

Abstract
The membrane fusion events which initiate human immunodeficiency virus type 1 (HIV-1) infection and promote cytopathic syncytium formation in infected cells commence with the binding of the HIV envelope glycoprotein (Env) to CD4 and an appropriate coreceptor. Here, we show that HIV Env-coreceptor interactions activate Rac-1 GTPase and stimulate the actin filament network reorganizations that are requisite components of the cell fusion process. Disrupting actin filament dynamics with jasplakinolide or latrunculin A arrested fusion at a late step in the formation of Env-CD4-coreceptor complexes. Time-lapse confocal microscopy of living cells revealed vigorous activity of actin-based, target cell membrane extensions at the target cell-Env-expressing cell interface. The expression of dominant-negative forms of actin-regulating Rho-family GTPases established that HIV Env-mediated syncytium formation relies on Rac-1 but not on Cdc42 or Rho activation in target cells. Similar dependencies were found when cell fusion was induced by Env expressed on viral or cellular membranes. Additionally, Rac activity was specifically upregulated in a coreceptor-dependent manner in fusion reaction cell lysates. These results define a role for HIV Env-coreceptor interactions in activating the cellular factors essential for virus-cell and cell-cell fusion and provide evidence for the participation of pertussis toxin-insensitive signaling pathways in HIV-induced membrane fusion.
Human immunodeficiency virus (HIV) utilizes the fusion peptide of Env to induce the merger of viral or infected-cell membranes with target cell membranes (14). Conformational changes in Env, which expose the fusion peptide, are triggered by the sequential binding of Env to CD4 and one of two primary coreceptors, CCR5 or CXCR4. The signaling functions of CD4 (27) and the coupling of coreceptors to heterotrimeric G proteins are dispensable for HIV-mediated membrane fusion and infection (1, 2, 9), which suggests that binding alone regulates the fusion event. As such, present strategies to prevent HIV-induced membrane fusion block the interaction of Env with CD4 or with a coreceptor or the formation of Env structural intermediates (3). HIV-1 Env binding to CCR5 or CXCR4 essentially mimics the ligation of these receptors by chemokines, stimulating responses such as chemotaxis, gene transcription, and phosphorylation, in some cases independent of heterotrimeric G protein activation (28). Therefore, coreceptor-mediated signal transduction in HIV Env-dependent fusion remains an open question and may provide a new focus for therapeutic interventions aiming to inhibit HIV coreceptor, but not chemokine receptor, function.
One target of the chemokine receptor signal transduction pathways is the actin filament network (26). During chemotaxis the actin cytoskeleton controls the polarization, orientation, and forward motility of cells, primarily by responding to the regulatory Rho-family GTPases Cdc42, Rac, and Rho, which are differentially activated through chemokine receptor ligation (31). Accumulating evidence from diverse experimental systems indicates a central role for actin cytoskeletal remodeling and Rho GTPases in regulating the fusion of biological membranes (10). Actin filament reorganization can exert diverse effects, depending on the stage and type of membrane fusion, which can sometimes be discerned through pharmacological manipulation (34). During HIV entry and syncytium formation, the actin cytoskeleton may play a role in formation and/or localization of Env-CD4-coreceptor complexes, a process which is sensitive to the action of the actin filament-capping drug, cytochalasin D (CD) (15, 19). The effect of other actin-targeted drugs on HIV Env-induced cell fusion and the participation of the actin cytoskeleton in later stages have not been investigated.
We studied the role of the actin filament network during HIV Env-dependent and virus-dependent syncytium formation, utilizing the actin filament stabilizing agent jasplakinolide (JP) and the actin monomer-sequestering drug latrunculin A (LA) (10). The data presented demonstrate actin cytoskeletal reorganizations that are a critical feature of HIV-induced cell-cell fusion. Additionally, the specific actin regulatory pathway, activated following the interaction of Env and a coreceptor, is identified. Together, these results suggest that HIV Env regulates the activity of cellular factors to facilitate virus-cell and cell-cell fusion.
MATERIALS AND METHODS
Cells. U87.CD4.CCR5 and U87.CD4.CXCR4 are astroglioma cell lines engineered to express CD4 and either CCR5 or CXCR4 constructs that are tagged at their C termini with green fluorescent protein (GFP). The maintenance of cell lines and peripheral blood lymphocytes has been described (25). Quail QT6 cells were maintained in M-199 Earle's medium containing 1% chicken serum, 5% fetal bovine serum, 10% tryptose phosphate broth, penicillin, streptomycin, sodium pyruvate, and l-glutamine. Unless noted, tissue culture supplies were obtained from the Tissue Culture Support Center, Washington University School of Medicine (St. Louis, Mo.).
Reagents.
JP, LA, and BODIPY-630/650 were purchased from Molecular Probes (Eugene, Oreg.). Red fluorescent protein (RFP) expression plasmid pDS-RFP-N1 was supplied by Clontech. A p24 antigen enzyme-linked immunosorbent assay kit was obtained from Beckman-Coulter. All other reagents were purchased from Sigma Aldrich (St. Louis, Mo.) unless otherwise noted.
Viruses.
Modified vaccinia Ankara expressing T7 polymerase (MVA-T7) (35) was a gift from Andrew Pekosz. Wild-type (WT) vaccinia (WR strain) and recombinant vaccinia viruses expressing β-galactosidase (vCB21R), T7 polymerase (vPT7-3), CD4, or HIV-1 Env proteins were obtained as reported (25). Recombinant vaccinia viruses encoding the dominant-negative mutant GTPases Cdc42 N17, Rac N17, and Rho N19 were generated and characterized as described (36). HIV stocks were prepared by the lipofection of plasmid DNA encoding full-length proviral molecular clones, which contain the Env gene of the R5 YU2 strain or the X4 HXB2 strain in the HIVNL4-3 backbone (33). Transfected 293T cell supernatants were harvested 48 h postlipofection, filtered, and assayed for p24 antigen content by enzyme-linked immunosorbent assay as described (25).
Env-dependent fusion assay.
The HIV Env-dependent cell fusion assay was performed as reported (25), with modifications for drug treatments and alternate detection methods. For the enzymatic quantitation of fusion, BSC40 cells were coinfected with vaccinia viruses encoding an HIV Env protein and with vPT7-3, and target cells were infected with the virus vCB21R, which expresses the lacZ gene in the presence of T7 polymerase. Because the recombinant vaccinia viruses expressing the GTPase mutants also express the lacZ gene, β-galactosidase activity was used as an indicator of viral protein expression rather than cytoplasmic mixing in some experiments. Cells were infected for 1 h (multiplicity of infection [MOI], 10) at 37°C and cultured overnight prior to mixing. Unless noted, drugs were added to the cells as fusion partners were combined. The concentrations of the drugs used were as follows: CD, 1 μM; LA, 2 μM; and JP, 3 μM. To allow fusion, 105 cells in triplicate wells were combined with a fusion partner at a ratio of 1:1 and incubated at 37°C for 3 h unless otherwise noted. For enzymatic detection, fusion was stopped by the addition of NP-40 to a final concentration of 1% and β-galactosidase activity was determined (13). For microscopic analysis, fusion was stopped by the addition of formaldehyde to a final concentration of 3.7%. For each sample, 10 fields of equal area were selected at random and scanned. Area measurements were calculated by outlining the syncytia within each scan by using the overlay function of the laser scanning microscope LSM 510 software. Syncytia were defined by the presence of dye within the cytoplasm of GFP-illuminated cells.
Confocal microscopy.
For time-lapse confocal microscopy of living cells, QT6 cells were infected with MVA-T7 (MOI, 5) for 1 h at 37°C prior to overnight lipofection of pTM3.HXB2env or pTM3.YU2env, which encode the HIV env gene under the control of the T7 polymerase promoter. QT6 cells were cotransfected with pDS-RFP-N1 or were loaded with BODIPY-630/650. QT6 cells were layered on target cells and allowed to settle at 25°C. Once a potential fusion event was located (i.e., Env-expressing and target cells in proximity to each other), cells were warmed to 37°C, and sequential scans were acquired for 15 to 30 min. Some scans were acquired after the cells had incubated for up to 1 h at 37°C. Images were collected by using a Zeiss LSM 510 microscope. Measurements were made by using the overlay function of the LSM 510 software. Certain images of fixed-cell preparations were viewed with a Zeiss axiovert microscope equipped with a Bio-Rad laser scanning system. For the supplemental material, time-lapse confocal microscopy was performed as described above. The resulting scans, acquired approximately every 9 s, are displayed as QuickTime movies, played in series at 6 to 10 frames per s in the supplemental material.
Rac activation assay.
A total of 1.5 × 107 U87.CD4.CCR5 cells were mixed at a ratio of 1:1 with BSC40 cells which were infected with vCB-39 (HIVADA Env), vSP-5 (HIVYU2 Env), vSC60 (HIVHXB2 Env), or WT vaccinia virus as described above. Where indicated, TAK-779 (1 μM) was added. Reactions were incubated at 37°C for 10 or 30 min, washed two times, and lysed. GTPγS- and GDP-loaded cell lysates were generated by using 0.5 × 107 U87.CD4.CCR5 cells mixed at a ratio of 1:1 with vCB-39-infected BSC40 cells and incubated at 37°C for 30 min. Lysates were immediately analyzed by using a Rac activation assay kit according to the manufacturer's instructions (Upstate Biotechnology, N.Y.), with equal amounts of protein for column loading.
Virus-dependent fusion assay.
Virus-induced cell fusion was monitored by β-galactosidase activity or confocal microscopy as described above, modifying “fusion-from-without” assays previously reported by other groups (6, 11, 22). For enzymatic detection, U87.CD4.CCR5.GFP cells were infected with vCB21R or vPT7-3 and cultured overnight. Cells were harvested by trypsin treatment, washed, and mixed (1:1) in triplicate wells of a 96-well microtiter plate. Drugs were added, where noted, prior to the addition of virus and DEAE-dextran (20 μg/ml). Fusion reactions were incubated for 3 h at 37°C unless reported otherwise. Reactions were assayed for β-galactosidase activity as described above. For microscopic analysis, U87.CD4.CCR5.GFP cells were infected with WT vaccinia or vaccinia viruses encoding the Rho-family GTPase dominant-negative mutants and cultured overnight in chamber slides. Virus stocks were added to the chambers and incubated for 3 h at 37°C prior to fixation with 3.7% formaldehyde and viewing by confocal microscopy as described above.
RESULTS
Late event(s) in HIV-induced cell fusion are dependent on cytoskeletal reorganization. We studied the effects of JP and LA in a quantitative Env-dependent cell fusion assay (Fig. 1). When added at the time of cell mixing, LA and JP inhibited cell-cell fusion mediated by either CCR5- or CXCR4-utilizing (R5 or X4 HIV strains, respectively) Env glycoproteins in a dose-dependent manner (Fig. 1A). Unlike the action of CD and LA, the effects of JP are irreversible (4), which allowed for treatment of individual cell populations prior to mixing. Pretreatment of target or Env-expressing cells with JP for 10 min inhibited fusion by 85 and 40%, respectively, while pretreating both cell populations abolished fusion completely (Fig. 1B). Inhibition of syncytium formation was complete at concentrations of 1 to 3 μM for both JP and LA regardless of Env coreceptor preference or the cell type used for Env expression or as fusion target (Table 1). These results demonstrate a general dependency of HIV Env-mediated cell fusion on the ability of the actin cytoskeleton to reorganize, especially in the target cell population.
FIG. 1.
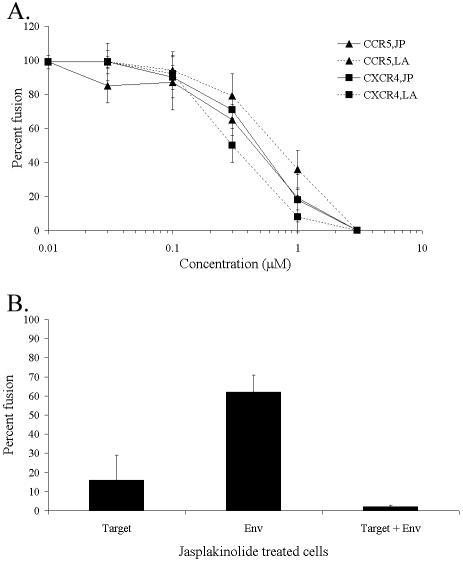
Actin-dependent cell fusion. Average fusion compared to untreated control reactions and detected by β-galactosidase activity ± standard deviation are shown. (A) CCR5- or CXCR4-expressing U87.CD4 cells were incubated with HIVADA or HIVHXB2 Env-expressing cells, respectively, in the presence of JP or LA at the indicated concentrations. (B) U87.CD4.CCR5 cells and/or HIVADA Env-expressing cells were treated with JP for 10 min and washed extensively prior to mixing. In each case, representative data from 1 of 4 experiments are shown.
TABLE 1.
Cell types and HIV Env strains found to be sensitive to the inhibitory action of JP and LA in the HIV Env-dependent cell fusion assaya
| Cell lines and strains used |
|---|
| Target cells |
| U87.CD4.CCR5.GFP |
| U87.CD4.CXCR4.GFP |
| MAG1.CCR5 |
| Human peripheral blood lymphocytes |
| Env-expressing cells (species) |
| BSC40 (monkey) |
| 293T (human) |
| QT6 (quail) |
| HIV Env strains (coreceptor preference) |
| ADA (R5) |
| BaL (R5) |
| JR-FL (R5) |
| YU2 (R5) |
| SF162 (R5) |
| SF2 (R5X4) |
| 89.6 (R5X4) |
| HXB2 (X4) |
| UG92046 (X4) |
Percent inhibition of fusion was determined for drug-treated fusion reactions compared to reactions in untreated controls and ranged from 90 to 100% inhibition by either JP or LA for all fusion partners and HIV Env strains tested.
To see whether there was any time at which JP was ineffective at blocking fusion, JP or NP-40 was added to stop the fusion reaction at several time points within the 37°C incubation period (Fig. 2A). The amount of fusion obtained following the addition of JP closely approximated the amount observed after the addition of detergent at each time point. This result suggested that the JP-sensitive step is temporally related to the fusion event, perhaps occurring after binding of Env to CD4 and/or the coreceptor. Because the formation and/or accumulation of these complexes are sensitive to the action of CD (15, 30), we compared the effects of CD with JP or LA. The fusion assay was modified to include incubation at 25°C prior to warming, which primes the reaction by allowing early events, such as Env-CD4 binding, to occur while preventing membrane fusion. In these experiments, the results of which are shown in Fig. 2B, JP and LA inhibited membrane fusion by 80 to 100% whether added before or after the 25°C incubation. In contrast, CD was an effective inhibitor only when added before the 25°C incubation. This result indicated that the differential effects of CD, JP, and LA on the actin cytoskeletal network blocked the HIV-induced syncytium formation at distinct points in the cell fusion pathway. Whether JP or LA inhibits the same actin-mediated step during HIV Env-induced cell fusion has not been determined.
FIG. 2.
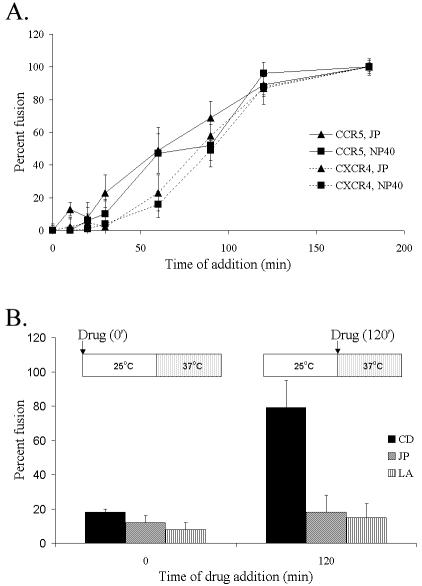
Actin filament network involvement late in the HIV Env-induced cell fusion pathway. Average fusion compared to untreated control reactions and detected by β-galactosidase activity ± standard deviation are shown. (A) CCR5- or CXCR4-expressing U87.CD4 cells were incubated with HIVADA or HIVHXB2 Env-expressing cells, respectively, with JP or NP-40 added at the indicated times. (B) U87.CD4.CCR5 cells were incubated with HIVADA Env-expressing cells at 25°C for 2 h and then were warmed to 37°C for 1 h. As indicated, drugs were added before or after the 25°C incubation. Each graph is representative of data from 1 of 4 experiments.
Confocal imaging supports a role for actin-based membrane structures in HIV Env-mediated syncytium formation.
The morphology of cells treated with JP was altered, characterized by the collapse of the plasma membrane and the loss of cell polarity at the highest concentration (Fig. 3A). Some collapse of the membrane at the cell periphery was noted in cells treated with JP at lower dosages, although cells maintained spread or elongated phenotypes. No gross differences in cell surface localizations of CD4 or a coreceptor were observed by confocal microscopy following JP treatment of CCR5-expressing cells (Fig. 3B). In cells fixed before or after JP treatment (upper and lower panels, respectively), a positive signal for CD4 staining at the cell surface was obtained and found to partially overlap with the CCR5.GFP signal. JP treatment failed to increase or decrease the area of colocalization witnessed when the signals were overlaid.
FIG. 3.
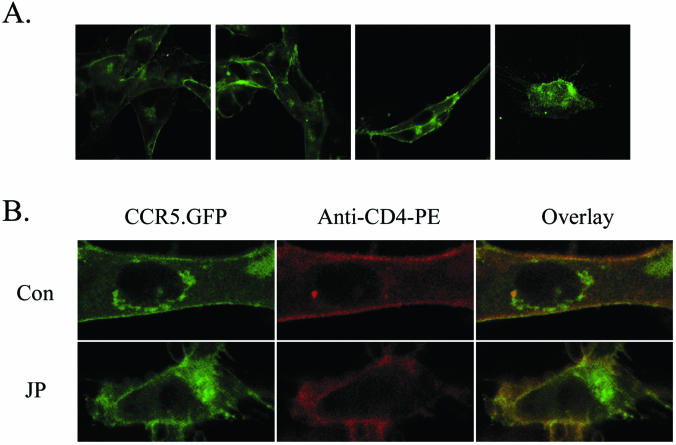
Effect of JP treatment on cell morphology and surface localization of CD4 and CCR5.GFP cells. (A) Confocal micrographs of U87.CD4.CCR5 cells fixed after 30 min of incubation with JP at final concentrations of (from left to right) 0, 0.1, 1.0, and 3.0 μM. (B) Confocal micrographs of U87.CD4.CCR5 cells fixed before (upper panels) or after (lower panels) JP treatment (3 μM; 10 min) and stained with anti-CD4-phycoerythrin antibodies. The green GFP signal and red phycoerythrin signal have been merged to show areas of colocalization (yellow). Note the collapsed appearance of the JP-treated cell. Data are representative of results from 3 experiments. Images were collected by using an oil objective (magnification, ×63). Con, control.
To observe the effect of JP on cells undergoing HIV Env-induced syncytium formation, we developed a system to study the behavior of living target cells, with membranes illuminated by CCR5.GFP, by mixing target cells with HIV Env-expressing cells loaded with fluorescent cytoplasmic marker and using time-lapse confocal microscopy. Selected scans are shown in Fig. 4, and each full series can be viewed in movie format (see Videos S1 and S2 in the supplemental material). The effect of laser scanning on living U87.CD4.CCR5.GFP cells is shown in Fig. 4A (see Video S1A in the supplemental material). In this series, which is representative of five similar series, the field was scanned 132 times in 23.5 min while the cell cultures were held at 37°C. Photobleaching is apparent, with the GFP fluorescent signal losing intensity with successive scans (Fig. 4A, left to right). Cells remained adherent, spread, and in contact with neighboring cells throughout the collection period, with no net movement of cells occurring. Plasma membrane ruffles and projections were observed, and their activity may have been stimulated by exposure to the laser. However, these structures were also evident in U87.CD4.CCR5.GFP cells which were fixed prior to scanning (Fig. 3). Overall, these cells exhibited few symptoms of phototoxicity during scan acquisition, with a general maintenance of cell morphology.
FIG. 4.
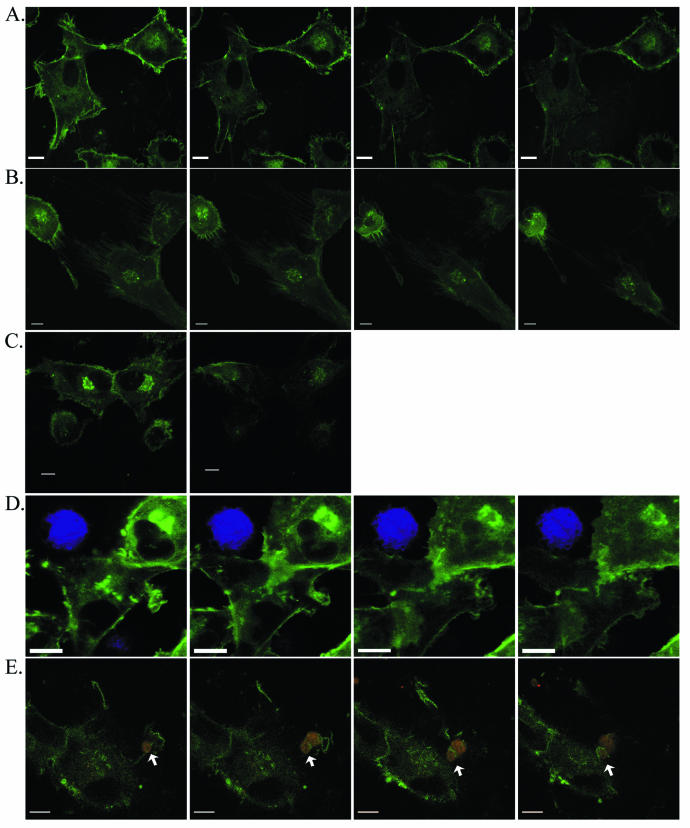
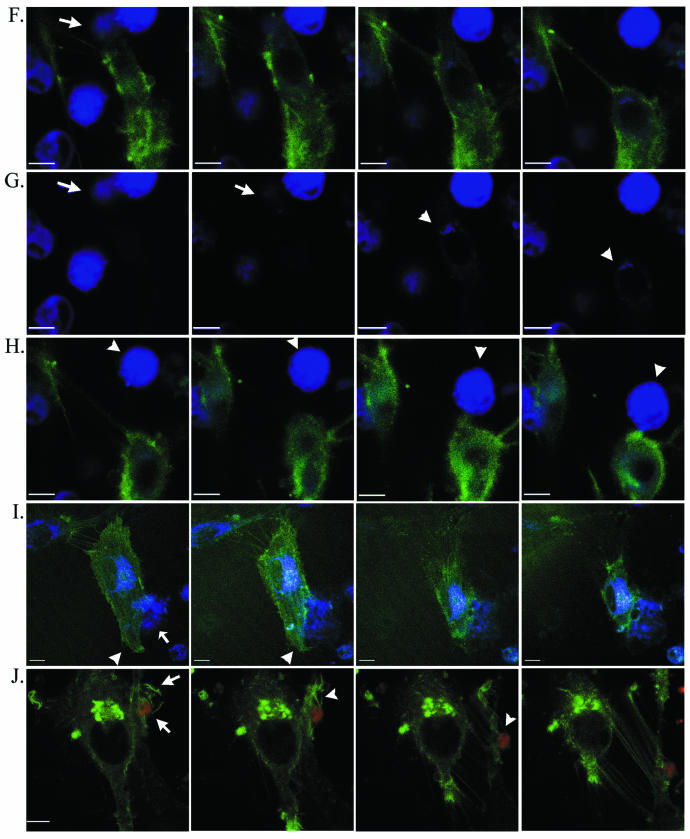
Time-lapse confocal microscopy. Selected images from sequential scans (see Videos S1 and S2 in supplemental material) of living U87.CD4.CCR5.GFP target cells (green) with and without Env-expressing cells (panel D, HIVHXB2 Env; panels E to J, HIVYU2 Env) combined for HIV-induced fusion. Env-expressing cells are blue in panel D and panels F to I and red in panels E and J. Cells with which targets fuse are indicated with an arrow in panels F, G, and I. An Env-expressing cell is indicated with an arrow in E and an arrowhead in H. Arrowheads in I highlight the dynamic membrane structures formed at the leading edge of the target cell. Panels in G are single-channel (blue) renditions of panels in F to facilitate visualization of dye transfer (arrowheads). Scans were captured at (from left to right) 0, 6.2,13.8, and 23.5 min (A); at 0.8, 2.1, 3.0, and 6.2 min, with JP added at 1 min (B); at 0 and 20 min, with JP added at 0.2 min and no intervening scans acquired (C); at 0.5, 2.3, 8.7, and 13.8 min (D); at 0, 0.8, 7.0, and 15.6 min (E); at 7.6, 9.6, 9.8, and 9.9 min (F and G); at 10.2, 10.6, 12.0, and 17.3 min (H); at 0, 6.7, 16.9, and 19.5 min (I); or at 9, 12.3, 14.3, and 17.8 min, with JP added at 10 min (J). For panels A to H and J, time zero indicates the time when cells were warmed to 37°C. For panel I, time zero is 60 min after the cells were warmed to 37°C. Bar = 10 μm.
Dramatic changes in cell architecture were evident following JP treatment (Fig. 4B; see Video S1B in the supplemental material). Within minutes of JP application, the characteristic spread morphology collapsed, leaving behind retraction microspikes or a filamentous membrane that failed to maintain adhesions with neighboring cells. Cells became rounded but did not detach from the culture dish. The time at which JP was added after laser scanning commenced was varied from 0 to 10 min, with no change in time required for a loss of spreading following JP addition (<2 min) (data not shown), which indicated that the changes were due to JP and not to phototoxicity. Additionally, cells scanned once before and once after the addition of JP exhibited the morphological effects of drug treatment indistinguishable from JP-treated cells subjected to the time-lapse laser scanning (Fig. 4C).
Few changes in cellular architecture were noted when cells expressing Env from X4 strain HXB2 were layered on U87.CD4.CCR5.GFP cells. As shown in Fig. 4D (see Video S1C in supplemental material), and in each similar series acquired, contact between the mismatched Env-expressing cells and target cells was evident, but no instances of cell migration, engulfment, or fusion were recorded (0 of 8 series collected). Consistent with the results from biochemical assays (15, 19), no fusion events were observed when fusion reactions were maintained at 25°C (0 of 7 series collected).
In contrast, vigorous interactions at the plasma membrane, rapid cell movements, and dye transfer following cell fusions were recorded when R5 Env-expressing cells were incubated with U87.CD4.CCR5.GFP cells. In the example shown in Fig. 4E (see Video S2A in supplemental material), target cell ruffles, lamellipodia, and filopodial extensions wrapped around the Env-expressing cell, possibly facilitating its movement, which was calculated at an average velocity of 1.19 μm min−1. This series did not reveal a fusion event. Capturing pre- and postfusion events in a single series was relatively rare, achieved in 6 out of 46 series acquired under permissive conditions.
In each sequence leading to a cell fusion event, a similarity to chemotaxis was noted, with target cells orienting and moving towards Env-expressing cells by extending leading-edge lamellipodia and retracting trailing edges, events that are absolutely dependent on actin filament assembly and disassembly (12). Two series were chosen to illustrate this point. In the first (Fig. 4F and G; see Video S2B in the supplemental material), an Env-expressing cell was rapidly engulfed by and fused with a target cell, which retracted following incorporation of the cytoplasmic dye. This series of events continued, as shown in Fig. 4H, where the same target cell can be observed to orient towards and move forward to contact a second Env-expressing cell, which appeared to be pulled close to the target cell. In Fig. 4I (see Video S2C in the supplemental material), a target cell which has previously fused with an Env-expressing cell can be seen to extend leading edge lamellipodia and filopodia around an Env-expressing cell prior to fusion and to simultaneously retract from a neighboring target cell. In this series, Env-expressing cell membrane extensions can also be observed at the target cell-Env-expressing cell interface. If such projections facilitate the cellular or molecular interactions leading to HIV-induced cell fusion, they may provide an explanation for the sensitivity of the fusion reaction to the JP treatment of the Env-expressing cell (Fig. 1B). In most cases, the cytoplasmic markers used to illuminate the Env-expressing cell did not allow plasma membrane structures to be observed. Target cell membrane structures observed at the interface between target and Env-expressing cells included ruffles, filopodia, and lamellipodia (Fig. 4F and G). Although present at the surface of all target cells, these actin-based protrusions exhibited vigorous and directed motion towards Env-expressing cells. The activity of target cell filopodia and lamellipodia suggested that localized reorganization of the actin cytoskeleton may be stimulated when target and Env-expressing cells interact.
The response of interacting target and Env-expressing cells to JP treatment is shown in Fig. 4J (see Video S2D in the supplemental material). Changes in target cell morphology occurred within minutes of JP application. Most notable was the complete cessation of movement in the membrane structures engulfing the Env-expressing cell shortly after JP treatment. Before the addition of JP, membrane protrusions extended and curved around the Env-expressing cell (Fig. 4J, panel 1). After treatment these same extensions locked into straight, rigid structures which remained attached to the Env-expressing cell, even as the target cell retracted and lost junctions with a neighboring cell (Fig. 4J, panels 2 to 4).
Rac-1 GTPase dominant-negative mutant prevents HIV Env-induced cell fusion.
The actin filament-based structures observed at the target cell-Env-expressing cell interface are dependent on the differential activity of Rho GTPase regulatory pathways. Specifically, Cdc42 and Rac1 mediate formation of filopodia and lamellipodia, respectively, while Rho1 stimulates retraction through the activation of actomyosin contractility (24). Dominant-negative point mutants Cdc42N17, RacN17, and RhoN19 selectively suppress specific actin-based functions (12). In the HIV Env-dependent fusion assay, RacN17 essentially eliminated syncytium formation and transfer of cytoplasmic dye when it was expressed in target cells (Fig. 5). The expression of Cdc42N17, RacN17, or RhoN19 in Env-expressing cells (data not shown) or Cdc42N17 or RhoN19 in target cells (Fig. 5A and C) had little effect on cell fusion. No fusion events were observed when both partners expressed RacN17, and an analysis of RacN17 cell lysates for PAK-1 binding demonstrated that Rac activity was inhibited in RacN17-expressing cells (data not shown). Similar results were obtained with U87.CD4.CXCR4 cells, and flow cytometry confirmed that Rho family mutants did not alter the cell surface expression of CD4 (data not shown). Under high magnification, target cells expressing RacN17 lacked the prominent membrane extensions visualized in control cells and syncytia (Fig. 5B). The inability of RacN17 target cells to undergo fusion appeared to reflect the failure of engulfing lamellar membrane structures to form.
FIG. 5.
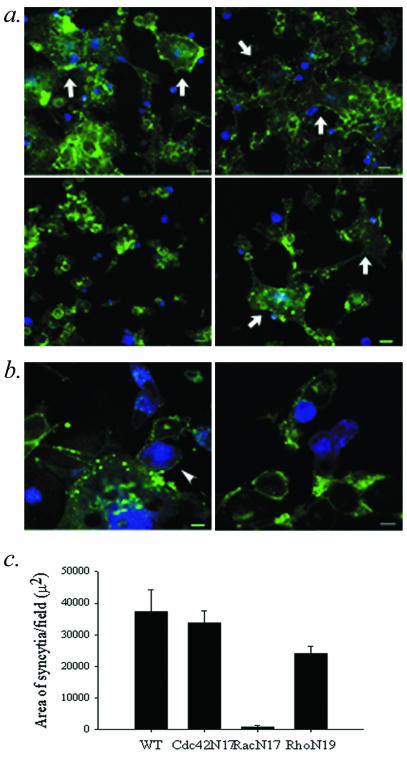
Rho-GTPases and cell-cell fusion. (a and b) Confocal micrographs of target cells (green) incubated for 2.5 h with Env-expressing cells (blue) to induce fusion. (c) The average area per field occupied by syncytia. Shown is the average of 10 fields ± standard error of the means. Target cells were infected with WT vaccinia (a, upper left frame; b, left frame) or with recombinant vaccinia viruses expressing Cdc42N17 (a, upper right frame), RacN17 (a, lower left frame; b, right frame) or RhoN19 (a, lower right frame). Syncytia are indicated with arrows. Target cell membrane extension is indicated with an arrowhead (b, left frame). Bars, 30 μm (a) and 10 μm (b). Representative data from 1 of 3 experiments are shown.
The Env-coreceptor interaction stimulates Rac activity.
The activation of Rac GTPase through ligation has been documented for CXCR4 (23) but has not been reported for CCR5. The activation state of Rac-1 in reaction lysates was assessed by utilizing PAK-1 binding to separate Rac-GTP (active Rac) from Rac-GDP (inactive Rac). As shown in Fig. 6, specific 3.4- and 3.6-fold increases in activated Rac were achieved when U87.CD4.CCR5 cells were mixed with cells expressing Env from R5 HIVADA (lane 5) and HIVYU2 (lane 7) strains, respectively, but not with cells lacking Env (lane 3) or expressing Env from the X4 HIVHXB2 strain (lane 8). The increase in Rac activation was absent in the presence of TAK-779 (lane 6), a small-molecule inhibitor of CCR5 that prevents gp120 binding (3). These data indicate that HIV Env stimulates Rac-1 activity through interaction with a coreceptor rather than CD4. The kinetics of Env-dependent Rac activation, which was observed by 30 min of incubation (lane 5), coincide with the kinetics of fusion in our assay system (Fig. 2C). The lack of detectable increases in Rac activity after 10 min (lane 4) is consistent with reports that Env-coreceptor binding occurs late in the sequence leading to membrane fusion (14).
FIG. 6.
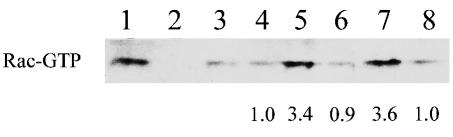
Rac activation and cell-cell fusion. Western blot analysis of PAK-1 binding fractions from lysates of U87.CD4.CCR5 cells mixed with BSC40 cells expressing no Env (lane 3) or Env from HIV-1 strains ADA (lanes 1, 2 and 4 to 6), YU2 (lane 7), or HXB2 (lane 8) at 37°C for 10 min (lane 4) or 30 min (lanes 1 to 3 and 5 to 8). TAK-779 was included to inhibit CCR5-Env binding (lane 6). Positive (lane 1) and negative (lane 2) controls were generated by GTPγS- and GDP-loading of reaction lysates, respectively. Increases (n-fold) in the amount of Rac-GTP compared to lane 3 were determined by densitometry and are indicated below the blots. Data represent results from 1 of 2 experiments with similar results.
Virus-dependent cell fusion is dependent on the actin cytoskeleton and Rac-1 activation.
Postentry steps in the HIV life cycle are susceptible to the actions of Rho GTPases (7, 21, 32), and preliminary experiments revealed that overnight infection of U87.CD4.CCR5.GFP cells by luciferase reporter viruses was inhibited when cells were first infected with vaccinia viruses expressing Cdc42N17, RacN17, or Rho N19 but not WT protein (data not shown). Therefore, we monitored the induction of fusion between permissive U87.CD4.CCR5.GFP cells by an R5 virus particle, HIVYU2, in a rapid assay of fusion from without (6, 11, 22). Virus-dependent cell fusion provides the fusogenic Env protein exogenously and does not require viral replication or cellular synthesis of Env (6). As shown in Fig. 7A, productive cell fusion was dependent on the concentration of virus. Virus-dependent fusion also required coreceptor expression in both cell populations and was inhibited when vector control cells replaced CCR5-expressing cells as a fusion partner (Fig. 7B, pBABE). Productive fusion was dependent on appropriately matched virus and coreceptor (i.e., fusion occurred in the presence of R5 HIVYU2 but not X4 HIVHXB2) and proceeded at 37°C but not at 4°C or 25°C (Fig. 7B). Like Env-dependent cell fusion, virus-induced fusion was sensitive to increasing concentrations of JP, with a maximal effect at a concentration of 3 μM, and was inhibited by both CD and LA at a concentration of 1 μM (Fig. 7C).
FIG. 7.
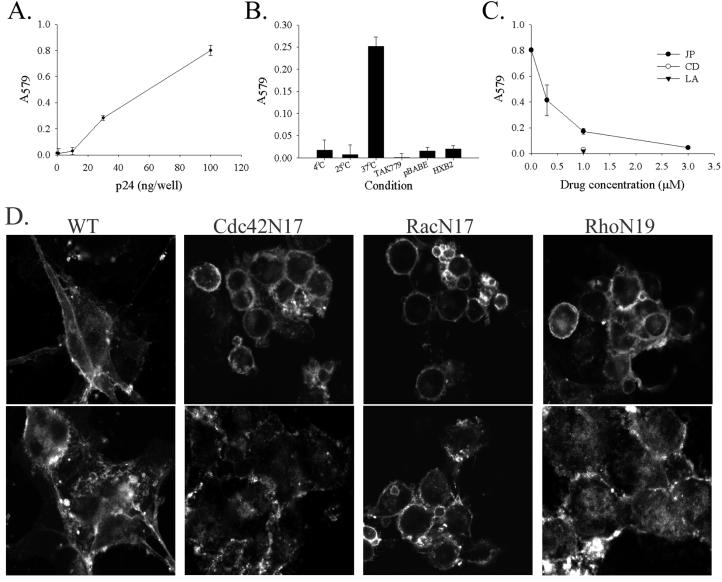
The actin cytoskeleton and Rac activation in virus-dependent cell fusion. Fusion of U87.CD4.CCR5.GFP cells incubated with increasing amounts (A) or 100 ng of HIVYU2 per well (B to D) at 37°C (except where noted in B). In panels A to C, relative fusion is indicated by β-galactosidase activity (average A579 of triplicate wells ± standard deviation; data are representative of results from three similar experiments). (D) U87.CD4.CCR5.GFP cells were infected with WT vaccinia or vaccinia viruses encoding the dominant-negative point mutants Cdc42N17, RacN17, or RhoN19 as indicated, prior to incubation with HIVHXB2 (upper panels) or HIVYU2 (lower panels), fixation, and confocal microscopy. Images were collected by using an oil objective (magnification, ×63). The experiment was performed twice with similar results.
Virus-dependent cell fusion was similarly inhibited by the expression of RacN17 but not Cdc42N17 or RhoN19. The upper panels in Fig. 7D show cells and HIV mismatched for coreceptor usage, whereas the bottom panels include cells and virus which are appropriately matched. The single (unfused) cells shown after incubation of CCR5-expressing cells with X4 strain HIVHXB2 reveal the morphological changes that occur when Cdc42N17, RacN17, and RhoN19 are expressed in these cells, namely, a smaller, rounded appearance lacking peripheral extensions as opposed to the angular, spread morphology of WT vaccinia-infected cells. Although the gross morphology of WT vaccinia-infected U87 cells is similar to that of uninfected cells (Fig. 3A, 1st panel), infection with this virus does have effects on the actin cytoskeleton (17). Changes in morphology did not coincide with changes in the ability to undergo virus-dependent cell fusion, however. Large syncytia were formed when Cdc42N17- and RhoN19-expressing U87.CD4.CCR5.GFP cells were exposed to HIVYU2, comparable to control syncytia formed by WT vaccinia-infected cells (Fig. 7D, lower panels). In contrast, RacN17-expressing cells were resistant to virus-induced fusion, maintaining their single-cell phenotype in the presence of HIVYU2.
DISCUSSION
Data presented here indicate that HIV-1 Env-coreceptor interactions stimulate an intracellular signaling cascade that promotes reorganization of the actin filament network and facilitates membrane fusion. Critical dependencies on the plasticity of the actin cytoskeleton and the activation of Rac-1 GTPase in the target cell suggest that formation of specific actin filamentous structures is an obligatory step in HIV-mediated cell fusion. Taken together, the data indicate that both depolymerization and nucleation of actin filament polymerization may be required during virus-cell and cell-cell fusion. The observation that JP and LA, but not CD, are inhibitory when added after incubation at 25°C suggests that the actin filament network is required in the period between receptor binding and membrane fusion. CD, JP, and LA treatment can produce differential effects as a result of their unique modes of action (31). The possibility remains that JP and LA disrupt the formation and maintenance of Env-CD4-coreceptor complexes more efficiently than CD and that receptor and coreceptor localization is the sole function of the actin cytoskeleton in HIV-induced fusion. However, the participation of the Rac regulatory pathway and the microscopic analyses support the views that HIV-induced membrane fusion and syncytium formation contain an active component (14, 19, 20) that is related to cellular motility and changes in cellular architecture (29).
The evidence that CCR5 transduces a signal from HIV Env, leading to Rac1 activation, provides a novel mechanism whereby Env regulates host factors critical to the process of membrane fusion. This regulation appears to be a general feature of HIV Env-dependent syncytium formation, and the data from virus-dependent cell fusion assays indicate that a similar mechanism operates during virus-cell fusion. Virus-dependent cell fusion can proceed along two pathways (6). In one scenario, virus binding bridges two cells, allowing each cell to fuse with the shared viral membrane. Alternatively, virus could fuse with a single cell, which would in turn utilize the acquired Env to direct fusion with another cell. While it is likely that both types of fusion occur at some level during virus-dependent cell fusion, we favor the former mechanism as an explanation for the bulk of the cell fusion detected in our assay. First, Env that is restricted in the viral membrane has the potential to be more highly concentrated than that released into the cell membrane following virus-cell fusion. This would favor the occurrence of virus-cell fusion over secondary cell-cell fusions. Second, Env is capable of a single round of fusion (14), and any Env participating in the virus-cell fusion would be useless for subsequent membrane fusion events, further decreasing the effective concentration of Env in the target cell membrane following virus-cell fusion. Regardless, the complete absence of cell fusion in the presence of inhibitors of cytoskeletal function indicates that neither pathway is operational.
Ligation of CCR5 or CXCR4 by HIV gp120 stimulates activities such as Pyk2 phosphorylation (5, 8), which can lead to Rac activation independent of G protein signaling (16). Env-mediated activation of Rac-1 likely occurs through such a pathway. G protein coupling is clearly dispensable during HIV infection and syncytium formation (1, 2, 9). Furthermore, attempts to stimulate G protein-mediated calcium flux through chemokine ligation of CCR5 or CXCR4 expressed in our U87 cell lines have failed, suggesting that these coreceptors are functionally uncoupled from G proteins in the cells (unpublished observations). Efforts are under way to determine the upstream regulators of Rac that participate in HIV-induced cell fusion, as they are potential targets for novel antiviral strategies. Of equal or greater importance is the identification of fusion-specific effectors of Rac to provide information on the fusion regulatory mechanism and additional pathways for therapeutic intervention. Of particular interest is whether Rac activation directly influences the fusogenic potential of lipid bilayers through the activation of effectors such as phospholipase D, facilitating membrane fusion independent of, or in conjunction with, the actin regulatory pathway (18).
The function of actin cytoskeletal rearrangements in HIV-induced syncytium formation remains unclear. One possibility is that protrusive membrane activity allows interacting surfaces to pull together, overcoming electrostatic forces at the virus-cell or cell-cell interface to allow fusion peptide insertion and/or lipid bilayer mixing. The involvement of Rac, and presumably of lamellar membrane structure formation, suggests that the actin cytoskeleton may serve to polarize the fusion process, allowing for the accumulation or restriction of Env-CD4-coreceptor complexes at the interface between fusion partners. A third possibility is that actin polymerization may drive the final steps of lipid mixing, perhaps by producing mechanical strain on the lipid bilayer (10). The evidence that the actin cytoskeleton and Rac activation participate similarly in virus-cell and cell-cell fusion suggests that the mechanism may be a generalized feature underlying membrane fusion events occurring in other systems.
Supplementary Material
Acknowledgments
The authors thank Andrew Pekosz for MVA-T7 and QT6 cells and John Cooper and Daniel Rauch for critical reading of the manuscript.
This work was supported by PHS grants to L.R.
Footnotes
The supplemental material for this article may be found at http://jvi.asm.org/.
REFERENCES
- 1.Alkhatib, G., M. Locati, P. E. Kennedy, P. M. Murphy, and E. A. Berger. 1997. HIV-1 coreceptor activity of CCR5 and its inhibition by chemokines: independence from G protein signaling and importance of coreceptor downmodulation. Virology 234:340-348. [DOI] [PubMed] [Google Scholar]
- 2.Amara, A., A. Vidy, G. Boulla, K. Mollier, J. Garcia-Perez, J. Alcami, C. Blanpain, M. Parmentier, J.-L. Virelizier, P. Charneau, and F. Arenzana-Seisdedos. 2003. G protein-dependent CCR5 signaling is not required for efficient infection of primary T lymphocytes and macrophages by R5 human immunodeficiency virus type 1 isolates. J. Virol. 77:2550-2558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Biscone, M. J., T. C. Pierson, and R. W. Doms. 2002. Opportunities and challenges in targeting HIV entry. Curr. Opin. Pharmacol. 2:529-533. [DOI] [PubMed] [Google Scholar]
- 4.Bubb, M. R., I. Spector, B. B. Beyer, and K. M. Fosen. 2000. Effects of jasplakinolide on the kinetics of actin polymerization. An explanation for certain in vivo observations. J. Biol. Chem. 275:5163-5170. [DOI] [PubMed] [Google Scholar]
- 5.Cicala, C., J. Arthos, M. Ruiz, M. Vaccarezza, A. Rubbert, A. Riva, K. Wildt, O. Cohen, and A. S. Fauci. 1999. Induction of phosphorylation and intracellular association of CC chemokine receptor 5 and focal adhesion kinase in primary human CD4+ T cells by macrophage-tropic HIV envelope. J. Immunol. 163:420-426. [PubMed] [Google Scholar]
- 6.Clavel, F., and P. Charneau. 1994. Fusion from without directed by human immunodeficiency virus particles. J. Virol. 68:1179-1185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cook, J. A., L. Albacker, A. August, and A. J. Henderson. 2003. CD28-dependent HIV-1 transcription is associated with Vav, Rac, and NF-κB activation. J. Biol. Chem. 278:35812-35818. [DOI] [PubMed] [Google Scholar]
- 8.Del Corno, M., Q. H. Liu, D. Schols, E. de Clercq, S. Gessani, B. D. Freedman, and R. G. Collman. 2001. HIV-1 gp120 and chemokine activation of Pyk2 and mitogen-activated protein kinases in primary macrophages mediated by calcium-dependent, pertussis toxin-insensitive chemokine receptor signaling. Blood 98:2909-2916. [DOI] [PubMed] [Google Scholar]
- 9.Doranz, B. J., M. J. Orsini, J. D. Turner, T. L. Hoffman, J. F. Berson, J. A. Hoxie, S. C. Peiper, L. F. Brass, and R. W. Doms. 1999. Identification of CXCR4 domains that support coreceptor and chemokine receptor functions. J. Virol. 73:2752-2761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Eitzen, G. 2003. Actin remodeling to facilitate membrane fusion. Biochim. Biophys. Acta 1641:175-181. [DOI] [PubMed] [Google Scholar]
- 11.Esser, M. T., T. Mori, I. Mondor, Q. J. Sattentau, B. Dey, E. A. Berger, M. R. Boyd, and J. D. Lifson. 1999. Cyanovirin-N binds to gp120 to interfere with CD4-dependent human immunodeficiency virus type 1 virion binding, fusion, and infectivity but does not affect the CD4 binding site on gp120 or soluble CD4-induced conformational changes in gp120. J. Virol. 73:4360-4371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Etienne-Manneville, S., and A. Hall. 2002. Rho GTPases in cell biology. Nature 420:629-635. [DOI] [PubMed] [Google Scholar]
- 13.Feng, Y., C. C. Broder, P. E. Kennedy, and E. A. Berger. 1996. HIV-1 entry cofactor: functional cDNA cloning of a seven-transmembrane, G protein-coupled receptor. Science 272:872-877. [DOI] [PubMed] [Google Scholar]
- 14.Gallo, S. A., C. M. Finnegan, M. Viard, Y. Raviv, A. Dimitrov, S. S. Rawat, A. Puri, S. Durell, and R. Blumenthal. 2003. The HIV Env-mediated fusion reaction. Biochim. Biophys. Acta 1614:36-50. [DOI] [PubMed] [Google Scholar]
- 15.Gallo, S. A., A. Puri, and R. Blumenthal. 2001. HIV-1 gp41 six-helix bundle formation occurs rapidly after the engagement of gp120 by CXCR4 in the HIV-1 Env-mediated fusion process. Biochemistry 40:12231-12236. [DOI] [PubMed] [Google Scholar]
- 16.Gismondi, A., J. Jacobelli, R. Strippoli, F. Mainiero, A. Soriani, L. Cifaldi, M. Piccoli, L. Frati, and A. Santoni. 2003. Proline-rich tyrosine kinase 2 and Rac activation by chemokine and integrin receptors controls NK cell transendothelial migration. J. Immunol. 170:3065-3073. [DOI] [PubMed] [Google Scholar]
- 17.Guerra, S., L. A. Lopez-Fernandez, A. Pascual-Montano, M. Munoz, K. Harshman, and M. Esteban. 2003. Cellular gene expression survey of vaccinia virus infection of human HeLa cells. J. Virol. 77:6493-6506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Humeau, Y., M. R. Popoff, H. Kojima, F. Doussau, and B. Poulain. 2002. Rac GTPase plays an essential role in exocytosis by controlling the fusion competence of release sites. J. Neurosci. 22:7968-7981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Iyengar, S., J. E. Hildreth, and D. H. Schwartz. 1998. Actin-dependent receptor colocalization required for human immunodeficiency virus entry into host cells. J. Virol. 72:5251-5255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jernigan, K. M., R. Blumenthal, and A. Puri. 2000. Varying effects of temperature, Ca2+ and cytochalasin on fusion activity mediated by human immunodeficiency virus type 1 and type 2 glycoproteins. FEBS Lett. 474:246-251. [DOI] [PubMed] [Google Scholar]
- 21.Lu, X., X. Wu, A. Plemenitas, H. Yu, E. T. Sawai, A. Abo, and B. M. Peterlin. 1996. CDC42 and Rac1 are implicated in the activation of the Nef-associated kinase and replication of HIV-1. Curr. Biol. 6:1677-1684. [DOI] [PubMed] [Google Scholar]
- 22.Murakami, T., S. Ablan, E. O. Freed, and Y. Tanaka. 2004. Regulation of human immunodeficiency virus type 1 Env-mediated membrane fusion by viral protease activity. J. Virol. 78:1026-1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nishita, M., H. Aizawa, and K. Mizuno. 2002. Stromal cell-derived factor 1α activates LIM kinase 1 and induces cofilin phosphorylation for T-cell chemotaxis. Mol. Cell. Biol. 22:774-783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nobes, C. D., and A. Hall. 1999. Rho GTPases control polarity, protrusion, and adhesion during cell movement. J. Cell Biol. 144:1235-1244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pontow, S., and L. Ratner. 2001. Evidence for common structural determinants of human immunodeficiency virus type 1 coreceptor activity provided through functional analysis of CCR5/CXCR4 chimeric coreceptors. J. Virol. 75:11503-11514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rossi, D., and A. Zlotnick. 2000. The biology of chemokines and their receptors. Annu. Rev. Immunol. 18:217-242. [DOI] [PubMed] [Google Scholar]
- 27.Salzwedel, K., E. D. Smith, B. Dey, and E. A. Berger. 2000. Sequential CD4-coreceptor interactions in human immunodeficiency virus type 1 Env function: soluble CD4 activates Env for coreceptor-dependent fusion and reveals blocking activities of antibodies against cryptic conserved epitopes on gp120. J. Virol. 74:326-333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Stantchev, T. S., and C. C. Broder. 2001. Human immunodeficiency virus type-1 and chemokines: beyond competition for common cellular receptors. Cytokine Growth Factor Rev. 12:219-243. [DOI] [PubMed] [Google Scholar]
- 29.Sylwester, A., D. Wessels, S. A. Anderson, R. Q. Warren, D. C. Shutt, R. C. Kennedy, and D. R. Soll. 1993. HIV-induced syncytia of a T cell line form single giant pseudopods and are motile. J. Cell Sci. 106:941-953. [DOI] [PubMed] [Google Scholar]
- 30.Viard, M., I. Parolini, M. Sargiacomo, K. Fecchi, C. Ramoni, S. Ablan, F. W. Ruscetti, J. M. Wang, and R. Blumenthal. 2002. Role of cholesterol in human immunodeficiency virus type 1 envelope protein-mediated fusion with host cells. J. Virol. 76:11584-11595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Vicente-Manzanares, M., D. Sancho, M. Yanez-Mo, and F. Sanchez-Madrid. 2002. The leukocyte cytoskeleton in cell migration and immune interactions. Int. Rev. Cytol. 216:233-289. [DOI] [PubMed] [Google Scholar]
- 32.Wang, L., H. Zhang, P. A. Solski, M. J. Hart, C. J. Der, and L. Su. 2000. Modulation of HIV-1 replication by a novel RhoA effector activity. J. Immunol. 164:5369-5374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Westervelt, P., H. E. Gendelman, and L. Ratner. 1991. Identification of a determinant within the human immunodeficiency virus 1 surface envelope glycoprotein critical for productive infection of primary monocytes. Proc. Natl. Acad. Sci.USA 88:3097-3101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wickner, W. 2002. Yeast vacuoles and membrane fusion pathways. EMBO J. 21:1241-1247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wyatt, L. S., B. Moss, and S. Rozenblatt. 1995. Replication-deficient vaccinia virus encoding bacteriophage T7 RNA polymerase for transient gene expression in mammalian cells. Virology 210:202-205. [DOI] [PubMed] [Google Scholar]
- 36.Zhong, B., K. Jiang, D. L. Gilvary, P. K. Epling-Burnette, C. Ritchey, J. Liu, R. J. Jackson, E. Hong-Geller, and S. Wei. 2003. Human neutrophils utilize a Rac/Cdc42-dependent MAPK pathway to direct intracellular granule mobilization toward ingested microbial pathogens. Blood 101:3240-3248. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.