

Abstract
M11L, a 166-amino-acid antiapoptotic protein of myxoma virus, was previously shown to bind to the peripheral benzodiazepine receptor by hydrophobic interactions at the outer mitochondrial membrane. Here we demonstrate that an additional property of M11L is the ability to constitutively form inhibitory complexes with the proapoptotic Bcl-2 family member Bak in human cells. This binding interaction was identified by both FLAG-tagged pull-down assays and tandem affinity purification from transfected and virus-infected human cells. M11L binds constitutively to human Bak and, under some inducible conditions, to human Bax as well, but not to the other Bcl-2 family members (Bad, Bid, Bcl-2). When stably expressed in human embryonic kidney (HEK293) cells, M11L effectively protects these cells from Fas ligand-induced apoptosis, thereby blocking release of cytochrome c, activation of caspase 9, and cleavage of poly(ADP-ribose) polymerase. We also demonstrate in coexpression studies that M11L can interact with Bak independently of any involvement with Bax. Furthermore, cells stably expressing M11L function to prevent apoptosis that is induced by overexpression of Bak. We conclude that M11L inhibits, in a species-independent fashion, apoptotic signals mediated by activation of Bak.
Apoptosis is a controlled cell death program that is designed to eliminate unwanted cells, including those that are virus infected (1, 22, 30). Viruses, in turn, often need to prevent or delay cellular apoptosis following infection and thereby safeguard their continued propagation (4, 6). Myxoma virus, a member of the Leporipoxvirus genus of poxviruses, is the etiologic agent that causes myxomatosis in European rabbits (Oryctolagus cuniculus) (17, 28). Like all poxviruses, myxoma virus encodes multiple proteins that function to disrupt various classes of apoptotic signals (16, 47). The antiapoptotic factors M-T2, M-T4, M-T5, and M11L have been described previously; to date, however, investigation of their role in preventing apoptosis has been limited to myxoma virus-infected rabbit cells (3, 16). The virulence factor M11L specifically acts to prevent cell death signals that are transduced via mitochondria (14, 15). Mitochondria are key coordinating centers for specific classes of intrinsic and extrinsic apoptotic signals and undergo marked morphological and biochemical changes during cell death (36). One of these mitochondrial changes is the redistribution of proapoptotic proteins from the mitochondrial intermembrane space into the cytosol. This redistribution affects the key apoptotic regulator cytochrome c, which acts as a cofactor in activation of caspase 9 within apoptosomes (36, 45). Another notable physiological alteration is manifested by dissipation of the mitochondrial membrane potential (MMP) normally maintained across the mitochondrial inner membrane (55). It has been shown previously that M11L can prevent both cytochrome c redistribution and MMP loss following apoptosis induction (14, 15). M11L has been shown by cross-linking studies to form inhibitory complexes with the peripheral benzodiazepine receptor (PBR) component of the permeability transition port (PTP) complex that spans the inner and outer mitochondrial membranes (15). M11L expression could specifically block apoptosis induced by ligands of PBR, and furthermore, M11L could also provide some measure of protection against MMP loss in PBR-deficient cells (15). This raised the possibility that M11L has additional modes of action, in addition to binding and inhibiting PBR, that might regulate the mitochondrion-dependent apoptosis pathway.
The Bcl-2 family of proteins are important regulators of mitochondrially mediated cell death and are identified by the presence of at least one of four characterized Bcl-2 homology (BH) domains (5, 9). This family can be divided functionally into two groups with opposing activities: the antiapoptotic proteins, such as Bcl-2 and Bcl-xL, and the proapoptotic proteins, which can be further subdivided into the Bax subfamily, including Bax and Bak, and the BH3-only family, including Bid and Bad. Bak and Bax play a central role in the mitochondrion-dependent cell death pathway (13, 53). The target of the Bcl-2 family of regulators appears to be centered on mitochondria, because many cell death stimuli, such as signaling by the tumor necrosis factor receptor/Fas death receptors, converge on this organelle (36, 41, 54). The precise mechanistic role of these Bcl-2 family proteins in the regulation of apoptotic signals is still controversial.
Interestingly, many viruses encode Bcl-2 homologs that are thought to function as Bcl-2 family mimics and prevent mitochondrion-mediated cell death by sequestering host Bcl-2 family members (6, 11, 22). For example, the BHRF-1 protein of Epstein-Barr virus protects cells from a variety of proapoptotic stimuli through heterodimerization with Bcl-2 and Bak (11, 34) or by preventing the disruption of mitochondrial integrity (12). Similarly, E1B 19K of adenovirus (49), another Bcl-2 homolog, interacts with Bax and Bak (11) and blocks Bax and Bak oligomerization (50) so as to inhibit downstream apoptotic events (39). In addition, other viral proteins with no obvious homology to members of the Bcl-2 family, such as vMIA of human cytomegalovirus, prevent apoptosis at the mitochondrial checkpoint through physical interaction with the adenine nucleotide transporter subunit of the PTP complex (19). Recently, a new poxvirus antiapoptotic protein has been identified in vaccinia virus (52). Like M11L, vvF1L localizes to other mitochondria and inhibits the loss of the inner MMP and the release of cytochrome c.
In this paper, we demonstrate that the antiapoptotic protein M11L, a protein known to bind and inhibit the PBR at the outer mitochondrial membrane (15), is also able to interact with Bak, an important proapoptotic Bcl-2 family member, and block Bak-mediated apoptosis. We propose that both of these binding interactions contribute to the antiapoptotic function of M11L and that the inhibitory mechanism of M11L for the PBR and Bak is conserved across species barriers.
MATERIALS AND METHODS
Viruses and cells.
Recombinant viruses used in this study have been described previously and include vMyxlac, a control myxoma virus that expresses wild-type M11L, and vMyxM11LKO, which fails to express M11L due to a targeted gene disruption within open reading frame (ORF) M011L (38). Other myxoma knockout viruses described previously and used in this study include vMyxM-T2KO (44), vMyxM-T4KO (2), and vMyxM-T5KO (33). Infections were carried out at a multiplicity of infection (MOI) of 5. The Bax-deficient DU145 prostate cancer cells were provided by B. Li (University of Kansas). These cells, along with HEK293T, Cos-7, RK13, and human osteosarcoma (HOS) cells, were maintained in Dulbecco's modified Eagle medium containing 10% fetal bovine serum (FBS). HEK293 epithelial cells (a gift of Y. Ma, Kyoto University) were grown in RPMI 1640 supplemented with 10% FBS.
Plasmids.
M11L was amplified from myxoma virus genomic DNA (7) by PCR and then cloned into various expression vectors, including pcDNA3 (Invitrogen, San Diego, Calif.), pcDNA3HA (a gift of Jin Q. Cheng, University of South Florida), pFlag-CMV2 (Kodak), pIRESneo2, pEGFP-C1, and pDsRed2-C1 (BD Biosciences, San Jose, Calif.). Constructs containing M11L were designated pcDNA3-M11L, pcDNA3HA-M11L, pFlag-CMV2-M11L, pIRESneo2-M11L, pEGFP-M11L, and pDsRed2-M11L, respectively. Human Bak, amplified from HEK293T cDNA by reverse transcription-PCR (RT-PCR), was cloned into the following vectors: pcDNA3, pcDNA3HA, and pEGFP-C1. The resulting constructs were designated pcDNA3-huBak, pcDNA3HA-huBak, and EGFP-huBak (see Fig. 2A).
FIG. 2.
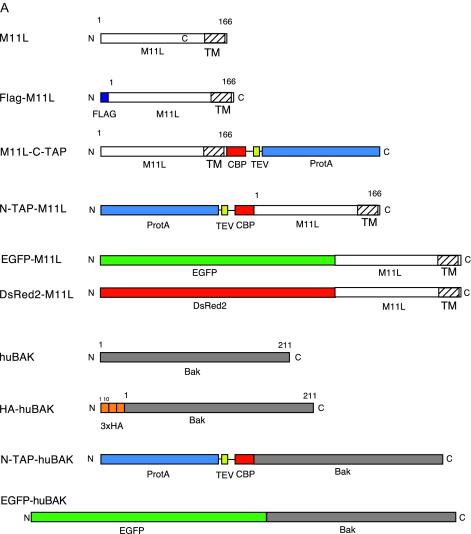

(A) M11L and huBak constructs utilized for this study. Constructs were either left untagged or fused to Flag, TAP, or HA epitope tags as indicated. The TAP cassette is composed of three components; the protein A (ProtA) IgG-binding sequence and the calmodulin binding protein (CBP) domain are separated by a TEV protease cleavage site. (B) Table listing the binding partners associated with M11L during Flag pull-down assays and identified by mass spectrometry. Interaction frequency, percentage of 20 independent transfections in which the protein was identified. (C) Immunoprecipitation of Flag-M11L and identification of associated proteins. Cell lysates were immunoprecipitated with anti-Flag agarose, and bound proteins were eluted with Flag peptide. Eluted proteins were resolved on a 4 to 15% Tris-HCl SDS-gel and stained with Coomassie blue. Lane 1, molecular weight markers; lane 2, immunoprecipitation from untransfected control HEK293T cells; lane 3, immunoprecipitation from cells transfected with Flag-M11L. Each of the bands in the second and third lanes was excised, digested with trypsin, and analyzed by mass spectrometry. (D) Typical output from mass spectrometry of peptide sequence run. A short, 10-amino-acid residue peptide within the complete Bak sequence was identified as a Bak peptide (lowercased letters).
Antibodies and reagents.
The following rabbit polyclonal antibodies were used: anti-M11L (38); anti-Bak NT (directed against amino acids 23 to 37 of human Bak) and anti-Bax NT (directed against amino acids 1 to 21 of human Bax) (both from Upstate Biotechnology, Lake Placid, N.Y.); anti-Bad (H-168; directed against amino acids 1 to 168 of human Bad); and anti-Bid (C-20; directed against the carboxy terminus of human Bid). We also used a rabbit anti-Bid polyclonal antibody that recognized full-length and cleaved human and mouse Bid (BD PharMingen, San Diego, Calif.). An anti-caspase 3 polyclonal antibody was used to detect endogenous full-length caspase 3 (35 kDa) as well as the cleaved fragments (19 and 17 kDa) (Cell Signaling Technology, Beverly, Mass.). Anti-actin (C-11) was a goat polyclonal antibody directed against the carboxy terminus of actin of human origin (identical to the corresponding mouse sequence) (Santa Cruz Biotechnology, Inc., Santa Cruz, Calif.). A rabbit peroxidase-anti-peroxidase antiserum (anti-PAP; Sigma) recognizing the protein A sequence of the tandem affinity purification (TAP) tag was utilized for monitoring TAP tag expression. The anti-M11L antibody was previously described (20). Mouse monoclonal antibodies against Bcl-2 (Santa Cruz) and against a peptide epitope derived from the hemagglutinin (HA) protein of human influenza virus (clone 12CA5) (Roche) were also used. A purified mouse anti-human caspase 9 antibody was employed to detect the 47-kDa proform and the 37-kDa cleaved form of human caspase 9 (BD PharMingen). For poly(ADP-ribose) polymerase (PARP) cleavage analysis, we employed two monoclonal antibodies. A mouse monoclonal PARP antibody (clone 4C10-5; Santa Cruz) directed against the human DNA binding domain was used to identify PARP cleavage. A mouse monoclonal cytochrome c antibody (clone 7H8.2C12) directed against amino acids 93 to 104 of pigeon cytochrome c (BD PharMingen) was used to monitor cytochrome c release. A mouse monoclonal antibody against cytochrome oxidase subunit IV (COX IV; Molecular Probes, Eugene, Oreg.) was used to confirm loading controls for the cytochrome c cleavage assay. Horseradish peroxidase-coupled goat anti-mouse and goat anti-rabbit secondary antibodies were obtained from Jackson Immunoresearch (West Grove, Pa.). CHAPS {3-[(3-cholamidopropyl)-dimethylammonio]-1-propanesulfonate} was obtained from Calbiochem (San Diego, Calif.). A complete protease inhibitor cocktail (Roche) was included in all lysis buffers. Trypan blue (0.4%) solution, used to determine cell death numbers, was purchased from Sigma.
Immunoprecipitation of Flag-M11L and identification of interacting proteins by mass spectrometry.
Transfection of HEK293T cells and identification of M11L and interacting proteins were performed as described elsewhere (18). Briefly, approximately 108 HEK293T cells were transfected with 80 μg of N-terminally Flag-tagged M11L by calcium phosphate precipitation (48). Cells were incubated for 44 h and then harvested in KLB lysis buffer (20 mM Tris-HCl [pH 7.5], 150 mM NaCl, 1% [vol/vol] NP-40, 0.5% sodium deoxycholate, 0.2 mM phenylmethylsulfonyl fluoride, 1.5 μg of aprotinin/ml, 1 μg of leupeptin/ml, 2 μM pepstatin). The lysate was clarified by centrifugation at 20,000 × g, precleared with Sepharose 4B (10 μl [packed volume]/ml of lysate; Sigma-Aldrich), and incubated with M2-Flag agarose (1 μl [packed volume]/ml of lysate; Sigma-Aldrich). The beads were washed three times with KLB lysis buffer and then eluted with 400 μM Flag peptide in 50 mM ammonium bicarbonate. The eluant was lyophilized and then resuspended in 1× sodium dodecyl sulfate (SDS) sample buffer. The proteins were resolved on a 4 to 15% Tris-HCl gradient gel (Bio-Rad) and then stained with colloidal Coomassie blue (Gel-Code Blue; Pierce). Bands were excised from the gel and subjected to in-gel digestion (18). Mass spectrometry was carried out on a quadrapole time-of-flight hybrid mass spectrometer (SCIEX QSTAR) equipped with a nanospray ion source.
Construction of TAP tag vectors and TAP-tagged protein purification.
The C- and N-terminal TAP dual epitope tags (40) were provided under a material transfer agreement by J. Walkenhorst (Cellzome GmbH and European Molecular Biology Laboratory). C-TAP and N-TAP cassettes were subcloned into pcDNA3, resulting in the pcDNA3.6 C-TAP and pcDNA3.4 N-TAP fusion vectors, respectively, which were used for further subcloning of M11L and human Bak (M11L-C-TAP, N-TAP-M11L, and N-TAP-huBak, respectively). Logarithmic-phase 293T cells were seeded in 100-mm-diameter tissue culture dishes (5 × 106 cells/dish) in fresh medium the day before transfection. Cells were transiently transfected or cotransfected with 20 μg of each of the expression plasmids by using a calcium phosphate mammalian transfection kit (BD Clontech) according to the manufacturer's recommended procedure. Cells expressing the TAP-tagged M11L protein or pcDNA3-M11L plus N-TAP-huBak were lysed 24 h after transfection in cell lysis buffer (50 mM Tris-HCl [pH 8.0], 137 mM NaCl, 2% CHAPS) supplemented with a protease inhibitor cocktail. Following centrifugation at 14,000 × g for 30 min at 4°C, the supernatant was recovered and added to rabbit immunoglobulin G (IgG) agarose beads (Sigma), which were then rotated for 2 h at 4°C to allow the binding of the fusion protein. The beads were washed three times with wash buffer (10 mM Tris-HCl [pH 8.0], 150 mM NaCl, 0.1% CHAPS) and resuspended in 1 ml of tobacco etch virus (TEV) cleavage buffer (10 mM Tris-HCl [pH 8.0], 0.5 mM EDTA, 1 mM dithiothreitol) containing 200 U of TEV protease (Invitrogen). The samples were mixed by inversion at room temperature for 2 h to cleave the tagged protein. Calmodulin binding buffer (10 mM β-mercaptoethanol, 10 mM Tris-HCl [pH 8.0], 150 mM NaCl, 1 mM magnesium acetate, 1 mM imidazole, 2 mM CaCl2, and 0.1% CHAPS) was added to the eluted product together with calmodulin affinity resin (Stratagene) and mixed for 1 h at 4°C to allow binding to the beads. Following three washes with calmodulin binding buffer, the samples were eluted with calmodulin elution buffer (10 mM β-mercaptoethanol, 10 mM Tris-HCl [pH 8.0], 150 mM NaCl, 1 mM magnesium acetate, 1 mM imidazole, 2 mM EGTA, 0.1% CHAPS). The eluted purified product was concentrated to an appropriate volume through a protein concentrator (NANOSEP 10K OMEGA; Pall Gelman Laboratory) and resolved by denaturing SDS-polyacrylamide gel electrophoresis (PAGE). Proteins were stained with a silver stain kit (Bio-Rad) or immunoblotted with the proper antibodies.
Coimmunoprecipitation assay of M11L-Bak complexes and Western blotting.
Cells, either transfected by the calcium phosphate method or infected with the appropriate virus, were lysed in 2% CHAPS lysis buffer (20 mM Tris-HCl [pH 8.0], 137 mM NaCl, 1.5 mM MgCl2, 1 mM EDTA, 2% CHAPS) containing a cocktail of protease inhibitors. Immunoprecipitation and immunoblot assays were performed with the indicated antibodies. Protein A-agarose beads (Upstate) were added and incubated overnight. The immunocomplexes with agarose beads were washed in a 0.5% CHAPS wash buffer (20 mM Tris-HCl [pH 8.0], 137 mM NaCl, 1.5 mM MgCl2, 1 mM EDTA, 0.5% CHAPS, 0.5% sodium deoxycholate, and cocktail inhibitors). The complexes were resolved by SDS-PAGE and then analyzed by Western blotting with the appropriate antibodies.
Stable expression of M11L in HEK293 cells.
Control pIRESneo2 and pIRESneo2M11L constructs were transiently transfected into HEK293 cells by using LipofectAMINE 2000 reagent (Invitrogen). Transfected cells were passaged into fresh culture medium 1 day after the start of transfection, and at 2 days, various amounts of Geneticin (G418; 200 to 1,000 μg/ml; Invitrogen) were added to select for expression of the transfected antibiotic-resistant gene. Several weeks of selection were required for stable expression. HEK293 cells stably expressing M11L and the neomycin empty vector were designated HEKM11L and HEKneo, respectively, and were grown in RPMI 1640 supplemented with 10% FBS and 500 μg of G418/ml. RT-PCR and immunoprecipitation verified the stable incorporation and expression of M11L and neomycin resistance.
35S-labeled protein-protein binding assay in vitro.
The pcDNA3-M11L and pcDNA3-huBak constructs were translated in vitro with the TNT transcription-translation system kit (Promega, Madison, Wis.). Briefly, 2 μg of pcDNA3-M11L was incubated with 20 μCi of [35S]methionine (ICN Pharmaceuticals, Inc., Irvine, Calif.) in TNT quick master mix for 90 min at 30°C; 2 μg of pcDNA3-huBak was incubated with an amino acid mixture including methionine instead of [35S]methionine for the TNT transcription-translation method. Equal amounts of 35S-labeled M11L and unlabeled Bak were mixed in the binding buffer (phosphate-buffered saline [PBS] with 0.2% CHAPS including protease inhibitors) and incubated for 2 h at 4°C. Four microliters of a rabbit polyclonal Bak antibody and 50 μl of protein A-agarose beads were added and incubated for 16 h at 4°C. Following four washes in PBS buffer, samples were boiled, resolved by SDS-PAGE, and visualized by autoradiography.
Subcellular fractionation.
Both the cytosolic fraction and the heavy membrane (HM) fraction, which included the mitochondria, were isolated at 4°C by using an established protocol (24) with some modifications. Briefly, cells were washed twice in PBS buffer, resuspended in a hypotonic buffer containing 20 mM HEPES-KOH (pH 7.5), 1.5 mM MgCl2, 5 mM KCl, 1 mM dithiothreitol, and a cocktail of protease inhibitors, and incubated on ice for 10 min. The cells were sheared four times through a 30-gauge 1/2-in. needle fitted on a 1-ml syringe, and the lysate was centrifuged at 1,000 × g for 10 min. The supernatant was collected and centrifuged again under the same conditions. The resulting supernatant was then centrifuged at 14,000 × g for 30 min, followed by collection of both the supernatant and pellet fractions. The pellet was washed twice with a buffer (210 mM mannitol, 70 mM sucrose, 5 mM Tris-HCl [pH 7.5], 1 mM EDTA, and a protease inhibitor cocktail). The pellet was resuspended in a lysis buffer (50 mM Tris-HCl [pH 7.5], 5 mM EDTA, 150 mM NaCl, and 0.5% NP-40, supplemented with a cocktail of protease inhibitors), and this represented the HM fraction including the mitochondria. The supernatant was further ultracentrifuged at 100,000 × g for 30 min, and the resulting supernatant was collected as the cytosolic fraction. To prepare a whole-cell extract, cells were lysed in the lysis buffer, and the lysate was centrifuged at 14,000 × g for 30 min. The supernatant was collected as the whole-cell lysate. Twenty micrograms of protein from the HM or cytosolic fraction was resolved by SDS-PAGE and blotted for cytochrome c release.
Induction and measurement of apoptosis.
HOS cells were infected with various myxoma virus knockouts, including vMyxM11LKO, vMyxM-T2KO, vMyxM-T4KO, and vMyxM-T5KO, at an MOI of 5. The percentage of dead cells was determined by a trypan blue exclusion assay as described by the manufacturer. Briefly, cells were incubated with 0.2% trypan blue dye for 5 min at room temperature and were then counted on a hemocytometer under a low-power microscope. The percentage of cell death was calculated as the number of dead cells (dye stained) divided by the total number of cells. Three hundred cells were counted for each infection time point. Four replicates were performed for each time point. Trypan blue exclusion was also used to monitor apoptosis induction with Fas ligand (FasL) in HEKM11L and HEKneo cells as a measure of protection. Data were expressed as means ± standard errors of the means. Statistical evaluation was performed by using the Student t test (P < 0.01).
HEKneo and HEKM11L cells were either left untreated or treated with recombinant FasL protein (4 or 10 ng/ml; Upstate) at various time points (0, 4, 8, 12, 24, or 48 h). Cells were collected following trypsinization and were either used for isolation of mitochondria or tested for PARP cleavage by a Western blot assay. Transfection of the HA-Bak plasmid was used to measure the ability of stable cells expressing M11L to protect against Bak-induced apoptosis. Transfections of various concentrations of HA-Bak plasmid DNA into HEKM11L and HEKneo cells were performed with LipofectAMINE 2000 in accordance with the manufacturer's instructions. Cells were transfected either with pcDNA3HA-Bak at different concentrations (0, 0.25, 0.5, 1.0, 1.5, or 2.0 μg of plasmid/106 cells) or with the pcDNA3HA vector alone (2.0 μg). Apoptosis was determined 24 h posttransfection. Apoptotic cells were determined by flow cytometric analysis. Briefly, cells were washed twice with cold PBS and then resuspended in 1× binding buffer containing 10 mM HEPES-NaOH (pH 7.4), 140 mM NaCl, and 2.5 mM CaCl2 at a concentration of 106 cells/ml. One hundred microliters of the suspension (105 cells) was taken, mixed with 5 μl of fluorescein isothiocyanate-conjugated annexin V and 10 μl of propidium iodide (PI) (BD PharMingen), and incubated for 15 min at room temperature in the dark. Binding buffer (1×) was added to each tube (400 μl) and analyzed with a FACScan (BD Biosciences) within 60 min. Activation of caspase 3 and cleavage of Bid and PARP were also used as measures of apoptosis. Each time point represents the mean percent apoptosis ± the standard error of the mean from three independent experiments.
RESULTS
Myxoma virus infection blocks apoptosis in productively infected human cells.
Previous work has demonstrated that multiple myxoma virus proteins block virus-induced apoptosis in rabbit RL-5 CD4+ lymphocytes (16). Infection of RL-5 cells, but not rabbit fibroblasts, with deletion mutants of M-T2, M-T4, M-T5, or M11L resulted in apoptosis and aborted virus replication (2, 31, 33). To evaluate these inhibitor proteins in human cells, we tested the abilities of these knockout constructs to replicate in activated HOS cells, which are permissive for myxoma virus replication. The most dramatic phenotype was observed by trypan blue staining in HOS cells infected with vMyxM11LKO (Fig. 1A). The absence of M11L resulted in an increase in cell death, starting between 8 and 12 h postinfection and increasing dramatically over time until 48 h postinfection, when approximately 76% of HOS cells were unable to exclude trypan blue (Fig. 1A). In contrast, wild-type virus or constructs with targeted deletions of M-T2, M-T4, or M-T5 did not result in a significant increase in this parameter of cell death.
FIG. 1.
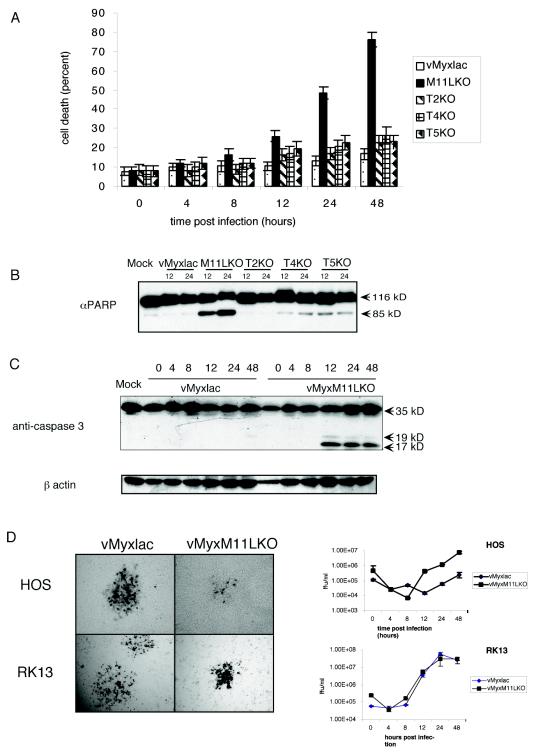
Myxoma virus infection of HOS cells blocks apoptosis. (A) Determination of cell death of HOS cells following infection with vMyxlac or the deletion mutant vMyxM-T2KO, vMyxM-T4KO, vMyxM-T5KO, or vMyxM11LKO (MOI, 5). At various times postinfection, the cells were stained with trypan blue, and the percentage of blue cells within the total cell population was determined. Values in the graph represent four replicates of 300 cells each. (B) PARP activation in infected HOS cells was measured at 12 and 24 h postinfection. Thirty micrograms of total protein was resolved on an 8% SDS-PAGE gel and probed with anti-PARP. (C) Cleavage of caspase 3 was examined in HOS cells infected with either vMyxlac or the M11L deletion mutant vMyxM11LKO at various times postinfection. Caspase 3 cleavage results in the production of three cleaved products. Normally only the 19- and 17-kDa fragments are picked up by the antibody. (D) Near-confluent monolayers of HOS or RK13 cells were infected with either vMyxlac or vMyxM11LKO. The monolayers were stained with 5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside (X-Gal) at 48 h postinfection and photographed under visible light with a Leica DMR-5 microscope. HOS and RK13 cells were infected at an MOI of 5, and infected cells were collected at the times indicated. Recovered virus was titrated on BGMK cells.
To further characterize this virus-induced death, we examined two classic markers of apoptosis. During early stages of apoptosis, activated caspases cleave a variety of cellular substrates, including PARP. PARP is a 116-kDa molecule involved in DNA repair and other cellular events, and it is frequently cleaved to a characteristic 85-kDa fragment. To establish the cause of the cell death observed in HOS cells infected with vMyxM11LKO, we probed with anti-PARP at 12 and 24 h postinfection. Although infection of HOS cells with the parental virus, vMyxlac, resulted in only minimal levels of PARP cleavage that ware indistinguishable from those in uninfected cells, infection with vMyxM11LKO was sufficient to induce significant PARP cleavage at 12 h postinfection (Fig. 1B). Furthermore, we tested for cleavage by the initiator caspase 3 during a time course infection. The presence of cleaved caspase 3, first detectable between 8 and 12 h postinfection (Fig. 1C), confirms that infection of HOS cells with myxoma virus lacking M11L is sufficient to induce apoptosis through a caspase-dependent pathway. Cleaved caspase 3 was detected at 48 h postinfection in the M-T2, M-T4, and M-T5 knockout viruses (data not shown) at levels similar to those observed for vMyxlac-infected HOS cells at 48 h postinfection and significantly lower than those observed for vMyxM11LKO (Fig. 1C). To examine the consequences for productive virus replication, we first examined focus formation of the parental virus vMyxlac and of vMyxM11LKO on HOS cells (Fig. 1D). Replication of vMyxlac in HOS cells appears to be similar to that observed in the control rabbit kidney (RK13) cells, based on the morphology of focus formation (Fig. 1D). The focus formation of vMyxM11LKO on RK13 cells appears more compact, for unknown reasons; however, single-step growth curves confirm that the loss of M11L does not result in a significant difference in the ability of the virus to replicate in either RK13 or HOS cells (Fig. 1D).
The antiapoptotic role of M11L has been demonstrated previously in rabbit T lymphocytes and monocytes (14, 31), and loss of M11L results in a profound reduction of virulence in European rabbits (38). More recently, it has been demonstrated that M11L localizes to mitochondria and that the antiapoptotic properties of M11L are directed at the mitochondrial checkpoint (14, 15). Specifically, the PBR component of the PTP complex was identified by cross-linking under hydrophobic conditions, and in vivo inhibition assays demonstrated that the PBR was a biologically functional target for M11L inhibition (15). Importantly, it was also demonstrated that M11L, unlike Bcl-2, blocks cell death signals that are relayed by ligands that specifically interact with the PBR (15). However, the observation that M11L has antiapoptotic properties against diverse cell death signals in PBR-deficient cells suggested that this protein may have additional inhibitory targets. Accordingly, we used M11L as bait to identify other cellular binding partners in human cells under experimental conditions that would favor non-membrane mediated interactions as opposed to the hydrophobic cross-linking protocol that originally identified the PBR.
M11L pull-down experiments identify Bak as a specific binding partner.
In pilot experiments, an N-terminally Flag-tagged M11L construct (Fig. 2A) was used to screen for binding partners to M11L via pull-down assays in transiently transfected 293T cells. Several putative partners were identified by mass spectrometry (Fig. 2B), and the most frequent (>60% of all transfections) pull-down partner detected was cellular Bak (Fig. 2C and D). However, Baxβ, another member of the Bcl-2 family, was also detected in a minority (15%) of these isolations. Because of the identified role of M11L as an inhibitor of apoptosis (14, 15, 31) and the established proapoptotic roles of Bak and Bax, we initially set out to confirm and then characterize these possible interactions. Therefore, we used the TAP system, originally designed for yeast (40), and constructed TAP-tagged expression vectors suitable for identifying interactions between poxvirus and host proteins. Two TAP-based fusion expression vectors were constructed and designated pcDNA3.6 C-TAP (C-terminal tag) and pcDNA3.4 N-TAP (N-terminal tag). We cloned M11L into these two vectors to generate constructs designated N-TAP-M11L and M11L-C-TAP in order to allow the expression of M11L with either an N-terminal or a C-terminal TAP tag appended (Fig. 2A). We then used the TAP method (Fig. 3A) to isolate M11L-associated proteins in transiently transfected 293T cells and confirmed that Bak routinely copurified with either N-TAP-M11L (data not shown) or M11L-C-TAP (Fig. 3B). Controls showing expression of the intact 34-kDa M11L-C-TAP fusion protein prior to the TAP procedure and recovery of the 17-kDa TEV-cleaved M11L after TAP are shown in Fig. 3C and D, respectively.
FIG. 3.
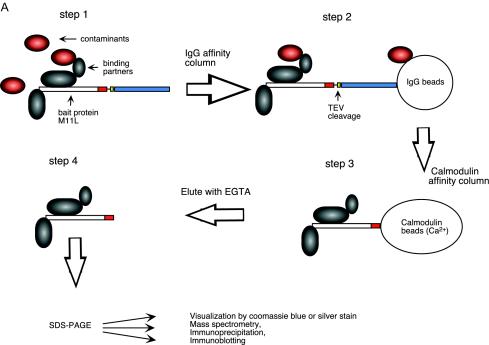
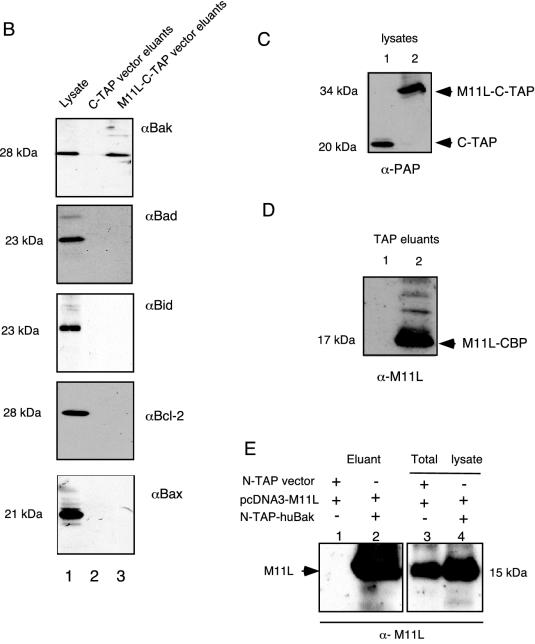
M11L specifically binds to Bak but not to other Bcl-2 family members by use of the TAP method. (A) TAP method for isolating M11L binding partners. The plasmid (M11L-C-TAP) was transiently transfected into 293T cells by the calcium phosphate method (step 1). The lysate with putative TAP M11L binding partner complexes was incubated with rabbit agarose IgG beads in order to bind the protein A of the TAP cassette. The beads were washed to remove unbound proteins and then incubated with TEV protease (step 2) to release bound complexes. The protein complex from cleaved products was allowed to bind via calmodulin binding protein (CBP) resin, and then the purified complex was eluted with calmodulin elution buffer containing EGTA (step 3). The eluants, containing proteins interacting with M11L, were separated by SDS-PAGE, and the proteins were analyzed (step 4). (B) Bak, but not Bad, Bid, Bax/p21, or Bcl-2, was detected as a binding partner of M11L in the M11L-C-TAP eluted products (lane 3), but not in control eluants of C-TAP alone (lane 2), following lysis with CHAPS buffer. Lane 1 confirms endogenous expression levels of the various Bcl-2 family members in 293T cell lysates. (C) Cell lysates of the C-TAP vector alone (lane 1) or the M11L-C-TAP bait (lane 2) were probed with anti-PAP before the TAP procedure to confirm the expression and size of the M11L-TAP fusion protein (34 kDa). (D) Eluants of the C-TAP vector alone (lane 1) or M11L-C-TAP (lane 2) were immunoblotted with anti-M11L, indicating that the M11L bait tagged with CBP (17 kDa) was recovered following TEV cleavage and elution. (E) huBak binds to untagged M11L. Shown are TAP eluants from lysates of cotransfections of pcDNA3-M11L plus N-TAP vector alone (lane 1) or pcDNA3-M11L plus N-TAP-huBak (lane 2) in 293T cells. Lane 3 (M11L and TAP vector) and lane 4 (M11L and N-TAP-huBak) represent corresponding cell lysates, confirming the expression of the appropriate cotransfected plasmids.
Depending on the precise lysis conditions, we also occasionally (in 25% of the transfections) identified Bax (21 kDa) as a binding partner for M11L-C-TAP (data not shown). Although we tested for M11L-Bax complexes in both transfected and myxoma virus-infected cells, the results were variable under our experimental conditions and suggested that M11L-Bax complexes could be inducibly formed, whereas M11L-Bak complexes were routinely copurified. In the original FLAG pull-downs, we had utilized a lysis buffer containing 1% NP-40 detergent during the extraction procedure. It has been noted that nonionic detergents such as NP-40 and Triton X-100 are able to induce dimerization and conformational changes of some Bcl-2 family members, which may cause artificial binding during cell lysis (25). Therefore, for the TAP method, we performed the M11L binding partner analysis by utilizing a lysis buffer containing either 1% NP-40 or 2% CHAPS, a detergent that minimizes artificial oligomerization (25). For lysis either with CHAPS (Fig. 3B) or with NP-40 (data not shown), endogenous cellular Bak was always found to copurify with either M11L-C-TAP or N-TAP-M11L, but not with the control TAP protein alone, suggesting that Bak interacted constitutively with TAP-tagged M11L.
We next analyzed whether, in addition to Bak, other Bcl-2 family proteins could be copurified with M11L by use of the TAP method following CHAPS lysis. Aside from the variable isolations of Bax with M11L, none of the other Bcl-2 family members tested, including Bad, Bid, and Bcl-2, were ever observed in the eluted purified product, although these endogenous proteins were routinely detectable in the cell lysates before purification (Fig. 3B). These results showed that the TAP method is effective for identifying specific binding partners of M11L under isolation conditions that favor the detection of long-lived (>4 h) complexes following detergent extraction of proteins. The TAP method also has an experimental advantage over the FLAG method in that it routinely exhibits low nonspecific background, as assessed by silver staining (data not shown).
To confirm that the interaction also occurs between untagged M11L and Bak, we performed reciprocal pull-down assays using TAP-tagged Bak. Bak was expressed from the pcDNA N-TAP vector (N-TAP-huBak [Fig. 2A]) in transfected 293T cells. Following transfection with the N-TAP control vector alone (pcDNA3.4 N-TAP), pcDNA3-M11L alone, or pcDNA3.4 N-TAP-huBak and pcDNA3-M11L together, lysates were subjected to the TAP procedure. Eluted products were immunoblotted with an anti-M11L antibody, which confirmed that N-TAP-huBak associated with transfected M11L and that the two molecules copurified efficiently (Fig. 3E, lanes 1 and 2). Expression of M11L (by using anti-M11L), N-TAP vector alone, and the N-TAP-huBak fusion protein (by using an anti-PAP antibody) was confirmed by immunoblotting of control cell lysates (Fig. 3E, lanes 3 and 4).
Thus, we conclude that M11L constitutively associates with Bak (whether tagged with N-TAP, C-TAP, or N-Flag, or untagged) under all conditions tested, whereas M11L-Bax interactions could be detected only variably under certain conditions, such as following NP-40 lysis or transient overexpression of both M11L and Bax in transfected cells.
M11L constitutively binds to Bak in both transfected and virus-infected human cells.
To address the question of whether M11L and huBak also interact in virus-infected cells, and whether this interaction requires other partners, we used several approaches. First, to confirm that native untagged M11L would complex with non-TAP-tagged Bak, cotransfection of pcDNA3-M11L (untagged) with pcDNA3HA-huBak (Fig. 2A) was carried out in transfected 293T cells. Twenty-four hours following transfection, immune complexes were coprecipitated with antibodies against either M11L (anti-M11L) or HA-huBak (anti-HA) and detected with either anti-HA or anti-M11L, respectively. These reciprocal coimmunoprecipitations confirmed that M11L and huBak complexes can be isolated following cotransfection and are precipitated with either anti-M11L or anti-HA Bak (Fig. 4A).
FIG. 4.

M11L interacts directly with Bak. (A) M11L interacts with ectopically expressed huBak. 293T cells were transfected with pcDNA3-M11L and pcDNA3HA-huBak or an empty vector. Coimmunoprecipitations (CO-IP) were performed in 2% CHAPS lysis buffer by using anti-M11L or anti-HA (12CA5); immunoblot (WB) analysis detected the resulting immune complexes with anti-HA (upper left panel) or anti-M11L (lower left panel), respectively. Expression of the transfected plasmids is shown in the panels on the right. (B) M11L interacts with endogenous Bak in infected HOS cells. HOS cells were infected with vMyxlac (MOI, 10), and at 12 h postinfection, immune complexes were pulled down with anti-Bak and immunoblotted with anti-M11L (top). Expression of M11L (center) and endogenous human Bak (bottom) is also shown. (C) M11L binds to Bak in the absence of Bax. DU145 human prostate cancer cells (deficient in Bax) were infected with vMyxlac (MOI, 10) for 12 h. Immune complexes were precipitated with anti-Bak and immunoblotted with anti-M11L (top panel). Immunoblotting of lysates confirmed that M11L was expressed only in infected cells (second panel) and that endogenous Bak was detected in the cells (third panel) but endogenous huBax was not (bottom panel). (D) In vitro-translated M11L can bind directly to huBak. Translated products of unlabeled huBak alone (pcDNA3-huBak [lane 1]) or unlabeled huBak plus 35S-radiolabeled M11L (pcDNA3-M11L [lane 2]) were incubated together with anti-Bak-protein A agarose beads. The unlabeled translated huBak alone was precipitated with anti-Bak (lane 3).
To confirm the ability of M11L to interact with endogenous levels of untagged huBak following virus infection, we examined HOS cells following productive infection with myxoma virus strain vMyxlac (38) at an MOI of 10. Twelve hours postinfection, the cells were collected and lysed with 2% CHAPS buffer, and immune complexes were precipitated with an anti-Bak antibody. Immunoblotting with anti-M11L indicated that anti-Bak was able to coprecipitate native M11L from virus-infected human cells in vivo (Fig. 4B).
As proapoptotic members of the Bcl-2 family, both Bak and Bax play a significant role in the regulation of mitochondrion-mediated apoptosis. Although the essential biological functions of Bak and Bax are similar and appear to be at least partly redundant (53), Bak and Bax may also function independently or in a cooperative manner (8, 32, 49). Our initial transfection experiments suggested that, at least under certain experimental conditions (e.g., NP-40 lysis), Bax could be induced to enter into complexes with M11L (data not shown). To test whether M11L can function with these two proapoptotic proteins individually, we examined the potential interaction between M11L and Bak in a cellular environment in the absence of Bax. For this purpose, we infected human prostate tumor cells (DU145) that are deficient in Bax expression (23) with vMyxlac (at an MOI of 10) for 12 h. The cells were collected and lysed with CHAPS buffer, and the lysates were immunoprecipitated with anti-Bak-protein A beads. Western blotting with anti-M11L revealed that M11L still coimmunoprecipitated efficiently with endogenous cellular Bak, even in the complete absence of Bax (Fig. 4C).
Direct interaction between M11L and Bak proteins was demonstrated by using an in vitro binding assay in which translated huBak (unlabeled) and 35S-labeled M11L were mixed and incubated at 4°C for 2 h, and then Bak was immunoprecipitated with anti-huBak-protein A beads. 35S-labeled M11L clearly coprecipitated with Bak and was detected in the immunoprecipitate by autoradiography (Fig. 4D), indicating that the M11L-Bak complex detected in vivo could also be reproduced in vitro.
Bak localizes predominantly in the mitochondria (10, 21, 35), although it has recently been demonstrated that a small proportion of Bak and other Bcl-2 family members can also regulate apoptosis from the endoplasmic reticulum (37, 46). M11L has previously been localized to the mitochondria in both transfected and virus-infected cells (14). When EGFP-M11L or DsRed2-huBak was individually transfected into Cos-7, each colocalized with Mitotracker, and when EGFP-huBak and DsRed2-M11L were cotransfected into Cos-7 cells, they were confirmed to colocalize, as indicated by consistent yellow staining in the merged images (data not shown). Taken together with the previous coimmunoprecipitation data, this localization pattern suggests that the M11L-Bak complex forms constitutively at the mitochondrial outer membrane, even in the absence of proapoptotic signals.
M11L blocks FasL-induced apoptosis in stably expressing HEK293 cells.
Previous research has shown that stable expression of M11L in Rat2 fibroblasts or HeLa cells could block induction of apoptosis by staurosporine and PBR ligands such as protoporphyrin IX (14, 15). To confirm and extend these observations, a stable HEK293 epithelial cell line constitutively expressing M11L (HEKM11L) was constructed along with the neomycin control line (HEKneo). RNA analysis by RT-PCR (data not shown) and immunoprecipitation with anti-M11L (Fig. 5A) confirmed constitutive expression of M11L in this cell line. FasL, a member of the tumor necrosis factor superfamily, can rapidly activate the caspase cascade in Fas receptor-expressing cells such as HEK293 (27, 29). Both HEKneo and HEKM11L cells were treated with FasL (at 4 and 10 ng/ml) to assess the antiapoptotic properties of M11L. As expected, FasL induced rapid cell death in the control HEKneo cells, whereas there was little or no increased cell death phenotype in the stably expressed M11L line (Fig. 5B).
FIG. 5.

M11L efficiently blocks the release of cytochrome c and PARP cleavage induced by FasL in HEKM11L cells. (A) Expression of M11L was shown in stably expressing HEK293 cells (HEKM11L) by immunoprecipitation. (B) HEKneo and HEKM11L cells were either left untreated or treated with FasL (at 4 or 10 ng/ml) to determine cell death numbers. At various times posttreatment, the stable cell population was stained with trypan blue. The percentage of blue cells within the total cell population was determined from four replicates of 300 cells each. (C) M11L expressed from HEKM11L cells inhibits PARP cleavage in response to FasL. Total cell lysates (50 μg/lane) were analyzed by immunoblotting with anti-PARP, which indicated that M11L protected most of the PARP from cleavage, unlike controls exposed to FasL. (D) (Top panel) Analysis of cytochrome c release in HEKM11L and control HEKneo cells following FasL treatment. The HM fraction, enriched for mitochondria, was separated from the soluble cytosolic fraction. Cytochrome c cleavage was not detectable in the cytosolic fraction in either cell line; this is considered the uninduced basal level (lanes 1 and 2). However, upon induction with FasL, there was a significant loss of cytochrome c from the HM fractions of control cells (lane 7) relative to that in stable cells (lane 8). This result corresponds with an increased appearance of cytochrome c in the cytosolic fraction for controls (lane 3) versus cells expressing M11L (lane 4). (Bottom panels) Loading controls (actin and COX IV) for each fraction. (E) Measurement of pro-caspase 9 decrease in stable cells. (Right) Lanes 1, 3, and 5, HEKneo cells; lanes 2, 4, and 6, HEKM11L cells. Samples were either left untreated (lanes 1 and 2) or treated with FasL at 10 ng/ml (lanes 3 and 4) or 4 ng/ml (lanes 5 and 6). (Left) Differences in pro-caspase 9 band intensity were calculated after normalization against the actin loading control and plotted on a bar graph as a representation of protection by M11L from FasL-mediated apoptosis.
Control HEKneo cells induced with FasL underwent classic PARP cleavage, as expected (Fig. 5C). In contrast, PARP cleavage was reduced to near-basal levels in HEKM11L cells induced with FasL. These results indicate that M11L blocks FasL-induced signaling prior to the release of cytochrome c from mitochondria and the subsequent activation of the downstream effector caspases that normally cleave PARP.
Cytochrome c is normally sequestered within the mitochondrial intermembrane space, but following the introduction of a death receptor-mitochondrion-dependent apoptotic stimulus such as Fas/FasL, a cascade of events occurs at the outer mitochondrial membrane, resulting in the rapid release of cytochrome c (4, 6). The presence of cytochrome c in the cytosol is often used as a marker of loss of mitochondrial membrane integrity and an indicator that proapoptotic signaling has been amplified via the mitochondrial pathway. M11L expression in HEKM11L cells effectively reduced the redistribution of cytochrome c from the mitochondria to the cytosol induced by FasL (Fig. 5D). In contrast, FasL-treated control cells (HEKneo) were subject to extensive release of cytochrome c into the cytosol fraction.
Following an apoptotic stimulus, cytochrome c is released from the mitochondria and activates the apoptasome, resulting in cleavage of pro-caspase 9 into its processed active subunit (35 or 17 kDa). Treatment of HEKneo and HEKM11L cells with FasL (10 ng/ml) resulted in a decrease in pro-caspase 9 levels in HEKneo cells, but the same decrease was not observed in treated HEKM11L cells (Fig. 5E), suggesting that expression of M11L indirectly blocks caspase 9 cleavage because the release of cytochrome c is blocked. Taken together, these results indicate that M11L blocks Fas-induced apoptosis through a mitochondrion-dependent pathway in HEK293 cells.
M11L specifically inhibits Bak-induced apoptosis in stable HEKM11L cells.
As with Bax, overexpression of Bak is sufficient to induce apoptosis in HEK293 (51) and 293T (43) cells. In order to demonstrate that M11L functions to block Bak-mediated apoptosis through its interaction with Bak, we chose a direct approach using HEKM11L cells transfected with an HA Bak plasmid as the proapoptotic stimulus. Stable expression of M11L dramatically protected HEK293 cells from Bak-induced apoptosis, as monitored by annexin V staining 24 h posttransfection, relative to control cells (Fig. 6A). The percentage of control cells killed by Bak-induced apoptosis increased until it reached a plateau at 1.0 μg of transfected plasmid DNA per 106 cells. In contrast, transfection of Bak plasmid DNA had no measurable effect on HEKM11L cells at any concentration tested (Fig. 6A).
FIG. 6.

M11L specifically inhibits Bak-induced apoptosis. HEKM11L and HEKneo cells were transfected with increasing amounts of the HA-Bak expression plasmid as indicated. Cells were harvested after 24 h posttransfection. (A) Apoptotic cells were counted by annexin V-PI staining as measured by fluorescence-activated cell sorter analysis. (B) Western blot analysis of HA-Bak expression, decrease in full-length Bid levels, and cleavage of caspase-3 and PARP in total-cell extracts (50 μg/lane) at various time points after transient transfection of HEKM11L and HEKneo cells with either a vector encoding HA-Bak or the empty control vector. Actin was used as a loading control. HA-Bak has an NH2-terminal HA tag, providing an additional molecular mass of approximately 3 kDa relative to that of endogenous Bak.
To dissect the ability of M11L to functionally protect against Bak-induced apoptosis, we examined a series of cellular markers. Both Bid and caspase 3 exhibited signs of cleavage following Bak overexpression in control HEKneo cells (Fig. 6B). There was also a decrease in the amount of full-length Bid in HEKneo cells; however, the steady-state level of full-length Bid appeared unchanged with increasing levels of transfected Bak in HEKM11L cells. The cleavage of both caspase 3 and PARP was also induced in a Bak-concentration-dependent manner only in transfected HEKneo cells, not in HEKM11L cells (Fig. 6B).
DISCUSSION
In this paper, we propose an expanded, antiapoptotic role for M11L above and beyond its previously documented role as an inhibitor of the PBR (15), as demonstrated through its interaction with Bak in human cells. Many of the myxoma virus antiapoptotic factors were first discovered to function specifically in rabbit leukocytes (47). Here we show that at least one of these viral proteins, M11L, can also act in human tumor cells to prevent apoptosis via the mitochondrial pathway. In this study we demonstrate that M11L constitutively binds to proapoptotic Bak in both transfected and virus-infected human cells.
Apoptosis represents an important innate cellular mechanism for curtailing virus infection, and many viruses have, in turn, developed strategies for inhibiting or delaying this cellular response (4). In particular, the existence of many diverse classes of poxvirus antiapoptotic proteins suggests that manipulating apoptotic cascades at multiple points is an important survival strategy for these viruses (47). Evidence is accumulating to suggest that mitochondria act to amplify and coordinate diverse cell death signals, and many viruses have focused on this particular checkpoint in order to modulate cell death (6, 19). Some of these viral mitochondrially targeted proteins are Bcl-2 homologs and are thought to antagonize the effects of cellular proapoptotic Bcl-2 family proteins (11). Previously, by use of a cross-linking technique that favors the identification of hydrophobic interactions, it had been shown that M11L forms inhibitory complexes with the PBR (15). To investigate the possibility that M11L might also target the Bcl-2 family in a nonhydrophobic fashion, Flag-tagged M11L was used in a pull-down assay that favors hydrophilic interactions, and several putative cellular binding partners were visualized on silver-stained SDS-PAGE gels and identified by mass spectrometry. The binding partner that most commonly copurified with M11L was identified as Bak. With this preliminary information, we then exploited the TAP method, which is associated with high purity of isolated complexes (40), to confirm and extend these observations. The M11L-TAP binding partners purified by this method were probed with antibodies directed against various members of the Bcl-2 family to show that Bak consistently copurified as a constitutive complex with M11L (Fig. 3). Interestingly, Bax was also observed to occasionally enter into complexes with M11L, but this interaction was dependent on experimental conditions (e.g., overexpression of both M11L and Bax). At present, we favor the model that Bak is a constitutive binding partner of M11L, whereas Bax might inducibly enter into inhibitory complexes under certain conditions that remain to be better defined.
The present study using Flag- or TAP-tagged M11L vectors required an initial detergent lysis step (with NP-40 or CHAPS) to optimize hydrophilic interactions that require short domains. This experimental approach therefore complemented the previous cross-linking method that detected the PBR and revealed that M11L can bind constitutively to Bak and, depending on isolation conditions, inducibly to Bax. Interestingly, the hydrophobic cross-linking approach used to identify the PBR as a binding partner for M11L did not identify either Bak or Bax, whereas the hydrophilic pull-downs reported here did not reveal the PBR. This serves to emphasize the importance of exploiting multiple technologies to characterize proteomic interactions of complex viral inhibitors such as M11L.
A human cell line deficient in Bax expression was employed to examine the functional consequence of M11L-Bak interaction in the absence of Bax. Because Bak resides predominantly in the outer membrane of the mitochondria (5) and M11L is also targeted to the PTP complex of the mitochondria (14), we employed fluorescence microscopy to confirm that M11L indeed colocalizes with Bak at the mitochondria (data not shown). Importantly, functional assays show that stably expressed M11L can block FasL-induced apoptosis in M11L-expressing HEK293 cells as needed by PARP cleavage, cytochrome c release, and activation of caspase 9 (Fig. 5).
We were unable to detect M11L binding directly to Bid (Fig. 3B), but we did observe that HEKM11L cells transfected with increasing concentrations of Bak were protected against the Bid cleavage observed in HEKneo control cells (Fig. 6B). We interpret this observation to indicate that M11L prevents Bak-triggered Bid cleavage by directly binding and sequestering Bak, but we cannot rule out the possibility that M11L might inactivate both Bak and Bax under these conditions. It is also not known whether M11L might block the translocation of Bid or prevent Bid-mediated conformational changes of Bak and/or Bax as an inhibitory complex of the mitochondria. Mechanistically, the expression of M11L does not affect the level of endogenous Bak protein (data not shown) and therefore does not enhance the degradation of this protein, in contrast to the Bak degradation induced by the E6 protein from human papillomavirus (26). Our results thus indicate that M11L prevents cell death downstream of Bid cleavage but upstream of mitochondrial cytochrome c release.
M11L clearly is capable of interacting with Bak independently of Bax. An in vitro binding assay also indicated that M11L and Bak can form complexes in the absence of other Bcl-2 family members (Fig. 4D). We postulate that M11L blocks cytochrome c release from mitochondria through the constitutive interaction with Bak, but we cannot rule out the possibility that under certain circumstances M11L forms inhibitory complexes with Bax.
Sequence comparisons between M11L and Bcl-2 family members suggest that there is a putative conserved BH3-like domain in M11L (Fig. 7), but whether this is the region of Bak binding is not known. Notably, at least two of the conserved residues (Bak Leu78 and Asp83) in the Bcl-2 family have been shown to be required for heterodimerization with Bcl-xL (42). Whether this strict conservation of critical residues is necessary for M11L structure, and thereby for M11L function as an inhibitor of apoptosis, is not clear. Previous analysis of the M11L protein sequence indicates that the charge distribution of the 26-amino-acid transmembrane domain shares some similarity with the Bcl-2 signal anchor sequence and is required for targeting to mitochondria (14). Taken together, the nature of M11L domains that interact with either the PBR or Bak needs to be clarified, but analysis of this interaction and structure may well shed light on a possible functional linkage between the PBR and Bak.
FIG. 7.
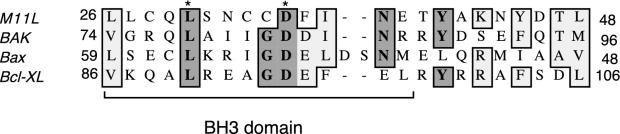
Sequence comparison of the BH3 domains from human Bak, Bax, and Bcl-xL and the related BH3-like region in M11L. Numbers indicate amino acid positions within the respective proteins. Dark shading, identical residues; light shading, conserved substitutions. Asterisks indicate residues necessary for Bak and Bax binding to Bcl-xL.
In conclusion, we demonstrate that M11L blocks diverse mitochondrial cell death signals through constitutive physical interaction with Bak. Our results indicate that M11L, like Bcl-2, interacts with multiple binding partners (including the PBR) that contribute to its antiapoptotic properties. The exact mechanism by which M11L functions as a modulator of the PBR, Bak, and possibly Bax under certain inducible conditions remains to be deduced.
Acknowledgments
We thank Y. Ma for invaluable discussions. Honggang Wang (H. Lee Moffitt Cancer Center and Research Institute, Tampa, Fla.) provided suggestions and critically reviewed the data before publication.
The work is supported by grants to G.M. from the CIHR of Canada. G.M. holds a Canada Research Chair in Molecular Virology.
REFERENCES
- 1.Adams, J. M. 2003. Ways of dying: multiple pathways of apoptosis. Genes Dev. 17:2481-2495. [DOI] [PubMed] [Google Scholar]
- 2.Barry, M., S. Hnatiuk, K. Mossman, S.-F. Lee, L. Boshkov, and G. McFadden. 1997. The myxoma virus M-T4 gene encodes a novel RDEL-containing protein that is retained within the endoplasmic reticulum and is important for the productive infection of lymphocytes. Virology 239:360-377. [DOI] [PubMed] [Google Scholar]
- 3.Barry, M., and G. McFadden. 2000. Regulation of apoptosis by poxviruses, p. 509-520. In M. Cunningham and R. Fujinami (ed.), Effects of microbes on the immune system. Lippincott-Raven Publishers, Philadelphia, Pa.
- 4.Benedict, C. A., P. S. Norris, and C. F. Ware. 2002. To kill or be killed: viral evasion of apoptosis. Nat. Immunol. 3:1013-1018. [DOI] [PubMed] [Google Scholar]
- 5.Borner, C. 2003. The Bcl-2 protein family: sensors and checkpoints for life-or-death decisions. Mol. Immunol. 39:615-647. [DOI] [PubMed] [Google Scholar]
- 6.Boya, P., T. Roumier, K. Andreau, R. A. Gonzalez-Polo, N. Zamzami, M. Castedo, and G. Kroemer. 2003. Mitochondrion-targeted apoptosis regulators of viral origin. Biochem. Biophys. Res. Commun. 304:575-581. [DOI] [PubMed] [Google Scholar]
- 7.Cameron, C., S. Hota-Mitchell, L. Chen, J. Barrett, J.-X. Cao, C. Macaulay, D. Willer, D. Evans, and G. McFadden. 1999. The complete DNA sequence of myxoma virus. Virology 264:298-318. [DOI] [PubMed] [Google Scholar]
- 8.Cartron, P.-F., P. Juin, L. Oliver, S. Martin, K. Meflah, and F. M. Vallette. 2003. Nonredundant role of Bax and Bak in Bid-mediated apoptosis. Mol. Cell. Biol. 23:4701-4712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cory, S., and J. M. Adams. 2002. The Bcl2 family: regulators of the cellular life-or-death switch. Natl. Rev. Cancer 2:647-656. [DOI] [PubMed] [Google Scholar]
- 10.Cory, S., D. C. S. Huang, and J. M. Adams. 2003. The Bcl-2 family: roles in cell survival and oncogenesis. Oncogene 22:8590-8607. [DOI] [PubMed] [Google Scholar]
- 11.Cuconati, A., and E. White. 2002. Viral homologs of BCL-2 role of apoptosis in the regulation of virus infection. Genes Dev. 16:2465-2478. [DOI] [PubMed] [Google Scholar]
- 12.Dumont, A., S. P. Hehner, T. G. Hofmann, M. Ueffing, W. Droge, and M. L. Schmitz. 1999. Hydrogen peroxide-induced apoptosis is CD95-independent, requires the release of mitochondria-derived reactive oxygen species and the activation of NF-κB. Oncogene 18:747-757. [DOI] [PubMed] [Google Scholar]
- 13.Esposti, M. D., and C. Dive. 2003. Mitochondrial membrane permeabilisation by Bax/Bak. Biochem. Biophys. Res. Commun. 304:455-461. [DOI] [PubMed] [Google Scholar]
- 14.Everett, H., M. Barry, S. F. Lee, X. J. Sun, K. Graham, J. Stone, R. C. Bleackley, and G. McFadden. 2000. M11L: a novel mitochondria-localized protein of myxoma virus that blocks apoptosis in infected leukocytes. J. Exp. Med. 191:1487-1498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Everett, H., M. Barry, X. Sun, S. F. Lee, C. Frantz, L. G. Berthiaume, G. McFadden, and R. C. Bleackley. 2002. The myxoma poxvirus protein, M11L, prevents apoptosis by direct interaction with the mitochondrial permeability transition pore. J. Exp. Med. 196:1127-1139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Everett, H., and G. McFadden. 2002. Poxviruses and apoptosis: a time to die. Curr. Opin. Microbiol. 5:395-402. [DOI] [PubMed] [Google Scholar]
- 17.Fenner, F., and F. N. Ratcliffe. 1965. Myxomatosis. Cambridge University Press, Cambridge, United Kingdom.
- 18.Figeys, D., L. D. McBroom, and M. F. Moran. 2001. Mass spectrometry for the study of protein-protein interactions. Methods 24:230-239. [DOI] [PubMed] [Google Scholar]
- 19.Goldmacher, V. S. 2002. vMIA, a viral inhibitor of apoptosis targeting mitochondria. Biochimie 84:177-185. [DOI] [PubMed] [Google Scholar]
- 20.Graham, K. A., A. Opgenorth, C. Upton, and G. McFadden. 1992. Myxoma virus M11L ORF encodes a protein for which cell surface localization is critical for manifestation of viral virulence. Virology 191:112-124. [DOI] [PubMed] [Google Scholar]
- 21.Griffiths, G. J., L. Dubrez, C. P. Morgan, N. A. Jones, J. Whitehouse, B. M. Corfe, C. Divd, and J. A. Hickman. 1999. Cell damage-induced conformational changes of the pro-apoptotic protein Bak in vivo precede the onset of apoptosis. J. Cell Biol. 144:903-914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hardwick, J. M., and D. S. Bellows. 2003. Viral versus cellular BCL-2 proteins. Cell Death Differ. 10(Suppl. 1):S68-S76. [DOI] [PubMed] [Google Scholar]
- 23.Hemmati, P. G., B. Gillissen, C. von Haefen, J. Wendt, L. Starck, D. Guner, B. Dorken, and P. T. Daniel. 2002. Adenovirus-mediated overexpression of p14ARF induces p53 and Bax-independent apoptosis. Oncogene 21:3149-3161. [DOI] [PubMed] [Google Scholar]
- 24.Hockenberry, D. M., G. Nunez, R. D. Minniman, R. D. Schreiber, and S. J. Korsmeyer. 1990. Bcl-2 is an inner mitochondrial membrane protein that blocks programmed cell death. Nature 348:334-336. [DOI] [PubMed] [Google Scholar]
- 25.Hsu, Y. T., and R. J. Youle. 1997. Nonionic detergents induce dimerization among members of the Bcl-2 family. J. Biol. Chem. 272:13829-13834. [DOI] [PubMed] [Google Scholar]
- 26.Jackson, S., C. Harwood, M. Thomas, L. Banks, and A. Storey. 2000. Role of Bak in UV-induced apoptosis in skin cancer and abrogation by HPV E6 proteins. Genes Dev. 14:3065-3073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kavurma, M. M., and L. M. Khachigian. 2003. Signaling and transcriptional control of Fas ligand gene expression. Cell Death Differ. 10:36-44. [DOI] [PubMed] [Google Scholar]
- 28.Kerr, P., and G. McFadden. 2002. Immune responses to myxoma virus. Viral Immunol. 15:229-246. [DOI] [PubMed] [Google Scholar]
- 29.Larregina, A. T., A. E. Morelli, R. A. Dewey, M. G. Castro, A. Fontana, and P. R. Lowenstein. 1998. FasL induces Fas/Apo1-mediated apoptosis in human embryonic kidney 293 cells routinely used to generate E1-deleted adenoviral vectors. Gene Ther. 5:563-568. [DOI] [PubMed] [Google Scholar]
- 30.Lawen, A. 2003. Apoptosis—an introduction. Bioessays 25:888-896. [DOI] [PubMed] [Google Scholar]
- 31.Macen, J. L., K. A. Graham, S. F. Lee, M. Schreiber, L. K. Boshkov, and G. McFadden. 1996. Expression of the myxoma virus tumor necrosis factor receptor homologue (T2) and M11L genes is required to prevent virus-induced apoptosis in infected rabbit T lymphocytes. Virology 218:232-237. [DOI] [PubMed] [Google Scholar]
- 32.Mikhailov, V., M. Mikhailova, K. Degenhardt, M. A. Venkatachalam, E. White, and P. Saikumar. 2003. Association of Bax and Bak homo-oligomers in mitochondria. Bax requirement for Bak reorganization and cytochrome c release. J. Biol. Chem. 278:5367-5376. [DOI] [PubMed] [Google Scholar]
- 33.Mossman, K., S. F. Lee, M. Barry, L. Boshkov, and G. McFadden. 1996. Disruption of M-T5, a novel myxoma virus gene member of the poxvirus host range superfamily, results in dramatic attenuation of myxomatosis in infected European rabbits. J. Virol. 70:4394-4410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nava, V. E., E. H. Cheng, M. Veliuona, S. Zou, R. J. Clem, M. L. Mayer, and J. M. Hardwick. 1997. Herpesvirus saimiri encodes a functional homolog of the human bcl-2 oncogene. J. Virol. 71:4118-4122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nechushtan, A., C. L. Smith, I. Lamensdorf, S. H. Yoon, and R. J. Youle. 2001. Bax and Bak coalesce into novel mitochondria-associated clusters during apoptosis. J. Cell Biol. 153:1265-1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Newmeyer, D. D., and S. Ferguson-Miller. 2003. Mitochondria: releasing power for life and unleashing the machineries of death. Cell 112:480-490. [DOI] [PubMed] [Google Scholar]
- 37.Nutt, J. K., A. Pataer, A. Pahler, B. Fang, J. Roth, D. J. McConkey, and S. G. Swisher. 2002. Bax and Bak promote apoptosis by modulating endoplasmic reticular and mitochondrial Ca2+ stores. J. Biol. Chem. 277:9219-9225. [DOI] [PubMed] [Google Scholar]
- 38.Opgenorth, A., K. Graham, N. Nation, D. Strayer, and G. McFadden. 1992. Deletion analysis of two tandemly arranged virulence genes in myxoma virus, M11L and myxoma growth factor. J. Virol. 66:4720-4731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Perez, D., and E. White. 2000. TNF-α signals apoptosis through a bid-dependent conformational change in Bax that is inhibited by E1B 19K. Mol. Cell 6:53-63. [PubMed] [Google Scholar]
- 40.Rigaut, G., A. Shevchenko, B. Rutz, M. Wilm, and B. Seraphin. 1999. A generic protein purification method for protein complex characterization and proteome exploration. Nat. Biotechnol. 17:1030-1032. [DOI] [PubMed] [Google Scholar]
- 41.Ruffolo, S. C., D. G. Breckenridge, M. Nguyen, J. S. Goping, A. Gross, S. J. Korsmeyer, H. Li, J. Yuan, and G. C. Shore. 2000. BID-dependent and BID-independent pathways for BAX insertion into mitochondria. Cell Death Differ. 7:1101-1108. [DOI] [PubMed] [Google Scholar]
- 42.Sattler, M., H. Liang, D. Nettesheim, R. P. Meadows, J. E. Harlan, M. Eberstadt, H. S. Yoon, S. B. Shuker, B. S. Chang, A. J. Minn, C. B. Thompson, and S. W. Fesik. 1997. Structure of Bcl-xL-Bak peptide complex: recognition between regulators of apoptosis. Science 275:983-986. [DOI] [PubMed] [Google Scholar]
- 43.Sawada, M., W. Sun, P. Hayes, K. Leskov, D. A. Boothman, and S. Matsuyama. 2003. Ku70 suppresses the apoptotic translocation of Bax to mitochondria. Nat. Cell Biol. 5:281-283. [DOI] [PubMed] [Google Scholar]
- 44.Schreiber, M., L. Sedger, and G. McFadden. 1997. Distinct domains of M-T2, the myxoma virus tumor necrosis factor (TNF) receptor homolog, mediate extracellular TNF binding and intracellular apoptosis inhibition. J. Virol. 71:2171-2181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Scorrano, L., and S. J. Korsmeyer. 2003. Mechanisms of cytochrome c release by proapoptotic BCL-2 family members. Biochem. Biophys. Res. Commun. 304:437-444. [DOI] [PubMed] [Google Scholar]
- 46.Scorrano, L., S. A. Oakes, J. T. Opferman, E. H. Cheng, M. D. Sorcinelli, T. Pozzan, and S. J. Korsmeyer. 2003. BAX and BAK regulation of endoplasmic reticulum Ca2+: a control point for apoptosis. Science 300:135-139. [DOI] [PubMed] [Google Scholar]
- 47.Seet, B. T., J. B. Johnston, C. R. Brunetti, J. W. Barrett, H. Everett, C. Cameron, J. Sypula, S. H. Nazarian, A. Lucas, and G. McFadden. 2003. Poxviruses and immune evasion. Annu. Rev. Immunol. 21:377-423. [DOI] [PubMed] [Google Scholar]
- 48.Stone, J. C., M. F. Moran, and T. Pawson. 1991. Construction and expression of linker insertion and site-directed mutants of v-fps protein-tyrosine kinase. Methods Enzymol. 200:673-692. [DOI] [PubMed] [Google Scholar]
- 49.Sundararajan, R., A. Cuconati, D. Nelson, and E. White. 2001. Tumor necrosis factor-alpha induces Bax-Bak interaction and apoptosis, which is inhibited by adenovirus E1B 19K. J. Biol. Chem. 276:45120-45127. [DOI] [PubMed] [Google Scholar]
- 50.Sundararajan, R., and E. White. 2001. E1B 19K blocks Bax oligomerization and tumor necrosis factor alpha-mediated apoptosis. J. Virol. 75:7506-7516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Thomas, M., and L. Banks. 1998. Inhibition of Bak-induced apoptosis by HPV-18 E6. Oncogene 17:2943-2954. [DOI] [PubMed] [Google Scholar]
- 52.Wasilenko, S. T., T. L. Stewart, A. F. Meyers, and M. Barry. 2003. Vaccinia virus encodes a previously uncharacterized mitochondrial-associated inhibitor of apoptosis. Proc. Natl. Acad. Sci. USA 100:14345-14350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wei, M. C., W.-X. Zong, E. H.-Y. Cheng, T. Lindsten, V. Panoutsakopoulou, A. J. Ross, K. A. Roth, G. R. MacGregor, C. B. Thompson, and S. J. Korsmeyer. 2001. Proapoptotic BAX and BAK: a requisite gateway to mitochondrial dysfunction and death. Science 292:727-730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yi, X., X. M. Yin, and Z. Dong. 2003. Inhibition of Bid-induced apoptosis by Bcl-2. tBid insertion, Bax translocation, and Bax/Bak oligomerization suppressed. J. Biol. Chem. 278:16992-16999. [DOI] [PubMed] [Google Scholar]
- 55.Zamzami, N., and G. Kroemer. 2003. Apoptosis: mitochondrial membrane permeabilization—the (w)hole story? Curr. Biol. 13:R71-R73. [DOI] [PubMed] [Google Scholar]