

Abstract
Drug-resistant viruses may be present as minority variants during early treatment failures or following discontinuation of failed antiretroviral regimens. A limitation of the traditional direct PCR population sequencing method is its inability to detect human immunodeficiency virus type 1 (HIV-1) variants present at frequencies lower than 20%. A drug resistance genotyping assay based on the isolation and DNA sequencing of minority HIV protease variants is presented here. A multiple-codon-specific heteroduplex generator probe was constructed to improve the separation of HIV protease genes varying in sequence at 12 codons associated with resistance to protease inhibitors. Using an RNA molecule as probe allowed the simple sequencing of protease variants isolated as RNA/DNA heteroduplexes with different electrophoretic mobilities. The protease gene RNA heteroduplex generator-tracking assay (RNA-HTA) was tested on plasma quasispecies from 21 HIV-1-infected persons in whom one or more protease resistance mutations emerged during therapy or following initiation of salvage regimens. In 11 of 21 cases, RNA-HTA testing of virus from the first episode of virologic failure identified protease resistance mutations not seen by population-based PCR sequencing. In 8 of these 11 cases, all of the low-frequency drug resistance mutations detected exclusively by RNA-HTA during the first episode became detectable by population-based PCR sequencing at the later time point. Distinct sets of protease mutations could be linked on different genomes in patients with high-frequency protease gene lineages. The enhanced detection of minority drug resistance variants using a sequencing-based assay may improve the efficacy of genotype-assisted salvage therapies.
A frequent cause of treatment failure in human immunodeficiency virus type 1 (HIV-1)-infected persons is the emergence of viruses resistant to antiretroviral (ARV) drugs. A number of studies have shown that viral drug resistance genotyping can improve virologic outcome (6, 9, 10, 22, 74). Resistance to ARV drugs can be determined by identifying primary drug resistance mutations known to confer increased resistance to specific ARV drugs and secondary drug resistance mutations that further increase resistance and can improve the replicative fitness of viruses carrying primary drug resistance mutations (25). Recent studies have also indicated that the presence of minority drug-resistant variants may also be an independent predictor of virological failure (37, 40). This may be particularly relevant in persons in whom drug-resistant variants are only beginning to emerge or who have discontinued treatment and whose drug-resistant variants become displaced by preexisting fitter wild-type variants (14, 40).
Sequence-based genotyping can be performed either by direct PCR product sequencing (also called population-based or bulk sequencing) or by sequencing multiple subclones derived from a PCR product. Direct PCR sequencing is primarily used in the clinical setting, but one of its major limitations is its inability to consistently detect minority variants present at frequencies below 10 to 25% (47, 49, 64, 76). The presence of mixed bases in clinical samples is also largely responsible for discordant results when the same samples are analyzed in different laboratories or using different nonsequencing methods (20, 28, 36, 38, 66). The laborious nature of sequencing multiple plasmid subclones, where the major variant may be resequenced multiple times (50), largely restricts this approach to research settings (3, 11, 30, 36, 38, 42, 48, 54, 56, 57, 62).
To increase the sensitivity of current sequencing-based genotyping methods, we developed a method for the separation and sequencing of minority drug-resistant variants. We present here this method and its application, using clinical samples from persons in whom HIV-1 developed new drug resistance mutations while on a failing treatment regimen(s), and we compare the results to direct PCR population sequencing.
MATERIALS AND METHODS
Synthesis of the HIV-1 protease gene universal heteroduplex generator (UHG).
The DNA template used for synthesis of the RNA probe was synthesized by assembling 18 oligonucleotides (each 30 to 48 nucleotides long) into a highly mutated version of the HIV-1 protease gene. Gene assembly was carried out as described elsewhere, with minor modifications (70). A 250 μM concentration of each oligonucleotide was mixed, and the mixture was subsequently diluted 100-fold in 50 μl of a PCR buffer containing 10 mM Tris-HCl (pH 9.0), 50 mM KCl, 2.5 mM MgCl2, 0.1% Triton X-100, a 2.5 mM concentration of each deoxynucleoside triphosphate, 3.5 U of Taq polymerase, and 0.05 U of Pfu polymerase (Promega, Madison, Wis.). The PCR program consisted of 50 cycles of 94°C for 30 s, 50°C for 30 s, and 72°C for 30 s. The oligonucleotides used were the following (5′ to 3′): PF1, GAAGCAGGAGCCGATAGACAAGGAACTGTATCCTTTAACT; PF2, TCCCTCAGATCACTCTTTGGCAACGACCGCTCGTCACAAT; PF3, AAAGATAGGGGGGCAACTAAAGGAAGCTCTATTAGATACA; PF4, GGAGCAGATCGATACTGTATTAGAACAAATGAATTTGCC; PF5, AGGAAGATGGAAACCAAAAAAGATAGGCGGGAAATGGA; PF6, GGTTTTAATCAAAGTAAGACAGTATGATCAGATACTCATA; PF7, GAAATCTGTGGACATAAAGCATTAGGTACAGTATTAGTAG; PF8, GACCTACACCTGATCAACAATAATTGGAAGTAATCTGTCTGACTC; PF9, AGATTGGTTGCACTTTAAATTTTCCCATTAGCCCTATTGAGACTGTACCAG; PR1, CTGGTACAGTCTCAATAGGGCTAATGGGA; PR2, AAATTTAAAGTGCAACCAATCTGAGTCAGACAGATTACTTCCAA; PR3, TTATTGTTGATCAGGTGTAGGTCCTACTAATACTGTACCTAATGCTTTATG; PR4, TCCACAGATTTCTATGAGTATCTGATCATACT; PR5, GTCTTACTTTGATTAAAACCTCCATTTCCCGCCTATCTTTTT; PR6, TGGTTTCCATCTTCCTGGCAAATTCATTTCTTCTAATACA; PR7, GTATCGATCTGCTCCTGTATCTAATAGAGCTTCCTTTAG; PR8, TTGCCCCCCTATCTTTATTGTGACGAGCGGTCGTTG; and PR9, CCAAAGAGTGATCTGAGGGAAGTTAAAGGATACAGTTCCTTGTCTATCGGCTCCTGCTTC. After the initial gene assembly PCR, the reaction mixture was diluted 40-fold in 100 μl of the same PCR buffer, with deoxynucleoside triphosphates plus 10 pmol of each flanking primer: EDPR3, GAAGCAGGAGCCGATAGACAAGG (HXB2 positions 2211 to 2233); EDPR4, CTGGTACAGTTTCAATAGGACTAATGG (HXB2 positions 2551 to 2577). The second PCR program consisted of three cycles of 94°C for 45 s, 57°C for 45 s, and 72°C for 45 s, followed by 34 cycles of 94°C for 30 s, 57°C for 30 s, and 72°C for 30 s, and final extension at 72°C for 5 min. A 100-μl aliquot of the PCR product was run in a 1.5% agarose gel, and the 360-bp band was purified (QIAQuick gel extraction kit; QIAGEN Inc.). The mutated protease gene DNA fragment was subcloned into the pGEM-T plasmid vector (Promega), and 10 subclones were sequenced using M13 reverse primer. One subclone with the expected sequence was used in all subsequent experiments. The resulting plasmid (pAK1-pro1UHG), necessary for generating the protease RNA probe, is available upon request.
RNA probe synthesis.
The mutated protease gene insert of pAK1-pro1UHG was amplified from 10 ng of plasmid as above but for only 20 PCR cycles with primers EDPR3 and T7-EDPR4 (T7 RNA polymerase promoter region [TAATACGACTCACTATAGGG] added to the 5′ end of primer EDPR4). A 100-μl aliquot of the PCR product was run in a 2% agarose gel, and the 370-bp band was purified from the gel. One microliter of the purified PCR product was added to 100 μl of transcription mixture containing 20 μl of 5× transcription buffer, 10 μl of 0.1 M dithiothreitol, 20 μl of 2.5 mM (each) ribonucleoside triphosphate, 100 U of human placental RNase inhibitor, and 45 U of T7 RNA polymerase. The mixture was incubated for in vitro transcription at 37°C for 90 min, followed by 70°C for 15 min to inactivate the enzyme.
Virus isolates.
The plasma samples of persons undergoing direct PCR population sequencing at Stanford University Hospital Diagnostic Virology Laboratory meeting the following criteria were chosen for further evaluation: (i) two genotypes were performed within the course of 1 year, each following virologic failure on a protease inhibitor-containing regimen; and (ii) the second genotype contained at least one major protease inhibitor resistance mutation that was not observed in the direct PCR-based sequence at the time of the first virologic failure. Further analyses were performed on the first virologic failure sample of all patients meeting this criteria and on the second virologic failure sample if that sample was also available. The study was approved by the Stanford University Hospital and University of California San Francisco human subjects committees. All testing was performed anonymously.
Viral RNA isolation and reverse transcription-nPCR.
Viral RNA was extracted from plasma and reverse transcribed as previously described (21). PCR primers used for the first round of nested PCR protease gene amplification were EDPR1 (GAGCAGACCAGAGCCAACAGCCCCA [HXB2 positions 2139 to 2163]) and EDPR2 (TTGTTTAACTTTTGGGCCATCC [HXB2 positions 2597 to 2618]). Second-round primers were EDPR3 and EDPR4. Each of the nested PCR (nPCR) programs consisted of three cycles of 94°C for 45 s, 57°C for 45 s, and 72°C for 45 s, followed by 27 cycles of 94°C for 30 s, 57°C for 30 s, and 72°C for 30 s, and final extension at 72°C for 5 min. Using pNL4-3 dilutions, the sensitivity of this nPCR was determined to be between 1 and 10 copies (21).
RNA/DNA heteroduplex formation and gel electrophoresis.
For hybridization reactions, 2 μl of the UHG-pro1 RNA probe was mixed with 3 μl of patients' nPCR products and 0.5 μl of 10× heteroduplex annealing buffer (100 mM NaCl, 10 mM Tris-HCl [pH 7.5], and 2 mM EDTA), incubated at 95°C for 2 min, and rapidly transferred to ice for hybridization at 4°C. The RNA/DNA heteroduplexes were then separated in a 7.5% gel (29:1 acrylamide-bis solution) with 1.25× Tris-acetate-EDTA electrophoresis buffer at 250 V in a temporal temperature gradient of 26 to 45°C over 6 h (ramp time of 3.5°C increase per h) using the Decode Universal mutation detection system electrophoretic gel apparatus (Bio-Rad Laboratories). Temporal temperature gradient electrophoresis exploits the principle of sequence-dependent differences in DNA melting and slightly improves the separation of protease sequence variants.
Isolation of RNA/DNA heteroduplexes and DNA sequencing.
After electrophoresis, polyacrylamide gels were stained with ethidium bromide (0.5 μg/ml) and placed on a UV illuminator. Following photographic documentation using a charged-coupled device camera, five small gel pieces of approximately 10 μl were taken from each gel lane by using a 1,000-μl PCR pipette tip whose opening was slightly widened by cutting it with a razor blade. In RNA-HTA gels where multiple distinct RNA/DNA heteroduplex bands were visible, small gel pieces were taken from all bands. In patient samples where only one broad RNA/DNA heteroduplex band was visible, one gel piece below the center of the fluorescent signal, one from the center of the signal, and three immediately above it were taken. A distance of approximately 3 mm was maintained between sampled gel pieces. The gel pieces were ejected into a 50-μl PCR mix with second-round primers EDPR3 and EDPR4 for reamplification of the protease gene. An initial melting step of 5 min at 95°C was added to the second-round primer PCR amplification cycles. Since the RNA probe is not PCR amplified from the RNA/DNA heteroduplex, only the annealed DNA variant strand was reamplified. PCR products were then purified and subjected to dideoxy cycle sequencing using 10 pmol of EDPR3 primer and an ABI Prism 3700 capillary sequencer with ABI Prism BIG-DYE terminators.
Measurement of variant frequencies.
The percentage of mutated base at a nucleotide position in any single sequencing electropherogram was estimated using the formula [MPH/(MPH + WPH)] × 100 (where MPH and WPH are the sequencing electropherogram peak heights of mutant and wild-type bases at the same position). The program used for the analysis of multiple aligned sequence electropherograms derived from the same sample was SeqMan from DNASTAR version 5.0.
RESULTS
HTA with an RNA probe based on the UHG concept.
To improve heteroduplex tracking analysis (HTA) so that electrophoretically separated sequence variants differing by only one or a few nucleotide substitutions could be distinguished and then purified for sequencing, we designed an HTA probe based on the UHG concept (60). To be able to sequence amplified protease genes and avoid sequencing the HTA probe present in a DNA heteroduplex, we changed the probe from DNA to a single-stranded RNA molecule. The use of a UHG-based RNA probe resulted in RNA/DNA heteroduplexes exhibiting greatly reduced electrophoretic mobilities with no overlap with the much-faster DNA/DNA heteroduplexes formed between the quasispecies variants themselves. The DNA/DNA heteroduplexes contained only a few nucleotide mismatches and comigrated with the DNA homoduplexes (data not shown). The use of a single-stranded RNA probe also allowed the extraction of RNA/DNA heteroduplexes from gels and their direct sequencing, as only the DNA strand of the RNA/DNA heteroduplexes was reamplified following PCR. The assay was named RNA-HTA for RNA UHG tracking assay.
Design of the UHG-pro1 sequence.
UHGs are DNA molecules designed to increase the electrophoretic separation of sequence variants with which they form DNA heteroduplexes (7, 12, 60, 63, 79, 80, 82, 83). The protease UHG probe used here (pro1) was based on the HIV-1 subtype B protease gene consensus sequence and was constructed using a series of 18 overlapping oligomers (see Materials and Methods). The pro1 RNA-HTA probe was designed based on the multiple-site-specific protease UHG approach described by Resch et al. (60) with the following modifications. The pro1 probe was made of RNA, it included all 99 codons of the HIV protease, and its mutation-detecting features consisted of nucleotide insertions as well as substitutions. Single-nucleotide insertions were introduced into or immediately flanking six targeted primary drug resistance codons (protease amino acids 30, 48, 54, 82, 84, and 90), while single- or double-nucleotide substitutions were introduced into or immediately flanking six targeted accessory drug resistance codons (protease amino acids 10, 32, 46, 50, 71, and 88) (Fig. 1A). The pro1 probe was therefore designed to improve electrophoretic separation of coamplified variants within PCR products that differed at 1 or more of the 12 codons where mutations are most often selected by protease inhibitor treatment (46).
FIG. 1.
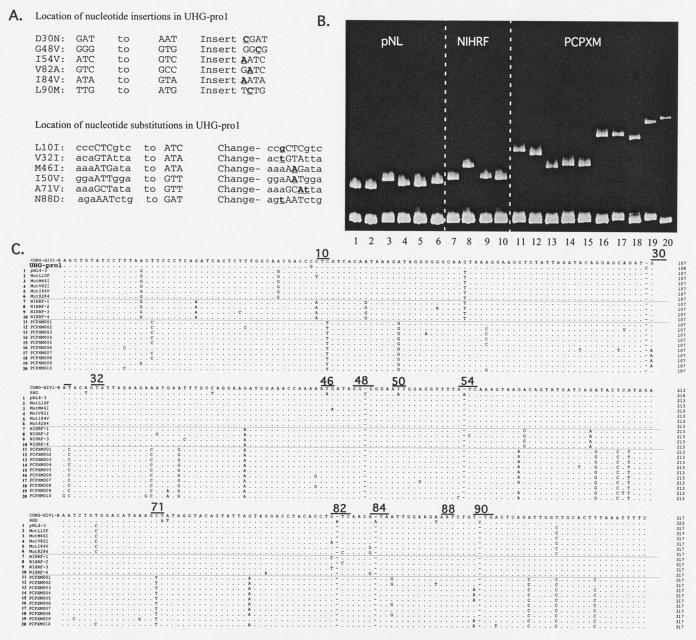
RNA-HTA pro1 probe design and its use with protease variants. (A) Mutations engineered in the protease subtype B consensus sequence. Targeted codons are shown in capital letters. Six primary and six accessory drug resistance mutations were targeted by nucleotide insertion and substitutions, respectively (underlined). (B) RNA-HTA using protease variants from three different isolates (pNL, NIHRF, and PCPXM variants). The protease variants used in each lane correspond to the sequence numbers in panel C. The gel is shown from the loading wells to the DNA homoduplexes. (C) Alignment of the subtype B consensus, UHG-pro1 probe, and protease variants of pNL, NIHRF, and PCPXM used in panel B. Drug resistance codons targeted by the UHG-pro1 probe are underlined.
When RNA/DNA heteroduplexes are generated with patients' protease variants, the six extra inserted nucleotides in the RNA strand are accommodated by looping them out of the heteroduplex, causing a highly kinked double-stranded structure whose electrophoretic mobility through polyacrylamide is greatly retarded (Fig. 1B) (39). The exact structure of the heteroduplex molecule at each of the nucleotide insertion sites is highly dependent on the regional nucleotide sequence. Different sequences in the six codons targeted by insertions therefore result in different heteroduplex structures, which in turn lead to different electrophoretic mobilities. For example, a wild-type codon at position 30 will form a structure with an unmatched cytosine looping out of the RNA/DNA heteroduplex. A D30N mutant will form a structure that will also include a flanking mismatched nucleotide base pair.
Nucleotide substitutions were also introduced into the consensus sequence of the RNA probe such that the presence of an accessory drug resistance mutation would increase by 1 bp an already-present region of heteroduplex melting. For example, a wild-type protease variant annealed to the pro1 RNA probe would have a single mismatched base pair at codon 10, while the L10I mutant will increase the region of mismatch to two neighboring base pairs. Such mismatched nucleotides result in heteroduplex “bubbles” (39) that also reduce electrophoretic mobility through polyacrylamide.
RNA-HTA using cloned sequence variants from the same patients.
To confirm the ability of RNA-HTA to separate variants with point mutations, we reannealed the pro1 RNA probe to PCR products derived from sequenced plasmid subclones and fractionated the RNA/DNA heteroduplexes by polyacrylamide gel electrophoresis (Fig. 1B). Collectively distorting the structure of the RNA/DNA heteroduplexes and slowing their electrophoretic mobilities were, therefore, (i) unmatched (i.e., inserted) nucleotides introduced in the six targeted primary resistance codons, (ii) mismatched base pairs resulting from the substitutions introduced at the six targeted accessory resistance codons, and (iii) other mismatched base pairs due to differences between the viral strain being analyzed and the subtype B consensus. Lanes 1 to 6 show the mobility of variants of pNL4-3 (the pNL4-3 protease differs from the subtype B consensus by five nucleotides). Lanes 7 to 10 show the mobility of variants of RF (the RF protease consensus differs from the subtype B consensus by seven mutations with a range 8 to 12 differences). Lanes 11 to 20 show the mobility of variants from patient PCPXM (PCPXM consensus protease differs from the subtype B consensus by 15 mutations with a range 17 to 24 differences). As expected from prior studies of DNA/DNA heteroduplexes (18, 19, 21), mobility retardation generally increased with greater divergence between the reannealed target DNA and RNA probe strands. The higher level of divergence between PCPXM and the subtype consensus is more typical of currently circulating HIV-1 strains than the lower level of divergence seen with the pNL4-3 and RF strains, both collected early in the subtype B epidemic (26, 32, 51).
Different pNL4-3-derived mutants showed minor but reproducible mobility differences, except for V82I and I84V, whose mobilities were undistinguishable, and L10F, whose mobility was identical to that of pNL4-3. Among RF-derived mutants, the mobility of variants NIHRF-1, -3, and -4 were also only slightly different. These three RF variants' sequence differences were a single base pair difference in either primary resistance codon 82 or 84 and three to five substitutions in nontargeted (i.e., non- drug resistance-associated) codons. The highly distinct mobility of NIHRF-2 was associated with its unique mutation in targeted primary resistance codon 54 (Fig. 1B and C).
A set of patient-derived variants (PCPXM) was also compared to further analyze the effect of sequence differences at targeted versus nontargeted codons. The PCPXM clones were divided into five groups according to their differences in targeted primary drug resistance codons (amino acids 30, 54, and 90) (Fig. 1C). Variants within each group (PCPXM01 and PCPXMO2, D30, I54, and L90; PCPXMO3, PCPXMO4, and PCPXMO5, D30, I54, and L90M; PCPXMO6, PCPXMO7, and PCPXMO8, D30N, I54, and L90; PCPXMO9, D30N, I54, and L90M; PXM10, D30, I54L, and L90M) displayed electrophoretic mobilities that were more similar than those of variants from the other groups (Fig. 1B). Electrophoretic mobility differences were also observed within the five PCPXM groups. Group 1 variants (PCPXMO1 and PCPXMO2) differed by one substitution in codon 87 immediately upstream of a targeted accessory codon (at position 88) and at four nontargeted sites. Group 2 variants with different mobilities (PCPXMO3, PCPXMO4, and PCPXMO5) differed by single nucleotide differences in nontargeted sites. Group 3 variants PCPXMO6, PCPXMO7, and PCPXMO8 differed by four to seven nucleotides in nontargeted sites.
We conclude that differences in primary drug resistance-targeted codons result in a stronger effect on mobility than mutations in nontargeted regions of the protease gene. Nonetheless, some targeted mutations (e.g., L10F in the pNL4-3 background) did not result in noticeable mobility changes, and mutations in nontargeted regions also influenced the mobility of RNA/DNA heteroduplexes (e.g., PCPXM variants with identical targeted codon sequences).
Isolation and sequencing of HIV protease drug-resistant variants present at low frequencies.
The sensitivity of RNA-HTA for the detection of minority drug-resistant protease variants was measured using mixtures of two clones with different primary drug resistance mutations. The PCR products were mixed such that one variant ranged in frequency from 50 to 0.1% of the PCR product. Following RNA-HTA, the presence of the minority variant was readily detected by ethidium bromide staining when it was present at a frequency of ≥5% (Fig. 2A). Using a pipette tip, small pieces of the gel were taken over the location of the minority RNA/DNA heteroduplex (Fig. 2A). The DNA strands of the RNA/DNA heteroduplexes were then reamplified by PCR (see Materials and Methods). The PCR products were then hybridized with the RNA probe and analyzed in another RNA-HTA (Fig. 2B). The resulting RNA-HTA mobility indicated which of the two variants had been amplified from the gel and showed that the minority variant was readily purified even when originally present at a frequency of ≥0.5%. At the 0.5% concentration, a 50:50 mixture of both the minority and majority variant was amplified by RNA-HTA, a likely consequence of a fraction of the faster RNA/DNA heteroduplex trailing (streaking) over the position of the slower-mobility minority variant. At a 1% frequency, the purified minority variant was amplified pure (Fig. 2B).
FIG. 2.
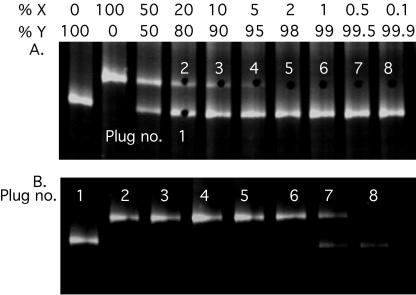
Detection of low-frequency variants using RNA-HTA. (A) Mixtures of two protease variant PCRs (PCPXM002 and PCPXM003) were separated by RNA-HTA, and gel pieces (plugs) were taken from the gel. Plug holes are visible in the gel. (B) RNA-HTA of the PCR product generated from the gel plugs.
Persons receiving protease inhibitors.
The treatment histories and plasma viremia levels of 21 persons in whom new primary protease inhibitor resistance mutations emerged in between two episodes of virologic failure are shown in Fig. 3. The plasma samples at the two time points shown by vertical lines are referred to here as the baseline and the follow-up samples. The mean interval between the paired plasma samples tested was 31.4 weeks (4 to 51 weeks). Thirteen persons were receiving their first protease inhibitor at the time of the baseline sample; eight persons had received two or more protease inhibitors before the baseline sample. Fourteen persons were treated with a new protease inhibitor between the baseline and follow-up samples; seven persons continued receiving their initial protease inhibitor.
FIG. 3.
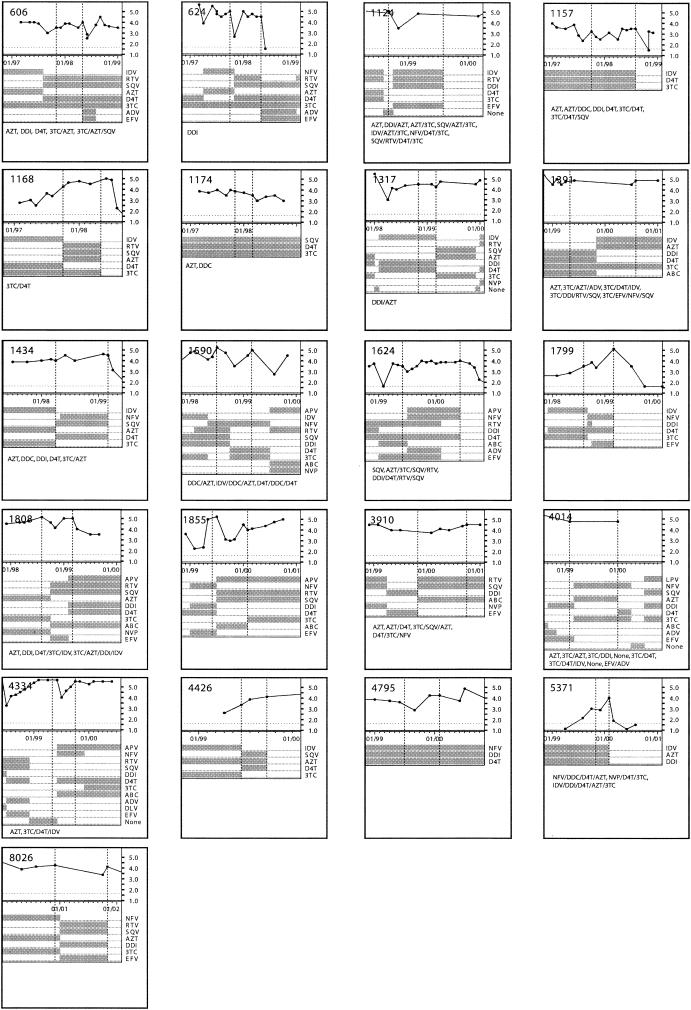
Viral load and ARV drug history of 21 patients analyzed by RNA-HTA. The baseline and follow-up time points are shown with vertical lines. ARV drug history is presented in each box.
Fractionation of protease variants within patients' quasispecies.
The protease locus was amplified by reverse transcription-nPCR from the baseline and when available from the follow-up plasma samples and directly sequenced (see Materials and Methods) (Table 1). The drug resistance genotypes derived by direct PCR population sequencing were identical to those originally determined at Stanford University within the range of interlaboratory reproducibility of drug resistance genotyping (<1% difference for all base calls, with 90% of these difference being due to mixed base differences) (20, 34, 66). The same PCR amplicons were then analyzed by RNA-HTA (Fig. 4). When both the baseline and follow-up plasma sample from the same subject were available, different RNA-HTA mobility variants were seen at both time points (Fig. 4A). Such mobility differences were expected following the acquisition of at least one primary drug resistance mutation detected by direct PCR population sequencing (Table 1). RNA-HTA from patients for which only the baseline plasma sample was available for analysis are also shown (Fig. 4B). In contrast to the sharp bands produced by clonal PCR products, the PCR products of these clinical samples generated RNA-HTA patterns consisting of one major band flanked by a diffuse smear of DNA or two or more distinct RNA-HTA bands (Fig. 4A and B).
TABLE 1.
Comparison of protease gene drug resistance genotypes obtained for baseline and follow-up samples by using direct PCR and RNA-HTA enhanced sequencinga
| Patient | Baseline genotype from direct PCR (population) sequencing | Baseline RNA-HTA genotype | Follow-up genotype from direct PCR (population) sequencing | Interval (wks) |
|---|---|---|---|---|
| 606 | L10I, A71VAIT, L90M | L10I, A71VAIT, L90M | L10I, M46IM, A71TA, 184V, L90M | 27 |
| K20MI, N37DN, I62V, L63P, K70R, I72LI, G73S | K20MI, N37DN, I62V, L63P, K70R, I72TI, G73S | K20I, D29VD, N37D, R41KR, I62V, L63P, K70R, I72TI, G73S | ||
| 624 | D30N, N88D | D30N, N88D | D30N, I54V, A71TA, N88D, L90M | 29 |
| I13V, E35D, M36I, L63P | I13V, E35D, M36I, L63P | I13V, K20R, L33IL, E35D, M36I, L63P | ||
| 1124 | L10I | L101, I54VI, V82AV | L101, I54V, A71V, V82A, L90M | 46 |
| I62V, L63P, V77I, 193L | D60ED, Q61EQ, I62V, L63P, V77I, I93L | K43T, D60E, Q61E, I62V, L63P, V77I, I93L | ||
| 1157 | L101, M46L, A71V, L90M | L101, M46LI, A71V, I84VI, L90M | L101, M461, A71V, I84V, L90M | 43 |
| I13V, K14R, N37D, L63P, G73S, V77I, I85V | I13V, K14R, N37D, L63P, G73S, V77I, I85V | I13V, K14R, N37D, L63P, I72LI, G73S, V77I, 185V | ||
| 1168 | L90M | V82AV, L90M | I84VI, L90M | 30 |
| N37H, R41K, I64V, G73CG, V77I | N37H, R41K, I64V, G73CG, V77I | N37H, R41K, I64V, H69RH, G73CG, V77I | ||
| 1174 | A71VA, L90ML | A71VA, L90ML | 16 | |
| L63PL, V77I, I93L | L63PL, V77I, I93L | L63PL, V77I, I93L | ||
| 1317 | N88S | N88S | A71X, N88S, L90M | 21 |
| I15V, K20T, E35D, M36I, R57K, L63P, I93LI | I15V, K20T, E35D, M36I, R57K, L63P, I93LI | I15V, K20T, E35D, M36I, R41KR, R57K, I62V, L63P, C67YC | ||
| 1391 | I84VI, L90ML | A71V, I84V, L90M | 45 | |
| R57K, L63P | R57K, L63P | R57K, L63P | ||
| 1434 | V32I, M46IM, V82A | V32IV, M46IM, I54VI, A71VA, V82A | L10IL, M46L, I54VI, A71V, V82A, L90ML | 46 |
| T12S, N37S, R41K, I47VI, L63P, I93L | T12S, N37S, R41K, I47VI, L63P, I93L | T12S, N37S, R41K, L63P, T74ST, I93L | ||
| 1590 | L10I, I54V, A71V, L90M | L10I, I54V, A71V, V82AV, L90M | L10I, I54V, A71V, V82AV, L90M | 34 |
| T12E, L19IL, L63P, G73S, V77I, I93L | T12E, L19IL, L63P, G73SG, V77IV, I93L | T12E, L19I, L63P, G73SG, I93L | ||
| 1624 | I54V, A71V | I54VI, A71V, I84VI | I54LI, A71V, I84VI | 51 |
| I15V, L24I, L33F, E34Q, M36L, N37SN, L38W, P39SP, F53L, R57K, D60E, I62V, L63P G73C, N83D | I15V, L24I, L33F, E34Q, M36L, N37SN, L38W, P39SP, F53L, R57K, D60E, I62V, L63P G73C, N83D | I15V, A22VA, L24I, L33F, E34Q, M36L, L38W, P39S, F53L, R57K, D60E, I62V, L63P, G73C, N83D | ||
| 1799 | V82AV | V32I, M46I, A71V, V82A | 27 | |
| K14R, R41K, L63P | K14R, R41K, L63P | K14R, R41K, L63P | ||
| 1808 | L10I, M46I, L90M | L10I, M46I, L90M | L10I, M46I, I84V, L90M | 28 |
| I15V, K20I, M36I, R41K, I62V, L63P, I72L, G73S, I93L | I15V, K20I, M36I, R41K, I62V, L63P, I72LI, G73S, I93L | I15V, K20I, M36I, R41K, I62V, L63P, I72L, G73S, I93L | ||
| 1855 | L10I, I54V, A71V, V82A | L10I, I54V, A71V, V82A | L10I, M46L, I54V, A71V, V82A, L90ML | 33 |
| I62V, L63P, G73S, V77I, I93L | I62V, L63P, G73S, V77I, I93L | I62V, L63P, G73S, V77I, I93L | ||
| 3910 | L10I, M46L, I54V, A71V, V82A, I84V, L90M | 50 | ||
| P39A, L63P | P39A, L63P | L23I, P39A, D60ED, I62VI, L63P, V77I, | ||
| 4014 | L10FL, M46IM, I54VI, V82AV, L90ML | M46I, I54V, V82A | 50 | |
| I64V | I64VI | R41SR, I64V | ||
| 4334 | A71T | L10IL, I54VI, A71IT, V82AV, I84VI, L90ML | L10IL, I54V, A71IT, I84V, L90ML | 24 |
| E35D, N37D, L63P, I66MI | E35D, M36IM, N37D, L63P, I66MI | L19IL, K20KIM, L33IL, E35D, M36IM, N37D, L63P, Q92KQ | ||
| 4426 | L10I, M46LM, I54AV, A7IV, V82A | L10I, M46LM, I54AV, A71V, V82A | L10I, I54V, A71V, V82A, L90M | 15 |
| L19T, N37S, F53LF, L63PL | L19T, N37S, F53LF, L63PL | L19T, N37S, F53LF, L63P | ||
| 4795 | D30N, N88D | 30 | ||
| L63PS | L63PS | L63P | ||
| 5371 | L10FL | L10LF | V82VAIT | 13 |
| I15MI, K20R, L24FL, L63P, I93L | I15MI, K20R, L24FL, L63P, I93L | K20R, L63P, I93L | ||
| 8026 | L10V, A71V, L90M | L10V, A71V, L90M | L10X, A71V, I84V, L90M | 49 |
| K20I, E35D, M36I, N37S, R41K, D60E, I62V, L63P, I64V, I93L | K20I, E35D, M36I, N37S, R41K, D60E, I62V, L63P, I64V, I93L | K20IR, E35D, M36I, N37S, R41K, D60E, I62V, L63P, I64V, P79A, I93L |
The first line of each genotype lists the detected drug resistance mutations targeted by the RNA-HTA probe. The second and third lines list all mutations from the subtype B consensus. Targeted drug resistance mutations in 11 patients that were not detected by direct PCR population sequencing in the baseline sample are underlined.
FIG. 4.
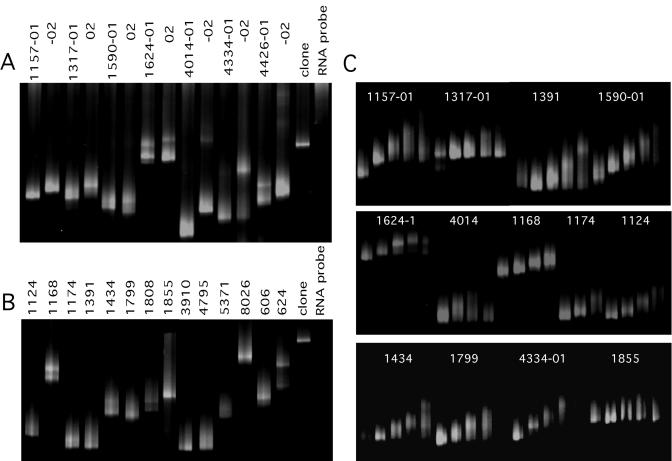
RNA-HTA of clinical samples. Patient identifications are similar to those in Fig. 3. (A) RNA-HTA of samples collected at baseline and follow-up are labeled −01 and −02, respectively. (B) RNA-HTA of samples from patient baseline only. (C) RNA-HTA analysis of the PCR products derived from RNA-HTA gel pieces. A clonal PCR target (PCPXM010) and RNA probe-alone lanes are included.
Protease gene variants with different RNA-HTA mobilities were isolated by taking five small gel plugs along the length of each RNA-HTA lane (see Materials and Methods) (Fig. 2A). The DNA strands of the RNA/DNA heteroduplexes in the gel plugs were amplified by PCR. For a few representative patients, the multiple PCR products derived from gel pieces were again reannealed with the pro1 RNA probe and separated using RNA-HTA (Fig. 4C). Some series of PCR products from the same gel lanes showed little or no RNA-HTA mobility differences (Fig. 4C, 1317 and 1855), while others showed clear mobility differences (Fig. 4C, 1157-01 and 1391). This result indicated that sequence variants within the same quasispecies could be isolated from different locations of an RNA-HTA gel lane even when it consisted of a single diffuse band.
Sequence analysis of RNA-HTA-isolated protease variants.
The five PCR products derived from each of the RNA-HTA lanes of the 21 baseline plasma samples were then directly sequenced. The sequencing electropherograms of each of the five RNA-HTA gel pieces-derived PCR products and the electropherogram of the original, unfractionated PCR product were aligned. Both complete and partial (i.e., mixed) nucleotide base differences (see Materials and Methods) were then identified. The presence of a drug resistance (or subtype B consensus-deviating) mutation was defined as its detection at a level of ≥25% in one or more of the five electropherograms obtained per sample (see Materials and Methods). Examples of differences seen between the direct (population) and the RNA-HTA-assisted sequencing electropherograms, showing the detection of low-frequency drug-resistant variants, are shown in Fig. 5. Newly detected drug resistance mutations were often seen in more than one of the RNA-HTA plugs (Fig. 5). Minority drug resistance and other mutations relative to subtype B consensus were typically seen in the PCR from the RNA-HTA gel pieces with the slowest mobilities (Fig. 5). The extra RNA/DNA heteroduplex structure distortions (and mobility retardation) induced by sequence differences between the subtype B consensus-based probe and annealed protease sequence variants likely account for this observation.
FIG. 5.
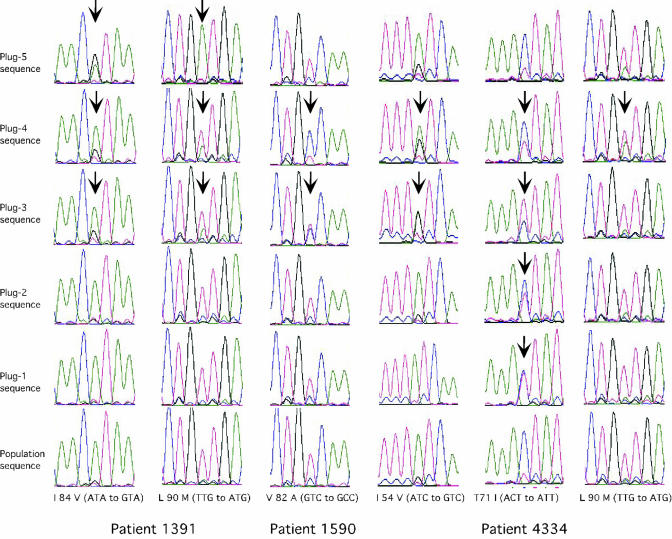
Comparison of direct PCR population sequencing and RNA-HTA-enhanced sequencing. Representative electropherograms show the detection of drug resistance mutations (arrows) in PCR products derived from RNA-HTA-derived gel plugs, while such mutations were not detected in the directly sequenced PCR product (population sequence).
Mixed base calls were often detected in the RNA-HTA gel piece-derived electropherograms. Four possible mechanisms may account for such mixed, rather than pure, nucleotide base calls. The size of the gel piece used and RNA/DNA heteroduplex gel streaking from electrophoretically faster bands (Fig. 2B, lane 7) may result in coamplification of distinct variants in the same gel piece. Two variants differing at a particular base may also comigrate on RNA-HTA due to the inability of the RNA probe and gel condition used here to cause sufficient mobility differences (Fig. 1B). Lastly, other protease gene differences may result in incidental comigration of different variants.
For each baseline plasma sample, the drug resistance mutations (and other mutations resulting in non-subtype B consensus amino acids) from each of the five RNA-HTA gel piece-derived electropherograms were assembled into a single composite RNA-HTA genotype (Table 1). When compared with the population sequencing-based genotype of the same plasma sample, previously undetected low-frequency drug resistance mutations were detected in 11 of 21 persons (Table 1). In 8 of these 11 persons, every drug resistance mutation detected at baseline using RNA-HTA was detected at the follow-up bleed (16 to 51 weeks later) using traditional direct PCR population sequencing (Table 1). This result indicates that emerging drug-resistant variants could be identified earlier using RNA-HTA than by direct population sequencing. In 4 of 11 persons in whom primary drug resistance mutations were detected only using RNA-HTA, the baseline samples tested were from persons who had temporarily discontinued either all protease inhibitors (persons 1391 and 4014) or all ARV drugs (patients 1124 and 4334) in their regimens (Fig. 3).
Absence of minority drug-resistant variants in drug-naive subjects.
Using RNA-HTA, we analyzed the plasma quasispecies from nine HIV-infected persons with no previous history of ARV therapy that were identified during testing of voluntary blood donations. No primary drug resistance mutations were detected as either majority or minority variants. Only common polymorphisms (K20R, L63P, and A71V), which can also serve as accessory drug-resistant mutations, were detected as majority variants by both direct PCR population and RNA-HTA sequencing (data not shown).
Linkage between different drug resistance mutations.
Direct PCR population sequencing provides a quasispecies consensus sequence of all the coamplified HIV variants. When multiple drug resistance mutations are detected as mixed base peaks in direct PCR population sequencing, it is not possible to determine if they are present on the same or different genomes. Samples from seven persons whose plasma quasispecies generated distinct RNA-HTA bands were further analyzed (Fig. 6A). The distinct RNA-HTA bands were first quantified by fluorescence gel scanning to determine the relative frequencies of these protease gene variants in plasma (Table 2). RNA-HTA bands were then isolated from gels, reamplified by PCR, and sequenced. The different RNA-HTA band-derived protease variants from the same patients all showed one or more base differences at one or more of the 12 targeted drug resistance codons as well as at other amino acid positions (Table 2). In sample PXM01, primary protease inhibitor-resistant mutations D30N and L90M appeared to be present on different genomes, in keeping with a report that these two mutations may evolve along independent pathways (72), while in sample 624-2 D30N and L90M mutations were both present on each of the three major linkage groups. Mixed bases were also occasionally detected when distinct RNA-HTA bands were isolated and sequenced, indicating that different variants may comigrate (for example, PXM01 linkage group II K20TK and D30ND [Table 2]). The presence of mixed bases in a distinct RNA-HTA band may therefore still require analysis of plasmid subclones to establish linkage, but the process can be made more efficient using RNA-HTA.
FIG. 6.
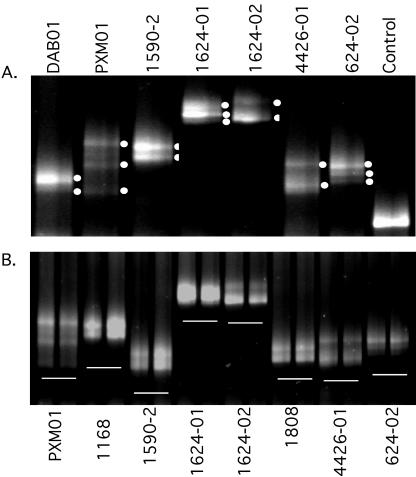
Mutation linkage analysis and quasispecies sampling reproducibility. (A) Analysis of samples with distinct RNA-HTA bands. RNA/DNA heteroduplex bands labeled by a white dot were separately purified and sequenced. (B) Independently generated PCR products were analyzed by RNA-HTA in neighboring gel lanes to test for reproducible viral population sampling.
TABLE 2.
Linkage analysis of protease mutations on different genomes
| Patient | Linkage groupsa | % Variants |
|---|---|---|
| DAB01 | I. I15V, M36I, N37E, L63P, I64V, T74S, L90M | 32 |
| II. I15V, M36I, N37E, L63P, A71T, N88D, L90M | 68 | |
| PXM01 | I. I15V, K20T, E35D, N37S, R41K, R57K, I62V, L63P, I64V, A71V, V77I, L90M, I93L | 17 |
| II. I15V, K20TK, D30ND, E35D, N37S, R41K, R57K, I62V, L63P, I64V, A71V, V77I, I93L | 21 | |
| III. I15V, D30N, E35D, N37S, R41K, R57K, I62V, L63P, I64V, A71V, V77I, I85VI, I93L | 44 | |
| 1590-2 | I. L10I, T12E, L19I, I54V, L63P, A71V, V82A, L90M, I93L | 43 |
| II. L10I, T12E, L19I, I54V, L63P, A71V, G73S, V77I, L90M, I93L | 57 | |
| 1624-1 | I. V11I, I15V, L24I, L33F, E34Q, M36L, L38W, P39S, F53L, R57K, D60E, I62V, L63P, A71V, G73CS, N83D, I84VI | 12 |
| II. I15V, L24I, L33F, E34Q, M36L, L38W, P39S, F53L, I54V, R57K, D60E, I62V, L63P, A71V, G73C, N83D | 52 | |
| III. I15V, L24I, L33F, E34Q, M36L, N37S, L38W, F53L, I54V, R57K, D60E, I62V, L63P, A71V, G73C, N83D | 34 | |
| 1624-2 | I. I15V, A22V, L24I, L33F, E34Q, M36L, L38W, P39S, F53L, R57K, D60E, I62V, L63P, A71V, G73C, N83D, I84V | 70 |
| II. I15V, L24I, L33F, E34Q, M36L, N37SN, L38W, P39S, F53L, I54L, R57K, D60E, I62V, L63P, A71V, G73C, N83D | 30 | |
| 4426-1 | I. L10I, L19T, N37S, M46LM, F53LF, I54VI, A71V, V82A | 61 |
| II. L10I, L19T, N37S, I54AV, L63P, A71V, V82A | 39 | |
| 624-2 | I. I13V, K20R, D30N, E35D, M36I, I54VI, L63P, A71TA, I84IV, N88D, L90M | 8 |
| II. I13V, K20R, D30N, E35D, M36I, L63P, A71V, I84V, N88D, L90M | 20 | |
| III. I13V, K20R, D30N, L33IL, E35D, M36I, I54V, L63P, A71T, N88D, L90M | 71 |
Differences between linkage groups are underlined.
Reproducible quasispecies population sampling.
An accurate genetic analysis of virus populations requires that enough genomes be sampled (i.e., reverse transcribed and amplified by PCR) so that the resulting mixture of amplified variants reflects their original frequency in the blood sample. A consequence of insufficient population sampling is therefore the generation of different variant frequencies in independent PCRs generated from the same sample (16, 21, 33, 50). For example, if only a few genomes are PCR amplified from a highly diverse population because of its low viral load (i.e., low cDNA input into the PCR), then different genomes may be amplified in independent PCRs. In order to test the ability of the protocol used here to reproducibly sample persons' protease quasispecies, we generated and compared by RNA-HTA duplicate PCRs initiated with different aliquots of the same cDNA (i.e., independent samplings) (Fig. 6B). The RNA-HTA patterns from independent samplings were identical, indicating that the protease quasispecies samplings were sufficiently high to reproducibly generate the same mixed populations of genetic variants.
DISCUSSION
Techniques used for HIV drug resistance genotyping can be divided into sequencing and non-sequencing-based methods (13). Nonsequencing methods include the line probe assay, oligonucleotide ligation assay, microarrays, and PCR with mutation-specific primers (1, 2, 23, 27, 29, 37, 40, 41, 53, 61, 67, 71, 77). DNA sequencing methods include direct PCR sequencing and sequencing of multiple plasmids from subcloned PCR products.
Non-sequencing-based methods are generally highly sensitive for detecting minority drug resistance mutations (65, 76, 77), but they suffer from several limitations. The exact sequence context of any mutation cannot be determined without sequencing the entire gene. The growing number of drug resistance-related mutations requires the optimization and simultaneous use of an increasingly large number of highly sensitive single-nucleotide querying assays. Polymorphisms in the sequence immediately neighboring the queried nucleotide positions may decrease annealing of the probe or primer, potentially leading to decreased sensitivity or false-negative results (44). Linkage of mutations present at low frequency on the same genome is not possible using single-nucleotide querying assays.
Direct PCR population sequencing can overcome all but the last of these problems, but at the cost of decreased sensitivity for low-frequency variants. A PCR subcloning-sequencing approach allows the linkage of different mutations on the same genomes and, if sufficient numbers of subclones are analyzed, may be highly sensitive to low-frequency variants but is extremely labor-intensive (39, 43, 49, 55, 57, 58, 63). The RNA-HTA assay was developed to overcome the low sensitivity of direct PCR population sequencing without resorting to generating and sequencing large numbers of subclones.
Heteroduplex mobility assays and HTAs can provide information on the presence of specific low-frequency variants, overall genetic diversity, or changes in quasispecies variant composition within HIV quasispecies (5, 15, 17-19, 21, 33, 35, 43, 55, 58-60) and have been used for the study of other variable viral species (4, 8, 31, 35, 45, 52, 68, 69, 73, 75, 78, 81, 84). Using heteroduplex mobility assays or HTAs, the electrophoretic separation of variants differing by only single-nucleotide substitutions is not possible unless special electrophoretic conditions or HTA probes are used (21, 60), and these approaches have not been used to directly isolate low-frequency variants for sequencing.
The UHG RNA probe used was designed to enhance the electrophoretic separation of wild type from drug-resistant variants at six common primary codons (D30N, G48V, I54V, V82A, I84V, and L90M) and six secondary or accessory drug resistance codons (L10I, V32I, M46I, 150V, A71V, and N88D). Using a panel of subcloned protease variants and quasispecies from clinical samples, we showed that the pro1 RNA probe allowed the separation of sequence variants at these targeted sites. Reconstitution experiments indicated that variants could be quantified at frequencies of ≥5% and PCR amplified at frequencies of ≥0.5%.
For some quasispecies PCR amplicons, distinct RNA-HTA bands were detected. Such quasispecies appeared to consist of multiple high-frequency viral lineages, and their purification on RNA-HTA gels allowed mutation linkage groups to be generated (Fig. 4 and 6 and Table 2). Other quasispecies produced a single but diffuse RNA-HTA band. When different regions of such diffuse bands were isolated and the variants were reamplified and sequenced, different drug-resistant variants were detected. These viral populations therefore appeared to be less structured than those producing distinct RNA-HTA bands and to consist of a multitude of low-frequency variants distributed around a quasispecies consensus variant in a more typical theoretical quasispecies viral “cloud” fashion (24).
Using RNA-HTA-based sequencing on 21 patient samples, we were able to detect primary drug resistance mutations at an earlier date than was possible using direct PCR population sequencing. These results indicate that RNA-HTA, by virtue of identifying low-frequency drug resistance mutations, may reduce the likelihood of treatment failure by helping to exclude potentially inactive drugs from salvage regimens. This study may overestimate the actual frequency with which such low-frequency mutations occur in genotyped samples, as persons were selected on the basis of acquiring one or more new protease inhibitor resistance mutations at the follow-up time point. Further studies are under way to determine how often additional mutations may be detected by RNA-HTA in less-heavily treated persons who are not selected on the basis of a second virologic failure showing additional mutations. The approach used here for the study of the HIV-1 protease gene should be readily applicable to the sequence analysis of other highly variable loci.
Acknowledgments
Support for this study was provided to E.L.D. by NIAID (RO1-AI47320), a research grant from AMFAR (02840-31), and the Blood Systems Foundation.
REFERENCES
- 1.Abravaya, K., J. J. Carrino, S. Muldoon, and H. H. Lee. 1995. Detection of point mutations with a modified ligase chain reaction (Gap-LCR). Nucleic Acids Res. 23:675-682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Anderson, B. D., T. Shirasaka, E. Kojima, R. Yarchoan, and H. Mitsuya. 1994. Identification of drug-related genotypic changes in HIV-1 from serum using the selective polymerase chain reaction. Antivir. Res. 25:245-258. [DOI] [PubMed] [Google Scholar]
- 3.Bacheler, L. T., E. D. Anton, P. Kudish, D. Baker, J. Bunville, K. Krakowski, L. Bolling, M. Aujay, X. V. Wang, D. Ellis, M. F. Becker, A. L. Lasut, H. J. George, D. R. Spalding, G. Hollis, and K. Abremski. 2000. Human immunodeficiency virus type 1 mutations selected in patients failing efavirenz combination therapy. Antimicrob. Agents Chemother. 44:2475-2484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bachmann, M. H., C. Mathiason-Dubard, G. H. Learn, A. G. Rodrigo, D. L. Sodora, P. Mazzetti, E. A. Hoover, and J. I. Mullins. 1997. Genetic diversity of feline immunodeficiency virus: dual infection, recombination, and distinct evolutionary rates among envelope sequence clades. J. Virol. 71:4241-4253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Barlow, K. L., J. Green, and J. P. Clewley. 2000. Viral genome characterisation by the heteroduplex mobility and heteroduplex tracking assays. Rev. Med. Virol. 10:321-335. [DOI] [PubMed] [Google Scholar]
- 6.Baxter, J. D., D. L. Mayers, D. N. Wentworth, J. D. Neaton, M. L. Hoover, M. A. Winters, S. B. Mannheimer, M. A. Thompson, D. I. Abrams, B. J. Brizz, J. P. Ioannidis, T. C. Merigan, et al. 2000. A randomized study of antiretroviral management based on plasma genotypic antiretroviral resistance testing in patients failing therapy. AIDS 14:F83-F93. [DOI] [PubMed] [Google Scholar]
- 7.Belli, C., C. De Brasi, and I. Larripa. 2003. Rapid detection of exon 1 NRAS gene mutations using universal heteroduplex generator technology. Hum. Mutat. 21:132-137. [DOI] [PubMed] [Google Scholar]
- 8.Chezzi, C., and B. D. Schoub. 1996. Differentiation between vaccine-related and wild-type polioviruses using a heteroduplex mobility assay. J. Virol. Methods 62:93-102. [DOI] [PubMed] [Google Scholar]
- 9.Cingolani, A., A. Antinori, M. G. Rizzo, R. Murri, A. Ammassari, F. Baldini, S. Di Giambenedetto, R. Cauda, and A. De Luca. 2002. Usefulness of monitoring HIV drug resistance and adherence in individuals failing highly active antiretroviral therapy: a randomized study (ARGENTA). AIDS 16:369-379. [DOI] [PubMed] [Google Scholar]
- 10.Clevenbergh, P., J. Durant, P. Halfon, P. del Giudice, V. Mondain, N. Montagne, J. M. Schapiro, C. A. Boucher, and P. Dellamonica. 2000. Persisting long-term benefit of genotype-guided treatment for HIV-infected patients failing HAART. The Viradapt Study: week 48 follow-up. Antivir. Ther. 5:65-70. [PubMed] [Google Scholar]
- 11.Condra, J. H., D. J. Holder, W. A. Schleif, O. M. Blahy, R. M. Danovich, L. J. Gabryelski, D. J. Graham, D. Laird, J. C. Quintero, A. Rhodes, H. L. Robbins, E. Roth, M. Shivaprakash, T. Yang, J. A. Chodakewitz, P. J. Deutsch, R. Y. Leavitt, F. E. Massari, J. W. Mellors, K. E. Squires, R. T. Steigbigel, H. Teppler, and E. A. Emini. 1996. Genetic correlates of in vivo viral resistance to indinavir, a human immunodeficiency virus type 1 protease inhibitor. J. Virol. 70:8270-8276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Culpan, D., G. Standen, N. Wood, C. Mazurier, C. Gaucher, and J. Bidwell. 1997. Rapid mutation screening in type 2A von Willebrand's disease using universal heteroduplex generators. Br. J. Haematol. 96:464-469. [DOI] [PubMed] [Google Scholar]
- 13.D'Aquila, R. T. 2000. Limits of resistance testing. Antivir. Ther. 5:71-76. [PubMed] [Google Scholar]
- 14.Deeks, S. G., T. Wrin, T. Liegler, R. Hoh, M. Hayden, J. D. Barbour, N. S. Hellmann, C. J. Petropoulos, J. M. McCune, M. K. Hellerstein, and R. M. Grant. 2001. Virologic and immunologic consequences of discontinuing combination antiretroviral-drug therapy in HIV-infected patients with detectable viremia. N. Engl. J. Med. 344:472-480. [DOI] [PubMed] [Google Scholar]
- 15.Delwart, E. L., M. P. Busch, M. L. Kalish, J. W. Mosley, and J. I. Mullins. 1995. Rapid molecular epidemiology of human immunodeficiency virus transmission. AIDS Res. Hum. Retrovir. 11:1081-1093. [DOI] [PubMed] [Google Scholar]
- 16.Delwart, E. L., P. Heng, A. Neumann, and M. Markowitz. 1998. Rapid, transient changes at the env locus of plasma human immunodeficiency virus type 1 populations during the emergence of protease inhibitor resistance. J. Virol. 72:2416-2421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Delwart, E. L., H. Pan, H. W. Sheppard, D. Wolpert, A. U. Neumann, B. Korber, and J. I. Mullins. 1997. Slower evolution of HIV-1 quasispecies during progression to AIDS. J. Virol. 71:7498-7508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Delwart, E. L., H. W. Sheppard, B. D. Walker, J. Goudsmit, and J. I. Mullins. 1994. Human immunodeficiency virus type 1 evolution in vivo tracked by DNA heteroduplex mobility assays. J. Virol. 68:6672-6683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Delwart, E. L., E. G. Shpaer, F. E. McCutchan, J. Louwagie, M. Grez, H. Rübsamen-Waigmann, and J. I. Mullins. 1993. Genetic relationships determined by a heteroduplex mobility assay: analysis of HIV env genes. Science 262:1257-1261. [DOI] [PubMed] [Google Scholar]
- 20.Demeter, L. M., R. D'Aquila, O. Weislow, E. Lorenzo, A. Erice, J. Fitzgibbon, R. Shafer, D. Richman, T. M. Howard, Y. Zhao, E. Fisher, D. Huang, D. Mayers, S. Sylvester, M. Arens, K. Sannerud, S. Rasheed, V. Johnson, D. Kuritzkes, P. Reichelderfer, A. Japour, et al. 1998. Interlaboratory concordance of DNA sequence analysis to detect reverse transcriptase mutations in HIV-1 proviral DNA. J. Virol. Methods 75:93-104. [DOI] [PubMed] [Google Scholar]
- 21.Doukhan, L., and E. Delwart. 2001. Population genetic analysis of the protease locus of human immunodeficiency virus type 1 quasispecies undergoing drug selection, using a denaturing gradient-heteroduplex tracking assay. J. Virol. 75:6729-6736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Durant, J., P. Clevenbergh, P. Halfon, P. Delgiudice, S. Porsin, P. Simonet, N. Montagne, C. A. Boucher, J. M. Schapiro, and P. Dellamonica. 1999. Drug-resistance genotyping in HIV-1 therapy: the VIRADAPT randomised controlled trial. Lancet 353:2195-2199. [DOI] [PubMed] [Google Scholar]
- 23.Edelstein, R. E., D. A. Nickerson, V. O. Tobe, L. A. Manns-Arcuino, and L. M. Frenkel. 1998. Oligonucleotide ligation assay for detecting mutations in the human immunodeficiency virus type 1 pol gene that are associated with resistance to zidovudine, didanosine, and lamivudine. J. Clin. Microbiol. 36:569-572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Eigen, M. 1993. Viral quasispecies. Sci. Am. 269:42-49. [DOI] [PubMed] [Google Scholar]
- 25.Erickson, J. W., S. V. Gulnik, and M. Markowitz. 1999. Protease inhibitors: resistance, cross-resistance, fitness and the choice of initial and salvage therapies. AIDS 13(Suppl. A):S189-S204. [PubMed] [Google Scholar]
- 26.Foley, B., H. Pan, S. Buchbinder, and E. L. Delwart. 2000. Apparent founder effect during the early years of the San Francisco HIV type 1 epidemic (1978-1979). AIDS Res. Hum. Retrovir. 16:1463-1469. [DOI] [PubMed] [Google Scholar]
- 27.Frater, A. J., C. C. Chaput, S. Beddows, J. N. Weber, and M. O. McClure. 2001. Simple detection of point mutations associated with HIV-1 drug resistance. J. Virol. Methods 93:145-156. [DOI] [PubMed] [Google Scholar]
- 28.Frater, A. J., C. C. Chaput, J. N. Weber, and M. O. McClure. 2000. HIV-1 resistance genotyping by sequencing produces inconsistent results for mixed viral populations. AIDS 14:1473-1475. [DOI] [PubMed] [Google Scholar]
- 29.Frenkel, L. M., L. E. Wagner II, S. M. Atwood, T. J. Cummins, and S. Dewhurst. 1995. Specific, sensitive, and rapid assay for human immunodeficiency virus type 1 pol mutations associated with resistance to zidovudine and didanosine. J. Clin. Microbiol. 33:342-347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Frenkel, L. M., Y. Wang, G. H. Learn, J. L. McKernan, G. M. Ellis, K. M. Mohan, S. E. Holte, S. M. De Vange, D. M. Pawluk, A. J. Melvin, P. F. Lewis, L. M. Heath, I. A. Beck, M. Mahalanabis, W. E. Naugler, N. H. Tobin, and J. I. Mullins. 2003. Multiple viral genetic analyses detect low-level human immunodeficiency virus type 1 replication during effective highly active antiretroviral therapy. J. Virol. 77:5721-5730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Garcia, F., Jr., F. Garcia, M. C. Bernal, G. Piedrola, and M. C. Maroto. 2000. Genomic variability of hepatitis G virus/GBV-C at the NS3 region: clinical implications. Microbios 102:17-25. [PubMed] [Google Scholar]
- 32.Gaschen, B., J. Taylor, K. Yusim, B. Foley, F. Gao, D. Lang, V. Novitsky, B. Haynes, B. H. Hahn, T. Bhattacharya, and B. Korber. 2002. Diversity considerations in HIV-1 vaccine selection. Science 296:2354-2360. [DOI] [PubMed] [Google Scholar]
- 33.Gordon, C. J., and E. L. Delwart. 2000. Genetic diversity of primary HIV-1 isolates and their sensitivity to antibody-mediated neutralization. Virology 272:326-330. [DOI] [PubMed] [Google Scholar]
- 34.Grant, R. M., D. R. Kuritzkes, V. A. Johnson, J. W. Mellors, J. L. Sullivan, R. Swanstrom, R. T. D'Aquila, M. Van Gorder, M. Holodniy, R. M. Lloyd Jr., Jr., C. Reid, G. F. Morgan, and D. L. Winslow. 2003. Accuracy of the TRUGENE HIV-1 genotyping kit. J. Clin. Microbiol. 41:1586-1593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Greenier, J. L., C. J. Miler, D. Lu, P. J. Dailey, F. X. Lu, K. J. Kunstman, S. M. Wolinsky, and M. L. Marthas. 2001. Route of simian immunodeficiency virus inoculation determines the complexity but not the identity of viral variant populations that infect rhesus macaques. J. Virol. 75:3753-3765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gunthard, H. F., J. K. Wong, C. C. Ignacio, D. V. Havlir, and D. D. Richman. 1998. Comparative performance of high-density oligonucleotide sequencing and dideoxynucleotide sequencing of HIV type 1 pol from clinical samples. AIDS Res. Hum. Retrovir. 14:869-876. [DOI] [PubMed] [Google Scholar]
- 37.Hance, A. J., V. Lemiale, J. Izopet, D. Lecossier, V. Joly, P. Massip, F. Mammano, D. Descamps, F. Brun-Vezinet, and F. Clavel. 2001. Changes in human immunodeficiency virus type 1 populations after treatment interruption in patients failing antiretroviral therapy. J. Virol. 75:6410-6417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hanna, G. J., V. A. Johnson, D. R. Kuritzkes, D. D. Richman, J. Martinez-Picado, L. Sutton, J. D. Hazelwood, and R. T. D'Aquila. 2000. Comparison of sequencing by hybridization and cycle sequencing for genotyping of human immunodeficiency virus type 1 reverse transcriptase. J. Clin. Microbiol. 38:2715-2721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hsieh, C. H., and J. D. Griffith. 1989. Deletions of bases in one strand of duplex DNA, in contrast to single-base mismatches, produce highly kinked molecules: possible relevance to the folding of single-stranded nucleic acids. Proc. Natl. Acad. Sci. USA 86:4833-4837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Izopet, J., C. Souyris, A. Hance, K. Sandres-Saune, M. Alvarez, C. Pasquier, F. Clavel, J. Puel, and P. Massip. 2002. Evolution of human immunodeficiency virus type 1 populations after resumption of therapy following treatment interruption and shift in resistance genotype. J. Infect. Dis. 185:1506-1510. [DOI] [PubMed] [Google Scholar]
- 41.Kaye, S., C. Loveday, and R. S. Tedder. 1992. A microtitre format point mutation assay: application to the detection of drug resistance in human immunodeficiency virus type-1 infected patients treated with zidovudine. J. Med. Virol. 37:241-246. [DOI] [PubMed] [Google Scholar]
- 42.Kijak, G. H., V. Simon, P. Balfe, J. Vanderhoeven, S. E. Pampuro, C. Zala, C. Ochoa, P. Cahn, M. Markowitz, and H. Salomon. 2002. Origin of human immunodeficiency virus type 1 quasispecies emerging after antiretroviral treatment interruption in patients with therapeutic failure. J. Virol. 76:7000-7009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kitrinos, K. M., N. G. Hoffman, J. A. Nelson, and R. Swanstrom. 2003. Turnover of env variable region 1 and 2 genotypes in subjects with late-stage human immunodeficiency virus type 1 infection. J. Virol. 77:6811-6822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Koch, N., N. Yahi, P. Colson, J. Fantini, and C. Tamalet. 1999. Genetic polymorphism near HIV-1 reverse transcriptase resistance-associated codons is a major obstacle for the line probe assay as an alternative method to sequence analysis. J. Virol. Methods 80:25-31. [DOI] [PubMed] [Google Scholar]
- 45.Kreis, S., and T. Whistler. 1997. Rapid identification of measles virus strains by the heteroduplex mobility assay. Virus Res. 47:197-203. [DOI] [PubMed] [Google Scholar]
- 46.Kuiken, C., B. Korber, and R. W. Shafer. 2003. HIV sequence databases. AIDS Rev. 5:52-61. [PMC free article] [PubMed] [Google Scholar]
- 47.Larder, B. A., A. Kohli, P. Kellam, S. D. Kemp, M. Kronick, and R. D. Henfrey. 1993. Quantitative detection of HIV-1 drug resistance mutations by automated DNA sequencing. Nature 365:671-673. [DOI] [PubMed] [Google Scholar]
- 48.Lech, W. J., G. Wang, Y. L. Yang, Y. Chee, K. Dorman, D. McCrae, L. C. Lazzeroni, J. W. Erickson, J. S. Sinsheimer, and A. H. Kaplan. 1996. In vivo sequence diversity of the protease of human immunodeficiency virus type 1: presence of protease inhibitor-resistant variants in untreated subjects. J. Virol. 70:2038-2043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Leitner, T., E. Halapi, G. Scarlatti, P. Rossi, J. Albert, E. M. Fenyo, and M. Uhlen. 1993. Analysis of heterogeneous viral populations by direct DNA sequencing. BioTechniques 15:120-127. [PubMed] [Google Scholar]
- 50.Liu, S. L., A. G. Rodrigo, R. Shankarappa, G. H. Learn, L. Hsu, O. Davidov, L. P. Zhao, and J. I. Mullins. 1996. HIV quasispecies and resampling. Science 273:415-416. [DOI] [PubMed] [Google Scholar]
- 51.Lukashov, V. V., and J. Goudsmit. 2002. Recent evolutionary history of human immunodeficiency virus type 1 subtype B: reconstruction of epidemic onset based on sequence distances to the common ancestor. J. Mol. Evol. 54:680-691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mattick, K. L., J. Green, P. Punia, F. J. Belda, C. I. Gallimore, and D. W. Brown. 2000. The heteroduplex mobility assay (HMA) as a pre-sequencing screen for Norwalk-like viruses. J. Virol. Methods 87:161-169. [DOI] [PubMed] [Google Scholar]
- 53.Myint, L., K. Ariyoshi, H. Yan, A. J. Frater, W. Auwanit, P. Pathipvanith, K. Yamada, M. Matsuda, T. Chiba, K. Fujita, M. McClure, J. N. Weber, and W. Sugiura. 2002. Mutagenically separated PCR assay for rapid detection of M41L and K70R zidovudine resistance mutations in CRF01_AE (subtype E) human immunodeficiency virus type 1. Antimicrob. Agents Chemother. 46:3861-3868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Najera, I., A. Holguin, M. E. Quinones-Mateu, M. A. Munoz-Fernandez, R. Najera, C. Lopez-Galindez, and E. Domingo. 1995. pol gene quasispecies of human immunodeficiency virus: mutations associated with drug resistance in virus from patients undergoing no drug therapy. J. Virol. 69:23-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Nelson, J. A., S. A. Fiscus, and R. Swanstrom. 1997. Evolutionary variants of the human immunodeficiency virus type 1 V3 region characterized by using a heteroduplex tracking assay. J. Virol. 71:8750-8758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Nijhuis, M., C. A. Boucher, P. Schipper, T. Leitner, R. Schuurman, and J. Albert. 1998. Stochastic processes strongly influence HIV-1 evolution during suboptimal protease-inhibitor therapy. Proc. Natl. Acad. Sci. USA 95:14441-14446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Paolucci, S., F. Baldanti, G. Campanini, M. Zavattoni, E. Cattaneo, L. Dossena, and G. Gerna. 2001. Analysis of HIV drug-resistant quasispecies in plasma, peripheral blood mononuclear cells and viral isolates from treatment-naive and HAART patients. J. Med. Virol. 65:207-217. [DOI] [PubMed] [Google Scholar]
- 58.Quinones-Mateu, M. E., S. C. Ball, A. J. Marozsan, V. S. Torre, J. L. Albright, G. Vanham, G. van Der Groen, R. L. Colebunders, and E. J. Arts. 2000. A dual infection/competition assay shows a correlation between ex vivo human immunodeficiency virus type 1 fitness and disease progression. J. Virol. 74:9222-9233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Quinones-Mateu, M. E., Y. Gao, S. C. Ball, A. J. Marozsan, A. Abraha, and E. J. Arts. 2002. In vitro intersubtype recombinants of human immunodeficiency virus type 1: comparison to recent and circulating in vivo recombinant forms. J. Virol. 76:9600-9613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Resch, W., N. Parkin, E. L. Stuelke, T. Watkins, and R. Swanstrom. 2000. A multiple-site-specific heteroduplex tracking assay as a tool for the study of viral population dynamics. Proc. Natl. Acad. Sci. USA 98:176-181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Richman, D. D., J. C. Guatelli, J. Grimes, A. Tsiatis, and T. Gingeras. 1991. Detection of mutations associated with zidovudine resistance in human immunodeficiency virus by use of the polymerase chain reaction. J. Infect. Dis. 164:1075-1081. [DOI] [PubMed] [Google Scholar]
- 62.Rouzine, I. M., and J. M. Coffin. 1999. Search for the mechanism of genetic variation in the pro gene of human immunodeficiency virus. J. Virol. 73:8167-8178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Saribas, Z., T. Kocagoz, A. Alp, and A. Gunalp. 2003. Rapid detection of rifampin resistance in Mycobacterium tuberculosis isolates by heteroduplex analysis and determination of rifamycin cross-resistance in rifampin-resistant isolates. J. Clin. Microbiol. 41:816-818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Schuurman, R., L. Demeter, P. Reichelderfer, J. Tijnagel, T. de Groot, and C. Boucher. 1999. Worldwide evaluation of DNA sequencing approaches for identification of drug resistance mutations in the human immunodeficiency virus type 1 reverse transcriptase. J. Clin. Microbiol. 37:2291-2296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Servais, J., C. Lambert, E. Fontaine, J. M. Plesseria, I. Robert, V. Arendt, T. Staub, F. Schneider, R. Hemmer, G. Burtonboy, and J. C. Schmit. 2001. Comparison of DNA sequencing and a line probe assay for detection of human immunodeficiency virus type 1 drug resistance mutations in patients failing highly active antiretroviral therapy. J. Clin. Microbiol. 39:454-459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Shafer, R. W., K. Hertogs, A. R. Zolopa, A. Warford, S. Bloor, B. J. Betts, T. C. Merigan, R. Harrigan, and B. A. Larder. 2001. High degree of interlaboratory reproducibility of human immunodeficiency virus type 1 protease and reverse transcriptase sequencing of plasma samples from heavily treated patients. J. Clin. Microbiol. 39:1522-1529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Shafer, R. W., M. A. Winters, D. L. Mayers, A. J. Japour, D. R. Kuritzkes, O. S. Weislow, F. White, A. Erice, K. J. Sannerud, A. Iversen, F. Pena, D. Dimitrov, L. M. Frenkel, P. S. Reichelderfer, et al. 1996. Interlaboratory comparison of sequence-specific PCR and ligase detection reaction to detect a human immunodeficiency virus type 1 drug resistance mutation. J. Clin. Microbiol. 34:1849-1853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Sitki-Green, D., R. H. Edwards, J. Webster-Cyriaque, and N. Raab-Traub. 2002. Identification of Epstein-Barr virus strain variants in hairy leukoplakia and peripheral blood by use of a heteroduplex tracking assay. J. Virol. 76:9645-9656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Sodora, D. L., F. Lee, P. J. Dailey, and P. A. Marx. 1998. A genetic and viral load analysis of the simian immunodeficiency virus during the acute phase in macaques inoculated by the vaginal route. AIDS Res. Hum. Retrovir. 14:171-181. [DOI] [PubMed] [Google Scholar]
- 70.Stemmer, W. P., A. Crameri, K. D. Ha, T. M. Brennan, and H. L. Heyneker. 1995. Single-step assembly of a gene and entire plasmid from large numbers of oligodeoxyribonucleotides. Gene 164:49-53. [DOI] [PubMed] [Google Scholar]
- 71.Stuyver, L., A. Wyseur, A. Rombout, J. Louwagie, T. Scarcez, C. Verhofstede, D. Rimland, R. F. Schinazi, and R. Rossau. 1997. Line probe assay for rapid detection of drug-selected mutations in the human immunodeficiency virus type 1 reverse transcriptase gene. Antimicrob. Agents Chemother. 41:284-291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Sugiura, W., Z. Matsuda, Y. Yokomaku, K. Hertogs, B. Larder, T. Oishi, A. Okano, T. Shiino, M. Tatsumi, M. Matsuda, H. Abumi, N. Takata, S. Shirahata, K. Yamada, H. Yoshikura, and Y. Nagai. 2002. Interference between D30N and L90M in selection and development of protease inhibitor-resistant human immunodeficiency virus type 1. Antimicrob. Agents Chemother. 46:708-715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Trincado, D. E., G. M. Scott, P. A. White, C. Hunt, L. Rasmussen, and W. D. Rawlinson. 2000. Human cytomegalovirus strains associated with congenital and perinatal infections. J. Med. Virol. 61:481-487. [DOI] [PubMed] [Google Scholar]
- 74.Tural, C., L. Ruiz, C. Holtzer, J. Schapiro, P. Viciana, J. Gonzalez, P. Domingo, C. Boucher, C. Rey-Joly, and B. Clotet. 2002. Clinical utility of HIV-1 genotyping and expert advice: the Havana trial. AIDS 16:209-218. [DOI] [PubMed] [Google Scholar]
- 75.Valas, S., C. Benoit, C. Guionaud, G. Perrin, and R. Z. Mamoun. 1997. North American and French caprine arthritis-encephalitis viruses emerge from ovine maedi-visna viruses. Virology 237:307-318. [DOI] [PubMed] [Google Scholar]
- 76.Van Laethem, K., K. Van Vaerenbergh, J. C. Schmit, S. Sprecher, P. Hermans, V. De Vroey, R. Schuurman, T. Harrer, M. Witvrouw, E. Van Wijngaerden, L. Stuyver, M. Van Ranst, J. Desmyter, E. De Clercq, and A. M. Vandamme. 1999. Phenotypic assays and sequencing are less sensitive than point mutation assays for detection of resistance in mixed HIV-1 genotypic populations. J. Acquir. Immune Defic. Syndr. 22:107-118. [DOI] [PubMed] [Google Scholar]
- 77.Van Vaerenbergh, K., L. Debaisieux, N. De Cabooter, C. Declercq, K. Desmet, K. Fransen, B. Maes, D. Marissens, K. Miller, G. Muyldermans, S. Sprecher, L. Stuyver, D. Vaira, C. Verhofstede, G. Zissis, M. Van Ranst, E. De Clercq, J. Desmyter, and A. M. Vandamme. 2001. Prevalence of genotypic resistance among antiretroviral drug-naive HIV-1-infected patients in Belgium. Antivir. Ther. 6:63-70. [PubMed] [Google Scholar]
- 78.White, P. A., Z. Li, X. Zhai, G. Marinos, and W. D. Rawlinson. 2000. Mixed viral infection identified using heteroduplex mobility analysis (HMA). Virology 271:382-389. [DOI] [PubMed] [Google Scholar]
- 79.Williams, D. L., T. L. Pittman, T. P. Gillis, M. Matsuoka, and Y. Kashiwabara. 2001. Simultaneous detection of Mycobacterium leprae and its susceptibility to dapsone using DNA heteroduplex analysis. J. Clin. Microbiol. 39:2083-2088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Williams, D. L., L. Spring, T. P. Gillis, M. Salfinger, and D. H. Persing. 1998. Evaluation of a polymerase chain reaction-based universal heteroduplex generator assay for direct detection of rifampin susceptibility of Mycobacterium tuberculosis from sputum specimens. Clin. Infect. Dis. 26:446-450. [DOI] [PubMed] [Google Scholar]
- 81.Wilson, J. J., S. J. Polyak, T. D. Day, and D. R. Gretch. 1995. Characterization of simple and complex hepatitis C quasispecies by heteroduplex gel shift analysis: correlation with nucleotide sequencing. J. Gen. Virol. 76:1763-1771. [DOI] [PubMed] [Google Scholar]
- 82.Wood, N., and J. Bidwell. 1996. Genetic screening and testing by induced heteroduplex formation. Electrophoresis 17:247-254. [DOI] [PubMed] [Google Scholar]
- 83.Wood, N., G. Standen, J. Hows, B. Bradley, and J. Bidwell. 1993. Diagnosis of sickle-cell disease with a universal heteroduplex generator. Lancet 342:1519-1520. [DOI] [PubMed] [Google Scholar]
- 84.Zou, S., C. Stansfield, and J. Bridge. 1998. Identification of new influenza B virus variants by multiplex reverse transcription-PCR and the heteroduplex mobility assay. J. Clin. Microbiol. 36:1544-1548. [DOI] [PMC free article] [PubMed] [Google Scholar]