes.
Spleens or all small intestinal PP (typically 8 to 10) were harvested from CD-1 and CF-1 outbred female mice and 129, C57BL/6, and BALB/c inbred female mice (all obtained from Charles River Breeding Laboratories; body weight, 22 to 25 g) or βδ T-cell receptor homozygous knockout (TCRKO) male mice on a C57BL/6 background (TCRKO; Jackson Laboratories, Bar Harbor, Maine). Spleens were placed in complete RPMI medium consisting of RPMI 1640 medium (BioWhittaker, Walkersville, Md.) supplemented with 10% fetal bovine serum (Summit Biotechnology, Fort Collins, Colo.), 200 mM l-glutamine, 10,000-U/ml penicillin and streptomycin, 50 mM 2-β-mercaptoethanol, and 5% NCTC-109 medium (BioWhittaker). Tissues were mechanically disrupted by pressing through a 70-μm-pore-size cell strainer (Fisher Scientific, Pittsburgh, Pa.). Erythrocytes were lysed with ACK lysing buffer (BioWhittaker), and cells were washed with complete RPMI medium. Cells were stained with trypan blue to assess viability and quantified by using a hemocytometer or a Z1 Coulter Counter (Beckman Coulter, Hialeah, Fla.).
Magnetic bead depletion of specific lymphocyte populations.
Manufacturer's instructions were followed to deplete CD90+ cells from splenic single-cell suspensions by using positive selection of CD90+ cells with MACS microbeads (Miltenyi Biotec, Auburn, Calif.). Purity was assessed by flow cytometry (data not shown).
In vitro activation of B or T lymphocytes.
Two million lymphocytes in 100 μl of complete RPMI medium were plated in 96-well polypropylene nonpyrogenic U-bottom plates (Corning Incorporation, Corning, N.Y.). Lymphocytes were stimulated overnight at 37°C and 5% CO2 with 0.01- to 10,000-ng/ml cesium chloride-purified rotavirus, Escherichia coli serotype 0111:B4 LPS (Sigma Chemical Company, St. Louis, Mo.), bovine serum albumin (Calbiochem, LaJolla, Calif.), or VLPs. To test for inhibition of B-cell activation, the following high-performance liquid chromatography-purified monoclonal antibodies (0.75 μg each) specific for VP7, VP4, and VP6 were used (49): for VP7, YO-1E3 (G3), KU-4 (G1), 159 (G3), and ST-2G7 (G4); for VP4 (11, 62), 9F6 (P5B2[2]), 954/23 (P5B), and 10G6 (P5B[2]); and for VP6 (28), 255 (subgroup I). These antibodies were preincubated with 10-ng/ml cesium chloride-purified virus for 2 h at room temperature prior to addition to the cells. As a control, RRV was treated with 0.75 μg of purified mouse IgG1, IgG2a, IgG2b, or IgG3 (Sigma).
To demonstrate that B-cell activation was not due to LPS contamination of reagents, both LPS and RRV were incubated in a 100°C heat block for 2 h prior to addition to the cells. A second aliquot of each reagent was incubated with a ratio of 10:1 polymyxin B (Sigma) to virus for 1 h at room temperature prior to addition to cells. Control wells received an equal amount of polymyxin B or boiled medium alone. All reagents were diluted in 5 μl of complete RPMI medium. Cells were harvested by being transferred to a 1-ml Eppendorf tube, washed with 1 ml of complete RPMI, and pelleted by centrifugation at 400 × g. The percentages of activated B lymphocytes were assessed as described below.
Flow cytometry.
Two million lymphocytes harvested directly from murine PP or after overnight stimulation as described above were suspended in 100 μl of complete RPMI medium containing phycoerythrin-labeled CD69 (0.03 μg/106 cells) and fluorescein isothiocyanate (FITC)-labeled CD19 (0.06 μg/106 cells) both obtained from Becton Dickinson (BD Biosciences, San Diego, Calif.). Cells were incubated for 30 min at room temperature, washed with 1 ml of complete RPMI, and pelleted at 400 × g. The resulting pellet was suspended in 500 μl of 4% paraformaldehyde (pH 7; Fisher Scientific) and stored at 4°C until analyzed. CD69+ CD19+ and total CD19+ cell numbers were determined by using a Coulter EPICS XL-MCL (Beckman Coulter, Hialeah, Fla.) flow cytometer. The percentage of activated B lymphocytes out of total B lymphocytes was calculated. Significant differences between groups were determined by Student's t test. All experiments were repeated a minimum of three times. A representative experiment is shown.
RESULTS
Homologous and heterologous strains of rotavirus induce PP B-cell activation in vivo.
Inoculation of mice with the highly infectious homologous rotavirus strain ECwt results in massive upregulation of the activation marker CD69 on B-cell populations in PP as early as 2 days after inoculation (7). To test whether a less infectious homologous murine strain (EDIM) or a heterologous rhesus strain (RRV) also induced elevated levels of B-cell activation in the PP, naïve mice were orally inoculated with ECwt, EDIM, or RRV. PP B cells were examined 2 days after oral inoculation for activation marker (CD69) expression by flow cytometry. Both EDIM infection and RRV infection resulted in a significantly higher percentage of B cells expressing CD69 than in uninfected mice, although the percentage of activated B cells was less than that in the ECwt group (Fig. 1A). The amount of rotavirus antigen present in fecal samples at the time of PP B-cell analysis was determined (Fig. 1B), and there was a significant positive correlation between the level of B-cell activation in the PP and the amount of virus shedding from the intestine as determined by Spearman's test (correlation coefficient = 0.825).
FIG. 1.
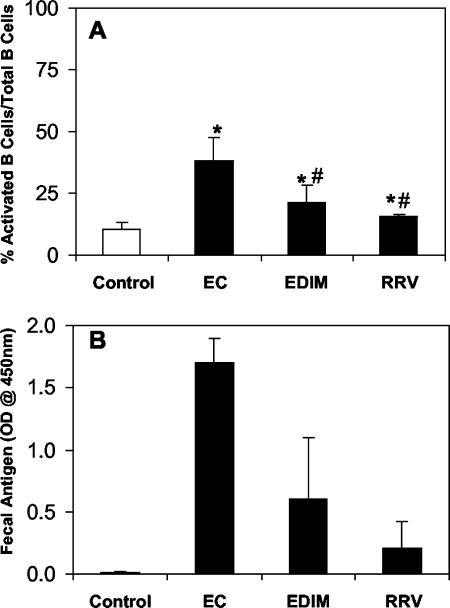
Both homologous and heterologous rotavirus strains induce PP B-cell activation in mice. CD-1 mice were orally inoculated with uninfected gut homogenate and cell lysate, gut homogenate containing 105 ID50 of ECwt, 104 ID50 of EDIM, or cell lysate containing 108 PFU of RRV. Single-cell suspensions of the PP from each mouse (n = 5) were collected 2 days after inoculation and analyzed by flow cytometry for activated B lymphocytes (CD19+ CD69+). (A) Average percentage of activated B cells (CD19+ CD69+) out of total B cells (CD19+) in the PP ± standard deviation for each group. *, P < 0.05 by Student's paired t test compared to control mice. #, P < 0.05 by Student’s paired t test compared to ECwt-inoculated mice. (B) Amount of antigen present in a fecal sample taken at the time of analysis as determined by ELISA.
Rotavirus induces B-cell activation in vitro.
B-cell activation can be examined in vitro by a variety of different markers of activation (2, 20, 39, 40, 55). Although rotavirus induces activation marker CD69 expression on B cells in vivo, it is unknown whether rotavirus can induce CD69 expression on B cells in vitro. We determined whether rotavirus induced activation of naïve murine intestinal and systemic B lymphocytes in vitro by treating single-cell suspensions from the PP and spleen with RRV. RRV was utilized because, unlike the wild-type murine rotavirus strains EDIM and ECwt, RRV is easily cultivated in and purified from MA104 cells (14). Similar to the observation that RRV induced B-cell activation in vivo, RRV treatment of PP or splenic cells in vitro resulted in a significant increase in the percentage of B cells expressing CD69 compared to cells receiving medium alone (Fig. 2A, B, and D). Splenic B cells are activated by rotavirus in vitro, but unlike in the PP, increases in activated B cells are not detected in the spleens of rotavirus-infected mice (7). This could be due to the rapid exit of activated B cells from the spleen (33) or the aggregation of extraintestinal virus by antibody making it unavailable to induce B-cell activation.
FIG. 2.
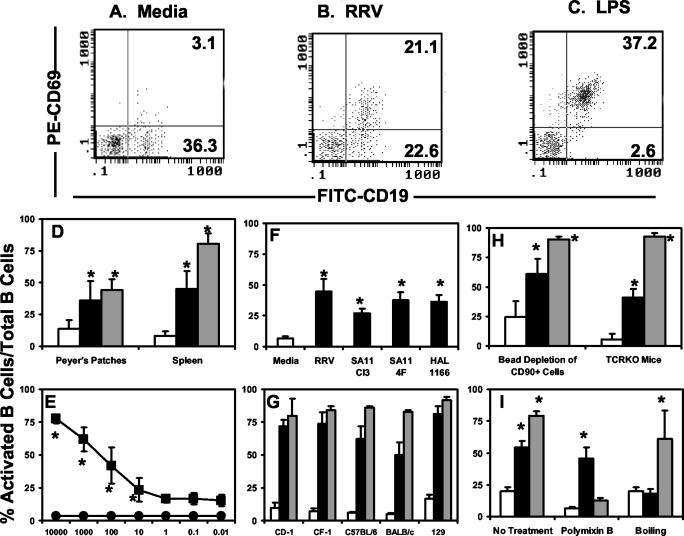
Rotavirus activates B cells in vitro. Single-cell suspensions (2 × 106), isolated from either the PP or spleens were stimulated in vitro with medium (white bars), rotavirus (black bars), or LPS (gray bars). Activation was quantified by flow cytometric analysis of CD69 expression on the B-cell populations. The data represent the average percentage of CD19+ CD69+ cells out of total CD19+ cells ± standard deviation (n = 3 to 5 mice). All cells were from CD-1 outbred mice unless otherwise noted. (A, B, and C) Representative flow cytometry dot plots of splenic lymphocytes harvested from one mouse and stimulated with either medium alone or 100-ng/ml RRV or LPS, respectively. Numbers in the upper-right-hand quadrant represent the CD19+ CD69+ population, and those in the bottom-right-hand quadrant represent the CD19+ CD69− population. (D) PP and splenic single-cell suspensions were stimulated overnight with medium or 100-ng/ml RRV or LPS. (E) Splenic cells were stimulated with either medium alone (circles) or decreasing amounts (nanograms per milliliter) of RRV (squares). (F) Splenic cells were stimulated in vitro with medium or 100-ng/ml each of the indicated strains of rotavirus. (G) Splenic cells from the indicated inbred and outbred strains of mice were stimulated with medium or 1,000-ng/ml RRV or LPS. (H) Splenic single-cell suspensions depleted of the CD90+ T-cell population by using magnetic beads or from TCRKO mice were stimulated with either medium or 100-ng/ml RRV or LPS. (I) Both RRV and LPS (100 ng/ml) were pretreated with 10 μg of polymyxin B (PB) for 1 h at room temperature or boiled for 2 h prior to addition to splenic single-cell suspensions overnight. *, P < 0.05 compared to medium alone by Student's paired t test.
Multiple preparations of RRV were tested, and all induced B-cell activation as measured by increased expression of CD69, but there was no correlation between infectious titer of the preparation (107 to 109 PFU) and induction of activation (data not shown), suggesting that infectivity is not required for the induction of B-cell activation. We estimated that, based on the PFU/microgram protein ratio of each virus preparation, the particle/cell ratio sufficient to induce B-cell activation was in the range of 1 to 10 virus particles per cell. To determine if the rotavirus-induced activation was concentration dependent, splenic single-cell suspensions were incubated with serial 10-fold dilutions of RRV (10,000 to 0.01 ng). RRV activation of B cells was dose dependent, with a threshold of activation occurring at a concentration of 1-ng/ml RRV (Fig. 2E). In vitro treatment of B cells with LPS, known to polyclonally activate B cells, also resulted in a large increase in the percentage of CD69+ B cells (Fig. 2C and D).
The finding that RRV induced dose-dependent B-cell activation in vitro raised the question as to whether other cultivatable strains of rotavirus that do not result in a robust infection in mice could induce B-cell activation. To address this question, splenic B cells were treated with purified preparations of simian strains SA11 Cl3 and SA11 4F (which contains a bovine-like VP4), and the human tissue culture-adapted strain HAL 1166. Both the simian and human strains of rotavirus induced mouse B-cell activation, as measured by upregulation of CD69 expression on the B-cell populations (Fig. 2F). We also tested whether B cells from both inbred and outbred strains of mice were responsive to rotavirus treatment in vitro. Splenic B cells from the outbred mouse strains CD-1 and CF-1 as well as B cells from inbred mouse strains C57BL/6, 129, and BALB/c all responded to rotavirus treatment with increased CD69 expression specifically on the B-cell population (Fig. 2G). Therefore, rotavirus-induced B-cell activation is a feature common to all rotavirus strains tested and is not limited by host genetic background.
We have previously demonstrated that rotavirus induces T-cell-independent B-cell activation in vivo as demonstrated by an increase in the percentage of CD19+ CD69+ B cells in the PP from rotavirus-infected βδ TCRKO mice (7). To determine whether rotavirus induces T-cell-independent B-cell activation in vitro, two approaches were used. First, magnetic beads labeled with CD90, a marker expressed on all T cells, were used to deplete T cells from splenic cell suspensions to less than 8% of the total cell population, as determined by flow cytometry (data not shown). Second, splenic cell suspensions from βδ TCRKO mice, which lack T cells, were stimulated with RRV. In both cases, rotavirus induced CD69 expression on B cells in the absence or reduction of T cells (Fig. 2H), further supporting the in vivo findings that rotavirus-induced B-cell activation does not require the presence of T cells.
We previously observed that rotavirus does not induce high levels of CD69 expression on T cells in vivo (7). To confirm that rotavirus does not induce CD69 expression on T cells in vitro, we measured CD69 expression on CD4+ and CD8+ T-cell populations after treatment with RRV. Rotavirus treatment did not significantly alter the percentage of T-cell activation compared to that with medium treatment alone (data not shown). The T cells maintained the ability to be activated in vitro, as treatment with nonspecific lymphocyte activators phorbol 12-myristate 13-acetate and ionomycin resulted in an increase in the percentage of CD4+ and CD8+ populations that were CD69+ (data not shown). These data support the in vivo findings that rotavirus does not induce T-cell activation.
Although all of the reagents used in the experiments described previously were screened for LPS contamination by using the Limulas amebocyte lysate assay (see Materials and Methods), two additional experiments were performed to confirm that the rotavirus-induced B-cell activation was not a result of LPS contamination of the RRV preparations. Boiling the RRV virus preparation for 2 h prior to addition to the cells completely abrogated the ability of rotavirus to induce B-cell activation. However, as expected, boiling LPS did not significantly affect the ability of LPS to induce B-cell activation (Fig. 2I). Conversely, pretreatment of LPS with polymyxin B, a known inhibitor of LPS-induced B-cell activation (30), abrogated the LPS-induced B-cell activation in vitro (Fig. 2I). In contrast, pretreatment of RRV with polymyxin B had no effect on the ability of the RRV preparation to induce activation (Fig. 2I). These results indicate that B-cell activation induced by in vitro treatment with purified rotavirus is not a result of LPS contamination of the virus preparation. Failure of boiled RRV to induce B-cell activation also suggested that rotavirus-induced B-cell activation was dependent on rotavirus structure.
The VP7 outer capsid protein is required for B-cell activation.
To determine whether replicating or infectious virus is required for B-cell activation, splenic single-cell suspensions were stimulated with 100-ng/ml of either live purified RRV (infectious and replication competent), an equivalent amount of psoralen- and UV light-inactivated RRV from the same preparation (not infectious and not replication competent), or double-layered RRV particles (replication competent but not infectious). Both live and inactivated RRV treatments resulted in an increase in the percentage of activated B cells (Fig. 3A). This finding explained our previous observation of a lack of correlation between the infectious virus titer of the RRV preparation and the ability to induce B-cell activation. Double-layered particles and bovine serum albumin (BSA), a protein control, failed to induce significant increases in B-cell activation (Fig. 3A). These findings indicated that replication and RNA were not required for B-cell activation. The finding that double-layered RRV particles did not induce B-cell activation also suggested that one or both of the outer capsid proteins VP4 and VP7 were required for the induction of B-cell activation.
FIG. 3.
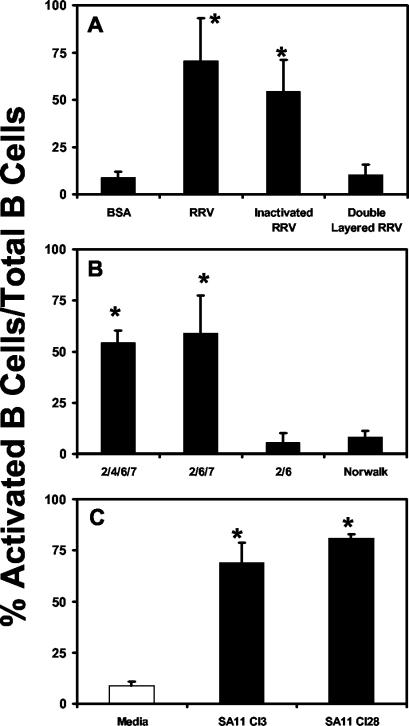
VP7 is necessary for the induction of B-cell activation by rotavirus in vitro. Splenic single-cell suspensions (n = 3 to 5) containing 2 × 106 lymphocytes were stimulated overnight at 37°C in vitro with the indicated virus or VLPs. Cells were incubated with antibodies to CD19 and CD69. Double-positive cells were analyzed by flow cytometry. The data show the average percentage of activated B cells out of total B cells ± standard deviation. (A) Cells were treated with 100 ng/ml of RRV, inactivated RRV, or double-layered rotavirus particles. *, P < 0.05 compared to BSA control by paired Student's t test. (B) Cells were stimulated overnight with 100-ng/ml 2/4/6/7-, 2/6/7-, or 2/6-VLPs or Norwalk virus VLPs. *, P < 0.05 compared to both 2/4/6/7- and 2/6/7-VLP-treated cells. (C) Cells were stimulated overnight with 1,000-ng/ml SA11 Cl3 with glycosylated VP7 or an SA11 Cl28 mutant without glycosylated VP7. *, P < 0.05 compared to medium alone by Student's paired t test.
To determine if either VP4 or VP7 was responsible for inducing B-cell activation, we utilized VLPs. VLPs retain the structural integrity of native rotavirus virions (17) and the different combinations (2/4/6/7-, 2/6/7-, and 2/6-VLPs) provided unique reagents for determining which viral component was responsible for inducing B-cell activation. When B cells were stimulated with 100-ng/ml 2/4/6/7- or 2/6/7-VLPs, a significant increase in the percentage of B cells expressing CD69 was observed (Fig. 3B). However, neither rotavirus 2/6-VLPs nor Norwalk virus VLPs (32) increased the percentage of activated B cells compared to that in medium alone (Fig. 3B). These results clearly implicated the VP7 outer capsid protein as a key mediator of B-cell activation.
Glycosylation of surface proteins has been previously demonstrated to be important for B-cell activation (25). To test whether glycosylation of the rotavirus VP7 outer capsid protein is an important factor for the induction of B-cell activation by the virus, we treated splenic cells with 1,000-ng/ml SA11 Cl28, a variant of SA11 Cl3 that expresses a nonglycoslyated VP7. SA11 Cl28 induced levels of B-cell activation comparable to that induced by SA11 Cl3, indicating that glycosylation of VP7 is not required for B-cell activation by rotavirus (Fig. 3C).
Antibodies to VP7 inhibit rotavirus-induced B-cell activation.
To confirm the role of VP7 in virus-induced B-cell activation, infectious and replication-competent RRV was preincubated with high-performance liquid chromatography-purified monoclonal antibodies to VP7, VP4, or VP6 prior to stimulation of B cells. IgG isotype-specific antibodies were also incubated with RRV to exclude the possibility of Fc-mediated nonspecific inhibition of B-cell activation. Rotavirus-induced B-cell activation was inhibited with several monoclonal antibodies to different serotypes of VP7 but not with an antibody to VP6 (Table 1). A VP4 antibody that decapsidates the virus (10G6) (62) as well as a VP4 antibody specifically known to bind the RRV strain of rotavirus (956/23) both inhibited rotavirus-induced B-cell activation, while a VP4 antibody (9F6) recognizing a different epitope on VP4 did not inhibit B-cell activation. These data clearly confirmed that VP7 induces B-cell activation but do not rule out that VP4 may play a role.
TABLE 1.
Antibody inhibition of rotavirus-induced B-cell activation
| Protein specificity | Antibody | Virus antigen | Isotype | Neutralizing antibody | % Activated B cells |
|---|---|---|---|---|---|
| Anti-VP7 | KU-4 | KU | IgG3 | Yes | 4.7b |
| ST-2G7 | ST-3 | IgG2a | Yes | 5.2b | |
| 159 | RRV | IgG1 | Yes | 5.1b | |
| YO-1E2 | YO | IgG2b | Yes | 4.4b | |
| Anti-VP4 | 9F6 | SA11 CI3 | IgG1 | Yes | 21.2 |
| 954/23 | RRV | IgG1 | Yes | 9.6b | |
| 10G6a | SA11 CI3 | IgG1 | Yes | 9.9b | |
| Anti-VP6 | 255 | RRV | IgG | No | 23.0 |
| NAc | Control | NA | IgG1 | NA | 20.2 |
| Control | NA | IgG2a | NA | 22.6 | |
| Control | NA | IgG2b | NA | 15.6 | |
| Control | NA | IgG3 | NA | 21.8 |
Decapsidates virus (62).
P < 0.05 versus isotype control (Student's t test).
N/A, not applicable.
DISCUSSION
It has been well established that B cells are important factors in protection from rotavirus infection (45), although there is a lack of understanding of the molecular mechanisms that lead to the generation of rotavirus-specific antibody in the intestine. We recently reported that rotavirus induces rapid T-cell-independent B-cell activation in intestinal immune tissues (7). Antibody production is a downstream effect of B-cell activation; therefore, we focused our current efforts on determining the mechanisms through which rotavirus induces B-cell activation. It is difficult to assess the effect of individual viral components on B-cell activation in vivo. Therefore, we developed an assay to measure rotavirus-induced B-cell activation in vitro. We used this assay to demonstrate that neither replication-competent virus nor viral RNA is required for the induction of B-cell activation. Instead, rotavirus-induced B-cell activation occurs in part due to the structural conformation of the outer capsid protein VP7.
We and others have demonstrated that B-cell responses to rotavirus occur in the absence of T cells (7, 24). T-cell-independent antigens fall into two categories: thymus-independent type I (TI-1) and thymus independent type II (TI-2) (31). TI-1 antigens are B-cell mitogens and directly induce B-cell activation, irrespective of the specificity of the B cell. This property is known as polyclonal activation, and one of the most well-characterized TI-1 antigens is the bacterial product LPS, which induces activation of B cells through Toll-like receptor signaling. Based on our findings, we propose that rotavirus polyclonally activates B cells. Previous findings also support our conclusion that rotavirus polyclonally activates B cells. Khoury et al. reported that B cells from mice orally inoculated with RRV produced large quantities of total nonrotavirus-specific IgG, IgA, and IgM antibodies in vitro, a possible downstream consequence of polyclonal activation (36). Rotavirus has also been implicated in polyclonal proliferative responses as well as polyclonal antibody responses. In vitro stimulation of naïve murine peripheral lymph node and splenic lymphocytes with infectious, but not inactivated, bovine rotavirus (9) resulted in increased overall lymphocyte proliferation. We observed that inactivated rotavirus induced lymphocyte activation. However, although lymphocyte activation occurs prior to proliferation, proliferation is not always a consequence of activation (3, 43, 44, 52, 58). Taken together, there is strong evidence that rotavirus polyclonally activates B cells similar to LPS. This conclusion implies that the B-cell receptor is not required for the activation induced by rotavirus, and future experiments will pursue this hypothesis.
Many other viruses cause polyclonal activation of B cells either in vitro or in vivo (4, 15, 16, 35, 51, 53, 55). Some viruses, such as mouse mammary tumor virus (MMTV), are thought to induce B-cell activation by directly infecting the B cell (3); others, like Epstein-Barr virus, induce activation by inducing the secretion of soluble factors, such as interleukin-6 or tumor necrosis factor alpha, that are known activators of B cells (1, 6, 42, 54), while the glycoproteins of some viruses, such as vesicular stomatitis virus (VSV), induce T cell-independent B-cell responses (21, 37). VSV polyclonal T-cell-independent B-cell antibody production is directed toward the G glycoprotein of the VSV virion, and glycosylation appears to be required for B-cell activation (25). Three analogies can be drawn between the VP7 outer capsid structure of rotavirus and the VSV-G protein: both are glycoproteins, both proteins have highly organized tertiary structures (25, 48, 61), and the native conformation of both proteins is required for B-cell activation (25). However, unlike VSV, we observed that glycosylation does not appear to be a critical requirement for rotavirus to induce polyclonal B-cell activation (Fig. 3C). Several other viruses, including influenza virus and polyomavirus, contain highly repetitive densely packed antigenic determinants on their envelopes that induce T-cell-independent B-cell responses (51, 57). The VP7 outer capsid protein of rotavirus also is a highly repetitive densely packed moiety, suggesting that it is a likely candidate to induce polyclonal B-cell responses. Evidence for the structure of VP7 as a critical factor for the induction of B-cell activation is provided by the absence of B-cell activation after boiling the rotavirus preparation. Boiling of rotavirus reduces the trimeric VP7 capsid protein to its monomeric components, and Western blot analysis of boiled virus preparations demonstrated the presence of VP7 protein (data not shown), suggesting that it is not the mere presence of VP7 but rather the secondary structure that is responsible for B-cell activation. New methods to separate expressed VP7 into purified monomeric and multimeric forms will be required to more definitively determine the key structural components of VP7 required for induction of polyclonal B-cell activation.
To determine the critical rotavirus protein(s) necessary to induce B-cell activation, we utilized multiple approaches. Our data clearly show that VP7 in its native conformation is sufficient to induce B-cell activation. VP6, either on double-layered virus particles (Fig. 3A), on 2/6-VLPs (Fig. 3B), or baculovirus-expressed purified VP6 (data not shown) is not capable of inducing polyclonal B-cell activation. In addition, an antibody specific for VP6 was unable to inhibit rotavirus-induced B-cell activation (Table 1). A role for VP4 in B-cell activation is less clear. Results with 2/6/7-VLPs clearly indicate that VP4 is not required. Additionally, B-cell activation is not enhanced with 2/4/6/7-VLPs compared to 2/6/7-VLPs. Instead, we repeatedly observed a slight but not statistically significant increase in activation after treatment with 2/6/7-VLPs compared to 2/4/6/7-VLPs, suggesting that in the absence of VP4, VP7 is better able to signal B-cell activation. We found that growing the virus in the presence or absence of trypsin did not affect B-cell activation (data not shown), indicating that cleavage of VP4 into VP5 and VP8 (18) does not affect the induction of B-cell activation. Treatment with anti-VP4 antibodies resulted in conflicting results. Binding of monoclonal antibody 10G6 to virus is known to cause decapsidation (11, 62); therefore, a reduction in B-cell activation with this antibody was expected. However, treatment of virus with one (954/23) of two other neutralizing antibodies to VP4 inhibited B-cell activation (Table 1). Monoclonal antibodies 9F6 and 954/23 bind to the VP8 portion of VP4 at amino acids 136 and 194, both of which are in a region implicated in hemagglutination, and both antibodies bind to RRV (11). We postulate that inhibition by 954/23, and not 9F6, results from steric hindrance blocking recognition of VP7. Due to the lack of ability to test VP4 in its natural conformational state in the absence of VP7, we cannot fully exclude the possibility that VP4 induces B-cell activation. Expression of VP4 in the absence of VP7 will be required to fully rule out a role for VP4 in polyclonal B-cell activation. However, based on all our data, it seems unlikely that VP4 polyclonally activates B cells.
Our finding that VP7 can induce polyclonal B-cell activation raises questions concerning the interacting molecules expressed by either the B-cell population or the non-T-cell populations. The conformational requirement for polyclonal B-cell activation suggests that innate immune receptors are possible candidates. Pattern recognition receptors, such as the Toll-like receptors, play an important role in innate immune responses. Although previously thought to only recognize bacterial components, these receptors are now known to also recognize viruses. Recent work has shown that the MMTV, which induces polyclonal B-cell activation, binds to Toll-like receptor 4 (50). Other viruses, such as measles virus and respiratory syncytial virus, also induce Toll-like receptor signaling (5, 38). The role of Toll-like receptor family members in rotavirus-induced B-cell activation is currently being investigated.
In conclusion, we show that the rotavirus VP7 protein induces polyclonal B-cell activation independent of replication competence, the presence of viral RNA, or VP4. The specific cellular molecule(s) that interacts with VP7 to induce activation remains to be identified. B cells express pattern recognition receptors, such as Toll-like receptors, that have already been demonstrated to interact with polyclonal B-cell activators, such as LPS (60) and MMTV (50). The identification of molecules that interact with VP7 and are expressed on the B-cell or non-T-cell surface will provide additional clues about the mechanism through which VP7 induces polyclonal B-cell activation.
Acknowledgments
We thank Richard Ward and Andrea Bertolotti-Ciarlet for generous provision of reagents; David Keeland, Jeff Scott, Erin Sargent, and Ann Robertson for technical assistance; and Manuel Franco for helpful suggestions.
This work was supported by CFAR AI36211 (D.E.L.), NIH AI10604 (S.E.B.), NIH AI24998 (S.E.C., K.L.W., M.K.E., and M.E.C.), NIH DK56338 (M.K.E.), and the Office of Research and Development, Medical Research Service, Department of Veterans Affairs (M.E.C.).
REFERENCES
- 1.Almeida, G. D., C. D. Porada, S. St. Jeor, and J. L. Ascensao. 1994. Human cytomegalovirus alters interleukin-6 production by endothelial cells. Blood 83:370-376. [PubMed] [Google Scholar]
- 2.Anders, E. M., A. A. Scalzo, and D. O. White. 1985. Mitogenic activity of influenza virus and haemagglutinin. Vaccine 3:241-244. [DOI] [PubMed] [Google Scholar]
- 3.Ardavin, C., F. Luthi, M. Andersson, L. Scarpellino, P. Martin, H. Diggelmann, and H. Acha-Orbea. 1997. Retrovirus-induced target cell activation in the early phases of infection: the mouse mammary tumor virus model. J. Virol. 71:7295-7299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bachmann, M. F., H. Hengartner, and R. M. Zinkernagel. 1995. T helper cell-independent neutralizing B cell response against vesicular stomatitis virus: role of antigen patterns in B cell induction? Eur. J. Immunol. 25:3445-3451. [DOI] [PubMed] [Google Scholar]
- 5.Bieback, K., E. Lien, I. M. Klagge, E. Avota, J. Schneider-Schaulies, W. P. Duprex, H. Wagner, C. J. Kirschning, V. ter Meulen, and S. Schneider-Schaulies. 2002. Hemagglutinin protein of wild-type measles virus activates Toll-like receptor 2 signaling. J. Virol. 76:8729-8736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Birx, D. L., R. R. Redfield, K. Tencer, A. Fowler, D. S. Burke, and G. Tosato. 1990. Induction of interleukin-6 during human immunodeficiency virus infection. Blood 76:2303-2310. [PubMed] [Google Scholar]
- 7.Blutt, S. E., K. L. Warfield, D. E. Lewis, and M. E. Conner. 2002. Early response to rotavirus infection involves massive B cell activation. J. Immunol. 168:5716-5721. [DOI] [PubMed] [Google Scholar]
- 8.Browning, G. F., D. R. Snodgrass, O. Nakagomi, E. Kaga, A. Sarasini, and G. Gerna. 1992. Human and bovine serotype G8 rotaviruses may be derived by reassortment. Arch. Virol. 125:121-128. [DOI] [PubMed] [Google Scholar]
- 9.Bruce, M. G., I. Campbell, Y. Xiong, M. Redmond, and D. R. Snodgrass. 1994. Recognition of rotavirus antigens by mouse L3T4-positive T helper cells. J. Gen. Virol. 75:1859-1866. [DOI] [PubMed] [Google Scholar]
- 10.Burns, J. W., D. Chen, M. K. Estes, and R. F. Ramig. 1989. Biological and immunological characterization of a simian rotavirus SA11 variant with an altered genome segment 4. Virology 169:427-435. [DOI] [PubMed] [Google Scholar]
- 11.Burns, J. W., H. B. Greenberg, R. D. Shaw, and M. K. Estes. 1988. Functional and topographical analyses of epitopes on the hemagglutinin (VP4) of the simian rotavirus SA11. J. Virol. 62:2164-2172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Burns, J. W., M. Siadat-Pajouh, A. A. Krishnaney, and H. B. Greenberg. 1996. Protective effect of rotavirus VP6-specific IgA monoclonal antibodies that lack neutralizing activity. Science 272:104. [DOI] [PubMed] [Google Scholar]
- 13.Casola, A., M. K. Estes, S. E. Crawford, P. L. Ogra, P. B. Ernst, R. P. Garofalo, and S. E. Crowe. 1998. Rotavirus infection of cultured intestinal epithelial cells induces secretion of CXC and CC chemokines. Gastroenterology 114:947-955. [DOI] [PubMed] [Google Scholar]
- 14.Ciarlet, M., M. K. Estes, C. Barone, R. F. Ramig, and M. E. Conner. 1998. Analysis of host range restriction determinants in the rabbit model: comparison of homologous and heterologous rotavirus infections. J. Virol. 72:2341-2351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Coutelier, J.-P., P. G. Coulie, P. Wauters, H. Heremans, and J. T. van der Logt. 1990. In vivo polyclonal B-lymphocyte activation elicited by murine viruses. J. Virol. 64:5383-5388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Coutelier, J. P., and J. Van Snick. 1985. Isotypically restricted activation of B lymphocytes by lactic dehydrogenase virus. Eur. J. Immunol. 15:250-255. [DOI] [PubMed] [Google Scholar]
- 17.Crawford, S. E., M. Labbé, J. Cohen, M. H. Burroughs, Y.-J. Zhou, and M. K. Estes. 1994. Characterization of virus-like particles produced by the expression of rotavirus capsid proteins in insect cells. J. Virol. 68:5945-5952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Crawford, S. E., S. K. Mukherjee, M. K. Estes, J. A. Lawton, A. L. Shaw, R. F. Ramig, and B. V. V. Prasad. 2001. Trypsin cleavage stabilizes the rotavirus VP4 spike. J. Virol. 75:6052-6061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Estes, M. K., D. Y. Graham, R. F. Ramig, and B. L. Ericson. 1982. Heterogeneity in the structural glycoprotein (VP7) of simian rotavirus SA11. Virology 122:8-14. [DOI] [PubMed] [Google Scholar]
- 20.Fehr, T., M. F. Bachmann, H. Bluethmann, H. Kikutani, H. Hengartner, and R. M. Zinkernagel. 1996. T-independent activation of B cells by vesicular stomatitis virus: no evidence for the need of a second signal. Cell Immunol. 168:184-192. [DOI] [PubMed] [Google Scholar]
- 21.Fehr, T., H. Y. Naim, M. F. Bachmann, A. F. Ochsenbein, P. Spielhofer, E. Bucher, H. Hengartner, M. A. Billeter, and R. M. Zinkernagel. 1998. T-cell independent IgM and enduring protective IgG antibodies induced by chimeric measles viruses. Nat. Med. 4:945-948. [DOI] [PubMed] [Google Scholar]
- 22.Feng, N., J. W. Burns, L. Bracy, and H. B. Greenberg. 1994. Comparison of mucosal and systemic humoral immune responses and subsequent protection in mice orally inoculated with a homologous or a heterologous rotavirus. J. Virol. 68:7766-7773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Franco, M. A., and H. B. Greenberg. 1995. Role of B cells and cytotoxic T lymphocytes in clearance of and immunity to rotavirus infection in mice. J. Virol. 69:7800-7806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Franco, M. A., and H. B. Greenberg. 1997. Immunity to rotavirus in T cell deficient mice. Virology 238:169-179. [DOI] [PubMed] [Google Scholar]
- 25.Freer, G., C. Burkhart, I. Ciernik, M. F. Bachmann, H. Hengartner, and R. M. Zinkernagel. 1994. Vesicular stomatitis virus Indiana glycoprotein as a T-cell-dependent and -independent antigen. J. Virol. 68:3650-3655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Glass, R. I., P. E. Kilgore, R. C. Holman, S. Jin, J. C. Smith, P. A. Woods, M. J. Clarke, M. S. Ho, and J. R. Gentsch. 1996. The epidemiology of rotavirus diarrhea in the United States: surveillance and estimates of disease burden. J. Infect. Dis. 174(Suppl. 1):S5-S11. [DOI] [PubMed] [Google Scholar]
- 27.Green, K. Y., and A. Z. Kapikian. 1992. Identification of VP7 epitopes associated with protection against human rotavirus illness or shedding in volunteers. J. Virol. 66:548-553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Greenberg, H., V. McAuliffe, J. Valdesuso, R. Wyatt, J. Flores, A. Kalica, Y. Hoshino, and N. Singh. 1983. Serological analysis of the subgroup protein of rotavirus, using monoclonal antibodies. Infect. Immun. 39:91-99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ho, M. S., R. I. Glass, P. F. Pinsky, N. C. Young-Okoh, W. M. Sappenfield, J. W. Buehler, N. Gunter, and L. J. Anderson. 1988. Diarrheal deaths in American children. Are they preventable? JAMA 260:3281-3285. [PubMed] [Google Scholar]
- 30.Jacobs, D. M., and D. C. Morrison. 1977. Inhibition of the mitogenic response to lipopolysaccharide (LPS) in mouse spleen cells by polymyxin B. J. Immunol. 118:21-27. [PubMed] [Google Scholar]
- 31.Janeway, C., P. Travers, M. Walport, and J. Shlomckik. 2001. Immunobiology: the immune system in health and disease, p. 356-358. Elsevier Science Ltd./Garland Publishing, London, United Kingdom.
- 32.Jiang, X., M. Wang, D. Y. Graham, and M. K. Estes. 1992. Expression, self-assembly, and antigenicity of the Norwalk virus capsid protein. J. Virol. 66:6527-6532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Johnson, B. J., E. O. Costelloe, D. R. Fitzpatrick, J. B. Haanen, T. N. Schumacher, L. E. Brown, and A. Kelso. 2003. Single-cell perforin and granzyme expression reveals the anatomical localization of effector CD8+ T cells in influenza virus-infected mice. Proc. Natl. Acad. Sci. USA 100:2657-2662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kapikian, A. Z., Y. Hoshino, and R. M. Chanock. 2001. Rotavirus, p. 1787-1833. In D. M. Knipe and P. M. Howley (ed.), Fields virology, 4th ed. Lippincott Williams & Wilkins, Philadelphia, Pa.
- 35.Karupiah, G., T. E. Sacks, D. M. Klinman, T. N. Fredrickson, J. W. Hartley, J. H. Chen, and H. C. Morse. 1998. Murine cytomegalovirus infection-induced polyclonal B cell activation is independent of CD4+ T cells and CD40. Virology 240:12-26. [DOI] [PubMed] [Google Scholar]
- 36.Khoury, C. A., K. A. Brown, J. E. Kim, and P. A. Offit. 1994. Rotavirus-specific intestinal immune response in mice assessed by enzyme-linked immunospot assay and intestinal fragment culture. Clin. Diagn. Lab. Immunol. 1:722-728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kizaka, S., G. Goodman-Snitkoff, and J. J. McSharry. 1983. Sendai virus glycoproteins are T cell-dependent B cell mitogens. Infect. Immun. 40:592-600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kurt-Jones, E. A., L. Popova, L. Kwinn, L. M. Haynes, L. P. Jones, R. A. Tripp, E. E. Walsh, M. W. Freeman, D. T. Golenbock, L. J. Anderson, and R. W. Finberg. 2000. Pattern recognition receptors TLR4 and CD14 mediate response to respiratory syncytial virus. Nat. Immunol. 1:398-401. [DOI] [PubMed] [Google Scholar]
- 39.Lopez, D. M., V. Charyulu, and M. M. Sigel. 1981. Subset of spleen lymphocytes from BALB/cCrgl mice stimulated by mouse mammary tumor virus. Cancer Res. 41:813-818. [PubMed] [Google Scholar]
- 40.Lucas, A. H., and S. M. Asser. 1986. The type-specific capsular carbohydrate of Hemophilus influenzae B is a potent mitogen for murine B lymphocytes. J. Immunol. 137:3130-3134. [PubMed] [Google Scholar]
- 41.Matson, D. O., M. L. O'Ryan, L. K. Pickering, S. Chiba, S. Nakata, P. Raj, and M. K. Estes. 1992. Characterization of serum antibody responses to natural rotavirus infections in children by VP7-specific epitope-blocking assays. J. Clin. Microbiol. 30:1056-1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Matsuda, K., H. Tsutsumi, Y. Okamoto, and C. Chiba. 1995. Development of interleukin 6 and tumor necrosis factor alpha activity in nasopharyngeal secretions of infants and children during infection with respiratory syncytial virus. Clin. Diagn. Lab. Immunol. 2:322-324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Melchers, F., and C. Corbel. 1983. Studies on B-cell activation in vitro. Ann. Immunol. (Paris) 134D:63-73. [DOI] [PubMed] [Google Scholar]
- 44.Muraguchi, A., J. H. Kehrl, J. L. Butler, and A. S. Fauci. 1984. Regulation of human B-cell activation, proliferation, and differentiation by soluble factors. J. Clin. Immunol. 4:337-347. [DOI] [PubMed] [Google Scholar]
- 45.Offit, P. A. 2001. Correlates of protection against rotavirus infection and disease. Novartis Found. Symp. 238:106-113. [DOI] [PubMed] [Google Scholar]
- 46.O'Neal, C. M., S. E. Crawford, M. K. Estes, and M. E. Conner. 1997. Rotavirus virus-like particles administered mucosally induce protective immunity. J. Virol. 71:8707-8717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Parashar, U. D., E. G. Hummelman, J. S. Bresee, M. A. Miller, and R. I. Glass. 2003. Global illness and deaths caused by rotavirus disease in children. Emerg. Infect. Dis. 9:565-572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Prasad, B. V., G. J. Wang, J. P. Clerx, and W. Chiu. 1988. Three-dimensional structure of rotavirus. J. Mol. Biol. 199:269-275. [DOI] [PubMed] [Google Scholar]
- 49.Raj, P., D. O. Matson, B. S. Coulson, R. F. Bishop, K. Taniguchi, S. Urasawa, H. B. Greenberg, and M. K. Estes. 1992. Comparisons of rotavirus VP7-typing monoclonal antibodies by competition binding assay. J. Clin. Microbiol. 30:704-711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rassa, J. C., J. L. Meyers, Y. Zhang, R. Kudaravalli, and S. R. Ross. 2002. Murine retroviruses activate B cells via interaction with toll-like receptor 4. Proc. Natl. Acad. Sci. USA 99:2281-2286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rott, O., J. Charreire, K. Mignon-Godefroy, and E. Cash. 1995. B cell superstimulatory influenza virus activates peritoneal B cells. J. Immunol. 155:134-142. [PubMed] [Google Scholar]
- 52.Sacco, R. E., M. Hagen, J. E. Donelson, and R. G. Lynch. 1994. B lymphocytes of mice display an aberrant activation phenotype and are cell cycle arrested in G0/G1A during acute infection with Trypanosoma brucei. J. Immunol. 153:1714-1723. [PubMed] [Google Scholar]
- 53.Scalzo, A. A., and E. M. Anders. 1985. Influenza viruses as lymphocyte mitogens. II. Role of I-E molecules in B cell mitogenesis by influenza A viruses of the H2 and H6 subtypes. J. Immunol. 135:3524-3529. [PubMed] [Google Scholar]
- 54.Spender, L. C., G. H. Cornish, B. Rowland, B. Kempkes, and P. J. Farrell. 2001. Direct and indirect regulation of cytokine and cell cycle proteins by EBNA-2 during Epstein-Barr virus infection. J. Virol. 75:3537-3546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Stevenson, P. G., and P. C. Doherty. 1999. Non-antigen-specific B-cell activation following murine gammaherpesvirus infection is CD4 independent in vitro but CD4 dependent in vivo. J. Virol. 73:1075-1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Stuker, G., L. S. Oshiro, and N. J. Schmidt. 1980. Antigenic comparisons of two new rotaviruses from rhesus monkeys. J. Clin. Microbiol. 11:202-203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Szomolanyi-Tsuda, E., J. D. Brien, J. E. Dorgan, R. L. Garcea, R. T. Woodland, and R. M. Welsh. 2001. Antiviral T-cell-independent type 2 antibody responses induced in vivo in the absence of T and NK cells. Virology 280:160-168. [DOI] [PubMed] [Google Scholar]
- 58.Tough, D. F., S. Sun, and J. Sprent. 1997. T cell stimulation in vivo by lipopolysaccharide (LPS). J. Exp. Med. 185:2089-2094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ward, R. L., M. M. McNeal, and J. F. Sheridan. 1990. Development of an adult mouse model for studies on protection against rotavirus. J. Virol. 64:5070-5075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wright, S. D. 1995. CD14 and innate recognition of bacteria. J. Immunol. 155:6-8. [PubMed] [Google Scholar]
- 61.Yeager, M., K. A. Dryden, N. H. Olson, H. B. Greenberg, and T. S. Baker. 1990. Three-dimensional structure of rhesus rotavirus by cryoelectron microscopy and image reconstruction. J. Cell Biol. 110:2133-2144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zhou, Y.-J., J. W. Burns, Y. Morita, T. Tanaka, and M. K. Estes. 1994. Localization of rotavirus VP4 neutralization epitopes involved in antibody-induced conformational changes of virus structure. J. Virol. 68:3955-3964. [DOI] [PMC free article] [PubMed] [Google Scholar]