

Abstract
The serine/cysteine protease inhibitor SCCA1 (Serpin B3) is upregulated in many advanced cancers with poor prognosis, but there is limited information about whether it makes functional contributions to malignancy. Here we show that SCCA1 expression promoted oncogenic transformation and epithelial-mesenchymal transition (EMT) in mammary epithelial cells, and that SCCA1 silencing in breast cancer cells halted their proliferation. SCCA1 overexpression in neu+ mammary tumors increased the unfolded protein response (UPR), IL-6 expression, and inflammatory phenotypes. Mechanistically, SCCA1 induced a prolonged non-lethal increase in the UPR that was sufficient to activate NF-κB and expression of the pro-tumorigenic cytokine IL-6. Overall, our findings established that SCCA1 contributes to tumorigenesis by promoting EMT and a UPR-dependent induction of NF-κB and IL-6 autocrine signaling that promotes a pro-tumorigenic inflammation.
Introduction
Squamous cell carcinoma antigens (SCCAs) belong to the clade B subset of serpins that inhibit lysosomal proteases including cathepsins via the irreversible interaction between its carboxyl-terminal reactive site loop (RSL) and the target proteases (1,2). The first variant of the SCCAs, SCCA1 (SerpinB3), an inhibitor of cathepsins L, S, and K, was initially found to be elevated in squamous cell carcinoma (SCC) of the uterine cervix (3), and was later found to be highly expressed in squamous cell carcinomas of the lung, head and neck, and liver (4,5). Its functional connection with tumorigenesis has been mainly appreciated for its anti-cell death role against lysosomal membrane permeability transition in response to various stresses such as UV, radiation, chemotherapy, TNFα, and natural killer cells (5–9). Nevertheless, accumulating evidence including that from our own group has indicated that elevated SCCA1 expression is associated with poorly differentiated and more inflammatory and aggressive human malignancies including breast cancer (10–12), pointing to additional molecular functions. We have recently reported that ectopic expression of SCCA1 leads to the inhibition of both proteasomal and lysosomal protein degradation (13), suggesting that elevated SCCA1 expression may lead to an increased unfolded protein response (UPR).
UPR is a complex signaling event that is activated by the disturbance of cellular protein homeostasis. While it is well appreciated that excessive misfolded protein stress triggers apoptosis, UPR signaling under more physiological conditions plays an important role in helping cells to cope with stress and to restore homeostasis. The connection between UPR and cancer has been well appreciated in light of cancer cells’ highly increased growth rate and exposure to growth limiting conditions such as nutrient deprivation and hypoxia (14,15). While over-activating UPR in cancer cells can lead to cell death and has been regarded as a therapeutic opportunity using proteotoxic agents such as the proteasome inhibitor Velcade (Bortezomib) (16,17), the UPR signaling pathway has been implied in promoting tumorigenesis by increasing tumor cell survival and proliferation (15,18). However, it remains elusive how specific cell intrinsic lesions lead to increased UPR that functions as a driving factor in tumorigenesis. In this study, we report a previously unidentified pro-tumorigenic role of SCCA1, which is via the induction of a non-lethal level of UPR that activates NF-κB and expression of the pro-tumorigenic cytokine IL-6.
Materials and Methods
Cell lines and culture
MCF10A, MDA-MB-231, MDA-MB-468, SKBR3, HEK293T cells were obtained from ATCC. BMK cells were obtained from Dr. Eileen White’s laboratory, and HMLE cells were obtained from Dr. Robert Weinberg’s laboratory. All cell lines have been tested and authenticated as bacteria and mycoplasma free following ATCC’s instructions on a routine basis within 6 month of experiments.
Retroviral and lentiviral infection
For retroviral infections, the three plasmid system (gene of interest + helper virus + VSVG at the ratio of 4:3:1) used to generate virus particles after transfection into HEK 293T cells using Lipofectamine 2000 (Invitrogen), filtered viral supernatant along with 10 μg/ml polybrene (Sigma) was used to infect the target cells. Lentiviral infections were carried using the above mentioned protocol by replacing the helper virus with the ΔR8.91 plasmid.
IL-6 ELISA, conditioned medium, and neutralization experiment
The concentration of IL-6 secreted into the media was measured using IL-6 ELISA kit (R&D systems D6050), as per the manufacturer’s instructions.
Subcellular fractionation
Subcellular fractionation was carried out using the subcellular proteome extraction kit (Calbiochem). The fractions were quantified using the BCA assay, and equal amount of protein was then used for precipitation using four times the volume of ice cold acetone. The samples were incubated for 120 min at −20°C and centrifuged at 13,000 g for 15 min at 4°C. The pellets were air dried for 10 min and resuspended in 2X SDS sample buffer and boiled at 95°C for 5 min. The proteins in the cytosolic and nuclear fractions were then detected by western blotting.
Luciferase reporter assay
The dual-luciferase activity kit (Promega E1910) was employed to examine the luciferase activity. The luciferase assay is performed following the common protocols. The NF-κB binding site of IL-6 promoter was fused to a luciferase reporter gene.
Wound healing assay
Equal number of vector and SCCA1 expressing cells were plated and allowed to grow to sub-confluency. Two perpendicular wounds were made with the help of western loading tips across the culture dish, washed 3 times to remove cell debris, and imaged 6 h later at the junction of the wound. The cell front was marked out and the area of the gap was measured using the NIS elements software (Nikon Instruments Inc.).
Soft agar assay
5 × 104 cells were resuspended in 1.5 ml MCF10A complete media with 0.5% Noble agarose and overlaid onto 1.5 ml complete medium with 0.7% agarose in each well of a 6-well plate in triplicates. After 3 weeks, colonies larger than 100 μm were counted.
Orthotopic mouse tumor experiment and SCCA1 conditional transgenic mice
Female beige nude XID mice (Hsd:NIHS-Lystbg-JFoxn1nuBtkxid), age 6–8 wk, were obtained from Harlan Laboratories. SCCA1 knock-in transgenic mice with 129Ola/C57Bl/6 mixed background were developed by Genoway. Mice were housed and monitored at the Division of Laboratory Animal Resources at Stony Brook University. All experimental procedures and protocols were approved by the institutional animal care and use committee.
Image processing and densitometry measurements
Images captured by deconvolution microscope were viewed and processed by AxioVision LE image browser. Images were processed in Adobe Photoshop to enhance the brightness and contrast. Densitometry of immunoblot bands was determined by ImageJ software.
Statistics
The longitudinal data analyses were performed to assess the growth curves under different treatments. The Fisher’s exact test was used to test the difference between tumor incidence rates in two groups. The independent two-sample and one sample t-tests were used to make comparisons between groups and to evaluate whether fold changes are different from one, respectively. The analyses were carried out using PROC MIXED, PROC FREQ, PROC MEANS and PROC TTEST in the SAS 9.4 (SAS institute, Cary, NC). Statistical significance level was set at p ≤ 0.05.
Additional details can be found online in Supplemental Materials and Methods.
Results
SCCA1 expression results in EMT associated oncogenic transformation
Based on our recent report that SCCA1 is associated with poorly differentiated high grade breast carcinoma (11), we chose to study the biological function of SCCA1 in the non-neoplastic human mammary epithelial cell line MCF10A that has a low level of endogenous SCCA1 (11). Interestingly, ectopic expression of SCCA1 in MCF10A cells led to morphological changes from a cobblestone-like monolayer to a dispersed spindle-shaped morphology that resembles epithelial-mesenchymal transition (EMT) (Fig. 1A). This is reminiscent of the EMT-like changes in HepG2, a hepatocellular carcinoma cell line, upon SCCA1 overexpression (19). Consistent with the EMT-like morphological change, a change of transcriptional profile which included down-regulation of E-cadherin (CDH1), EpCam (EPCAM), and claudin4 (CLDN4), as well as up-regulation of Zeb1 (ZEB1), vimentin (VIM), and fibronectin1 (FN1) was observed in SCCA1-expressing MCF10A cells (Fig. 1B). The decreased E-cadherin and increased vimentin were also observed at the protein level by immunofluorescence (Fig. 1C) and immunoblotting (Fig. 1D). It is important to note that the morphological changes induced by SCCA in MCF10A cells did not occur instantaneously, rather at approximately 2 weeks of continued passaging, accompanied by a progressive loss of E-cadherin and the reciprocal gain of vimentin (Fig. 1D). This is consistent with the notion that EMT is a multi-step progressive event (20). Importantly, two catalytic-inactive mutants of SCCA1, the F352A point mutant that has decreased protease inhibitory activity (21) and the SCCA1 Δ340–345 hinge deletion mutant that renders the reactive site loop inflexible and therefore incapable of protease inhibition (22), failed to induce the EMT-like changes with regard to the expression levels of E-cadherin and vimentin (Fig. 1E) and cell morphology (Fig. 1F), indicating that the EMT-promoting function requires the anti-protease activity of SCCA1.
Figure 1. Ectopic expression of SCCA1 leads to EMT-like phenotype that is dependent on its protease inhibitory activity.

(A–D) MCF10A cells were retrovirally infected with vector control or Flag-SCCA1, and continuously passaged. (A–C) at passage #8, phase contrast images were taken (A). Total RNA were analyzed for indicated genes via qRT-PCR which was normalized against that in vector control cells. Data shown are the mean fold change +/− SEM of two independent experiments performed in triplicates. *p<0.05, **p < 0.01, ****p < 0.0001. (B) Immunofluorescence was performed for E-cadherin and vimentin, and counter stained with DAPI. Images were taken by a deconvolution fluorescence microscope. Note the loss of cell-surface E-cadherin and increase in vimentin expression in SCCA1-expressing cells (C). (D) Whole cell lysates were harvested at indicated passage numbers, and analyzed by immunoblotting. Graph on the right shows the expression level of E-cadherin and Vimentin in SCCA1-expressing cells, which was analyzed by densitometry and normalized to that of Passage #1. (E and F) MCF10A cells stably expressing indicated SCCA1 mutants were analyzed by immunoblotting (E), and the phase contrast images were taken (F).
EMT is functionally associated with increased cell mobility and oncogenic transformation. Indeed, expression of SCCA1 in MCF10A cells conferred increased wound closure associated with individually scattered cells at the migratory front (Fig. 2A). These cells were also resistant to cell death induced by loss of adherence (anoikis) (Fig. 2B), and displayed colony formation in soft agar (Fig. 2C). Furthermore, while MCF10A cells are dependent on growth factors and hormones (epidermal growth factor (EGF), insulin, hydrocortisone, and cholera toxin), SCCA1 expression rendered them independent of EGF (Fig. 2D) and enabled them to proliferate even when deprived of all four hormonal factors (Suppl. Fig. S1). The independence of EGF was accompanied by increased steady-state levels of phospho-ERK and phospho-Akt, which did not decrease upon EGF deprivation (Fig. 2E). We then examined the tumor-promoting function of SCCA1 in an orthotopic mouse tumor setting by injecting SCCA1-expressing MCF10A cells into the mammary fat pad of the immune-compromised XID mice. While MCF10A cells are non-tumorigenic, nine out of nine mice implanted with SCCA1-expressing cells developed tumors as shown by GFP imaging (Fig. 3A and 3B) where the ectopic expression of SCCA1 was confirmed by immunohistochemistry (Fig. 3C). Taken together, our results indicate that SCCA1 can promote an EMT-like phenotype and oncogenic transformation in a manner that is dependent on its anti-protease activity.
Figure 2. Ectopic expression of SCCA1 in MCF10A cells leads to oncogenic transformation.

(A) Vector and SCCA1-expressing MCF10A cells were assessed by the wound healing assay 6 h after the wound scratch on cell culture monolayer. Representative images are shown. Wound closure was quantified by the percent change in cell migration. Data shown are the mean of two independent experiments performed in triplicates +/− SEM. **p < 0.01. (B) Indicated cells were cultured in non-adherence culture dishes for indicated periods of time. Cell viability was measured by PI exclusion. Data shown are the mean +/− S.D. of an experiment performed in triplicate. Error bars are too small to be visible compared to the physical size of the symbol. ***p < 0.001. (C) Cells were cultured in soft agar for 3 weeks. Images of cell colonies were taken. Shown is the average number of colony spheres ≥ 100 μm per field from triplicate experiment + S.D. Five randomly selected fields were counted for each replicate. (D) Cells were cultured in complete or EGF-deprived medium. Relative cell growth was measured by crystal violet staining and normalized to that of the cells at time zero. Data shown are the mean +/− S.D. of two experiments measured in triplicate. ***p < 0.001. (E) Cells were cultured in EGF-deprived medium for indicated times and analyzed by immunoblotting. The densitometric ratios of pERK/tERK and pAkt/tAkt are normalized to that of vector cells at time zero.
Figure 3. SCCA1 promotes orthotopic breast tumor formation.
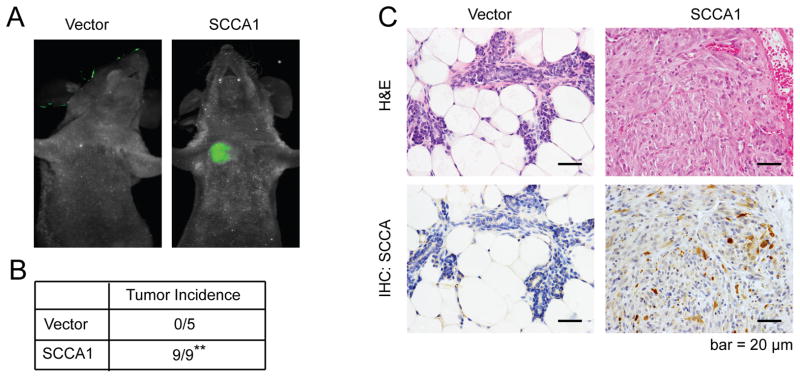
7.5 × 105 MCF10A cells expressing either GFP alone (n=5) or GFP plus SCCA1 (n=9) were implanted into the mammary fat pad of NOD-SCID mice. (A) Mice were imaged for tumor formation 60 days post implantation. (B) The formation of palpable tumors are summarized. Significance was judged based on Fischer’s exact test. **p < 0.01. (C) Tumors were sectioned and stained using H&E (top panels) or by IHC for SCCA (bottom panels).
SCCA1 expression induces IL-6 expression
Since the SCCA1-expressing cells displayed an EMT-like phenotype, EGF independence, and oncogenic transformation (Fig. 1 and 2), we speculated that SCCA1 might promote the expression of autocrine factors that are implicated in EMT and/or EGFR signaling and oncogenic transformation. We performed quantitative real-time PCR (qRT-PCR) to determine the expression of well-characterized EMT-associated factors including transforming growth factor beta (TGFβ), interleukin-6 (IL6), IL-8, IL1β, and (C-X-C motif) ligand 1 (CXCL1) (23–26), as well as the EGF family members EGF, TGFA, heparin-binding EGF-like growth factor (HBEGF), amphiregulin (AREG), betacellulin (BTC), and epiregulin (EPR) (27). Among these, IL6 transcription was significantly up-regulated in SCCA1-expressing cells (Fig. 4A). Correlating with the increased IL-6 transcript level, a dramatically increased amount of IL-6 was detected in the culture medium of SCCA1-expressing cells (Fig. 4B). The secreted IL-6 is functionally active since the culture medium collected from SCCA1-expressing but not vector control cells induced STAT3 phosphorylation, a known downstream consequence of IL-6 signaling, and this effect was abrogated when the conditioned medium was pre-incubated with an IL-6 blocking antibody (Fig. 4C). It is important to note that a slight yet apparent up-regulation of IL-6 transcript was detected in the early passages post SCCA1 expression, which progressively increased in subsequent passages resulting in over a hundred-fold elevation (Suppl. Fig. S2). Silencing IL-6 at the early passage post SCCA1 expression led to a drastic increase in E-cadherin expression at both the transcript and protein levels (Fig. 4D–F), delayed the change to spindle-like morphology (Fig. 4G), and decreased proliferation (Fig. 4H). These results, while they do not rule out the possible involvement of other cytokines, indicate that expression of SCCA1 leads to increased IL-6 production, which is likely a causative factor rather than the consequence of the SCCA-induced EMT-like phenotype and cell transformation.
Figure 4. SCCA1 promotes IL-6 expression by activating NF-κB.

(A) Total RNA isolated from vector control or SCCA1-expressing MCF10A cells were analyzed for the expression of indicated hormonal ligands by qRT-PCR. Data shown is the mean transcript level normalized to that of vector control cells + S. D. of triplicate experiment. *EGF was below detectable level. (B) Cell culture medium of vector control and SCCA1-expressing cells from overnight cultures was collected and measured for the level of secreted IL-6 by ELISA. *the absolute concentration of IL-6 in the control cells was below the sensitivity of the kit (3.25 pg/ml). Data shown are the mean + S.D. of two independent experiments performed in triplicates. (C) Vector control or SCCA1-expressing cells were cultured in EGF-free medium for 24 h. The medium from vector control cells was incubated with control IgG, and that from SCCA1 cells was incubated with IgG or an IL-6 neutralizing antibody. These media were added to EGF-starved MCF10A cells. Whole cell lysates were collected at indicated times and probed for phospho-Stat3 (Y705) and total Stat3. (D–H) SCCA1-expressing MCF10A cells at early passage (passage #3) were infected with control (shNTC) and IL-6 (shIL-6) shRNA for 4 days. (D and E) Total RNA was analyzed by qRT-PCR for IL-6 (D) and E-cadherin (E) transcript levels. Data shown is the average +/− SEM of three independent experiments performed in triplicate. *p<0.05. (F) Whole cell lysates were analyzed by immunoblotting. (G) Phase-contrast images of the cell culture was taken. (H) Relative cell proliferation was determined by crystal violet staining. Data shown is the mean +/− S.D. of three independent experiments in duplicates. ***p < 0.001. (I) Indicated cells were transfected with the NF-κB luciferase reporter and a renilla luciferase construct. 24 h post-transfection, NF-κB luciferase activity was calculated by normalizing against the renilla luciferase activity. Data shown is a representative graph of three independent experiments performed in triplicates, showing the mean + S.D. ****p<0.0001. (J) Indicated cells were subjected to subcellular fractionation. The cytosol (C) and nuclear (N) fractions were analyzed by immunoblotting. (K) Left panel: Cells were treated with either DMSO or 5 μM BAY-117082 for 4 h. Right panel: SCCA1-expressing cells were retrovirally infected with vector control or IκBαM (S32A, S36A) for 4 d. Expression of IκBαM was verified by immunoblotting. IL-6 transcript level was analyzed by qRT-PCR. Data shown are the mean +/− SEM of three independent experiments performed in triplicate. ****p<0.0001. (L and M) SCCA1-expressing MCF10A cells cultured in non-adherent culture dish, in the presence of DMSO or 5 μM BAY-117082, for 4 d. Phase contrast image of the cell culture was taken (L) and indicated proteins were probed by immunoblotting (M).
SCCA1-induced IL-6 expression is mediated by the activation of NF-κB
Expression of IL-6 is largely driven by the NF-κB transcription factors, which have been shown to be essential for EMT in breast cancer (28,29). Since we detected increased IL-6 expression in SCCA1-expressing cells, we determined whether SCCA1 could promote the activation of NF-κB. Indeed, a significant increase of NF-κB activity was detected in SCCA1-expressing cells using an NF-κB luciferase reporter assay (Fig. 4I), which was accompanied by a marked increase of nuclear RelA/p65 (Fig. 4J). The increased expression of IL-6 in SCCA1-expressing cells was abrogated by a pharmacological inhibitor of NF-κB, BAY11-7082, at a concentration tolerated by the parental MCF10A cells (24), and by the ectopic expression of the IκBαM (S32A, S36A) super-repressor mutant (Fig. 4K). Accordingly, treatment with BAY11-7082 resulted in the abrogation of anchorage-independent sphere formation (Fig. 4L) and increased apoptosis (Fig. 4M) in SCCA1-transformed cells. These results indicate that activation of NF-κB plays an essential role in SCCA1-mediated IL-6 production and cell transformation.
SCCA1 induces pro-inflammatory signaling by activating a low level chronic UPR
We have recently reported that expression of SCCA1, through its protease inhibitory activity, leads to impaired lysosomal and proteasomal protein turnover (13). This impaired protein degradation caused by elevated SCCA1 expression is non-lethal and may lead to UPR that has been implicated in pro-inflammatory response in numerous human diseases including cancer (30). Hence, we speculated that SCCA1 could promote NF-κB activation and subsequently IL-6 production by inducing a non-lethal yet prolonged UPR.
There are three arms of the UPR signaling pathway which are mediated by PERK, ATF6α, and IRE1α/β (31). It is generally accepted that while the three arms are closely interconnected, each of them has predominant molecular signatures: activation of PERK leads to increased translation of ATF4 transcriptional factor and its transcriptional targets such as CHOP; activation of the ATF6α arm is characterized by the proteolytic cleavage of the 90 kDa full-length pro-form of ATF6α into an N-terminal 50 kDa fragments (ATF6α-p50) and a 36 kDa fragment (ATF6α-p36); and activation of IRE1 leads to the splicing of XBP1 mRNA (32,33).
Activation of the PERK arm of UPR was detected as SCCA1-expressing cells displayed a slight increase in ATF4 expression, which was further enhanced upon treatment with the ER stress inducer tunicamycin (Fig. 5A). The ATF6 arm was also apparent since SCCA1-expressing cells displayed an elevated ATF6α-p50 (Fig. 5A). In addition, CHOP, a synergistic transcriptional target of ATF4 and ATF6 (34), was more robustly induced by tunicamycin in SCCA1-expressing cells (Fig. 5A). Both ATF6α-p50 and ATF6α-p36 translocate to the peri-nuclear and nuclear regions where p50 is believed to function as a transcription factor (35) while the function of p36 remains unclear (36). An increased nuclear and peri-nuclear ATF6α was detected by subcellular fractionation (Fig. 5B) and immunofluorescence (Fig. 5C) in SCCA1-expressing cells. Furthermore, the elevated ATF6α transcriptional activity was revealed by the induction of its transcriptional target the unspliced form of XBP1 (XBP1u) (Fig. 5D), which was distinguished from the spliced form XBP1s by restriction digestion with PstI that cuts XBP1u but not XBP1s (37). With regard to the IRE1 arm, SCCA1-expressing cells exhibited a decreased basal level of spliced XBP1s (Fig. 5D and 5E). This supports our notion that SCCA1 induces a prolonged low level UPR, which has been shown to suppress the IRE1α signaling (38). Moreover, upon tunicamycin treatment, the SCCA1-expressing cells were able to convert this increased amount of XBP1u to XBP1s (Fig. 5E), indicating that while the steady-state level of XBP1 splicing (as an indicator of IRE1α activity) is low, the signaling machinery is still intact in these cells. The UPR-promoting function of SCCA1 is dependent on its protease inhibitory activity as the SCCA1 F352A and SCCA1Δ340–345 inactive mutants did not alter the levels of ATF6α-p50 (Suppl. Fig. S3). These data indicate that increased SCCA1 expression leads to a chronic UPR with the activation of the PERK and ATF6 arms and slight inhibition of the IRE1 arm.
Figure 5. SCCA1 induces chronic UPR that is essential for IL-6 production and transformation.

(A) Vector control or SCCA1-expressing MCF10A cells were treated with 5 μM tunicamycin and analyzed by immunoblotting. (B) Cells were subjected to subcellular fractionation. The cytosol (C) and nuclear (N) fractions were analyzed by immunoblotting. (C) Cells were probed for ATF6α by immunofluorescence, and counter stained with DAPI. Images were taken by a deconvolution fluorescence microscope. (D) Primers across the splice junction of XBP1 were used to amplify XBP1 by semi-Q PCR. The PCR product was divided into two halves. One was subjected to PstI digestion and the other was not, and then resolved on an agarose gel. PCR of GAPDH was used as a control for equal amplification. Note that SCCA1 cells had increased amount of XBP1u and decreased XBP1 splicing. (E) Vector and SCCA1-expressing cells were treated with 5 μg/ml tunicamycin for indicated times. XBP1 transcript was detected by semi-Q PCR. (F–N) SCCA1-expressing MCF10A cells were lentivirally infected with short hairpins of control (shNTC) and PERK (F–H), ATF6α (I–K), or XBP1 (L–N). Cells were harvested 4 days later and qRT-PCR was performed for the transcript level PERK, ATF6α, IL–6, and immunoblotting for XBP1. Data shown are the mean +/− SEM of three independent experiments performed in triplicate. Relative cell growth was determined by crystal violet staining normalized to the reading of cells at Day 1 (G, J, and M). Significance judged by longitudinal data analysis was ***p<0.001 for G and J, and ns for M. Cells were cultured in suspension for 10 days. Phase-contrast images of cell spheres were taken (H, K, and N).
Next, to determine whether activation of PERK and ATF6α are critical for SCCA1-mediated IL-6 expression and cell transformation, we silenced PERK or ATF6α in SCCA1 cells. Silencing either PERK or ATF6α led to down-regulation of IL-6, impaired cell proliferation, and decreased anchorage-independent sphere formation in SCCA1-expressing MCF10A cells (Fig. 5F–5K). In contrast, silencing XBP1 led to an increase of IL-6 and no decrease in cell proliferation or sphere formation (Fig. 5L–5N). Similarly, silencing PERK and ATF6α in MDA-MB-231 cells with the endogenous level of SCCA1 also led to decreased IL-6 expression and reduced cell proliferation (Suppl. Fig. S4A–S4D). The specific role of SCCA1 in mediating UPR, IL-6 production, and transformation was further tested by silencing the over-expressed SCCA1 in MCF10A cells (Suppl. Fig. S5A–S5C), as well as endogenous SCCA1 in MDA-MB-231 cells (Suppl. Fig. S5D–S5F) and MDA-MB-468 cells (Suppl. Fig. S5G–S5I), which resulted in decreased expression of ATF4, IL-6, and cell proliferation.
Taken together, the above data indicate that elevated SCCA1 expression leads to IL-6 production and oncogenic transformation by up-regulating a non-lethal level of UPR in mammary epithelial cells. The generality of this notion is strengthened by further observations that overexpression of SCCA1 led to increased IL-6 production and UPR response in the baby mouse kidney (BMK) epithelial cells (39) (Suppl. Fig. S6A–S6C), in human mammary epithelial (HMLE) cell line (Suppl. Fig. S6D and S6E), and in human breast cancer cell line SKBR3 (Suppl. Fig. S6F and S6G).
SCCA1 alters UPR and inflammatory conditions in vivo
To further study the role of SCCA1 in affecting UPR and tumorigenesis in vivo, we generated a conditional SCCA1 transgenic mouse strain with a 129Ola/C57Bl/6 mixed background. The human SCCA1 open reading frame cDNA was cloned into the housekeeping Hprt gene located on the X-chromosome. A stop cassette flanked by LoxP sites was placed upstream of SCCA1 to allow for Cre-specific tissue expression of SCCA1 (LSL-SCCA1; Fig. 6A). The LSL-SCCA1 mice were bred to the MMTV-Cre mice to induce SCCA1 expression in mammary glands, which was confirmed by immunoblotting and IHC (Fig. 6B and 6C). The SCCA1-expressing mice were fertile, sustained pregnancies to term, produced viable offspring, and displayed no apparent defects in ductal morphogenesis, indicating that elevated expression of SCCA1 is not sufficient to drive mammary tumorigenesis by itself. Since our earlier results showed that SCCA1 can promote orthotopic tumor development in MCF10A cell line, which harbors loss of p19Arf tumor suppressor gene, we are currently investigating the role of SCCA1 in tumorigenesis in spontaneous mouse tumor models with loss of tumor suppressor genes including the p19Arf flox/flox model (40). Separately, as our above results also showed that SCCA1 can promote chronic UPR and IL-6 production, we bred the LSL-SCCA1 mice to the MMTV-neu mice (with FVB background; Ref (41)) to determine whether expression of SCCA1 can modulate tumor microenvironment and accelerate tumorigenesis. We chose the MMTV-neu model because it has a relatively long tumor latency of 7–14 months with 50% incidence. A total of 8 neu+; SCCA+ mice and 10 neu+ litter mate control female mice were generated. Four mice from each group developed palpable tumors. The average latency of tumor formation was 407 days and 551 days for neu+; SCCA+ and neu+ only mice, respectively (p = 0.24). The prolonged tumor latency is reminiscent of that of the F1 virgin mice of the FVB and B6 crossing (42). Metastatic tumors were not observed in these mice at endpoint (tumor size 300–500 mm3). We were able to collect 8 tumors for histology and 4 tumors for western blot (2 tumors from each group). Strikingly, when compared with neu+ only tumors, all neu+; SCCA1+ tumor cells invaded the basement membrane into the stromal tissue (Fig. 6D). The SCCA+ tumors also displayed elevated UPR indicated by increased expression of ATF4 and ATF6α-p50 (Fig. 6E), as well as increased intratumoral IL-6 expression and the infiltration of F4/80-positive cells (Fig. 6F). These results indicate that while SCCA1 is not able to accelerate tumor development in the MMTV-neu background, it can clearly alter the tumor microenvironment and increase the potential of tumor invasiveness. Further characterization of the role of SCCA1 in tumorigenesis in tumor models with other genetic lesions is under way.
Figure 6. SCCA1 induces UPR and inflammatory conditions in MMTV-neu mice.

(A) Schematic representation of the LoxP-Stop-LoxP-Flag-SCCA1 (LSL-SCCA1) conditional knock-in strategy. (B and C) LSL-SCCA1 mice were bred to MMTV-Cre mice. SCCA1 expression in mammary gland epithelium of control (LSL-SCCA1; Cre−) or LSL-SCCA1; Cre+ mice was detected by western blot (B) and IHC (C). (D–F) LSL-SCCA1 mice were bred to MMTV-Cre, then to MMTV-neu mice. Tumors were isolated from neu+ only (n = 4) or neu+; SCCA1+ (n = 4) littermates at end points were compared. (D) Representative images of H&E and SCCA1 IHC are shown. H&E staining shows the junction areas of tumor and normal tissues. Note that in neu+;SCCA1+ tumors, tumor cells had invaded the basement membrane into surrounding stroma. (E) Total tumor lysates were made and indicated protein levels were probed by western blots. A non-specific (n.s.) band and Ponseau S staining were used as loading controls. Densitometric ratio of respective proteins to loading control is normalized to that of neu+ mouse #1. (F) IHC staining for IL-6 and F4/80 in neu+ only (n =4) and neu+; SCCA1+ (n =4) tumors. For the quantification of F4/80 staining, 3 or 4 most intensive areas (x200 magnification) from each tumor of all tumors were photographed. The number of positive cells from each picture was analyzed using Image J software. Shown are representative images. The graph shows the average number of positive cells from each tumor section + SEM. *p<0.05.
Discussion
In this study, we report that an intracellular serine/cysteine protease inhibitor SCCA1 promotes the production of IL-6 by inducing chronic UPR and subsequent activation of NF-κB. This leads to an EMT-like phenotype and oncogenic transformation in the non-tumorigenic mammary epithelial cell line MCF10A. Silencing of the ectopically expressed SCCA1 in MCF10A cells and the endogenous SCCA1 in MDA-MB-231 and MDA-MB-468 cells leads to decreased UPR, IL-6 signaling, and halted cell proliferation. We also show that overexpression of SCCA1 in the neu-induced mammary tumors resulted in increased UPR, IL-6 expression, and pro-inflammatory condition.
Our study uncovers a novel pro-tumorigenic role of SCCA1 and helps to explain the association between elevated SCCA1 expression and poorly differentiated and more aggressive human malignancies, namely by inducing IL-6 autocrine signaling and EMT-like phenotype. While we cannot rule out the possibility that other cytokines/growth factors may also be involved, our data strongly indicate that IL-6 autocrine signaling plays a critical role. This is in line with recent literature showing that IL-6 acts to sustain the positive feedback loop to achieve chronic inflammation (24,25), and that cell autonomous inflammation mediated by IL-6 is a key component in driving EMT that have been shown to account for oncogenic transformation and tumor cell de-differentiation (43,44).
Our work also provides SCCA1 as a molecular signature that connects protein homeostasis, UPR, NF-κB activation, and tumorigenesis. We have previously reported that expression of SCCA1 leads to a suppression of both lysosomal and proteasomal protein turnover, in a manner that requires its protease inhibitory activity (13). Although the precise mechanism underlying this SCCA1-mediated blockade of protein turnover remains to be determined, our finding is in line with a recent report that the cathepsin L-deficient cells display impaired lysosomal turnover, accumulation of ubiquitinated proteins and protein aggregates, and disrupted ER homeostasis (45). It is conceivable that the blockade of protein turnover can lead to UPR, which we show here promotes pro-tumorigenic IL-6 production. Our work hence establishes a case where cell intrinsic blockade of protein turnover promotes EMT and tumorigenesis via UPR, which has been implicated in cell de-differentiation and EMT (46,47), as well as for tumorigenesis in animal models of breast cancer (48,49).
The UPR-inducing and pro-tumorigenic function of SCCA1 suggests it may be a predictor and target for therapy. We previously reported that while SCCA1 protects cells from lysosomal disruption resulting from DNA alkylating damage and hypotonic shock, it sensitizes cells to ER stressors (13), suggesting a therapeutic opportunity of using proteotoxic agents such as proteasome inhibitors to treat cancers with elevated SCCA1. In addition, as we show here that the anti-protease activity of SCCA1 is essential for it pro-tumorigenic function, targeting the protease inhibitory function of SCCA1 may be another vital therapeutic approach.
Supplementary Material
Acknowledgments
We thank Drs. Yongjun Fan and Nancy Reich for insightful discussions and critical reading, Drs. Nancy Reich, Laurie Krug, and Jian Cao for reagents. JMC was supported by the NCI T32 training grant (T32CA009176). This work was supported by grants from NIH (R01CA129536 and R01GM97355) and the Carol Baldwin Breast Cancer Research Foundation to WXZ. JZ was supported by NIHR01-NS064090 and a VA merit award.
Footnotes
The authors declare that they have no conflict of interest.
References
- 1.Schick C, Pemberton PA, Shi GP, Kamachi Y, Cataltepe S, Bartuski AJ, et al. Cross-class inhibition of the cysteine proteinases cathepsins K, L, and S by the serpin squamous cell carcinoma antigen 1: a kinetic analysis. Biochemistry. 1998;37:5258–66. doi: 10.1021/bi972521d. [DOI] [PubMed] [Google Scholar]
- 2.Suminami Y, Kishi F, Sekiguchi K, Kato H. Squamous cell carcinoma antigen is a new member of the serine protease inhibitors. Biochem Biophys Res Commun. 1991;181:51–8. doi: 10.1016/s0006-291x(05)81380-4. [DOI] [PubMed] [Google Scholar]
- 3.Kato H, Torigoe T. Radioimmunoassay for tumor antigen of human cervical squamous cell carcinoma. Cancer. 1977;40:1621–8. doi: 10.1002/1097-0142(197710)40:4<1621::aid-cncr2820400435>3.0.co;2-i. [DOI] [PubMed] [Google Scholar]
- 4.Kato H. Expression and function of squamous cell carcinoma antigen. Anticancer Res. 1996;16:2149–53. [PubMed] [Google Scholar]
- 5.Vidalino L, Doria A, Quarta S, Zen M, Gatta A, Pontisso P. SERPINB3, apoptosis and autoimmunity. Autoimmun Rev. 2009;9:108–12. doi: 10.1016/j.autrev.2009.03.011. [DOI] [PubMed] [Google Scholar]
- 6.Katagiri C, Nakanishi J, Kadoya K, Hibino T. Serpin squamous cell carcinoma antigen inhibits UV-induced apoptosis via suppression of c-JUN NH2-terminal kinase. J Cell Biol. 2006;172:983–90. doi: 10.1083/jcb.200508064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Murakami A, Suminami Y, Hirakawa H, Nawata S, Numa F, Kato H. Squamous cell carcinoma antigen suppresses radiation-induced cell death. Br J Cancer. 2001;84:851–8. doi: 10.1054/bjoc.2000.1683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Suminami Y, Nagashima S, Murakami A, Nawata S, Gondo T, Hirakawa H, et al. Suppression of a squamous cell carcinoma (SCC)-related serpin, SCC antigen, inhibits tumor growth with increased intratumor infiltration of natural killer cells. Cancer Res. 2001;61:1776–80. [PubMed] [Google Scholar]
- 9.Suminami Y, Nagashima S, Vujanovic NL, Hirabayashi K, Kato H, Whiteside TL. Inhibition of apoptosis in human tumour cells by the tumour-associated serpin, SCC antigen-1. Br J Cancer. 2000;82:981–9. doi: 10.1054/bjoc.1999.1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Turato C, Buendia MA, Fabre M, Redon MJ, Branchereau S, Quarta S, et al. Over-expression of SERPINB3 in hepatoblastoma: a possible insight into the genesis of this tumour? Eur J Cancer. 2012;48:1219–26. doi: 10.1016/j.ejca.2011.06.004. [DOI] [PubMed] [Google Scholar]
- 11.Catanzaro JM, Guerriero JL, Liu J, Ullman E, Sheshadri N, Chen JJ, et al. Elevated expression of squamous cell carcinoma antigen (SCCA) is associated with human breast carcinoma. PLoS One. 2011;6:e19096. doi: 10.1371/journal.pone.0019096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Collie-Duguid ES, Sweeney K, Stewart KN, Miller ID, Smyth E, Heys SD. SerpinB3, a new prognostic tool in breast cancer patients treated with neoadjuvant chemotherapy. Breast Cancer Res Treat. 2012;132:807–18. doi: 10.1007/s10549-011-1625-9. [DOI] [PubMed] [Google Scholar]
- 13.Ullman E, Pan JA, Zong WX. Squamous cell carcinoma antigen 1 promotes caspase-8-mediated apoptosis in response to endoplasmic reticulum stress while inhibiting necrosis induced by lysosomal injury. Mol Cell Biol. 2011;31:2902–19. doi: 10.1128/MCB.05452-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang S, Kaufman RJ. The impact of the unfolded protein response on human disease. J Cell Biol. 2012;197:857–67. doi: 10.1083/jcb.201110131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lee AS. Glucose-regulated proteins in cancer: molecular mechanisms and therapeutic potential. Nat Rev Cancer. 2014;14:263–76. doi: 10.1038/nrc3701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Clarke R, Cook KL, Hu R, Facey CO, Tavassoly I, Schwartz JL, et al. Endoplasmic reticulum stress, the unfolded protein response, autophagy, and the integrated regulation of breast cancer cell fate. Cancer Res. 2012;72:1321–31. doi: 10.1158/0008-5472.CAN-11-3213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Verfaillie T, Garg AD, Agostinis P. Targeting ER stress induced apoptosis and inflammation in cancer. Cancer Lett. 2013;332:249–64. doi: 10.1016/j.canlet.2010.07.016. [DOI] [PubMed] [Google Scholar]
- 18.Diehl JA, Fuchs SY, Koumenis C. The cell biology of the unfolded protein response. Gastroenterology. 2011;141:38–41. e1–2. doi: 10.1053/j.gastro.2011.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Quarta S, Vidalino L, Turato C, Ruvoletto M, Calabrese F, Valente M, et al. SERPINB3 induces epithelial-mesenchymal transition. J Pathol. 2010;221:343–56. doi: 10.1002/path.2708. [DOI] [PubMed] [Google Scholar]
- 20.Jordan NV, Johnson GL, Abell AN. Tracking the intermediate stages of epithelial-mesenchymal transition in epithelial stem cells and cancer. Cell Cycle. 2011;10:2865–73. doi: 10.4161/cc.10.17.17188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schick C, Bromme D, Bartuski AJ, Uemura Y, Schechter NM, Silverman GA. The reactive site loop of the serpin SCCA1 is essential for cysteine proteinase inhibition. Proc Natl Acad Sci U S A. 1998;95:13465–70. doi: 10.1073/pnas.95.23.13465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Turato C, Calabrese F, Biasiolo A, Quarta S, Ruvoletto M, Tono N, et al. SERPINB3 modulates TGF-beta expression in chronic liver disease. Lab Invest. 2010;90:1016–23. doi: 10.1038/labinvest.2010.55. [DOI] [PubMed] [Google Scholar]
- 23.Miettinen PJ, Ebner R, Lopez AR, Derynck R. TGF-beta induced transdifferentiation of mammary epithelial cells to mesenchymal cells: involvement of type I receptors. J Cell Biol. 1994;127:2021–36. doi: 10.1083/jcb.127.6.2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Iliopoulos D, Hirsch HA, Struhl K. An epigenetic switch involving NF-kappaB, Lin28, Let-7 MicroRNA, and IL6 links inflammation to cell transformation. Cell. 2009;139:693–706. doi: 10.1016/j.cell.2009.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Korkaya H, Liu S, Wicha MS. Regulation of cancer stem cells by cytokine networks: attacking cancer’s inflammatory roots. Clin Cancer Res. 2011;17:6125–9. doi: 10.1158/1078-0432.CCR-10-2743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Scheel C, Eaton EN, Li SH, Chaffer CL, Reinhardt F, Kah KJ, et al. Paracrine and autocrine signals induce and maintain mesenchymal and stem cell states in the breast. Cell. 2011;145:926–40. doi: 10.1016/j.cell.2011.04.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yarden Y. The EGFR family and its ligands in human cancer. signalling mechanisms and therapeutic opportunities. Eur J Cancer. 2001;37 (Suppl 4):S3–8. doi: 10.1016/s0959-8049(01)00230-1. [DOI] [PubMed] [Google Scholar]
- 28.Huber MA, Azoitei N, Baumann B, Grunert S, Sommer A, Pehamberger H, et al. NF-kappaB is essential for epithelial-mesenchymal transition and metastasis in a model of breast cancer progression. J Clin Invest. 2004;114:569–81. doi: 10.1172/JCI21358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chua HL, Bhat-Nakshatri P, Clare SE, Morimiya A, Badve S, Nakshatri H. NF-kappaB represses E-cadherin expression and enhances epithelial to mesenchymal transition of mammary epithelial cells: potential involvement of ZEB-1 and ZEB-2. Oncogene. 2007;26:711–24. doi: 10.1038/sj.onc.1209808. [DOI] [PubMed] [Google Scholar]
- 30.Garg AD, Kaczmarek A, Krysko O, Vandenabeele P, Krysko DV, Agostinis P. ER stress-induced inflammation: does it aid or impede disease progression? Trends Mol Med. 2012;18:589–98. doi: 10.1016/j.molmed.2012.06.010. [DOI] [PubMed] [Google Scholar]
- 31.Zhang K, Kaufman RJ. From endoplasmic-reticulum stress to the inflammatory response. Nature. 2008;454:455–62. doi: 10.1038/nature07203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Walter P, Ron D. The unfolded protein response: from stress pathway to homeostatic regulation. Science. 2011;334:1081–6. doi: 10.1126/science.1209038. [DOI] [PubMed] [Google Scholar]
- 33.Hetz C. The unfolded protein response: controlling cell fate decisions under ER stress and beyond. Nat Rev Mol Cell Biol. 2012;13:89–102. doi: 10.1038/nrm3270. [DOI] [PubMed] [Google Scholar]
- 34.Okada T, Yoshida H, Akazawa R, Negishi M, Mori K. Distinct roles of activating transcription factor 6 (ATF6) and double-stranded RNA-activated protein kinase-like endoplasmic reticulum kinase (PERK) in transcription during the mammalian unfolded protein response. Biochem J. 2002;366:585–94. doi: 10.1042/BJ20020391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Haze K, Yoshida H, Yanagi H, Yura T, Mori K. Mammalian transcription factor ATF6 is synthesized as a transmembrane protein and activated by proteolysis in response to endoplasmic reticulum stress. Mol Biol Cell. 1999;10:3787–99. doi: 10.1091/mbc.10.11.3787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mao W, Fukuoka S, Iwai C, Liu J, Sharma VK, Sheu SS, et al. Cardiomyocyte apoptosis in autoimmune cardiomyopathy: mediated via endoplasmic reticulum stress and exaggerated by norepinephrine. Am J Physiol Heart Circ Physiol. 2007;293:H1636–45. doi: 10.1152/ajpheart.01377.2006. [DOI] [PubMed] [Google Scholar]
- 37.Calfon M, Zeng H, Urano F, Till JH, Hubbard SR, Harding HP, et al. IRE1 couples endoplasmic reticulum load to secretory capacity by processing the XBP-1 mRNA. Nature. 2002;415:92–6. doi: 10.1038/415092a. [DOI] [PubMed] [Google Scholar]
- 38.Lin JH, Li H, Yasumura D, Cohen HR, Zhang C, Panning B, et al. IRE1 signaling affects cell fate during the unfolded protein response. Science. 2007;318:944–9. doi: 10.1126/science.1146361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Degenhardt K, Sundararajan R, Lindsten T, Thompson C, White E. Bax and Bak independently promote cytochrome C release from mitochondria. J Biol Chem. 2002;277:14127–34. doi: 10.1074/jbc.M109939200. [DOI] [PubMed] [Google Scholar]
- 40.Gromley A, Churchman ML, Zindy F, Sherr CJ. Transient expression of the Arf tumor suppressor during male germ cell and eye development in Arf-Cre reporter mice. Proc Natl Acad Sci U S A. 2009;106:6285–90. doi: 10.1073/pnas.0902310106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Guy CT, Webster MA, Schaller M, Parsons TJ, Cardiff RD, Muller WJ. Expression of the neu protooncogene in the mammary epithelium of transgenic mice induces metastatic disease. Proc Natl Acad Sci U S A. 1992;89:10578–82. doi: 10.1073/pnas.89.22.10578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rowse GJ, Ritland SR, Gendler SJ. Genetic modulation of neu proto-oncogene-induced mammary tumorigenesis. Cancer Res. 1998;58:2675–9. [PubMed] [Google Scholar]
- 43.Gao SP, Mark KG, Leslie K, Pao W, Motoi N, Gerald WL, et al. Mutations in the EGFR kinase domain mediate STAT3 activation via IL-6 production in human lung adenocarcinomas. J Clin Invest. 2007;117:3846–56. doi: 10.1172/JCI31871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sansone P, Storci G, Tavolari S, Guarnieri T, Giovannini C, Taffurelli M, et al. IL-6 triggers malignant features in mammospheres from human ductal breast carcinoma and normal mammary gland. J Clin Invest. 2007;117:3988–4002. doi: 10.1172/JCI32533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sun M, Ouzounian M, de Couto G, Chen M, Yan R, Fukuoka M, et al. Cathepsin-L ameliorates cardiac hypertrophy through activation of the autophagy-lysosomal dependent protein processing pathways. J Am Heart Assoc. 2013;2:e000191. doi: 10.1161/JAHA.113.000191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yang L, Carlson SG, McBurney D, Horton WE., Jr Multiple signals induce endoplasmic reticulum stress in both primary and immortalized chondrocytes resulting in loss of differentiation, impaired cell growth, and apoptosis. J Biol Chem. 2005;280:31156–65. doi: 10.1074/jbc.M501069200. [DOI] [PubMed] [Google Scholar]
- 47.Tanjore H, Cheng DS, Degryse AL, Zoz DF, Abdolrasulnia R, Lawson WE, et al. Alveolar epithelial cells undergo epithelial-to-mesenchymal transition in response to endoplasmic reticulum stress. J Biol Chem. 2011;286:30972–80. doi: 10.1074/jbc.M110.181164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Dong D, Ni M, Li J, Xiong S, Ye W, Virrey JJ, et al. Critical role of the stress chaperone GRP78/BiP in tumor proliferation, survival, and tumor angiogenesis in transgene-induced mammary tumor development. Cancer Res. 2008;68:498–505. doi: 10.1158/0008-5472.CAN-07-2950. [DOI] [PubMed] [Google Scholar]
- 49.Bobrovnikova-Marjon E, Grigoriadou C, Pytel D, Zhang F, Ye J, Koumenis C, et al. PERK promotes cancer cell proliferation and tumor growth by limiting oxidative DNA damage. Oncogene. 2010;29:3881–95. doi: 10.1038/onc.2010.153. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.