

Abstract
Herpes simplex virus type 1 (HSV-1) encodes a portal protein that forms a large oligomeric structure believed to provide the conduit for DNA entry and exit from the capsid. Chaperone proteins often facilitate the folding and multimerization of such complex structures. In this report, we show that cellular chaperone proteins, components of the 26S proteasome, and ubiquitin-conjugated proteins are sequestered in discrete foci in the nucleus of the infected cell. The immediate-early viral protein ICP0 was shown to be necessary to establish these foci at early times during infection and sufficient to redistribute chaperone molecules in transfected cells. Furthermore, we found that not only is the portal protein, UL6, localized to these sites during infection, but it is also a substrate for ubiquitin modification. Our results suggest that HSV-1 has evolved an elegant mechanism for facilitating protein quality control at specialized foci within the nucleus.
Protein folding and oligomerization can be facilitated by molecular chaperones. Molecular chaperones recognize and interact with non-native proteins, preventing their premature or improper interaction with other polypeptides, and assist proper folding in an energy-dependent fashion (13, 14, 16). Chaperones can also facilitate oligomerization of protein complexes. For example, the Escherichia coli GroES/EL chaperone system is required for the folding and multimerization of the lambda (λ) bacteriophage connector complex during a viral infection (16, 17). The bacteriophage connector (or portal) is a ring-shaped structure utilized by many large double-stranded DNA bacteriophages as the docking site for DNA packaging (terminase) enzymes, the channel for DNA entry and exit, and the site for tail attachment (reviewed in reference 3). It is now known that herpes simplex virus type 1 (HSV-1) encodes an analogous structure, as the portal protein (UL6) has been visualized at a unique vertex of the capsid by immuno-electron microscopy of purified virus particles (29). The HSV-1 portal protein can be isolated in a soluble form from recombinant baculovirus-infected insect cell lysates as a 1-MDa dodecameric ring that is reminiscent of connector proteins of some large DNA bacteriophages (26). Moreover, the specific association of the HSV-1 terminase homologue, UL15, with the immature viral capsid is dependent on the portal vertex protein, UL6 (32, 34, 39).
It is unknown whether chaperone assistance is required for portal formation during an HSV-1 infection. Furthermore, the question of how misfolded viral proteins are handled within the HSV-1-infected cell has never been addressed. Given the similarities between bacteriophage connector proteins and the HSV-1 portal protein and the possibility that this complex structure may need assistance during formation, we wanted to determine whether the cellular chaperone and proteasomal machinery were relocated during HSV-1 infection. In this report, we provide evidence supporting the hypothesis that the host chaperone machinery facilitates the formation of the HSV-1 portal complex. Moreover, our observations suggest that terminally misfolded portal proteins may be targeted for degradation in a ubiquitin-dependent fashion and that this occurs within novel nuclear structures established during viral infection.
MATERIALS AND METHODS
Cells, viruses, and antibodies.
African green monkey kidney cells (Vero CCl81; American Type Culture Collection, Rockville, Md.) were propagated and maintained as described previously (37). The human osteosarcoma cell line U2OS (U2OS HTB96; American Type Culture Collection) is naturally permissive for the HSV-1 ICP0 mutant virus (38). The KOS strain of HSV-1 was used as the wild-type virus. The HSV-1 ICP0 mutant 0β, a deletion mutant in which exons 1 and 2 and the intervening intron of ICP0 were replaced by an insertion of the LacZ gene, was kindly provided by Neal DeLuca (University of Pittsburgh School of Medicine). Jay C. Brown (University of Virginia Health System) provided the anti-UL6 monoclonal antibodies 1C9 and 4G9. The monoclonal anti-ICP0 antibody was described previously (35). The anti-ICP8 polyclonal antibody was generously provided by William T. Ruyechan (University of New York at Buffalo). Rat monoclonal anti-Hsc70, mouse monoclonal anti-Hsp70, and rabbit polyclonal anti-Hsp40 antibodies were purchased from StressGen (Victoria, British Columbia, Canada). The rabbit polyclonal anti-20S catalytic core and monoclonal FK2 antibodies were purchased from Affiniti (Exeter, Devon, United Kingdom). Gary H. Cohen and Roselyn J. Eisenberg (University of Pennsylvania School of Dental Medicine) kindly provided polyclonal antibodies NC-1 (anti-VP5), NC2 (anti-VP19c), NC5 (anti-VP23), and NC7 (anti-VP26). Other antibodies used for these studies included a monoclonal anti-VP5 antibody purchased from Advanced Biotechnologies Inc. (Columbia, Md.) and MCA406 (monoclonal anti-VP21 antibody) purchased from Serotech (Raleigh, N.C.). Secondary antibodies were purchased from Molecular Probes (Eugene, Oreg.) and included AlexaFluor 488-conjugated goat anti-mouse, AlexaFluor 594-conjugated goat anti-rat, AlexaFluor 647-conjugated goat anti-rabbit, AlexaFluor 488-conjugated goat anti-rabbit, and AlexaFluor 594-conjugated goat anti-rabbit antibodies. It was necessary to use commercially available highly cross-adsorbed secondary antibodies to prevent cross-reactivities between rat and mouse primary antibodies.
Immunofluorescence confocal microscopy.
Subconfluent Vero or U2OS cells (105) were grown on sterile glass coverslips prior to infection or transfection. After infection or transfection, the medium was aspirated and the coverslips were washed three times with room temperature 1× phosphate-buffered saline (PBS). The cells were fixed with fresh 4% paraformaldehyde, pH 7.4, in PBS for 10 min at room temperature. The coverslips were washed three times and then permeabilized with 1% NP-40 in PBS for 10 min at the ambient temperature. After three additional washes with PBS, the cells were blocked with 10% normal goat serum (Sigma Chemical, Steinheim, Germany) in PBS for 1 h to overnight. The coverslips were inverted on a drop of diluted primary antibody for 1 h, and then they were returned to culture dishes and subjected to seven washes with PBS. The dilutions of primary antibodies used were as follows: ICP8, 1:700; UL6 4g9, 1:700; ICP0, 1:200; Hsc70, 1:200; Hsp40, 1:200; 20S, 1:200; FK2, 1:700. The coverslips were inverted once again onto a drop of diluted secondary antibody (1:200) for 1 h. They were then returned to culture dishes and subjected to seven washes with PBS. Warm gelatin containing 2.5% diazibicyclooctane (a photobleaching retardant; Sigma Chemical) was used to secure the glass slips onto glass slides, and clear nail enamel was used to seal the edges of the coverslips. Fluorescence microscopy was performed with a Zeiss LSM 410 confocal microscope using a 100× objective. Adobe Photoshop 5.0 was used for image preparation for the figures. Controls included mock-infected cells and infected cells treated with a single primary antibody and a secondary antibody to assess cross-reactivity.
Viral infections. (i) Time course infections for Western blot analysis.
Vero cells seeded to confluence in 60-mm2 dishes (106 cells) were infected with HSV-1 (strain KOS) at a multiplicity of infection (MOI) of 3 for 1 h at 37°C. The virus was then removed and fresh medium was added. At each time point, the medium was removed and the cells were washed twice with PBS at room temperature. The infected cell monolayer was collected in 200 μl of 1× sodium dodecyl sulfate (SDS)-polyacrylamide gel electrophoresis loading buffer. This material was sonicated in a bath sonicator (three times for 10 s each at 50% strength) and then boiled for 5 min. For Western blot analysis, 20 μl of this material was used. A heat-shocked HeLa cell lysate used as a control was purchased from StressGen.
(ii) Time course infections.
Cells seeded on sterile glass coverslips were infected at an MOI of 3 for 1 h at 37°C. After this time, the virus was removed, the cells were washed once with sterile PBS, fresh medium was added, and the cells were incubated at an appropriate temperature. At each time point, the coverslips were removed and fixed immediately as described above. After samples for all time points had been collected, the coverslips were permeabilized and stained as described above. Infections in the presence of the viral polymerase inhibitor phosphonoacetic acid (PAA) were conducted essentially as described by Lukonis and Weller, except that an MOI of 3 was used for our studies (23). Infections in the presence of the protein synthesis inhibitor cycloheximide (CHX) were conducted as follows. Vero cells seeded to 60% confluence (5 × 105 cells) were treated with 100 μg of CHX per ml in Dulbecco's modified Eagle medium (Sigma Chemical) for 1 h prior to and during infection. CHX-treated cells were infected at an MOI of 3 and the virus was allowed to adsorb for 1 h at 37°C. After infection, the virus was removed and fresh medium containing 100 μg of CHX per ml was added.
Immunoprecipitation.
Confluent Vero cells grown in 225-mm2 flasks (2 × 107 cells) were infected with HSV-1 KOS at an MOI of 5 for 1 h at 37°C. After virus adsorption, the infection medium was removed and replaced with fresh medium and the cells were allowed to incubate at 37°C. Infections used for the analysis of UL6 ubiquitination were harvested at 18 h postinfection. At harvest, the medium was aspirated and infected cells were washed twice with room temperature 1× PBS. Five milliliters of PBS was added to each flask, and the cells were collected by gentle scraping. Cells were pelleted by low-speed centrifugation, and the PBS was then removed. The cells were lysed on ice in 4 ml of VGLB buffer (10 mM HEPES [pH 7.5], 10 mM NaCl, 10 mM EDTA, and 0.1% Triton X-100, with 1 mM dithiothreitol and complete protease inhibitor [Roche] added just prior to use) for 30 min. The insoluble material was removed by centrifugation at 13,000 × g at 4°C for 15 min. After centrifugation, NaCl was added to a final concentration of 150 mM. For each immunoprecipitation reaction mixture, 0.5 ml (approximately 400 ng of total protein) of infected cell lysate was incubated with 2 μg of primary antibody for 2 h at 4°C. Twenty-five microliters of a protein A/G-agarose slurry (Santa Cruz Biotech, Santa Barbara, Calif.) was added to each sample and allowed to incubate at 4°C for 1 h. Beads were collected by gentle centrifugation (2,000 × g for 2 min) and washed three times with VGLB buffer. After the final wash, 25 μl of 2× SDS-polyacrylamide gel electrophoresis buffer was added and samples were boiled for 5 min. Controls included normal mouse immunoglobulin G (IgG) and a lysate incubated with the protein A/G slurry alone.
RESULTS
Hsc70, Hsp70, and Hsp40 are recruited to nuclear domains during HSV-1 infection.
HSV-1 virus replication, capsid assembly, and DNA packaging occur in large globular domains called replication compartments within the nucleus of the infected cell (21). The Hsp70/Hsc70 family of molecular chaperones is the only major family of chaperones found in the nucleus, and we asked whether these proteins could be detected at or near sites of virus assembly. This family of molecular chaperones includes a highly inducible form (Hsp70) and a constitutively expressed cognate form (Hsc70). Hsp70 and Hsc70 specifically bind hydrophobic amino acid stretches that are exposed in the denatured state of the target protein. The ATP-dependent chaperone activities of Hsp70 and Hsc70 require Hsp40 as a cochaperone for the acquisition of misfolded peptides (reviewed in reference 13). This chaperone system has also been implicated in the resolution of protein aggregates (28). We used confocal fluorescence microscopy to study the subcellular localization of the mammalian chaperone machinery in HSV-1-infected cells. In uninfected cells cultured at 37°C, the Hsc70 protein was detected as diffuse staining in both the nuclear and cytoplasmic regions, with some staining in the nucleolar compartment (Fig. 1A and C). We found a dramatic redistribution of the Hsc70 protein during HSV-1 infection. In cells infected with HSV-1, Hsc70 was reorganized into punctate foci in the nucleus (Fig. 1D and F). In uninfected cells, Hsp40, the Hsc70 cochaperone, was observed in a diffuse nuclear staining pattern, but in HSV-1 infected cells a different pattern of staining was observed (Fig. 1, compare panels B and E). Although the cochaperone Hsp40 was detected at the Hsc70 foci (Fig. 1E and F, white arrows), a large fraction of the protein was redistributed throughout the infected cell, with increased staining in the cytoplasm. It is possible that Hsp40 cycles in and out of the nucleus, suggesting that it could be involved in trafficking unfolded proteins into the nucleus for proper folding and/or functioning. Additional experiments will be needed to address this issue.
FIG. 1.

Subcellular localization of the cellular chaperone Hsc70/Hsp70 and cochaperone Hsp40 during HSV-1 infection. (A to C, G to I) Uninfected Vero cells cultured for 6 h at 37°C. (D to F, J to L) Vero cells infected with wild-type HSV-1 (strain KOS) for 6 h at 37°C. The staining profile for Hsc70 (red) is shown in panels A and D, the staining profile for Hsp70 (red) is shown in panels G and J, and the staining profile of Hsp40 (green) is shown in panels B, E, H, and K. Merged Hsc70/Hsp40 images are shown in panels C, F, I, and L. White arrows show Hsp40-enriched foci (E and K) that costain with Hsc70 or Hsp70 in the merged image (F and L, respectively).
Cellular Hsp70 was also observed as foci within the nucleus of the HSV-1 infected cell (Fig. 1J). Moreover, these foci costained with an antibody specific to Hsp40 (Fig. 1K and L, white arrows). Hsc70-enriched foci were observed for a broad range of temperatures (34 to 39.5°C) (data not shown). We thus conclude that chaperone sequestration is not a temperature-dependent phenomenon. However, we noted that these foci were observed at earlier times postinfection in cells that were incubated at an elevated temperature (39.5°C) (data not shown); this may reflect an acceleration of the viral infection. It is interesting that at this temperature Hsc70 is concentrated in the nucleus, but it is unclear if this localization assists the HSV-1 infection.
To determine whether viral infection results in an increase in the amount of chaperone proteins, we performed a Western blot analysis with HSV-1-infected cell lysates (Fig. 2). As shown in Fig. 2, the overall levels of Hsc70, Hsp70, and Hsp40 in HSV-1-infected cells did not change relative to those in uninfected Vero cells, suggesting that the redistribution observed upon infection was of existing chaperone molecules.
FIG. 2.
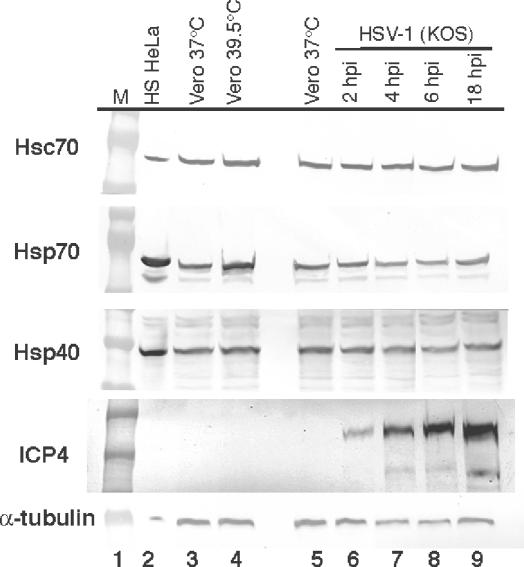
Western blot analysis of cellular chaperone molecules (Hsc70, Hsp70, and Hsp40) in HSV-1-infected cell lysates. Lane 1, molecular weight markers; lane 2, heat-shocked HeLa cell lysate; lane 3, uninfected Vero cells cultured at 37°C; lane 4, uninfected Vero cells heat shocked at 39.5°C; lane 5, uninfected Vero cells cultured at 37°C; lanes 6 to 9, HSV-1-infected cell lysates at 2, 4, 6, and 18 h postinfection, respectively. A Western blot of the viral protein ICP4 is shown as a positive control for infection, and a Western blot of α-tubulin was used as a control to ensure that similar amounts of total protein were loaded for all samples.
The immediate-early protein ICP0 is necessary to reorganize the host chaperone machinery at early times during HSV-1 infection and sufficient to alter the distribution of chaperone molecules in transfected cells.
The redistribution of Hsc70 corresponded to a time in the infection when relatively few viral proteins were abundant. To determine whether the formation of the foci required either protein synthesis or viral DNA synthesis, we treated cells with CHX, a global inhibitor of protein synthesis, or the viral polymerase inhibitor PAA and subsequently infected them with HSV-1 (Fig. 3). In uninfected cells treated with CHX (panel A) or PAA (panel B), Hsp40 and Hsc70 exhibited a similar staining pattern to that of untreated cells (Fig. 1C). In HSV-1-infected cells treated with CHX, no Hsc70-enriched foci were observed (Fig. 3C), indicating that de novo protein synthesis was required. On the other hand, in cells treated with PAA, foci enriched with Hsc70 and Hsp40 were still observed (Fig. 3D). Thus, the redistribution of Hsc70 and Hsp40 is independent of viral DNA synthesis but apparently requires the synthesis of either an immediate-early or early viral gene product.
FIG. 3.
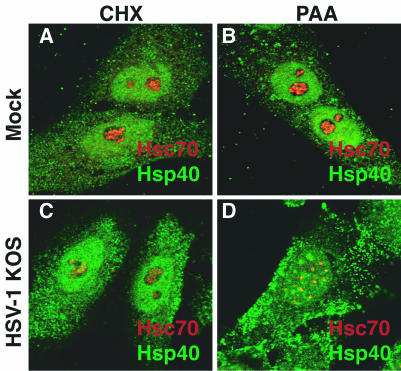
Analysis of chaperone localization in infected or uninfected cells treated with CHX or PAA. Merged images of cells stained with antibodies specific to Hsc70 (red) and Hsp40 (green) are shown. (A and B) Uninfected cells treated with CHX or PAA. (C and D) HSV-1-infected cells treated with CHX or PAA.
To identify the viral components responsible for redistribution, we examined the subcellular localization of the immediate-early viral proteins ICP0 and ICP4 at 1, 2, and 3 h postinfection at 37 and 39.5°C. At 1 and 2 h postinfection at 37°C, ICP0 was visualized as punctate dots within the nucleus of the infected cell (Fig. 4A and B, green areas). The host chaperone Hsc70 was observed as diffuse staining throughout the cell and was also concentrated within the nucleolus (Fig. 4A and B, red areas). At 3 h postinfection at this temperature, both the Hsc70 and ICP0 proteins were detected within the nucleus as punctate foci similar to those seen in Fig. 1D (Fig. 4C). Although most of the ICP0 foci stained with the ICP0 antibody alone (white arrow), some foci were observed which colocalized with Hsc70 (red arrow). It is likely that some ICP0 is recruited to other nuclear domains, such as ND10. Consistent with this observation, ICP0 has been observed to localize to ND10 at least at very early times postinfection (12). Moreover, it has been reported that only a fraction of the ICP0-containing foci contain components of ND10, such as promyelocytic leukemia protein (PML) (10, 25). Infected cells cultured at the higher temperature (39.5°C) revealed an even more efficient colocalization of ICP0 and Hsc70 (Fig. 4D to F). As early as 1 h postinfection, the ICP0 protein was colocalized with Hsc70 within the nucleolus of the infected cell (Fig. 4D, yellow areas). Subsequently, both proteins were detected within the nucleus as punctate dots (Fig. 4E and F). Many of these foci costained with antibodies specific to Hsc70 and ICP0 (panel F, red arrow) and others only stained with the antibody specific to ICP0 (panel F, white arrow). On the other hand, no colocalization was observed for the ICP4 protein and Hsc70-enriched foci (data not shown).
FIG. 4.

Merged images of HSV-1-infected cells stained with antibodies detecting Hsc70 (red) and the viral ICP0 protein (green). Times (in hours) postadsorption are indicated above each panel. (A to C) Infected cells cultured at 37°C; (D to F) infected cells cultured at 39.5°C. In panels C and F, the white arrows indicate ICP0-stained foci that did not costain with Hsc70 and likely represent ND10. The red arrows indicate foci that costained with antibodies recognizing either Hsc70 or ICP0. In panel D, note the colocalization (yellow) between ICP0 and Hsc70 within the nucleolus.
To determine whether the ICP0 protein was necessary to reorganize the Hsc70 protein, we analyzed cells infected with an ICP0 null mutant (0β) which had a deletion of the entire ICP0 gene (1). 0β can be propagated on U2OS cells, a human cell line that naturally supports the growth of HSV-1 ICP0 mutants (38). Vero and U2OS cells were infected at 39.5°C with the 0β mutant and analyzed at 6 h postinfection by confocal microscopy (Fig. 5A to F). As mentioned above, the kinetics of Hsc70-enriched focus formation are faster at this temperature. By 6 h after infection, we predicted that foci would be present. Vero cells infected with 0β exhibited a predominantly nucleolar pattern of Hsc70 staining that was identical to that of the uninfected control (compare Fig. 5B and Fig. 1A). To ensure that the cells were actually infected, we assessed ICP4 staining, since it is known that the expression of the ICP4 protein is independent of ICP0 expression (9). More than 90% of the cells analyzed were capable of expressing ICP4, indicating that the cells were indeed infected (data not shown).
FIG. 5.

ICP0 is necessary for chaperone sequestration at early times during infection and sufficient to redistribute the cellular chaperone machinery in transfected cells. Vero cells infected with the 0β mutant (ICP0 mutant) (A to C) at 39.5°C were stained with ICP4 (green) and Hsc70 (red). This temperature was chosen because of the faster kinetics of Hsc70-enriched foci. Thus, at 6 h postexposure to HSV-1, there would have been ample time for Hsc70 focus formation. Staining for the ICP4 protein (green) (A and D) was used as a control for successful infection. As described in the text, in the absence of the ICP0 protein, no redistribution of Hsc70 was observed at this time (B), indicating that ICP0 is necessary for this action early during infection. (D to F) 0β-infected U2OS cells (a human osteosarcoma cell line that naturally complements ICP0 HSV-1 mutants) stained for ICP4 (D) and Hsc70 (E). Merged images are shown in panels C and F. Merged images of HSV-1 (strain KOS)-infected (G) and 0β-infected (H) Vero cells collected at 20 h postinfection are shown as well. (I to K) Subcellular localization of ICP0 (green) and Hsc70 (red) in cells transfected with a plasmid carrying the ICP0 gene. A merged image of the transfected cell is shown in panel K.
In 0β-infected Vero cells at 6 h postinfection, ICP4 exhibited a diffuse nuclear staining pattern (Fig. 5A and C). On the other hand, in U2OS cells infected with 0β, ICP4 localized to replication compartments (Fig. 5D and F), indicating that the infection was permissive. Furthermore, Fig. 5E shows that 0β-infected U2OS cells exhibited Hsc70-containing foci, suggesting that the ability of U2OS cells to complement an ICP0 mutant correlates with their ability to redistribute Hsc70 during infection and raising the possibility that a cellular protein can substitute for the absence of ICP0. This concept is consistent with a report by Cai and Schaffer (7) suggesting that a cellular function can enhance the growth of an ICP0 mutant virus.
To determine whether Hsc70-enriched foci eventually formed in the ICP0 null viral infection at later times, we infected Vero cells with wild-type HSV-1 or 0β and analyzed them at 20 h postinfection. Twenty hours after infection with wild-type HSV-1, large replication compartments and Hsc70-enriched foci were detected (Fig. 5G). Figure 5H shows a merged image of 0β-infected Vero cells at 20 h postinfection. In these cells, Hsc70-enriched foci were detected adjacent to somewhat smaller replication compartments. These results indicate that an unidentified cellular factor can ultimately establish chaperone-enriched foci and suggest that ICP0 can accelerate this process.
We have previously shown that cells transfected with an ICP0-expressing plasmid form large nuclear and cytoplasmic inclusions (24). When cells were cotransfected with the ICP0 plasmid and plasmids encoding individual components of the trimeric viral helicase-primase or carrying the UL6 gene, the proteins were observed to be sequestered inside the ICP0 inclusions. We hypothesized that proteins that were difficult to fold or that lacked binding partners were sequestered within these sites, since other proteins which presumably could fold and function on their own, such as ICP8 and UL9, did not localize to these inclusions (24). In light of the results for infected cells described above, we decided to revisit the formation of these ICP0-stimulated inclusions. We were particularly interested in whether Hsc70, which might be expected to be present at sites of misfolded or aggregated proteins, would localize to the interior of the ICP0 transfection inclusions. Figure 5 (panels I to K) not only confirms the previous report that ICP0 forms large inclusions within transfected cells but also shows that the interiors of the inclusions stain brightly for Hsc70. ICP0 alone appears to be sufficient to reorganize Hsc70. We cannot rule out the possibility that, when overexpressed, ICP0 itself is misfolded and a substrate for the chaperone machinery; however, for reasons described below, we favor the hypothesis that ICP0 plays an active role in the redistribution of viral and cellular proteins. We suggest that the role of these Hsc70-enriched foci is to sequester aberrant or misfolded proteins from the rest of the cell.
Foci are enriched with components of the 26S proteasome and with ubiquitinated proteins.
It has been shown that ICP0 stimulates the degradation of specific cellular proteins in a ubiquitin-mediated proteasome-dependent fashion (11, 18, 30). Thus, we wanted to determine whether ubiquitin-conjugated proteins and components of the 26S proteasome were also localized to the Hsc70-enriched foci during infection. Ubiquitinated proteins (but not free ubiquitin) can be detected within cells by using the FK2 monoclonal antibody (15). In uninfected cells, ubiquitinated proteins were detected primarily in a punctate staining pattern on a diffuse nuclear background (Fig. 6B and C). In HSV-1-infected cells, staining of ubiquitinated proteins was observed in the cytoplasm and in punctate foci in the nucleus, while the diffuse nuclear background was not seen (Fig. 6E and F). The absence of the diffuse staining pattern is particularly obvious when the uninfected cell on the right (white arrow) is compared to the infected cell on the left (red arrow) (Fig. 6E). In infected cells, Hsc70 was redistributed from the predominantly nucleolar staining of uninfected cells (Fig. 6A) to a punctate pattern which colocalized with sites of ubiquitinated proteins (Fig. 6D and F). When stained with a polyclonal antibody directed against core catalytic components of the 26S proteasome and FK2, uninfected cells exhibited a diffuse nuclear staining pattern with a few punctate foci (Fig. 6G to I). In HSV-1-infected cells, we observed a remarkable redistribution of both proteasome subunits and ubiquitinated proteins into distinct foci (Fig. 6J to L) that were most likely equivalent to the Hsc70-enriched foci observed above (Fig. 6D and F).
FIG. 6.
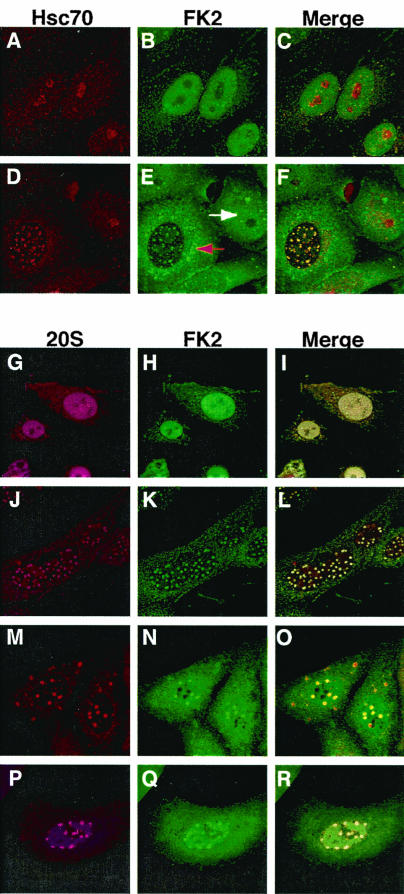
Localization of ubiquitinated proteins and components of the proteasomal machinery during HSV-1 infection. Uninfected (A to C) and HSV-1-infected (D to F) cells stained with antibodies specific to Hsc70 (A and D) and ubiquitinated proteins (FK2) (B and E) are shown. Merged images are shown in panels C and F. In panel E, the white arrow indicates the diffuse nuclear staining of ubiquitinated proteins in an uninfected cell and the red arrow indicates the dramatic redistribution of ubiquitinated proteins in an HSV-1-infected cell. The subcellular localization of the 20S core component of the 26S proteasome (G, J, M, and P) and of ubiquitin-conjugated proteins (H, K, N, and Q) is also shown. Merged images are shown in panels I, L, O, and R. (G to I) Mock-infected cells stained with α20S and αFK2 antibodies. (J to L) HSV-1-infected cells stained with the same antibodies. Infected cells treated with the proteasomal inhibitor MG132 throughout infection (M to O) or at 2 h postinfection (P to R) are also shown.
We next asked whether the formation of the nuclear foci was affected by the presence of the proteasomal inhibitor MG132 during infection. When MG132 was present before and during infection, nuclear foci were still observed, despite the fact that under these conditions no replication compartments would be expected to form since the drug inhibits ND10 disruption (Fig. 6M to O). In order to determine whether MG132 could inhibit 20S and FK2 sequestration under conditions in which replication compartments had formed, MG132 was added at 2 h postinfection. The results are shown in Fig. 6P to R. Again, discrete foci were able to form, but in addition to localization in discrete foci, staining was also observed in compartments that resembled replication compartments (Fig. 6Q, green areas). To our knowledge, this is the first time that ubiquitinated proteins have been localized to sites of viral replication and assembly. Furthermore, this is the first report of an HSV-1-induced formation of discrete foci containing Hsc70, Hsp70, Hsp40, 20S proteasomal subunits, and ubiquitinated proteins. The juxtaposition of the replication compartments and these foci is intriguing.
The HSV-1 portal protein UL6 is localized to nuclear domains enriched with Hsc70.
As mentioned in the introduction, many multimeric proteins require chaperone assistance during folding and assembly. Using newly generated monoclonal antibodies, 1C9 and 4G9, raised against the purified UL6 portal protein (kindly provided by Jay C. Brown, University of Virginia), we determined the intracellular localization of UL6 during infection. A time course study was conducted and cells were stained for the viral single-stranded DNA binding protein ICP8 (red) and with the 1C9 or 4G9 antibody (green). With the 1C9 antibody, the UL6 protein was detected in replication compartments (data not shown); this is consistent with previous reports indicating that cleavage and packaging occur within these large globular domains (22). When infected cells were stained at 2 h postinfection with 4G9, the UL6 protein was observed within the nucleus, and a fraction of the protein was observed colocalized with newly formed replication compartments that colocalized with ICP8 (Fig. 7C). At 3 h postinfection, the UL6 protein exhibited a globular staining pattern within the nucleus (Fig. 7D). Surprisingly, however, a different pattern was observed at later times postinfection (Fig. 7E and F). At 4 and 5 h postinfection, a substantial fraction of the UL6 protein was detected in foci adjacent to replication compartments (green). These foci costained with the Hsc70 antibody (Fig. 7G to I) and are thus likely to be equivalent to the Hsc70 foci observed in Fig. 1 to 6. Hsc70 foci were observed at all temperatures tested (34 to 39.5°C) and in cells infected with mutants of HSV-1 that were defective in assembly or DNA packaging (data not shown). Antisera raised against VP5, VP19c, VP23, and UL26 were used, and we found that no other viral structural proteins or cleavage and packaging proteins localized to the UL6- and Hsc70-enriched foci (data not shown). The Hsc70-enriched nuclear foci were also observed in cells infected with hr74, a previously described UL6 null virus (22), indicating that UL6 is not essential for their formation. The facts that the 1C9 antibody detected UL6 only within replication compartments and the 4G9 antibody detected UL6 primarily within the Hsc70 foci suggest that 4G9 detects a conformational epitope specific for misfolded, aggregated, or otherwise distinct populations of UL6. Interestingly, at 2 and 3 h postinfection, 4G9 detected UL6 in replication compartments (Fig. 7C and D). This may reflect that a fraction of misfolded UL6 proteins are present within replication compartments and that at later times this unfolded material is relocalized to chaperone-enriched foci. It is possible that viral or cellular proteins survey the replication compartments for misfolded proteins and can shuttle them to chaperone-enriched foci for remodeling or degradation. Given the multimeric nature of the portal structure, we propose that UL6 may require assistance during folding and/or oligomerization. The colocalization of a fraction of UL6 with components of the cellular machinery involved in protein folding and degradation may indicate that the sequestration of UL6 in Hsc70-enriched foci is necessary for proper folding and portal formation. Alternatively, it is possible that only misfolded UL6 is recruited to these foci for the purpose of refolding or protein degradation via the proteasomal pathway.
FIG. 7.

Localization of UL6 protein during HSV-1 infection. HSV-1-infected cells were collected at various times postinfection as described in Materials and Methods. The cells were stained with an antibody specific to the ICP8 protein (the single-stranded-DNA binding protein which is used as a marker for replication compartments) and the UL6 protein. (A to F) Merged images of ICP8 (red)- and UL6 (green)-stained cells. The times (in hours) postinfection are indicated in each panel. (A) Mock-infected cells stained with αICP8 and αUL6 antibodies. (G to I) Infected cells stained with αUL6 (G) and αHsc70 (H) antibodies. A merged image is shown in panel I.
The UL6 portal protein is a substrate for ubiquitination.
To investigate the possibility that a fraction of UL6 is degraded during infection, we performed a Western blot analysis to determine whether ubiquitinated UL6 proteins could be detected in cells infected with wild-type HSV-1. Cell lysates were analyzed for UL6 modification at various times postinfection. Figure 8A shows that at 39.5°C, the UL6 protein was detected as early as 1 h postinfection (lane 2), whereas at 34°C, UL6 was not detected until 3 h postinfection (lane 8), indicating that there are faster infection kinetics at higher temperatures. At the higher temperature, slow-migrating species of UL6 were detected at 6 h postinfection (Fig. 8A, lane 4). By 18 h postinfection, a smear of slow-migrating UL6-reactive products was detected at both temperatures (Fig. 8A, lanes 5 and 10). Slow-migrating bands could either represent modified, perhaps ubiquitinated, forms of UL6 or the presence of SDS-resistant multimers of UL6. In our hands, UL6 derived from virus particles or UL6 expressed in baculovirus-infected insect cell lysates resolves to a single band, suggesting that the slow-migrating bands are not likely to be oligomers of UL6. Thus, as described below, we think it is more likely that UL6 is modified by ubiquitination during infection.
FIG. 8.

The portal protein UL6 is a substrate for ubiquitination. (A) Posttranslational state of UL6 protein throughout HSV-1 infection at 39.5 and 34°C. Infected cell lysates were prepared as described in Materials and Methods. Uninfected cell lysates are shown in lanes 1 and 6. In lane 4 (6 h postadsorption; 39.5°C), note the presence of multiple slow-migrating UL6-reactive bands. At this time, fast-migrating UL6-reactive species were also apparent. In lanes 5 and 10, a smear of slow-migrating UL6-reactive products is detected. (B) UL6 Western blot analysis of immunoprecipitates recovered by using an αUL6 (lane 3) or αFK2 (lane 4; ubiquitin-conjugated proteins) antibody, normal mouse IgG (lane 5), or beads alone (lane 6). An infected cell lysate is shown in lane 2. (C) FK2 Western blot analysis of immunoprecipitates recovered by using an αUL6 (lane 3) or αFK2 (lane 4; ubiquitin-conjugated proteins) antibody, normal mouse IgG (lane 5), or beads alone (lane 6). Dots are used to indicate putative ubiquitin-conjugated species of UL6 recovered in the precipitation.
To determine whether UL6 was actually ubiquitinated, we precipitated infected cell lysates with the αFK2, α4G9, or αIgG antibody as described in Materials and Methods. Since the ubiquitin moiety is 7 kDa, the addition of one, two, or three moieties to the 75-kDa UL6 polypeptide chain would be predicted to result in proteins with molecular masses of approximately 83, 92, and 100 kDa, respectively. Immunoblots of immunoprecipitates probed with the αFK2 or α4G9 antibody indicated that UL6 can be detected in the total lysate as a 75-kDa band (Fig. 8B, lane 2). When the lysate was precipitated with an anti-UL6 antibody, the 75-kDa band and multiple slow-migrating UL6-reactive bands were detected (Fig. 8B, lane 3), with approximate molecular masses ranging from 78 to 100 kDa. The bands migrating at approximately 83, 92, and 100 kDa are marked with dots and may represent UL6 with one, two, and three ubiquitin conjugates, respectively (lane 3). It is possible that the 78- and 80-kDa species of UL6 found in lanes 3 and 4 are degradation products recovered from the lysate. Additional experiments will be required to confirm these assignments. The slow-migrating bands were not seen in the total lysate, perhaps because less UL6 is present in this sample or because slow-migrating species are enriched in the precipitate by using the 4G9 antibody. The hypothesis that these slow-migrating bands represent ubiquitinated forms of UL6 is supported by the observation that these species were also precipitated with an antibody specific for ubiquitinated proteins (Fig. 8B, lane 4). A Western blot analysis of these immunoprecipitates was also performed with an antibody specific to ubiquitinated proteins (FK2) (Fig. 8C). Figure 8C, lane 3, shows many slow-migrating UL6-reactive species that were recognized by the FK2 antibody. For lane 4, the lysates were immunoprecipitated and reacted with the FK2 antibody which, as expected, resulted in many reactive bands. When the αIgG antibody and beads alone were used to precipitate the lysates, none of the UL6-reactive bands were detected. Taken together, these results are consistent with the proposal that the slow-migrating species are ubiquitin-conjugated forms of UL6.
DISCUSSION
In this report, we have identified novel nuclear foci, enriched for cellular protein remodeling and degradation machinery, that are established during HSV-1 infection. The viral protein ICP0 was found to be necessary and sufficient for the establishment of these foci at early times during infection. The observation that a fraction of the UL6 portal protein was also redistributed to the Hsc70-enriched foci suggests that the function of the foci may be to facilitate protein folding and maturation. The colocalization of proteasomal and ubiquitination machinery with Hsc70 may indicate that terminally misfolded proteins are targeted for degradation within these foci. This suggestion is supported by the detection of ubiquitinated forms of the UL6 protein during infection. Collectively, our results indicate that the virus has evolved a mechanism to facilitate protein folding and degradation within specialized compartments in the nucleus of the cell.
Our results suggest that ICP0 or a cellular factor with ICP0-like functions can orchestrate the rearrangement of Hsc70 and Hsp70 during infection and that this relocalization occurs more efficiently at 39.5°C than at 37°C. Hsc70 is known to concentrate within the nucleolus during high-temperature stress (26). Interestingly, at early times after infection at 39.5°C, ICP0 itself colocalized with Hsc70 in the nucleolus (Fig. 4D). After the initial localization of ICP0 in the nucleolus, both Hsc70 and ICP0 were observed in discrete nuclear foci. The more efficient formation of these foci at higher temperatures may be due to the stress-induced concentration of the chaperone in the nucleolus or to the faster kinetics of viral infection. The significance of the nucleolar localization of host chaperones and ICP0 is not known. It was previously shown that HSV-1 infection induces a structural modification of the nucleolus (2). It will be interesting to determine the role played by ICP0 in the rearrangement of host chaperones and whether chaperone distribution is linked to the modification of the nucleolus during infection.
The principal mechanism of protein degradation in eukaryotic cells is the ubiquitin-proteasome pathway. This system requires that a protein targeted for degradation be covalently conjugated to ubiquitin by an E3-ubiquitin ligase. Client proteins conjugated to multiple ubiquitin moieties are recognized by the 26S proteasome and subsequently degraded (19, 36). It has previously been reported that ICP0 has ubiquitin ligase activity (6). During infection, ICP0 is known to target the degradation of PML, p53, and SP100 in a ubiquitin-dependent fashion (4, 5, 18). Although an ICP0-induced degradation of viral proteins has not been reported, the results presented in this paper suggest that ICP0 is necessary and sufficient for the redistribution of Hsc70 and the 26S proteasome to novel foci at early times during infection. These foci also contain a subfraction of the portal protein UL6; furthermore, we have shown that UL6 is ubiquitinated during infection. Thus, it is possible that ICP0 is either directly or indirectly involved in the remodeling and/or degradation of the viral portal protein.
It is likely that global protein quality control in cells involves cooperation between protein remodeling and degradation. A precedent exists for a connection between these two processes. For instance, protein cofactors such as CHIP (carboxyl terminus of Hsc70 interacting protein) interact directly with the chaperone machinery and exhibit ubiquitin ligase activity (10). It is possible that ICP0 performs a CHIP-like function to interact with Hsc70 and orchestrate the formation of Hsc70 and proteasome-enriched foci. The directed redistribution of the cellular chaperone and proteolytic machinery during infection may represent a novel mechanism to ensure viral protein quality control. Although Hsc70 can eventually be recruited to foci within the nucleus even in the absence of ICP0, the presence of ICP0 enhances the rate and efficiency of this process. The results presented in this paper thus extend the numerous functions already ascribed to ICP0 by suggesting that it may also link the cellular protein folding machinery to the 26S proteasome during infection.
We propose that viral proteins that are difficult to fold or that lack binding partners can be sequestered in specialized remodeling and degradation foci away from replication compartments in order to promote the appropriate maturation or degradation of terminally misfolded protein aggregates. As suggested for the mutant Huntingtin protein (33), nuclear aggregation into discrete foci may protect the cell against premature death. It is possible that the virus may take advantage of this state to prevent apoptosis and increase the virus yield. We previously showed that cells transfected with ICP0 and either UL6 or components of the helicase-primase displayed unusual inclusions observed by immunofluorescence microscopy in which ICP0 appeared to surround the potentially misfolded protein, whereas proteins which presumably could fold and function on their own, such as ICP8 and UL9, did not localize to these inclusions (24). Consistent with the notion that ICP0 may provide a link between misfolded proteins and the remodeling machinery, we have shown in this paper that transfection inclusions also contain Hsc70. We believe that similar structures are formed in the nuclei of infected cells. As seen in transfection studies, ICP8, UL9, and capsid proteins such as VP5, VP19C, and scaffold protein were not observed in the Hsc70 foci. Interestingly, foci outside of replication compartments containing the HSV-1 viral tegument protein VP22 have been observed in live and fixed herpesvirus-infected cells (8, 20, 27). In one study, these foci were juxtaposed with replication compartments colocalized with ICP0 in punctate dots (20). It is unclear whether these sites are analogous to the foci reported in this paper; however, we are intrigued by the possibility that a subset of viral proteins that require special attention during protein folding or, perhaps, oligomerization may be treated by the host folding machinery in either identical or very similar domains lying adjacent to the replication compartments.
It is clear that HSV-1 causes a dramatic nuclear redistribution of the chaperone and proteasomal machinery during infection, but whether this is essential for portal assembly and efficient viral production remains unknown. Attempts to express UL6 in heterologous systems to monitor its in vitro folding and oligomerization status have been hampered by the insolubility of the UL6 protein. When UL6 was expressed in insect cells infected with a recombinant baculovirus, the protein was insoluble unless 1 M arginine was added. Under those conditions, dodecameric rings of the UL6 protein were observed (29). It will be of considerable interest to determine if the overexpression of the chaperone machinery during UL6 production alleviates the need for the addition of arginine and thus allows for the purification of a protein that is suitable for folding and oligomerization studies. These experiments may indicate if and at what stage portal formation is facilitated by the cellular chaperone. In order to address dependence on the chaperone machinery vis-à-vis viral production, we will need to knock out the expression of Hsc70 and its cofactors and determine the effect on viral yield. We speculate that the partitioning of misfolded proteins in the nucleus will provide an antiapoptotic condition that will subsequently increase the virus yield.
Since HSV-1 assembly and maturation are restricted to the nucleus, it is not surprising that the virus has evolved a mechanism to manage proteins in this compartment. Although it is known that protein degradation can occur within both the nucleus and the cytoplasm, the dynamics of proteasomal distribution between compartments are poorly understood (31). Our findings suggest that during HSV-1 infection, the cellular chaperone and proteolytic machinery is sequestered, perhaps to facilitate viral protein folding, oligomerization, or disaggregation. Additional viral or cellular factors may play a role in shuttling misfolded proteins from replication compartments to the Hsp70/Hsp40 chaperone-enriched foci. Since the Hsp70/Hsp40 chaperone system is known to coordinate with the Hsp90 multichaperone system during the maturation of specialized proteins (reviewed in reference 13), it is possible that an even more complex operation is employed by the virus during infection. We propose that the sequestration of these processes represents a rudimentary compartmentalization of the nucleus and demonstrates that HSV may have evolved an elegant mechanism to facilitate protein quality control during infection.
Acknowledgments
We are grateful to all of the members of the Weller laboratory for their helpful discussions and constructive input. We are especially thankful to the laboratory of Pramod Srivastava at the UCHC Center for Immunotherapy for providing initial aliquots of various chaperone antibodies used during these studies.
This work was supported by NIH NRSA grant F32 AI50336 to A.D.B. and NIH AI37549 grant to S.K.W.
REFERENCES
- 1.Albrecht, M. A., N. A. DeLuca, R. A. Byrn, P. A. Schaffer, and S. M. Hammer. 1989. The herpes simplex virus immediate-early protein, ICP4, is required to potentiate replication of human immunodeficiency virus in CD4+ lymphocytes. J. Virol. 63:1861-1868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Besse, S., and F. Puvion-Dutilleul. 1996. Distribution of ribosomal genes in nucleoli of herpes simplex virus type 1 infected cells. Eur. J. Cell Biol. 71:33-44. [PubMed] [Google Scholar]
- 3.Black, L. W. 1989. DNA packaging in dsDNA bacteriophages. Annu. Rev. Microbiol. 43:267-292. [DOI] [PubMed] [Google Scholar]
- 4.Boutell, C., and R. D. Everett. 2003. The herpes simplex virus type 1 (HSV-1) regulatory protein ICP0 interacts with and ubiquitinates p53. J. Biol. Chem. 278:36596-36602. [DOI] [PubMed] [Google Scholar]
- 5.Boutell, C., A. Orr, and R. D. Everett. 2003. PML residue lysine 160 is required for the degradation of PML induced by herpes simplex virus type 1 regulatory protein ICP0. J. Virol. 77:8686-8694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Boutell, C., S. Sadis, and R. D. Everett. 2002. Herpes simplex virus type 1 immediate-early protein ICP0 and its isolated RING finger domain act as ubiquitin E3 ligases in vitro. J. Virol. 76:841-850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cai, W., and P. A. Schaffer. 1991. A cellular function can enhance gene expression and plating efficiency of a mutant defective in the gene for ICP0, a transactivating protein of herpes simplex virus type 1. J. Virol. 65:4078-4090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.del Rio, T., H. C. Werner, and L. W. Enquist. 2002. The pseudorabies virus VP22 homologue (UL49) is dispensable for virus growth in vitro and has no effect on virulence and neuronal spread in rodents. J. Virol. 76:774-782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.DeLuca, N. A., A. M. McCarthy, and P. A. Schaffer. 1985. Isolation and characterization of deletion mutants of herpes simplex virus type 1 in the gene encoding immediate-early regulatory protein ICP4. J. Virol. 56:558-570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Demand, J., S. Alberti, C. Patterson, and J. Hohfeld. 2001. Cooperation of a ubiquitin domain protein and an E3 ubiquitin ligase during chaperone/proteasome coupling. Curr. Biol. 11:1569-1577. [DOI] [PubMed] [Google Scholar]
- 11.Everett, R. D., P. Freemont, H. Saitoh, M. Dasso, A. Orr, M. Kathoria, and J. Parkinson. 1998. The disruption of ND10 during herpes simplex virus infection correlates with the Vmw110- and proteasome-dependent loss of several PML isoforms. J. Virol. 72:6581-6591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Everett, R. D., and G. G. Maul. 1994. HSV-1 IE protein Vmw110 causes redistribution of PML. EMBO J. 13:5062-5069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fink, A. L. 1999. Chaperone-mediated protein folding. Physiol. Rev. 79:425-449. [DOI] [PubMed] [Google Scholar]
- 14.Frydman, J. 2001. Folding of newly translated proteins in vivo: the role of molecular chaperones. Annu. Rev. Biochem. 70:603-647. [DOI] [PubMed] [Google Scholar]
- 15.Fujimuro, M., H. Sawada, and H. Yokosawa. 1994. Production and characterization of monoclonal antibodies specific to multi-ubiquitin chains of polyubiquitinated proteins. FEBS Lett. 349:173-180. [DOI] [PubMed] [Google Scholar]
- 16.Georgopoulos, C., and W. J. Welch. 1993. Role of the major heat shock proteins as molecular chaperones. Annu. Rev. Cell Biol. 9:601-634. [DOI] [PubMed] [Google Scholar]
- 17.Georgopoulos, C. P., R. W. Hendrix, S. R. Casjens, and A. D. Kaiser. 1973. Host participation in bacteriophage lambda head assembly. J. Mol. Biol. 76:45-60. [DOI] [PubMed] [Google Scholar]
- 18.Gu, H., and B. Roizman. 2003. The degradation of promyelocytic leukemia and Sp100 proteins by herpes simplex virus 1 is mediated by the ubiquitin-conjugating enzyme UbcH5a. Proc. Natl. Acad. Sci. USA 100:8963-8968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hershko, A., and A. Ciechanover. 1998. The ubiquitin system. Annu. Rev. Biochem. 67:425-479. [DOI] [PubMed] [Google Scholar]
- 20.Hutchinson, I., A. Whiteley, H. Browne, and G. Elliott. 2002. Sequential localization of two herpes simplex virus tegument proteins to punctate nuclear dots adjacent to ICP0 domains. J. Virol. 76:10365-10373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Knipe, D. M. 1989. The role of viral and cellular nuclear proteins in herpes simplex virus replication. Adv. Virus Res. 37:85-123. [DOI] [PubMed] [Google Scholar]
- 22.Lamberti, C., and S. K. Weller. 1996. The herpes simplex virus type 1 UL6 protein is essential for cleavage and packaging but not for genomic inversion. Virology 226:403-407. [DOI] [PubMed] [Google Scholar]
- 23.Lukonis, C. J., and S. K. Weller. 1997. Formation of herpes simplex virus type 1 replication compartments by transfection: requirements and localization to nuclear domain 10. J. Virol. 71:2390-2399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lukonis, C. J., and S. K. Weller. 1996. The herpes simplex virus type 1 transactivator ICP0 mediates aberrant intracellular localization of the viral helicase/primase complex subunits. Virology 220:495-501. [DOI] [PubMed] [Google Scholar]
- 25.Maul, G. G., H. H. Guldner, and J. G. Spivack. 1993. Modification of discrete nuclear domains induced by herpes simplex virus type 1 immediate early gene 1 product (ICP0). J. Gen. Virol. 74:2679-2690. [DOI] [PubMed] [Google Scholar]
- 26.Milarski, K. L., and R. I. Morimoto. 1989. Mutational analysis of the human HSP70 protein: distinct domains for nucleolar localization and adenosine triphosphate binding. J. Cell Biol. 109:1947-1962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Morrison, E. E., A. J. Stevenson, Y. F. Wang, and D. M. Meredith. 1998. Differences in the intracellular localization and fate of herpes simplex virus tegument proteins early in the infection of Vero cells. J. Gen. Virol. 79:2517-2528. [DOI] [PubMed] [Google Scholar]
- 28.Muchowski, P. J., G. Schaffar, A. Sittler, E. E. Wanker, M. K. Hayer-Hartl, and F. U. Hartl. 2000. Hsp70 and Hsp40 chaperones can inhibit self-assembly of polyglutamine proteins into amyloid-like fibrils. Proc. Natl. Acad. Sci. USA 97:7841-7846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Newcomb, W. W., R. M. Juhas, D. R. Thomsen, F. L. Homa, A. D. Burch, S. K. Weller, and J. C. Brown. 2001. The UL6 gene product forms the portal for entry of DNA into the herpes simplex virus capsid. J. Virol. 75:10923-10932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Parkinson, J., S. P. Lees-Miller, and R. D. Everett. 1999. Herpes simplex virus type 1 immediate-early protein vmw110 induces the proteasome-dependent degradation of the catalytic subunit of DNA-dependent protein kinase. J. Virol. 73:650-657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Reits, E. A., A. M. Benham, B. Plougastel, J. Neefjes, and J. Trowsdale. 1997. Dynamics of proteasome distribution in living cells. EMBO J. 16:6087-6094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Salmon, B., and J. D. Baines. 1998. Herpes simplex virus DNA cleavage and packaging: association of multiple forms of U(L)15-encoded proteins with B capsids requires at least the U(L)6, U(L)17, and U(L)28 genes. J. Virol. 72:3045-3050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Saudou, F., S. Finkbeiner, D. Devys, and M. E. Greenberg. 1998. Huntingtin acts in the nucleus to induce apoptosis but death does not correlate with the formation of intranuclear inclusions. Cell 95:55-66. [DOI] [PubMed] [Google Scholar]
- 34.Sheaffer, A. K., W. W. Newcomb, M. Gao, D. Yu, S. K. Weller, J. C. Brown, and D. J. Tenney. 2001. Herpes simplex virus DNA cleavage and packaging proteins associate with the procapsid prior to its maturation. J. Virol. 75:687-698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Showalter, S. D., M. Zweig, and B. Hampar. 1981. Monoclonal antibodies to herpes simplex virus type 1 proteins, including the immediate-early protein ICP 4. Infect. Immun. 34:684-692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Voges, D., P. Zwickl, and W. Baumeister. 1999. The 26S proteasome: a molecular machine designed for controlled proteolysis. Annu. Rev. Biochem. 68:1015-1068. [DOI] [PubMed] [Google Scholar]
- 37.Weller, S. K., D. P. Aschman, W. R. Sacks, D. M. Coen, and P. A. Schaffer. 1983. Genetic analysis of temperature-sensitive mutants of HSV-1: the combined use of complementation and physical mapping for cistron assignment. Virology 130:290-305. [DOI] [PubMed] [Google Scholar]
- 38.Yao, F., and P. A. Schaffer. 1995. An activity specified by the osteosarcoma line U2OS can substitute functionally for ICP0, a major regulatory protein of herpes simplex virus type 1. J. Virol. 69:6249-6258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yu, D., and S. K. Weller. 1998. Herpes simplex virus type 1 cleavage and packaging proteins UL15 and UL28 are associated with B but not C capsids during packaging. J. Virol. 72:7428-7439. [DOI] [PMC free article] [PubMed] [Google Scholar]