

Abstract
This study describes the physical and functional interactions between ICP0 of herpes simplex virus type 1 and class II histone deacetylases (HDACs) 4, 5, and 7. Class II HDACs are mainly known for their participation in the control of cell differentiation through the regulation of the activity of the transcription factor MEF2 (myocyte enhancer factor 2), implicated in muscle development and neuronal survival. Immunofluorescence experiments performed on transfected cells showed that ICP0 colocalizes with and reorganizes the nuclear distribution of ectopically expressed class I and II HDACs. In addition, endogenous HDAC4 and at least one of its binding partners, the corepressor protein SMRT (for silencing mediator of retinoid and thyroid receptor), undergo changes in their nuclear distribution in ICP0-transfected cells. As a result, during infection endogenous HDAC4 colocalizes with ICP0. Coimmunoprecipitation and glutathione S-transferase pull-down assays confirmed that class II but not class I HDACs specifically interacted with ICP0 through their amino-terminal regions. This region, which is not conserved in class I HDACs but homologous to the MITR (MEF2-interacting transcription repressor) protein, is responsible for the repression, in a deacetylase-independent manner, of MEF2 by sequestering it under an inactive form in the nucleus. Consequently, we show that ICP0 is able to overcome the HDAC5 amino-terminal- and MITR-induced MEF2A repression in gene reporter assays. This is the first report of a viral protein interacting with and controlling the repressor activity of class II HDACs. We discuss the putative consequences of such an interaction for the biology of the virus both during lytic infection and reactivation from latency.
The role of acetylation in the transcription pathway has been widely studied and is generally considered a major step toward gene expression (1). Two classes of enzymes with antagonist activities, called histone acetyltransferases (HATs) and deacetylases (HDACs), significantly contribute to the control of expression by the modification of the acetylation state of predominantly, but not exclusively, histone proteins (5, 70). Whereas HAT activity, and thus acetylation of histones, is generally associated with transcription, HDAC activity, and thus deacetylation, is responsible for repression (69). Three groups of HDACs, defined as classes I, II, and III, have been described in higher eukaryotes, according to sequence similarities with yeast RPD3, HDA1 and SIR2 proteins, respectively (reviewed in references 43 and 73). Class II HDACs have been classified as such mainly on the basis of amino acid sequence homologies with the deacetylation domain of members of the class I family. However, for most of them, and unlike class I HDACs, clear evidence concerning their direct implication in deacetylation of substrates is lacking. It is now thought that, with the exception of HDAC6, class II HDACs acquire their deacetylation activity only through the interaction of their carboxy-terminal end with at least the class I HDAC3 and the nuclear receptor corepressor SMRT (for silencing mediator of retinoid and thyroid receptor) (25, 26, 39, 80). Therefore, the contribution of class II HDACs to the regulation of the activity of gene promoters is suggested by their identification as components of multiprotein nuclear complexes known as Sin3/HDAC and NuRD/Mi2/NRD (nucleosome remodeling histone deacetylase complex), which are implicated in repression of gene expression (reviewed in reference 44). However, class II HDAC4, -5, and -7 possess an additional repressor domain in the amino-terminal extension of the sequence that is independent of the HDAC3/SMRT-associated catalytic deacetylase activity (42, 46, 56, 75, 82). This domain interacts with and represses the activity of the transcription factor myocyte enhancer factor 2 (MEF2), which is involved in the differentiation and/or survival of muscles and neurons (27, 54, 59, 77). It is now clear that class II HDACs play a significant role in the control of these processes by maintaining MEF-2 under an inactive form in the nucleus (reviewed in reference 73).
Herpes simplex virus type 1 (HSV-1) is a major human pathogen that is characterized by an outstanding mode of infection made of permanent switches between lytic cycles and latency. After multiplication of the virus in epithelial cells, HSV-1 infects neurons and delivers its 152-kb double-stranded linear DNA to the nucleus. Under certain conditions it starts an abortive acute infection involving limited replication of the viral genome and then enters latency until reactivation after appropriate stimuli. One of the major differences between lytic cycle and latency is encountered at the level of gene expression. Indeed, whereas during acute lytic infection the entire viral genome is expressed, during latency the genome undergoes an almost-complete gene expression shutoff (reviewed in references 61 and 74).
HSV-1 virion is composed of a capsid containing the genome surrounded by an envelope, both nucleocapsid and envelope being separated by the tegument. About 80 temporarily regulated genes are present in the viral genome. Genes and gene products are subdivided into three major families—immediate-early (IE), early, and late—depending on the time course of synthesis and requirement for prior viral gene expression and DNA replication (24). ICP0 is one of the five IE proteins encoded by the virus. It is a RING finger zinc-binding protein capable of activating gene expression, from viral and cellular promoters, without binding DNA (7, 12). Viruses unable to synthesize functional ICP0 are impaired in their ability to initiate a productive lytic infection, to reactivate from latently infected neurons, and to have a higher propensity to enter a nonreplicating quiescent state after infection of cultured cells at a low multiplicity (6, 8, 14, 17, 29, 33, 34, 45, 63, 67, 68). The tendency of ICP0-deficient viruses to be silenced suggests that a cellular repressor mechanism could control HSV-1 genome transcription and/or replication, and ICP0 could interfere with this mechanism. The control of the stability of a putative repressor protein by ICP0 became a growing postulate after the discovery that ICP0 was inducing the degradation of several cellular proteins through active ubiquitin-proteasome pathway (reviewed in reference 18). Several recent studies described an E3 ubiquitin ligase activity associated with ICP0, thus confirming the direct involvement of ICP0 in this degradation pathway (3, 4, 32, 71). However, ICP0 also interacts with several cellular components without inducing their degradation (reviewed in reference 18), and it is likely that these interactions will account for its contribution to lytic infection and reactivation from latency. Indeed, quiescent viruses can be reactivated by later expression of ICP0 from external sources or, but with less efficiency, by the addition of the inhibitor of HDACs, trichostatin A (TSA) (23, 35, 36, 61, 64, 67). Based on these data, we investigated the putative physical and functional interactions between ICP0 and class I or class II HDACs. We demonstrate that ICP0 specifically interacts with the amino-terminal region of class II HDAC4, -5, and -7, resulting in the offset of the repression activity associated with this domain. Consequently, the expression of ICP0 can counteract the repressor activity of HDAC5 and the class II HDACs-related MITR (for MEF2-interacting transcription repressor) protein on MEF2A, therefore enhancing significantly its transcriptional activity. However, unlike TSA, ICP0 does not inhibit the global deacetylation activity in the cell, which highlights a major difference between ICP0 and TSA concerning mechanisms implicated in the reactivation of quiescent viruses.
MATERIALS AND METHODS
Viruses, cells, and preparation of cell extracts.
The HSV-1 strain 17 syn+ (17+) is the parental viral strain. ICP0 mutant virus vFXE and the D30EBA virus were described previously (17, 60). HeLa cells were grown at 37°C in Glasgow modified Eagle medium containing 10 U of penicillin/ml and 100 μg of streptomycin/ml and supplemented with 10% fetal bovine serum. For cell extracts, cells were washed once with phosphate-buffered saline (PBS), scraped off the flask, and then centrifuged at 1,000 × g for 5 min. The cell pellet was resuspended in 200 μl of a lysis buffer containing 15 mM Tris-HCl (pH 7.5), 2 mM EDTA, 0.25 mM EGTA, 15 mM NaCl, 0.3 mM sucrose, 0.5% Triton X-100, and 300 mM KCl. Samples were incubated on ice for 30 min and centrifuged at 6,000 × g for 5 min at 4°C to remove all debris. For glutathione S-transferase (GST) pull-down assays, 400 μl of lysis buffer without KCl, to obtain a 100 mM KCl final concentration, was added to the supernatant. For immunoprecipitation, supernatants were flowed through Micro Bio-Spin 6 chromatography columns (Bio-Rad) preequilibrated with a buffer containing 20 mM HEPES (pH 7.9), 0.2 mM EDTA, 100 mM KCl, and 25% glycerol. The protease inhibitors phenylmethylsulfonyl fluoride and protease inhibitor cocktail tablets (Roche) were added to the lysis buffers. Protein concentrations were determined by the Bradford method.
Chromatin-associated protein extraction was done by scraping cells off the plates, resuspending them in PBS-phenylmethylsulfonyl fluoride, and sonicating them in a ultrasonic water bath for twice for 15 s each time. Nuclei and debris were centrifuged at 12,000 × g for 10 min at 4°C. Pellets were resuspended in HCl (250 mM) and incubated on ice for 30 min before centrifugation at 12,000 × g for 20 min at 4°C. Trichloroacetate was added to the supernatant to a final volume of 20%, and incubation was carried out for another 30 min on ice before centrifugation at 12,000 × g for 20 min at 4°C. Pellets were washed once with 500 μl of a mixed acetone-HCl (10 mM) solution and once with acetone alone and then dried in a SpeedVac (Savant) for 5 min before resuspension in water and dosage.
DNA constructs.
Plasmids pCi110 and pCiFXE express ICP0 and its RING finger mutant FXE, respectively, from a pCineo-based plasmid (21). Hemagglutinin (HA)- and FLAG-tagged HDACs, GAL4-HDAC5 fusion protein, pCMVβgal, pcDNAGAL4-DB, L8G5-Luc, and LEXA-VP16 expressing plasmids have already been described (46, 76). GAL4- and GST-HDAC3 fusion protein-expressing plasmids were constructed by inserting the EcoRI and BamHI fragment from the pCEP4F-HD3 plasmid (76) containing the entire HDAC3 gene in frame with the tag in the pcDNAGAL4 and pGEX4T1 (Pharmacia) plasmids, respectively.
Fusion protein GST-ICP0(1-241) expressing plasmid has been described earlier (55). GST-ICP0(1-109)- and GST-ICP0(106-241)-expressing plasmids were constructed by cloning PCR fragments obtained with the primers 5′-CGCGGATCCCCATGGAGCCCCGCCCCGGA-3′ and 5′-CGGAATTCTACCCCCCGTCCTCTCGAG-3′ for the former and 5′-CGCGGATCCCCGAGGACGGGGGGAGCGACGA-3′ and 5′-CGGAATTCTAGTCGTCCAGGTCGTCGTCAT-3′ for the latter. After digestion by BamHI and EcoRI, fragments were cloned into the BamHI and EcoRI site of the vector pGEX-3X (Pharmacia). Both constructs were verified by sequencing. GST-ICP0(245-518) and GST-ICP0(553-775) were constructed by cloning in pGEX-3X the fragment SnaBI-MluI for the former and AatII-HpaI for the latter, retrieved from the pJR3-ICP0 gene-containing plasmid (19). Constructs expressing GST-HDAC4(1-650), -HDAC5, and -HDAC7 have already been described (42, 46, 56). GST-HDAC4(569-1081) was constructed by cloning in pGEX-5X-2, a XhoI fragment obtained from the HA-tagged HDAC4 plasmid. GST-HDAC3(1-429) has been constructed by inserting a XhoI fragment from GAL4-HDAC3 in a pGEX-4T1 plasmid. Expression and purification of GST fusion proteins was done as described previously (65). All of the constructs were analyzed for protein expression by Coomassie staining and Western blotting with anti-GST (Sigma) or anti-GAL4 (Clontech) monoclonal antibodies (MAbs). HDAC4, -5, and -7 are of mouse origin, and HDAC3 is of human origin.
GST pull-down assays.
A total of 50 μg of GST fusion protein preparation was mixed with lysis buffer (100 mM KCl) containing 1 mg of protein from cellular extracts or 5 μl of the sample from in vitro-synthesized proteins. All extracts were first incubated for 1 h at 4°C with continuous mixing with beads linked to the GST protein expressed from vector plasmid pGEX-3X in order to reduce the background signal. The precleared extracts were then incubated with the appropriate GST fusion protein beads and a negative GST bead control for 2 h at room temperature with continuous mixing. The beads were harvested by brief centrifugation and then washed five times with 500 μl of the lysis buffer (100 mM KCl). Protein complexes were directly eluted from the beads by adding Laemmli buffer and then boiled.
Immunoprecipitations.
HeLa cells were seeded at 1.2 × 106 cells per 60-mm petri dish. The following day, cells were transfected with the adequate plasmids. Extracts from transfected HeLa cells were then prepared as described above and preincubated 1 h at 4°C with protein A-Sepharose beads (Amersham) to decrease nonspecific binding. Samples were then centrifuged, and the supernatants were incubated with the appropriate antibodies for 1 h at 4°C. Then, 50 μl of a 50% slurry protein A-Sepharose preequilibrated with the same extract-containing buffer was added to the samples, followed by incubation for another hour. The samples were then centrifuged briefly and washed five times with buffer before the beads were suspended in Laemmli buffer and then boiled.
siRNA experiments.
HeLa cells were seeded at 2 × 105 cells per well in six-well Linbro multiwell plates and transfected (Effectene transfection kit; Qiagen) the following day with 840 ng of small interfering RNA (siRNA). After 48 h, the cells were scraped off the plates in PBS buffer before protein dosage by the Bradford method, the addition of Laemmli buffer, and Western blotting. The characteristics of the siRNAs used in the present study are as follows: siRNAac4_337, 5′-GCAACAACAGGAGAUGCUGtt-3′ (sense) and 5′-CAGCAUCUCCUGUUGUUGCtt-3′ (antisense) (target sequence in human HDAC4 gene, nucleotides 337 to 357); siRNAac4_1513, 5′-CAAGAUCAUCCCCAAGCCAtt-3′ (sense) and 5′-UGGCUUGGGGAUGAUCUUGtt-3′ (antisense) (target sequence in human HDAC4 gene, nucleotides 1513 to 1533); and siRNAac4_2665, 5′-CGUCAACAUGGCUUUCACCtt-3′ (sense) and 5′-GGUGAAAGCCAUGUUGACGtt-3′ (antisense) (target sequence in human HDAC4 gene, nucleotides 2665 to 2685). The siRNA for human lamin A/C has been previously described (16).
Western blotting.
Samples from GST pull-down, immunoprecipitation, and siRNA assays were treated for Western blotting according to a protocol described earlier (48). The primary antibodies were MAbs 11060 anti-ICP0 (1/10,000), 12CA5 anti-HA-1 (Roche), or M5 anti-FLAG (Sigma) epitopes (1 μg/ml), Do-7 anti-p53 (1/100; Dako), and rabbit polyclonal sera, ML-19 anti-human HDAC4 (a generous gift from Sigma), anti-lamin A/C (Santa Cruz), anti-actin (Sigma), anti-acetylated histone H4 (AcH4; Santa Cruz), and anti-acetylated lysine (1 μg/ml; AcLys; Cell Signaling).
In vitro protein synthesis.
Plasmid pT7110 containing ICP0 encoding cDNA (22) was used for in vitro synthesis of [35S]methionine-labeled ICP0 by using a rabbit reticulocyte lysate system (TNT Quick-Coupled Transcription/Translation System; Promega) according to the manufacturer's recommendations. Of the 50-μl final volume, 5 μl was used per GST pull-down assay. Pull-down assays were performed as described above by adjusting sample volumes to 500 μl with the lysis buffer (100 mM KCl). Samples were then loaded onto a sodium dodecyl sulfate-polyacrylamide gel and electrophoresed. Gels were then incubated for 10 min in fixing solution (50% methanol, 10% acetic acid) and for 10 min in soaking solution (7% methanol, 7% acetic acid, 11% glycerol). Gels were dried at 80°C under vacuum for 30 min and then exposed to film.
GAL4 binding assay.
HeLa cells were seeded at 1.2 × 106 cells per 60-mm petri dish. The following day, cells were transfected with 400 ng of L8G5-Luc reporter and 100 ng of LexA-VP16 expression vector and a combination of empty pcineo plasmid or plasmid expressing ICP0 or parts of HDAC5 fused to GAL4 DNA-binding domain (GAL4-DB). A total of 100 ng of a β-galactosidase-expressing vector were also used in each transfection. At 24 h posttransfection, cells were washed with PBS, scraped out of the dish in 10 ml of PBS, and centrifuged at 1,000 × g for 4 min. Cell pellets were resuspended in 200 μl of lysis buffer compatible for the luciferase-β-galactosidase reporter gene assays (Roche), incubated at room temperature for 15 min, and centrifuged for 5 min at 3,000 × g. Supernatants were then transferred in new tubes and kept on ice until use. Then, 50 μl was used for each assay, and the luciferase activity was measured according to the protocol provided with the luciferase assay system kit (Promega) and normalized with respect to the activity of β-galactosidase measured according to the protocol provided with the β-galactosidase reporter gene assay kit (Roche).
Immunofluorescence and confocal microscopy.
HeLa cells were treated according to a protocol described previously (48). The data from channels were collected separately with eightfold averaging at a resolution of 1,024 × 1,024 pixels. The microscope was a Zeiss Axioplan utilizing a ×63 oil immersion objective lens (NA 1.4). Images were acquired by using the Zeiss LSM 510 META confocal microscopy software and then exported to Adobe Photoshop for processing.
The antibodies used for immunofluorescence analyses were MAb 11060 (1/1,000) and rabbit serum R190 (1/200) anti-ICP0 (20), MAb anti-ICP4 (1 μg/ml; U.S. Biologicals), MAb 12CA5 anti-HA (1 μg/ml; Boehringer Mannheim), MAb M5 anti-FLAG (1 μg/ml; Sigma), ML-19 rabbit anti-HDAC4 (1 μg/ml; Sigma), 4/21 rabbit anti-SMRT (1/200; a generous gift from Hung-Ying Kao, Case Western Reserve University, Cleveland, Ohio). The secondary antibodies used were Alexa 488-conjugated goat anti-mouse and anti-rabbit (1/200; Molecular Probes) and Cy5-conjugated goat anti-mouse and anti-rabbit (1/200; Amersham) antibodies.
RESULTS
ICP0 colocalizes with and redistributes overexpressed class I and II HDACs.
Immunofluorescence experiments were performed on cotransfected HeLa cells expressing ICP0 and an exogenous tagged version of either class I or class II HDACs. Overexpression of ICP0 alone results in the formation of ring-shaped structures in the nucleus (see Fig. 3Bi and ii, ICP0 panels). Coexpression of ICP0 with class II HA-tagged HDAC4, -5, or -7 showed a clear redistribution of these HDACs that colocalized with ICP0 (Fig. 1Ai to iii). The same experiments were done with exogenous expressed FLAG-tagged class I HDAC1, -2, or -3. The results obtained were slightly different from those seen with class II HDACs since class I HDACs preferentially localized inside the ICP0 rings (see Fig. 1Aiv, HDAC3, as an example). Hence, independent of the class they belong to, the distribution of HDACs (either diffused or in small dots, Fig. 1A small left panels) was reorganized in the presence of ICP0, a result not observed with other FLAG- or HA-tagged proteins (data not shown). These results, although obtained from HDAC overexpressing cells, at least suggested a physical interaction between ICP0 and class I and/or class II HDACs that is strong enough to modify the distribution of overexpressed HDACs.
FIG. 3.

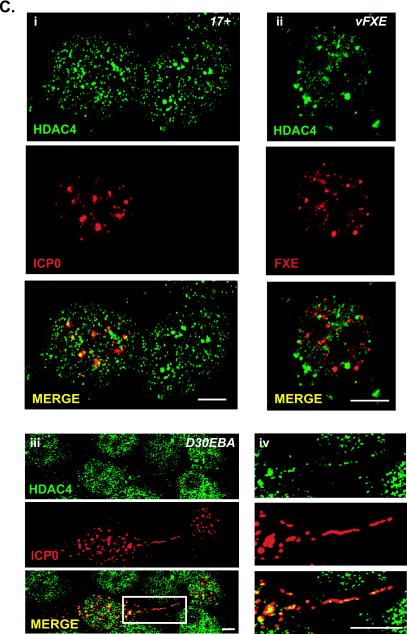
Colocalization of endogenous cellular proteins with ICP0. (A) Control of the specificity of the anti-HDAC4 ML-19 rabbit polyclonal serum by siRNA. (a) HeLa cells were transfected with three different siRNAs, ac4_334, ac4_1513, ac4_2665, targeting 5′, middle, and 3′ HDAC4 mRNA, respectively (for more details see Materials and Methods). After cell lysis, 10 μg of proteins was loaded for each sample, on an SDS-7.5% polyacrylamide gel and Western blots were performed with ML-19, anti-lamin A/C, and anti-actin rabbit polyclonal sera. Lamin siRNA was used as a control for specificity of the siRNA assay and for the detection of actin for protein loading. (b) HeLa cells were mock transfected (i) or transfected with siRNAac4_1513 (ii) or siRNAac4_2665 (iii) for 72 h and then treated for immunofluorescence with ML-19 to visualize the endogenous HDAC4. (B) HeLa cells were transfected with an ICP0-expressing plasmid and then analyzed at 24 h posttransfection by immunofluorescence for the detection of endogenous HDAC4 (i) and SMRT (ii) (upper right cell represents normal SMRT distribution). (C) Cells were infected for 4 h with wild-type 17+ (i) or vFXE ICP0 mutant virus (ii) or 5 h with D30EBA virus (iii and iv) at an MOI of 5 before immunofluorescence was performed. Panel iv shows a higher magnification of the region of panel iii marked by the white rectangle. ICP0, HDAC4, and SMRTwere detected by using MAb 11060, ML-19, and 4/21 rabbit anti-SMRT, respectively. The secondary antibodies used were Alexa 488-conjugated goat anti-rabbit antibody (1/200) and Cy5-conjugated goat anti-mouse immunoglobulin G (1/200). Images were collected as for Fig. 1. Bars, 5 μm.
FIG. 1.
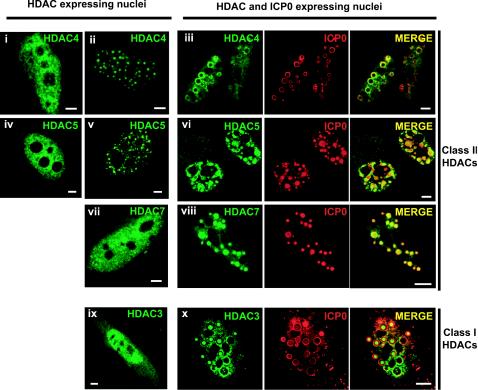
Nuclear redistribution of ectopically expressed epitope-tagged class I or II HDACs in HeLa cells expressing ICP0. Nuclear distribution of HA epitope-tagged class II HDAC4 (i to iii), 5 (iv to vi), 7 (vii and viii), and FLAG epitope-tagged class I HDAC3 (ix and x) expressed either alone (left images) or with ICP0 (right images). ICP0 and tagged HDACs were detected by using anti-ICP0 rabbit polyclonal antibody R190 and anti-HA (12CA5) or anti-FLAG (M5) MAbs. The secondary antibodies used were Alexa 488-conjugated goat anti-mouse (1/200) and Cy5-conjugated goat anti-rabbit (1/200) antibodies. Cell samples were examined with a Zeiss LSM 510 META confocal microscope with two lasers giving excitation lines at 488 and 633 nm as described in Materials and Methods. Bars, 5 μm.
ICP0 physically interacts with class II HDACs.
To check whether redistribution of exogenous HDACs by ICP0 is a consequence of their physical interaction, coimmunoprecipitation experiments were done on cotransfected HeLa cells expressing exogenous class I or II HDACs and ICP0. ICP0 was coimmunoprecipitated with class II HDAC4, -5, and -7 (Fig. 2A) but not class I HDAC1, -2, and -3 (data not shown). These interactions were confirmed by GST pull-down assays by using either GST-HDACs or GST-ICP0 fusion proteins (Fig. 2Cii). The noncatalytic amino-terminal regions of HDAC4 (amino acids 1 to 650), -5 (amino acids 123 to 673), and -7 (amino acids 1 to 506) were sufficient to interact with ICP0, whereas a fusion protein made of GST fused to the entire HDAC3 (amino acids 1 to 429) was unable to capture ICP0 (Fig. 2B). Similarly, the amino acid region from positions 106 to 241 of ICP0 contained the interaction domain with class II HDAC4, -5, and -7, but not class I HDAC1, -2, and -3 (HDAC3 is shown as an example) (Fig. 2Ci). To map the domain of interaction within HDAC sequences, we performed additional GST pull-down assays by using GST-HDAC5 fusion proteins. ICP0 strongly interacted with the amino acid region from 123 to 673, and regions 123 to 292 and 242 to 450 still possessed, although with a weaker affinity, the capacity to bind ICP0 (Fig. 2D). Finally, reverse GST pull-down assays performed with in vitro-translated proteins confirmed that the interaction between ICP0 (Fig. 2E) and class II HDACs (Fig. 2F, HDAC7 shown as an example) are likely to be direct. Interactions are specific for class II HDACs because in the same experimental conditions class I HDAC1, -2, and -3 never showed any binding to ICP0. This result was expected because ICP0 interacts with the amino-terminal region of class II HDACs, which is not conserved in class I HDACs. These results show that a class II-specific interaction between ICP0 and HDACs exists and confirm the observations made by confocal microscopy.
FIG. 2.
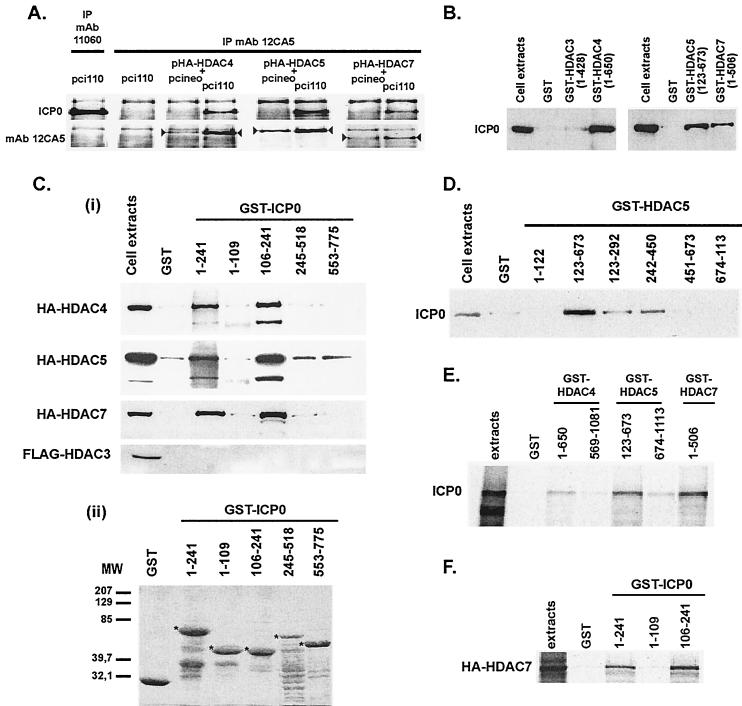
Physical interaction between ICP0 and class II HDACs and mapping of the interaction sites. (A) Coimmunoprecipitation of ICP0 with HA epitope-tagged class II HDAC4, -5, or -7. MAb 12CA5 was used for the immunoprecipitation (IP), and ICP0 was detected by Western blotting. Detection of the immunoprecipitated tagged-HDACs (arrowheads) was done as a control of HDAC expression. Immunoprecipitation of ICP0 with MAb 12CA5 (second upper left image) and detection of MAb 11060-immunoprecipitated ICP0 by MAb 12CA5 (first bottom left image) showed no cross-reactivity of ICP0 with this antibody. (B and Ci) GST pull-down experiments showing the capture of ICP0 (from ICP0-expressing cell extracts) (B) or tagged HDACs (from HDACs overexpressing cell extracts) (Ci) by GST-HDACs or GST-ICP0 fusion proteins, respectively. (Cii) Coomassie gel showing the expression of the GST-ICP0 fusion proteins (5 μg of proteins [✽]) used in panel Ci. (D) Mapping of the ICP0-interacting regions in HDAC5 protein by GST pull-down with GST-HDAC5 fusion proteins. (E and F) GST pull-down experiments with in vitro-synthesized ICP0 or HA-HDAC7, respectively. ICP0 and HA- or FLAG-tagged HDACs were detected in Western blots with MAbs 11060, 12CA5, or M5, respectively.
ICP0 redistributes and colocalizes with endogenous HDAC4.
To check whether ICP0 could affect the distribution of an endogenous class II HDAC, a rabbit polyclonal serum against HDAC4 was used to perform immunofluorescence analyses on ICP0-expressing HeLa cells. The specificity of the ML-19 anti-HDAC4 rabbit serum was checked by Western blot analyses of HeLa cell extracts transfected with several siRNAs targeting HDAC4 mRNA. One of three siRNAs, i.e., siRNAac4_2665, affected the signal of the HDAC4 135-kDa band detected by the serum (Fig. 3Aa). As a control, a siRNA targeting the lamin A/C mRNA showed no decrease of HDAC4. To confirm the accuracy of the anti-HDAC4 serum in immunofluorescence assays, we analyzed the endogenous HDAC4 staining in cells either mock transfected or transfected with siRNAac4_1513 or siRNAac4_2665 (Fig. 3Abi to iii). All of the data were collected by using strictly the same confocal parameters for the pinhole, optical slide, detector gain, amplifier offset, amplifier gain, contrast, and brightness. The decrease of the endogenous HDAC4 signal in cells transfected with siRNAac4_2665 corresponded to the observation made in Western blot and proved the accuracy of the anti-HDAC4 serum when used in immunofluorescence. Cells showed a relatively diffuse nuclear staining of HDAC4 (Fig. 3Abi), although high magnification highlighted a dotted phenotype diffusely distributed in the whole nucleus and some weak staining in the cytoplasm (not visible here due to contrast/brightness effect). Expression of ICP0 in transfected cells resulted in a change in HDAC4 distribution, which concentrated within the ICP0 rings (Fig. 3Bi). To determine whether the distribution of an HDAC4-interacting protein would also be affected, we similarly detected endogenous SMRT. As observed for HDAC4, SMRT distribution was also modified (Fig. 3Bii, compare upper right with lower left nuclei). Similar results were obtained in cotransfected cells expressing ICP0 and exogenous SMRT (data not shown). Staining with DAPI (4′,6′-diamidino-2-phenylindole) showed no DNA in these structures (data not shown), suggesting that ICP0 rings are composed of several proteins probably present at a high density. Such proteinated structures could attract, nonspecifically, any primary or secondary antibody, which could result in false interpretations. We therefore intensively looked at and ruled out any kind of artifacts that could come from experimental procedure (i) by using different anti-mouse and anti-rabbit secondary antibodies labeled with different fluorochromes; (ii) by using secondary antibodies highly cross-adsorbed on human, rabbit, and bovine antigens; (iii) by collecting data by sequential laser scanning; and finally (iv) by using Alexa 488- and Cy5-conjugated secondary antibodies to avoid any putative channel overlapping. In addition, the detection of several other proteins not supposed to interact with class II HDACs did not show such redistribution in ICP0-expressing cells (data not shown). These data indicate that ICP0 modify the overall distribution of HDAC4, probably as a result of their interaction, and suggest that other class II HDACs could behave in a similar manner. In addition, our results suggest that, similarly to SMRT, other class II HDAC-interacting proteins are likely to localize within the ICP0 structures.
To verify by immunofluorescence whether endogenous HDAC4 could colocalize with ICP0 during infection, cells were infected for 4 to 5 h with wild-type virus 17+, the ICP0 mutant virus vFXE, or the replication-defective HSV-1 virus D30EBA (60) at a multiplicity of infection (MOI) of 5 (Fig. 3C). D30EBA does not express the major viral transactivator ICP4 but overexpresses functional ICP0, which localizes 5 h postinfection in the nucleus and, unlike wild-type virus, in the cytoplasm of HeLa cells. Immunofluorescence labeling was done as described above. As expected, most nuclear and cytoplasmic ICP0 costained with HDAC4 (Fig. 3Ci, iii, and iv). Although class II HDACs are known to shuttle from the nucleus to the cytoplasm, the cytoplasmic signal of HDAC4 is too weak compared to the nuclear straining to be clearly visible in noninfected HeLa cells. Therefore, the accumulation of some HDAC4 within cytoplasmic ICP0 aggregates in cells infected with D30EBA highlights the stability of the interaction between the two proteins (Fig. 3Ciii and iv). Moreover, HDAC4, which we observed to form bigger aggregates in cells infected by vFXE, does not show a clear colocalization with FXE (Fig. 3Cii). This is in accordance with the GST pull-down data from Fig. 2, showing that the interaction site of ICP0 with class II HDACs lies in the amino acid domain of ICP0(106-241) that is partly deleted in the FXE mutant protein. Finally, no degradation of endogenous HDAC4 was detected by Western blotting, in cells infected with wild-type virus (data not shown), which suggests that ICP0 does not interact with class II HDACs to induce their degradation through the proteasome pathway.
ICP0 does not change the acetylation state of histone H4 and p53 proteins.
Because ICP0 and TSA have similar properties in regard to reactivation of quiescent viruses, we wanted to analyze whether expression of ICP0 could result in an overall increase of cellular acetylation activity. The acetylation of representative histone and nonhistone proteins, i.e., histone H4 and p53 was thus analyzed in infected and transfected cells, respectively. Cells were infected with the D30EBA replication-defective HSV-1 virus at an MOI of 10 so that all cells were infected by at least one virus. At 24 h postinfection, cells were harvested and treated to extract nuclear proteins. Unlike TSA-treated cells, infected cells did not show any modification of the amount of acetylated histone H4 either by immunoblotting (Fig. 4A) or by immunofluorescence performed at 2, 4, 6, and 24 h postinfection (data not shown). On the other hand, several nonhistone proteins, such as the proapoptotic transactivator p53, can be acetylated (30). To analyze whether a putative biological change could, similarly to TSA, nonspecifically affect the acetylation of nonhistone proteins in cells expressing ICP0, HeLa cells were cotransfected with plasmids expressing p53 and ICP0 or FXE, the inactive ICP0 mutant protein. Total acetylated proteins were immunoprecipitated and blotted against p53 antibodies to determine whether the level of acetylation of p53 changed in cells expressing ICP0. No specific increase of acetylated p53 proteins was observed in the presence of ICP0, unlike the CREB-binding protein (CBP), which is known to specifically acetylate p53 (Fig. 4B). The slight increase of acetylated p53 in cells coexpressing ICP0 correlated with an overall increase of the amount of p53 synthesized (as shown by Western blotting analysis of total cell extracts). This is most likely due to the unspecific and already described activation, by ICP0, of the cytomegalovirus IE promoter that drives the expression of the p53 ectopic gene. Taken together, these results suggest that the interaction of ICP0 with class II HDACs, both in infected and transfected cells, is unlikely to result in general cellular changes that would modify, at least nonspecifically and globally, the acetylation state of histone and nonhistone proteins. This major difference between ICP0 and TSA underlines two potentially different mechanisms by which the drug and the viral protein would affect reactivation of quiescent viruses.
FIG. 4.
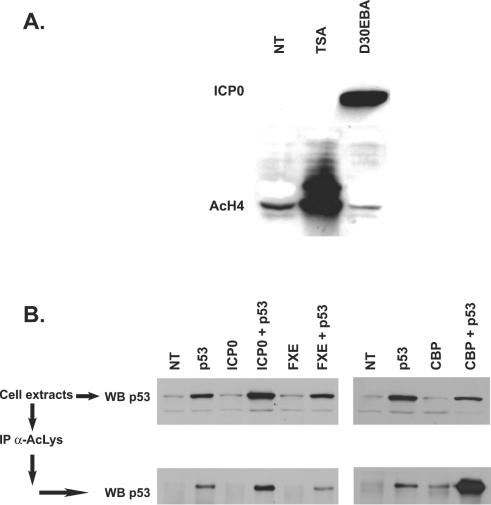
ICP0 does not modify the global acetylation activity in cells. (A) HeLa cells were infected at an MOI of 10 for 24 h by replication-defective D30EBA ICP0-overexpressing virus or treated with TSA (2 μM) for 12 h before chromatin-associated protein extraction (see Materials and Methods) and Western blotting for the detection of acetylated histone H4 (AcH4). (B) HeLa cells were transfected with indicated plasmids. At 24 h posttransfection, Western blotting was performed on cell extracts or, alternatively, acetylated proteins were immunoprecipitated with anti-acetylated lysines MAb (α-AcLys) before Western blotting. Then, 10 μg of protein was loaded for each sample on SDS-7.5% or 12.5% polyacrylamide gels for analysis. ICP0, p53, AcH4, and AcLys were detected by using MAbs 11060 and Do-7, and anti-AcH4 and anti-AcLys were detected by using rabbit polyclonal sera.
ICP0 counteracts the repressor activity of class II HDACs.
In addition to their HDAC3/SMRT-associated deacetylase domain, located in the carboxy-terminal part of the protein, HDAC4, -5, and -7 possess another repressor domain in their amino-terminal region. We thus performed GAL4-binding luciferase reporter gene assays to check whether ICP0 could affect this repressor activity (Fig. 5A). ICP0 was able to overcome, in a dose-dependent manner, the repressor activity associated to the amino-terminal part of HDAC5 (amino acids 123 to 673) (lanes 3 to 7). Conversely, ICP0 had no effect on the repression activity associated to the carboxy-terminal region of HDAC5 (amino acids 674 to 1113, lanes 8 and 9) or to HDAC3 (lanes 10 and 11), which do not interact with ICP0.
FIG. 5.
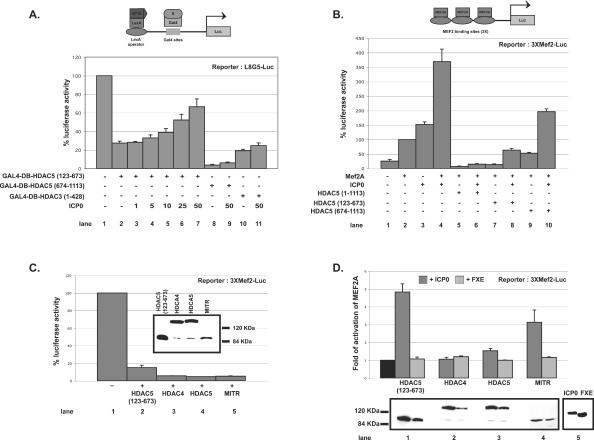
ICP0 counteracts the repressor activity associated with the amino-terminal region of class II HDACs. (A) HeLa cells were transfected with 400 ng of L8G5-Luc reporter and 100 ng of LexA-VP16 expression vector (46), and a combination of the indicated amount of plasmids expressing ICP0 (in nanograms) and 50 ng (+) of HDAC5 or HDAC3 proteins fused to GAL4 DNA-binding domain (GAL4-DB) (46) (see Materials and Methods). A value of 100% represents the luciferase activity calculated in the absence of HDAC5 coexpression but in the presence of 50 ng of vectors expressing the GAL4-DB; standard deviations are indicated. Lanes 2, 8, and 10 represent the controls with empty pcineo vectors. (B) HeLa cells were transfected with 1 μg of 3XMEF2-Luc reporter plasmid and as indicated (+): 200 ng of pMT2-MEF2A expressing plasmid (46) and/or plasmids expressing ICP0 (25 ng) and/or full-length or truncated HA-tagged HDAC5 (25 ng). A value of 100% represents the mean value of luciferase activity calculated in the absence of HDAC5 coexpression; standard deviations are indicated. Lanes 2, 5, 7, and 9 represent the controls with empty pcineo vectors. (C) Comparison of repression activities of truncated HDAC5(123-673), HDAC4, HDAC5, and MITR on MEF2A transcriptional activity. Experimental conditions were similar to those for panel B. A value of 100% represents the value of luciferase activity calculated in the absence of any repressor coexpression. A Western blot is shown to compare the relative amounts of each protein. (D) Same experiment as in panel C, but with the addition of 25 ng of plasmid expressing ICP0 or its RING finger mutant FXE. The black bar represents the normalized MEF2A activity in the presence of the repressor protein but in the absence of ICP0 or FXE. Western blots are shown to compare the relative amounts of each repressor protein expressed in the presence of ICP0 or FXE (lanes 1 to 4) and to show a representative amounts of ICP0 and FXE expression in all of the samples (lane 5). Western blots were performed by using 12CA5 or 11060 MAbs for the detection of HA-tagged HDACs and MITR or ICP0 and FXE, respectively. A total of 100 ng of β-galactosidase expression vector was used in each transfection for protein amount standardization. pcineo plasmid was used in all assays to keep the amount of transfected plasmid constant as a negative control. The luciferase activity was measured 24 h posttransfection and normalized with respect to that of β-galactosidase. All data resulted from at least three independently repeated assays; standard deviations are indicated.
The amino-terminal region of class II HDACs interacts with and inactivates MEF2 transcription factors. To confirm the results presented above, we investigated whether ICP0 could counterbalance the HDAC5-induced MEF2A repression. Luciferase reporter gene assays were performed with MEF2A as an activator in the presence of HDAC5 and/or ICP0 (Fig. 5B). As already described (46), full-length and amino-terminal (amino acids 1 to 673) HDAC5 strongly repressed MEF2A activity (lanes 5 and 7), unlike carboxy-terminal HDAC5 (amino acids 674 to 1113) (lane 9). The addition of ICP0 had a striking effect on the MEF2A activity. First, whereas ICP0 had little effect on its own on the MEF2 activated promoter (lane 3), it synergistically increased MEF2A activity in the absence of over expressed HDAC5 (lane 4) or in the presence of HDAC5 (amino acids 674 to 1113) (that does not bind MEF2A, lane 10). Second, ICP0 is capable to significantly release the HDAC5(123-673)-induced repression of MEF2A (compare lanes 7 and 8). All of these tests were standardized with an internal β-galactosidase control to avoid variation due to transfection efficiency and the level of protein expression. This ruled out any increase of MEF2A activity due to an increase of the protein amount when both ICP0 and MEF2A were coexpressed in the same cell. The two experiments above show that the repressor activity associated to the amino-terminal region of a class II HDACs could be overcome by ICP0. However, in our experimental conditions, ICP0 is unable to counterbalance the repressor activity of the HDAC5 carboxy-terminal domain that contains the deacetylase-associated catalytic activity (Fig. 5A, lane 9). This supports the results from Fig. 4B and confirms that rather than having a general effect on deacetylase-associated repressor activity, ICP0 is likely to specifically and locally (at the level of a promoter) counterbalance the repression induced by the amino-terminal region of class II HDACs on interacting partners such as MEF2.
We then analyzed the effect of ICP0 on the repressor activity of the MEF2-interacting transcription repressor (MITR) compared to truncated HDAC5 (amino acids 123 to 673) and full-length HDAC4 and HDAC5. Human MITR is a 590-amino-acid protein resulting from an alternative splicing of HDAC9 mRNA and shares high homologies with the amino-terminal region of HDAC4 and -5. It interacts with MEF2 but lacks any HDAC catalytic domain (66, 81, 82). We first compared their repression effect on MEF2A transcriptional activity. MITR, although expressed at a lower amount (Fig. 5C, Western blot data), repressed MEF2A at a level similar to that observed with HDAC4 or -5 (Fig. 5C, graph data). We then performed the same experiments in the presence of ICP0 or of its RING finger (amino acids 106 to 150) deletion mutant FXE. ICP0 enhanced MEF2A activity by about three- to fourfold in the presence of MITR compared to HDAC4 and -5 (Fig. 5D, graph data). As expected, the FXE mutant, which lacks the HDAC4, -5, and -7 binding site (see Fig. 2), is unable to counterbalance the repressive effect of any of the proteins. The increase in the amount of repressor proteins present in samples expressing ICP0 compared to FXE (Fig. 5D, Western blot data) is rather in favor of an underestimation of the capacity of ICP0 to counteract their repressive effect.
These last two sets of experiments confirm that ICP0 can specifically affect the repression activity associated to the amino-terminal region of class II HDACs and/or of putative short isoforms of these proteins. To that extent, it has been suggested that, similarly to HDAC9/MITR, shorter isoforms for other class II HDACs are likely to exist (72, 81).
DISCUSSION
Viruses have evolved so that they optimally divert cellular mechanisms to their own profit. It is now clear that the acetylation pathway is one of them. Indeed, several viral proteins are acetylated and/or recruit HATs on cellular or viral promoters and, in some cases, interfere with their enzymatic activity (reviewed in reference 10). Far fewer data are available that describe the interaction of viral proteins with class I HDACs (13, 31, 62, 79) and none with class II HDACs. The present study demonstrates for the first time the physical and functional interactions between a viral protein, HSV-1 ICP0, and several class II HDACs.
The recent description of the presence of another repressor activity, which is not sensitive to inhibitors such as TSA, within the amino-terminal region of class II HDACs raised the question of the role played by these nondeacetylase domains in the control of gene transcription. Suggestions were made about the need of additional mechanisms for repression by deacetylases or about a role of the nondeacetylase domains in recruiting other corepressors to promote the formation of large enzymatic repressor complexes. It was shown that ectopic overexpression of HDAC4, -5, and -7 could form nuclear aggregates named matrix-associated deacetylase (MAD) bodies (15) (see Fig. 1A, nuclear dots in HDAC4- and -5-expressing cells). Although the physiological significance of the MADs is still a matter of debate, the fact is that they contain several proteins clearly involved in repression of gene expression, such as SMRT. Our immunofluorescence data on transfected and/or infected cells showed that endogenous and exogenous class II HDACs and SMRT undergo an ICP0-dependent redistribution. It is therefore intriguing that in transfected cells ICP0 rings concentrate at least two of the components of the MADs. Given our observations of the similar accumulation of endogenous HDAC4 in ICP0 aggregates in infected cells, a compelling suggestion is that ICP0 could disrupt the activity of such protein complexes by interacting with the keystone proteins of these structures.
Could this contribute to the general effect of ICP0 on activation of viral and cellular promoters, as described in several studies (19, 36, 37)? Our results on the lack of changes in acetylation state of histone and nonhistone proteins in cells expressing ICP0 do not support the hypothesis that ICP0-induced gene activation could be a consequence of a global effect on acetylation. This finding is in accordance with the observations that, although many cellular genes are upregulated in defective or recombinant virus- infected cells expressing ICP0, a significant proportion are concomitantly downregulated (36, 37). In addition, the lack of interaction of ICP0 with class I HDACs does not support the idea that ICP0 would enable gene expression by the sole inhibition of deacetylation, as generally observed by the addition of TSA.
What could then be the significance of ICP0-induced overcome of class II HDAC repressor activity?
As mentioned above, the amino-terminal region of class II HDAC4, -5, and -7 interacts with and represses the activity of a family of transcription factors named MEF2 (46, 56, 75). MEF2 proteins have initially been described as regulators of muscle development (50, 53, 57). Recently, several studies attributed to MEF2 an important role in neuronal survival and differentiation by preventing apoptosis (27, 28, 47, 49, 58, 59). Consequently, several studies have been done that describe the antagonist effect of class II HDACs in MEF2-regulated developmental pathways (54, 77, 78). MEF2 transcriptional activity is also controlled by class II HDACs-related MITR, a protein sharing high homologies with the amino-terminal sequences of class II HDACs and resulting from alternative splicing of class II HDAC9 mRNA (66, 81). It is anticipated that shorter isoforms of other class II HDACs could also exist (72, 81). A striking finding obtained from our gene reporter assays is the efficacy of the counter effect of ICP0 on the HDAC5 amino-terminal- and MITR-induced repression of MEF2A. Interestingly, MITR transcripts were found among the dorsal root ganglia of embryonic mice, one of the sites of HSV-1 latency in experimental mouse models, as well as in the brains of adult mice (78). Because it is known that ICP0 also plays a central role in enhancing the efficiency of reactivation (33, 34) and given the growing importance attributed to class II HDACs (2, 11, 38) and MEF2 to neuronal survival, it is tempting to envisage a putative ICP0 control of the repressor activity of class II HDACs or any related proteins during reactivation from latency. This could boost MEF2 transcriptional activity therefore maintaining the stressed neuron in a physiological state compatible with reactivation. The results we obtained in Fig. 5B, showing the boost of MEF2A activity in the presence of ICP0, clearly support this hypothesis.
On the other hand, the lack of exhaustive data on the biological properties of class II HDACs makes the interpretation of their interaction with ICP0 difficult during HSV-1 lytic infection. However, a recent study by Kao et al. (41) highlighted the critical role of HDAC4 in favoring double-strand break repairs in response to DNA damage. What could then be the link with HSV-1 and ICP0? Until very recently, it was uniformly admitted that HSV-1 genomes circularized as soon as they entered the nucleus of the infected cell. However, in a recent breakthrough study, Jackson and DeLuca (40) demonstrated that most of HSV-1 genome accumulated under linear form during lytic infection. These authors also showed that ICP0 is required to prevent its circularization; this is probably due to the treatment of the ends of linear genomes as double-strand DNA breaks. It has been known for a while that the genomes of several DNA, as well as RNA, viruses localize to discrete nuclear domains, called nuclear domain 10 (ND10) or PML bodies, early in infection (52). Interestingly, it has recently been suggested that one of the properties of ND10 could be to control DNA double-stranded break repair (9). ICP0 induces both the destabilization of ND10 and the proteasome-dependent degradation of the catalytic subunit of the DNA-dependent protein kinase (implicated in nonhomologous end-joining DNA repair) (51). Jackson and DeLuca (40) then proposed that this could be related to a property of ICP0 to participate in a general mechanism devoted to preventing circularization of viral genomes and then to favor the onset of lytic infection. The interaction of ICP0 with at least HDAC4 and the redistribution of other proteins implicated in DNA double-stranded break repair such as Rad51, similarly to HDAC4 in transfected cells (P. Lomonte, unpublished observation), might reflect an additional level at which ICP0 could control mechanisms implicated in DNA repair.
Our data thus show that class II HDACs and related proteins might constitute, in addition to protein degradation through the ubiquitin-proteasome pathway, targets for ICP0 to create a favorable environment in infected cells and reactivating neurons that would enable the onset of viral gene expression.
Acknowledgments
Part of this study was done while P.L. was an EMBO Long-Term Fellow. This study was funded by the CNRS through an Action Thématique Incitative sur Programme Jeunes Chercheurs (ATIP) to P.L., by the French Ministère de l'Education et de la Recherche Technologique through a Programme de Recherche Fondamentale en Microbiologie et Maladies Infectieuses et Parasitaires (PRFMMIP) to A.L.E. and S.D., and by L'Association pour la Recherche sur le Cancer (ARC) to A.L.E. and P.L.
We thank Eric Morency for comments on the manuscript. 11060 anti-ICP0 MAb, fusion protein GST-ICP0 1-241, and several plasmids expressing ICP0 were provided by Roger Everett, Medical Research Council, Virology Unit, Glasgow, Scotland. ML-19 anti-HDAC4 and 4/21 anti-SMRT rabbit polyclonal antibodies are generous gifts from Sigma and Hung-Ying Kao, Case Western Reserve University, Cleveland, Ohio, respectively. Plasmids expressing p53 and CBP were provided by Pierre Hainaut, Unit of Molecular Carcinogenesis, International Agency for Research on Cancer (World Health Organization), Lyon, France, and Annick Harel-Bellan, Institut André Lwoff, Villejuif, France, respectively. Some of the immunofluorescence data were obtained with a confocal microscope kindly made available by the Ecole Normale Supérieure of Lyon.
REFERENCES
- 1.Aalfs, J. D., and R. E. Kingston. 2000. What does “chromatin remodeling” mean? Trends Biochem. Sci. 25:548-555. [DOI] [PubMed] [Google Scholar]
- 2.Ajamian, F., T. Suuronen, A. Salminen, and M. Reeben. 2003. Upregulation of class II histone deacetylases mRNA during neural differentiation of cultured rat hippocampal progenitor cells. Neurosci. Lett. 346:57-60. [DOI] [PubMed] [Google Scholar]
- 3.Boutell, C., and R. D. Everett. 2003. The herpes simplex virus type 1 (HSV-1) regulatory protein ICP0 interacts with and ubiquitinates p53. J. Biol. Chem. 278:36596-36602. [DOI] [PubMed] [Google Scholar]
- 4.Boutell, C., S. Sadis, and R. D. Everett. 2002. Herpes simplex virus type 1 immediate-early protein ICP0 and is isolated RING finger domain act as ubiquitin E3 ligases in vitro. J. Virol. 76:841-850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brownell, J. E., J. Zhou, T. Ranalli, R. Kobayashi, D. G. Edmondson, S. Y. Roth, and C. D. Allis. 1996. Tetrahymena histone acetyltransferase A: a homolog to yeast Gcn5p linking histone acetylation to gene activation. Cell 84:843-851. [DOI] [PubMed] [Google Scholar]
- 6.Cai, W., T. L. Astor, L. M. Liptak, C. Cho, D. M. Coen, and P. A. Schaffer. 1993. The herpes simplex virus type 1 regulatory protein ICP0 enhances virus replication during acute infection and reactivation from latency. J. Virol. 67:7501-7512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cai, W., and P. A. Schaffer. 1992. Herpes simplex virus type 1 ICP0 regulates expression of immediate-early, early, and late genes in productively infected cells. J. Virol. 66:2904-2915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cai, W. Z., and P. A. Schaffer. 1989. Herpes simplex virus type 1 ICP0 plays a critical role in the de novo synthesis of infectious virus following transfection of viral DNA. J. Virol. 63:4579-4589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Carbone, R., M. Pearson, S. Minucci, and P. G. Pelicci. 2002. PML NBs associate with the hMre11 complex and p53 at sites of irradiation induced DNA damage. Oncogene 21:1633-1640. [DOI] [PubMed] [Google Scholar]
- 10.Caron, C., E. Col, and S. Khochbin. 2003. The viral control of cellular acetylation signaling. Bioessays 25:58-65. [DOI] [PubMed] [Google Scholar]
- 11.Chawla, S., P. Vanhoutte, F. J. Arnold, C. L. Huang, and H. Bading. 2003. Neuronal activity-dependent nucleocytoplasmic shuttling of HDAC4 and HDAC5. J. Neurochem. 85:151-159. [DOI] [PubMed] [Google Scholar]
- 12.Chen, J., and S. Silverstein. 1992. Herpes simplex viruses with mutations in the gene encoding ICP0 are defective in gene expression. J. Virol. 66:2916-2927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chiocca, S., V. Kurtev, R. Colombo, R. Boggio, M. T. Sciurpi, G. Brosch, C. Seiser, G. F. Draetta, and M. Cotten. 2002. Histone deacetylase 1 inactivation by an adenovirus early gene product. Curr. Biol. 12:594-598. [DOI] [PubMed] [Google Scholar]
- 14.Clements, G. B., and N. D. Stow. 1989. A herpes simplex virus type 1 mutant containing a deletion within immediate-early gene 1 is latency-competent in mice. J. Gen. Virol. 70:2501-2506. [DOI] [PubMed] [Google Scholar]
- 15.Downes, M., P. Ordentlich, H. Y. Kao, J. G. Alvarez, and R. M. Evans. 2000. Identification of a nuclear domain with deacetylase activity. Proc. Natl. Acad. Sci. USA 97:10330-10335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Elbashir, S. M., J. Harborth, W. Lendeckel, A. Yalcin, K. Weber, and T. Tuschl. 2001. Duplexes of 21-nucleotide RNAs mediate RNA interference in cultured mammalian cells. Nature 411:494-498. [DOI] [PubMed] [Google Scholar]
- 17.Everett, R. D. 1989. Construction and characterization of herpes simplex virus type 1 mutants with defined lesions in immediate-early gene 1. J. Gen. Virol. 70:1185-1202. [DOI] [PubMed] [Google Scholar]
- 18.Everett, R. D. 2000. ICP0, a regulator of herpes simplex virus during lytic and latent infection. Bioessays 22:761-770. [DOI] [PubMed] [Google Scholar]
- 19.Everett, R. D. 1984. Trans activation of transcription by herpesvirus products: requirement for two HSV-1 immediate-early polypeptides for maximum activity. EMBO J. 3:3135-3141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Everett, R. D., A. Cross, and A. Orr. 1993. A truncated form of herpes simplex virus type 1 immediate-early protein Vmw110 is expressed in a cell type-dependent manner. Virology 197:751-756. [DOI] [PubMed] [Google Scholar]
- 21.Everett, R. D., M. Meredith, and A. Orr. 1999. The ability of herpes simplex virus type 1 immediate-early protein Vmw110 to bind to a ubiquitin-specific protease contributes to its roles in the activation of gene expression and stimulation of virus replication. J. Virol. 73:417-426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Everett, R. D., A. Orr, and M. Elliott. 1991. High level expression and purification of herpes simplex virus type 1 immediate-early polypeptide Vmw110. Nucleic Acids Res. 19:6155-6161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Everett, R. D., A. Orr, and C. M. Preston. 1998. A viral activator of gene expression functions via the ubiquitin-proteasome pathway. EMBO J. 17:7161-7169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fields, B. N., D. M. Knipe, and P. M. Howley (ed.). 1996. Fields virology, 3rd ed. Lippincott-Raven Publishers, Philadelphia, Pa.
- 25.Fischle, W., F. Dequiedt, M. Fillion, M. J. Hendzel, W. Voelter, and E. Verdin. 2001. Human HDAC7 histone deacetylase activity is associated with HDAC3 in vivo. J. Biol. Chem. 276:35826-35835. [DOI] [PubMed] [Google Scholar]
- 26.Fischle, W., F. Dequiedt, M. J. Hendzel, M. G. Guenther, M. A. Lazar, W. Voelter, and E. Verdin. 2002. Enzymatic activity associated with class II HDACs is dependent on a multiprotein complex containing HDAC3 and SMRT/N-CoR. Mol. Cell 9:45-57. [DOI] [PubMed] [Google Scholar]
- 27.Gaudilliere, B., Y. Shi, and A. Bonni. 2002. RNA interference reveals a requirement for myocyte enhancer factor 2A in activity-dependent neuronal survival. J. Biol. Chem. 277:46442-46446. [DOI] [PubMed] [Google Scholar]
- 28.Gong, X., X. Tang, M. Wiedmann, X. Wang, J. Peng, D. Zheng, L. A. Blair, J. Marshall, and Z. Mao. 2003. Cdk5-mediated inhibition of the protective effects of transcription factor MEF2 in neurotoxicity-induced apoptosis. Neuron 38:33-46. [DOI] [PubMed] [Google Scholar]
- 29.Gordon, Y. J., J. L. McKnight, J. M. Ostrove, E. Romanowski, and T. Araullo-Cruz. 1990. Host species and strain differences affect the ability of an HSV-1 ICP0 deletion mutant to establish latency and spontaneously reactivate in vivo. Virology 178:469-477. [DOI] [PubMed] [Google Scholar]
- 30.Gu, W., and R. G. Roeder. 1997. Activation of p53 sequence-specific DNA binding by acetylation of the p53 C-terminal domain. Cell 90:595-606. [DOI] [PubMed] [Google Scholar]
- 31.Gwack, Y., H. Byun, S. Hwang, C. Lim, and J. Choe. 2001. CREB-binding protein and histone deacetylase regulate the transcriptional activity of Kaposi's sarcoma-associated herpesvirus open reading frame 50. J. Virol. 75:1909-1917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hagglund, R., C. Van Sant, P. Lopez, and B. Roizman. 2002. Herpes simplex virus 1-infected cell protein 0 contains two E3 ubiquitin ligase sites specific for different E2 ubiquitin-conjugating enzymes. Proc. Natl. Acad. Sci. USA 99:631-636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Halford, W. P., and P. A. Schaffer. 2001. ICP0 is required for efficient reactivation of herpes simplex virus type 1 from neuronal latency. J. Virol. 75:3240-3249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Halford, W. P., and P. A. Schaffer. 2000. Optimized viral dose and transient immunosuppression enable herpes simplex virus ICP0-null mutants To establish wild-type levels of latency in vivo. J. Virol. 74:5957-5967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Harris, R. A., R. D. Everett, X. X. Zhu, S. Silverstein, and C. M. Preston. 1989. Herpes simplex virus type 1 immediate-early protein Vmw110 reactivates latent herpes simplex virus type 2 in an in vitro latency system. J. Virol. 63:3513-3515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hobbs, W. E., II, and N. A. DeLuca. 1999. Perturbation of cell cycle progression and cellular gene expression as a function of herpes simplex virus ICP0. J. Virol. 73:8245-8255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hobbs, W. E., D. E. Brough, I. Kovesdi, and N. A. DeLuca. 2001. Efficient activation of viral genomes by levels of herpes simplex virus ICP0 insufficient to affect cellular gene expression or cell survival. J. Virol. 75:3391-3403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hoshino, M., K. Tagawa, T. Okuda, M. Murata, K. Oyanagi, N. Arai, T. Mizutani, I. Kanazawa, E. E. Wanker, and H. Okazawa. 2003. Histone deacetylase activity is retained in primary neurons expressing mutant Huntingtin protein. J. Neurochem. 87:257-267. [DOI] [PubMed] [Google Scholar]
- 39.Hubbert, C., A. Guardiola, R. Shao, Y. Kawaguchi, A. Ito, A. Nixon, M. Yoshida, X. F. Wang, and T. P. Yao. 2002. HDAC6 is a microtubule-associated deacetylase. Nature 417:455-458. [DOI] [PubMed] [Google Scholar]
- 40.Jackson, S. A., and N. A. DeLuca. 2003. Relationship of herpes simplex virus genome configuration to productive and persistent infections. Proc. Natl. Acad. Sci. USA 100:7871-7876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kao, G. D., W. G. McKenna, M. G. Guenther, R. J. Muschel, M. A. Lazar, and T. J. Yen. 2003. Histone deacetylase 4 interacts with 53BP1 to mediate the DNA damage response. J. Cell Biol. 160:1017-1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kao, H. Y., M. Downes, P. Ordentlich, and R. M. Evans. 2000. Isolation of a novel histone deacetylase reveals that class I and class II deacetylases promote SMRT-mediated repression. Genes Dev. 14:55-66. [PMC free article] [PubMed] [Google Scholar]
- 43.Khochbin, S., A. Verdel, C. Lemercier, and D. Seigneurin-Berny. 2001. Functional significance of histone deacetylase diversity. Curr. Opin. Genet. Dev. 11:162-166. [DOI] [PubMed] [Google Scholar]
- 44.Knoepfler, P. S., and R. N. Eisenman. 1999. Sin meets NuRD and other tails of repression. Cell 99:447-450. [DOI] [PubMed] [Google Scholar]
- 45.Leib, D. A., D. M. Coen, C. L. Bogard, K. A. Hicks, D. R. Yager, D. M. Knipe, K. L. Tyler, and P. A. Schaffer. 1989. Immediate-early regulatory gene mutants define different stages in the establishment and reactivation of herpes simplex virus latency. J. Virol. 63:759-768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lemercier, C., A. Verdel, B. Galloo, S. Curtet, M. P. Brocard, and S. Khochbin. 2000. mHDA1/HDAC5 histone deacetylase interacts with and represses MEF2A transcriptional activity. J. Biol. Chem. 275:15594-15599. [DOI] [PubMed] [Google Scholar]
- 47.Li, M., D. A. Linseman, M. P. Allen, M. K. Meintzer, X. Wang, T. Laessig, M. E. Wierman, and K. A. Heidenreich. 2001. Myocyte enhancer factor 2A and 2D undergo phosphorylation and caspase-mediated degradation during apoptosis of rat cerebellar granule neurons. J. Neurosci. 21:6544-6552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lomonte, P., K. F. Sullivan, and R. D. Everett. 2001. Degradation of nucleosome-associated centromeric histone H3-like protein CENP-A induced by herpes simplex virus type 1 protein ICP0. J. Biol. Chem. 276:5829-5835. [DOI] [PubMed] [Google Scholar]
- 49.Mao, Z., A. Bonni, F. Xia, M. Nadal-Vicens, and M. E. Greenberg. 1999. Neuronal activity-dependent cell survival mediated by transcription factor MEF2. Science 286:785-790. [DOI] [PubMed] [Google Scholar]
- 50.Martin, J. F., J. M. Miano, C. M. Hustad, N. G. Copeland, N. A. Jenkins, and E. N. Olson. 1994. A Mef2 gene that generates a muscle-specific isoform via alternative mRNA splicing. Mol. Cell. Biol. 14:1647-1656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Maul, G. G., H. H. Guldner, and J. G. Spivack. 1993. Modification of discrete nuclear domains induced by herpes simplex virus type 1 immediate-early gene 1 product (ICP0). J. Gen. Virol. 74(Pt. 12):2679-2690. [DOI] [PubMed] [Google Scholar]
- 52.Maul, G. G., A. M. Ishov, and R. D. Everett. 1996. Nuclear domain 10 as preexisting potential replication start sites of herpes simplex virus type-1. Virology 217:67-75. [DOI] [PubMed] [Google Scholar]
- 53.McDermott, J. C., M. C. Cardoso, Y. T. Yu, V. Andres, D. Leifer, D. Krainc, S. A. Lipton, and B. Nadal-Ginard. 1993. hMEF2C gene encodes skeletal muscle- and brain-specific transcription factors. Mol. Cell. Biol. 13:2564-2577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.McKinsey, T. A., C. L. Zhang, J. Lu, and E. N. Olson. 2000. Signal-dependent nuclear export of a histone deacetylase regulates muscle differentiation. Nature 408:106-111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Meredith, M., A. Orr, M. Elliott, and R. Everett. 1995. Separation of sequence requirements for HSV-1 Vmw110 multimerisation and interaction with a 135-kDa cellular protein. Virology 209:174-187. [DOI] [PubMed] [Google Scholar]
- 56.Miska, E. A., C. Karlsson, E. Langley, S. J. Nielsen, J. Pines, and T. Kouzarides. 1999. HDAC4 deacetylase associates with and represses the MEF2 transcription factor. EMBO J. 18:5099-5107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Molkentin, J. D., and E. N. Olson. 1996. Combinatorial control of muscle development by basic helix-loop-helix and MADS-box transcription factors. Proc. Natl. Acad. Sci. USA 93:9366-9373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Okamoto, S., D. Krainc, K. Sherman, and S. A. Lipton. 2000. Antiapoptotic role of the p38 mitogen-activated protein kinase-myocyte enhancer factor 2 transcription factor pathway during neuronal differentiation. Proc. Natl. Acad. Sci. USA 97:7561-7566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Okamoto, S., Z. Li, C. Ju, M. N. Scholzke, E. Mathews, J. Cui, G. S. Salvesen, E. Bossy-Wetzel, and S. A. Lipton. 2002. Dominant-interfering forms of MEF2 generated by caspase cleavage contribute to NMDA-induced neuronal apoptosis. Proc. Natl. Acad. Sci. USA 99:3974-3979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Paterson, T., and R. D. Everett. 1990. A prominent serine-rich region in Vmw175, the major transcriptional regulator protein of herpes simplex virus type 1, is not essential for virus growth in tissue culture. J. Gen. Virol. 71(Pt. 8):1775-1783. [DOI] [PubMed] [Google Scholar]
- 61.Preston, C. M., and M. J. Nicholl. 1997. Repression of gene expression upon infection of cells with herpes simplex virus type 1 mutants impaired for immediate-early protein synthesis. J. Virol. 71:7807-7813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Radkov, S. A., R. Touitou, A. Brehm, M. Rowe, M. West, T. Kouzarides, and M. J. Allday. 1999. Epstein-Barr virus nuclear antigen 3C interacts with histone deacetylase to repress transcription. J. Virol. 73:5688-5697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Sacks, W. R., and P. A. Schaffer. 1987. Deletion mutants in the gene encoding the herpes simplex virus type 1 immediate-early protein ICP0 exhibit impaired growth in cell culture. J. Virol. 61:829-839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Samaniego, L. A., L. Neiderhiser, and N. A. DeLuca. 1998. Persistence and expression of the herpes simplex virus genome in the absence of immediate-early proteins. J. Virol. 72:3307-3320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Smith, D. B., and L. M. Corcoran. 1994. Expression and purification of glutathione S-transferase fusion proteins, p. 16.7.1-16.7.7. In F. M. Ausubel, R. Brent, R. E. Kingston, D. D. Moore, J. G. Seidman, J. A. Smith, and K. Struhl (ed.), Current protocols in molecular biology, vol. 3. John Wiley & Sons, Inc., New York, N.Y. [DOI] [PubMed] [Google Scholar]
- 66.Sparrow, D. B., E. A. Miska, E. Langley, S. Reynaud-Deonauth, S. Kotecha, N. Towers, G. Spohr, T. Kouzarides, and T. J. Mohun. 1999. MEF-2 function is modified by a novel corepressor, MITR. EMBO J. 18:5085-5098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Stow, E. C., and N. D. Stow. 1989. Complementation of a herpes simplex virus type 1 Vmw110 deletion mutant by human cytomegalovirus. J. Gen. Virol. 70:695-704. [DOI] [PubMed] [Google Scholar]
- 68.Stow, N. D., and E. C. Stow. 1986. Isolation and characterization of a herpes simplex virus type 1 mutant containing a deletion within the gene encoding the immediate-early polypeptide Vmw110. J. Gen. Virol. 67:2571-2585. [DOI] [PubMed] [Google Scholar]
- 69.Struhl, K. 1998. Histone acetylation and transcriptional regulatory mechanisms. Genes Dev. 12:599-606. [DOI] [PubMed] [Google Scholar]
- 70.Taunton, J., C. A. Hassig, and S. L. Schreiber. 1996. A mammalian histone deacetylase related to the yeast transcriptional regulator Rpd3p. Science 272:408-411. [DOI] [PubMed] [Google Scholar]
- 71.Van Sant, C., R. Hagglund, P. Lopez, and B. Roizman. 2001. The infected cell protein 0 of herpes simplex virus 1 dynamically interacts with proteasomes, binds and activates the cdc34 E2 ubiquitin-conjugating enzyme, and possesses in vitro E3 ubiquitin ligase activity. Proc. Natl. Acad. Sci. USA 98:8815-8820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Verdel, A., and S. Khochbin. 1999. Identification of a new family of higher eukaryotic histone deacetylases: coordinate expression of differentiation-dependent chromatin modifiers. J. Biol. Chem. 274:2440-2445. [DOI] [PubMed] [Google Scholar]
- 73.Verdin, E., F. Dequiedt, and H. G. Kasler. 2003. Class II histone deacetylases: versatile regulators. Trends Genet. 19:286-293. [DOI] [PubMed] [Google Scholar]
- 74.Wagner, E. K., and D. C. Bloom. 1997. Experimental investigation of herpes simplex virus latency. Clin. Microbiol. Rev. 10:419-443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Wang, A. H., N. R. Bertos, M. Vezmar, N. Pelletier, M. Crosato, H. H. Heng, J. Th'ng, J. Han, and X. J. Yang. 1999. HDAC4, a human histone deacetylase related to yeast HDA1, is a transcriptional corepressor. Mol. Cell. Biol. 19:7816-7827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Yang, W. M., Y. L. Yao, J. M. Sun, J. R. Davie, and E. Seto. 1997. Isolation and characterization of cDNAs corresponding to an additional member of the human histone deacetylase gene family. J. Biol. Chem. 272:28001-28007. [DOI] [PubMed] [Google Scholar]
- 77.Zhang, C. L., T. A. McKinsey, S. Chang, C. L. Antos, J. A. Hill, and E. N. Olson. 2002. Class II histone deacetylases act as signal-responsive repressors of cardiac hypertrophy. Cell 110:479-488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Zhang, C. L., T. A. McKinsey, and E. N. Olson. 2001. The transcriptional corepressor MITR is a signal-responsive inhibitor of myogenesis. Proc. Natl. Acad. Sci. USA 98:7354-7359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Zhang, Y., and C. Jones. 2001. The bovine herpesvirus 1 immediate-early protein (bICP0) associates with histone deacetylase 1 to activate transcription. J. Virol. 75:9571-9578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Zhang, Y., N. Li, C. Caron, G. Matthias, D. Hess, S. Khochbin, and P. Matthias. 2003. HDAC-6 interacts with and deacetylates tubulin and microtubules in vivo. EMBO J. 22:1168-1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Zhou, X., P. A. Marks, R. A. Rifkind, and V. M. Richon. 2001. Cloning and characterization of a histone deacetylase, HDAC9. Proc. Natl. Acad. Sci. USA 98:10572-10577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Zhou, X., V. M. Richon, R. A. Rifkind, and P. A. Marks. 2000. Identification of a transcriptional repressor related to the noncatalytic domain of histone deacetylases 4 and 5. Proc. Natl. Acad. Sci. USA 97:1056-1061. [DOI] [PMC free article] [PubMed] [Google Scholar]