

Abstract
Expression of human T-cell leukemia virus type 1 (HTLV-1) is regulated by the viral transcriptional activator Tax. Tax activates viral transcription through interaction with the cellular transcription factor CREB and the coactivators CBP/p300. In this study, we have analyzed the role of histone deacetylase 1 (HDAC1) on HTLV-1 gene expression from an integrated template. First we show that trichostatin A, an HDAC inhibitor, enhances Tax expression in HTLV-1-transformed cells. Second, using a cell line containing a single-copy HTLV-1 long terminal repeat, we demonstrate that overexpression of HDAC1 represses Tax transactivation. Furthermore, a chromatin immunoprecipitation assay allowed us to analyze the interaction of transcription factors, coactivators, and HDACs with the basal and activated HTLV-1 promoter. We demonstrate that HDAC1 is associated with the inactive, but not the Tax-transactivated, HTLV-1 promoter. In vitro and in vivo glutathione S-transferase-Tax pull-down and coimmunoprecipitation experiments demonstrated that there is a direct physical association between Tax and HDAC1. Importantly, biotinylated chromatin pull-down assays demonstrated that Tax inhibits and/or dissociates the binding of HDAC1 to the HTLV-1 promoter. Our results provide evidence that Tax interacts directly with HDAC1 and regulates binding of the repressor to the HTLV-1 promoter.
The human T-cell lymphotrophic virus type 1 (HTLV-1) is a human retrovirus that causes adult T-cell leukemia and the degenerative neuromuscular disease tropical spastic paraparesis or HTLV-1-associated myelopathy. The HTLV-1 proviral DNA encodes a 40-kDa protein, Tax, which is critical in HTLV-1 virus transformation. Tax not only regulates HTLV-1 gene expression, but also influences cellular gene expression (4, 5, 13, 29, 38, 39, 42, 43, 45, 53, 55). Tax potently activates HTLV-1 expression through three copies of a 21-bp Tax response element (TRE) found in the long terminal repeat (LTR). Because the viral genome is integrated into the host chromosome, understanding how the viral LTR is regulated in a chromatin-associated context is important for elucidating the life cycle of the provirus. The pattern of HTLV-1-associated disease progression suggests that while viral infection might be the initiating trigger, intracellular mechanisms may repress HLTV-1 LTR expression over an extended duration. Tax has been shown to interact with cellular factors such as CREB, transcriptional coactivators, and histone acetyltransferases p300, CBP, and PCAF (2, 16, 21, 22, 24, 27). Further, it has been shown that p300 can activate Tax-mediated HTLV-1 chromatin transcription through targeted acetylation of nucleosomal histones (18, 33). Recent studies suggest that histone deacetylases (HDACs) (19, 52), which play an important role in the repression of other genes, may be involved in this negative regulation (15, 30).
The deacetylase superfamily can be divided into three distinct classes based on structure (19). The HDACs comprise the first two classes and consist of class I (HDACs 1, 2, 3, 8, and 11) and class II (HDACs 4, 5, 6, 7, 9, and 10) enzymes. The class II enzymes are distinguished by a large NH2-terminal domain or a second catalytic site (e.g., HDAC 6). The class III enzymes, SIRTs (sirtuins) or Sir2-related proteins, deacetylate histones in yeast, while in mammalian cells they appear to involve deacetylation of other proteins or transcription factors, such as p53, rather than histones. In general, inhibition of HDAC activity by agents such as trichostatin A (TSA) or sodium butyrate leads to increased histone acetylation, correlating with increased mRNA expression (10, 52, 54). It is important to note that HDACs are found as components of multiprotein complexes containing DNA-histone binding proteins (e.g., NCoR, SMRT, MEF, MeCP2, and mSin3A) that use HDACs to repress transcription and block the function of cellular proteins such as MyoD, nuclear receptors, p53, NF-κB, and E2F (1, 9, 25, 26, 35, 56).
Conversely, histone acetylation has been correlated with transcriptionally active genes. The specific recruitment of a transcription factor complex with histone acetyltransferase activity to a promoter may play a critical role in overcoming the repressive effects of chromatin structure on transcription (8, 18, 33, 48). Transcriptional regulation, therefore, is a dynamic interplay between histone acetyltransferase and HDAC activity.
Although it has been shown that p300/CBP activates the HTLV-1 chromatin template through acetylation of nucleosomal histones, little is known about how HDACs regulate the transcriptional trans-acting function of Tax. Evidence from a recent study by Lemasson et al. suggests that HDACs are associated with the HTLV-1 LTR in vivo (30). It is important to note that these studies analyzed a heterogeneous population of active and inactive HTLV-1 LTR promoters. Thus, it is not clear whether the HDACs associated with the active or inactive templates. In a separate study, Ego et al. found that overexpression of HDAC1 could suppress activation from transiently transfected HTLV-1 LTR reporter constructs (15). There is, however, debate about the biological significance of such studies on transient templates. For example, it has been shown that the HDAC inhibitor TSA induces expression from the chromosomal CRE promoter but not from a transient template (37).
In this study, we have analyzed the role of HDACs on HTLV-1 gene expression from an integrated template. First, we show that TSA enhances Tax expression in HTLV-1-transformed cells. Second, using a cell line containing a single-copy HTLV-1 LTR, we demonstrate that overexpression of HDAC1 represses Tax transactivation. Finally, using an in vivo chromatin immunoprecipitation (ChIP) assay, we have analyzed the interaction of transcription factors, coactivators, and HDACs with the basal and activated HTLV-1 promoter. Our results provide the first experimental evidence that Tax negatively regulates the interaction of HDAC with the HTLV-1 promoter.
MATERIALS AND METHODS
Cell lines and TSA treatment.
HTLV-1-transformed C81 cells were maintained at 2 × 105 to 2 × 106 cells/ml in RPMI medium supplemented with 10% fetal bovine serum, 2 mM glutamine, and penicillin-streptomycin. pA-18G-BHK-21 cells were maintained in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum, 2 mM glutamine, penicillin-streptomycin, and 400 ng of G418/ml. C81 cells were treated with TSA (Sigma) at a concentration of 200 ng/ml for 0, 2, 4, 6, 8, or 24 h. After treatment, the cells were harvested and total RNA was extracted using RNAwiz (Ambion). mRNA levels of Tax were analyzed by reverse transcription-PCR (RT-PCR). The PCR primers (43) for Tax were as follows: 5′-TGTTTGGAGACTGTGTACAAGGCG-3′ and 5′-CAGGCTGTCAGCGTGACGG-3′. In addition, after 24 h of TSA treatment whole-cell extracts were prepared by lysis in buffer (50 mM Tris · HCl [pH 8.0], 150 mM NaCl, 10% glycerol, 0.5% Triton X-100) containing 1 mM AEBSF, 1 μg of aprotinin/ml, 1 μg of pepstatin/ml, and 1 μg of leupeptin/ml. Lysates were incubated at 4°C for 30 min and then centrifuged at 10,000 ×g for 15 min to obtain supernatants.
Transfection and β-Gal assay.
Tax expression vector pcTax and HDAC1 expression plasmids were described previously (23, 24). All plasmids were purified by using a CsCl gradient or QIAGEN kit. pA-18G-BHK-21 cells were seeded in six-well dishes, and the indicated amounts of plasmid were transfected into pA-18G-BHK-21 cells using Fugene 6 (Roche Applied Science) according to the manufacturer's instructions. Cells were harvested after 48 h, and β-galactosidase (β-Gal) assays were performed using Galacto-light (Tropix) according to the manufacturer's instructions.
Immunofluorescence.
For immunostaining, pA-18G-BHK-21 Tax− and pA-18G-BHK-21 Tax+ cells were cultured on coverslips, fixed in 4% paraformaldehyde, and permeabilized in cold methanol. The permeabilized cells then were incubated with 10% goat serum for 1 h, followed by immunostaining with an anti-Tax monoclonal antibody and an Alexa Fluor 488-conjugated anti-mouse immunoglobulin G (IgG) antibody. The cells then were mounted with medium containing 4′,6′-diamidino-2-phenylindole (Vectashield; Vector Labs) and were visualized by use of a Leica confocal microscope.
GST pull-down assay.
The glutathione S-transferase (GST)-HDAC1 expression plasmid (56) was provided by S. Ghosh's lab. The GST-Tax construct was described previously (24). The GST pull-down assay was performed by incubating 200 ng of GST or 600 ng of GST-HDAC1 with 15 μl of swollen glutathione-agarose (Sigma) in 400 μl of 0.5× Superdex buffer (12.5 mM HEPES [pH 7.9], 6.25 mM MgCl2, 75 mM KCl, 5 μM ZnSO4, 0.5 mM EDTA, 10% glycerol, 0.05% NP-40) at 4°C for 2 h. The beads were then washed twice with 0.5× Superdex buffer and incubated with 100 ng of Tax (in a total volume of 400 μl of 0.5× Superdex buffer) at 4°C for 4 h. After being washed twice with 0.5× Superdex, the beads were resuspended in sodium dodecyl sulfate (SDS) dye, boiled, and analyzed by SDS-polyacrylamide gel electrophoresis (SDS-PAGE; 4 to 20% polyacrylamide). Bound protein was detected by Western blot analysis with an antibody against Tax (Tab172).
In vitro and in vivo coimmunoprecipitations.
For the in vitro coimmunoprecipitation, 100 ng of Tax was incubated with 100 ng of Flag-HDAC1 protein in buffer A (50 mM Tris-HCl [pH 7.6], 50 mM NaCl, 0.5 mM EDTA, 1 mM dithiothreitol [DTT], 5 mM MgCl2, 0.2% Triton, 5% glycerol, 2.5 mg of bovine serum albumin [BSA]/ml) for 1 h at 4°C. One microliter of anti-Flag M2 monoclonal antibody preincubated with or without Flag peptide was added and incubated for 2 h. Complexes were bound to 25 μl of protein A-protein G-agarose beads (Calbiochem) by rocking for 2 h. The beads were then washed three times with buffer B (50 mM Tris-HCl [pH 7.6], 150 mM NaCl, 1% NP-40, 1 mM EDTA, 5% glycerol) and boiled in SDS loading buffer. SDS-PAGE and Western blotting were performed to analyze the proteins. For in vivo coimmunoprecipitation, HTLV-1-transformed T-cell C81 nuclear extracts were made in lysis buffer (20 mM HEPES [pH 7.3], 400 mM NaCl, 1 mM EDTA, 1 mM EGTA, 1 mM DTT, 1 mM phenylmethylsulfonyl fluoride). Aliquots of 250 μg of nuclear extract were incubated with 20 μl of antibody-bound protein A-G beads (beads were prebound with 1 μg each of anti-HDAC1 polyclonal or rabbit control IgG antibody) in IP buffer (25 mM HEPES [pH 7.3], 2 mM EDTA, 150 mM NaCl, 0.2% NP-40, 1 mM DTT, 1 mM AEBSF, 10 μg of leupeptin/ml, 2 μg of aprotinin/ml, and 10 μg of pepstatin A/ml) at 4°C overnight. Beads were washed with IP buffer four times and eluted in SDS-PAGE loading buffer.
Chromatin pull-down assay.
Oligonucleosomes were prepared from HeLa cells as before (7, 50) and used as competitor in the following protein-chromatin binding reaction. HeLa cell nuclear extract was incubated with 100 μg of chromatin template in the presence of 0, 100, or 200 ng of Tax protein in binding buffer (50 mM Tris [pH 7.6], 50 mM NaCl, 0.5 mM EDTA, 1 mM DTT, 5 mM MgCl2, 0.1% Triton, 5% glycerol, 2.5 mg of BSA per ml, 10 μg of poly(dI-dC) per ml, and 1 mg of oligonucleosome), and the reaction was continued for 30 min at 30°C. The beads were then washed four times with binding buffer without BSA or poly(dI-dC). Proteins were eluted by SDS loading buffer and loaded onto a 4-to-20% Tris-glycine gel (Invitrogen). Immunoblotting was performed using anti-HDAC1 antibody (Santa Cruz Biotechnology) and anti-Tax (Tab172) antibody.
Retroviral transduction.
Retroviral transductions were performed as described by Rivera-Walsh et al. (46). Briefly, 293 cells were seeded in a six-well dish. The next day, cells were transfected using Fugene 6 reagent (Roche Molecular Biochemicals) with 1 μg of the packaging plasmid pCL-Ampho, 1 μg of pCL-Tax or pCL-GFP, and 0.15 μg of pVSV-G. Viral supernatants were collected 48 h after transfection and filtered through a 0.45-μm-pore-size polysulfone filter, and titers were determined. pA-18G-BHK-21 cells were infected by adding 2 ml of viral supernatant per well of a six-well dish and adding 8 μg of Polybrene/ml. Cells were plated such that the multiplicity of infection was 1. Cells were stained with an in situ β-Gal staining kit (Stratagene) according to the manufacturer's instructions.
ChIP assay.
The ChIP assay was carried out using acetylated histone H3 or H4 antibody (Upstate Biotechnology), anti-CREB (Upstate Biotech), anti-HDAC1 (Affinity Bioreagent), anti-Tax (Tab 172), anti-RNA polymerase II (anti-RNAP II; Babco), and anti-CBP (Santa Cruz) antibodies following the methods previously described (33). After cross-linking proteins to DNA in pA-18G-BHK-21 cells (either Tax− or Tax+), chromatin was sonicated four times for 10 s each, generating DNA fragments with 200 to 800 bp. Then, the nucleosomes were precleared with salmon sperm DNA-protein A agarose beads. The supernatants were diluted 10-fold with ChIP dilution buffer, and the different antibodies indicated above were added. After overnight rotation at 4°C, the immune complexes were collected by addition of protein A-agarose beads. DNA was purified by proteinase K digestion, phenol extraction, and ethanol precipitation and amplified by PCR using primers specific for the HTLV-1 LTR (5′-CCACAGGCGGGAGGCGGCAGAA-3′ and 5′-TCATAAGCTCAGACCTCCGGGAAG-3′) and primers specific for the β-globin promoter (5′-AGGCTGCTGGTTGTCTACCCTTG-3′ and 5′-AGCTCACTGAGGCTGGCAAAGGTG-3′). The PCR products were analyzed by electrophoresis using 2% agarose gel and visualized with ethidium bromide staining.
RESULTS
The HDAC inhibitor TSA enhances Tax protein levels in HTLV-1-transformed T cells.
It has been estimated that approximately 2% of all genes are transcriptionally regulated through acetylation and deacetylation of nucleosomal histones (51). Previously, we and others have shown that p300 acetylation of histones facilitates HTLV-1 transcription on reconstituted chromatin templates (17, 18, 33). In the present studies, we have investigated the role of HDACs in HTLV-1 transcription. We first analyzed whether TSA, an HDAC inhibitor, could increase viral mRNA synthesis from an integrated viral promoter. HTLV-1-transformed C81 T cells were treated with TSA for up to 24 h. RNA was then purified and analyzed by RT-PCR. As shown in Fig. 1, treatment of cells with TSA increased the level of Tax mRNA by 4 h (Fig. 1A, lane 6, top panel). The mRNA level continued to increase and reached a maximum at 8 h. The effect was specific for Tax mRNA, since cellular glyceraldehyde-3-phosphate dehydrogenase (GAPDH) mRNA levels remained constant in the control and TSA-treated cells (Fig. 1A, bottom panel). Tax protein levels were also examined by Western blot analysis. Consistent with the RNA results, cells treated with TSA expressed higher levels of Tax protein than untreated cells (Fig. 1B). In contrast, GAPDH protein levels remained approximately the same after TSA treatment. Our transcription analysis is consistent with, and extends, a previous report that HDAC inhibitors sodium butyrate and TSA can increase nucleosomal histone acetylation levels within the HTLV-1 promoter (30).
FIG. 1.

Treatment with TSA enhances Tax mRNA and protein levels in HTLV-1 cells. After treatment with TSA (200 ng/ml), mRNA from C81 cells was prepared at various time points as described in Materials and Methods. Whole-cell extracts were prepared after 24-h treatment with TSA. (A) Tax mRNA levels were analyzed by RT-PCR. GAPDH was included as an internal control. PCR products were separated on 2% agarose. (B) Western blot assay of the Tax protein level. The GAPDH protein level was included as a control. C81 cell extracts were separated on 4-to-20% gels and transferred to polyvinylidene difluoride membranes, and Tax levels were determined by Western blot analysis with an anti-Tax monoclonal antibody.
HDAC1 represses Tax transactivation of the HTLV-1 LTR.
Next, we directly examined whether HDAC1 could suppress Tax transactivation of the HTLV-1 LTR. We chose HDAC1 because it is known to associate with corepressor complexes that regulate the expression of a number of genes, including retinoic acid receptor, thyroid hormone receptor, and estrogen receptor-responsive promoters (31, 32, 34, 49). The specific effect of HDAC1 on an integrated HTLV-1 promoter has not been established. We utilized the pA-18G-BHK-21 cell line, which contains a single-copy integrated HTLV LTR driving the lacZ gene, to investigate the effect of HDAC1 on Tax transactivation. Transfection of the HDAC1 expression plasmid into pA-18G-BHK-21 cells did not affect basal levels of HTLV-1 transcription (Fig. 2A, lanes 1 and 3). In contrast, HDAC1 reduced Tax transactivation up to 60% (Fig. 2A, lanes 2 and 4 to 6). To exclude the possibility that transcriptional repression by HDAC1 is due to alteration in Tax protein, we examined the Tax protein levels from the pA-18G-BHK-21 cells cotransfected with Tax and HDAC1 plasmids. Transfected cells were lysed, and Tax protein levels were determined by Western blot analysis. The cotransfection of HDAC1 did not alter Tax protein levels in pA-18G-BHK-21 cells (Fig. 2B).
FIG. 2.

Overexpression of HDAC1 represses Tax transactivation of the integrated HTLV-1 LTR reporter in vivo. (A) β-Gal activity from a single-copy HTLV-1 LTR-lacZ reporter in pA-18G-BHK-21 cells transfected with a Tax plasmid (50 ng) (lanes 2 and 4 to 6) and an HDAC plasmid (50, 100, and 200 ng; lanes 4, 5, and 6, respectively). Lane 3 contains 200 ng of HDAC1. Cotransfection of HDAC1 represses Tax-mediated transactivation. Data presented are the mean ± standard deviation (n = 3). Lanes 4 to 6 are significantly different (P < 0.01) from results in lane 2. (B) Cotransfection of HDAC1 did not alter Tax expression. Western blot assays of Tax levels in whole-cell extracts from BHK cells transfected with Tax (50 μg) and HDAC1 (200 μg) plasmids.
HDAC1 directly interacts with Tax in vivo and in vitro.
To determine whether purified Tax protein interacts directly with HDAC1, we first utilized coimmunoprecipitation experiments with purified proteins. We purified Tax protein from bacteria and Flag-tagged HDAC1 protein (23) from baculovirus-infected Sf9 cells. Following incubation of HDAC1 and Tax, the proteins were immunoprecipitated with anti-Flag antibody-conjugated agarose beads. Bound proteins were eluted and separated by SDS-PAGE, and the Tax protein level was determined by Western blot analysis using an anti-Tax antibody (Fig. 3A, left panel). The results of this experiment demonstrated that Tax coimmunoprecipitated with anti-Flag-HDAC1 (Fig. 3A, lane 2, left panel). Tax was not detectable by Western blotting when a blocking peptide was added to inhibit the interaction between the anti-Flag antibody and the Flag-HDAC1 protein (Fig. 3A, lane 1, left panel). The direct Tax-HDAC1 interaction was also demonstrated in a GST-HDAC pull-down assay in which bacterial GST-HDAC1, but not GST, interacted with purified Tax protein (Fig. 3A, right panel). Finally, in a GST pull-down assay GST-Tax, but not the control GST protein, was shown to interact with HDAC1 from HeLa cell extract (Fig. 3B), consistent with the report by Ego et al. (15).
FIG. 3.
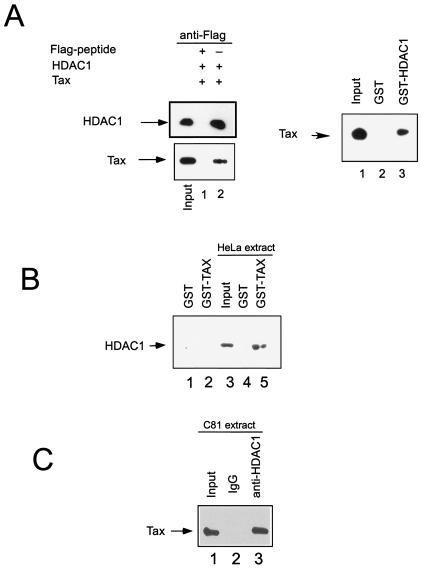
HDAC1 interacts with Tax directly in vivo and in vitro. (A) Left: Western blot assay of HDAC1 and Tax levels in the immunoprecipitates following incubation of purified Flag-tagged HDAC1 and Tax proteins and immunoprecipitation with anti-Flag antibody. Right: GST-HDAC1 pull-down experiment. Tax protein was detected by anti-Tax antibody by Western blotting. Lane 2, GST control; lane 3, GST-HDAC1 pull-down; lane 1, input representing one-third of the amount added to the binding reaction mixture. (B) GST-Tax associates with HDAC1 from HeLa cell extract. Western blot analysis of HDAC1 levels from GST-Tax pull-down after incubation of HeLa cell nuclear extract with GST-Tax (lane 5) and GST control (lane 4). (C) HDAC1 interacts with Tax in vivo, as shown in Western blot assays of Tax levels in immunoprecipitates from C81 nuclear extract with an anti-HDAC1 antibody (lane 3) or a control IgG (lane 2).
To demonstrate that Tax interacts with HDAC1 in vivo, HTLV-1-transformed C81 T cells that constitutively express Tax and HDAC1 were utilized. Nuclear extracts were prepared, and proteins were immunoprecipitated with either anti-HDAC1 or control IgG. The immunoprecipitates were washed three times with buffer containing 0.2% NP-40 and eluted in SDS-PAGE loading buffer. The results of the Western blot analysis demonstrated that Tax specifically coimmunoprecipitates with endogenous HDAC1 from HTLV-1-transformed cells (Fig. 3C). No Tax was detected in immunoprecipitates with the control IgG.
HDAC1 is associated with inactive HTLV-1 LTR chromatin in vivo.
The interaction between Tax and HDAC1 brings up an interesting question. Does the association with Tax recruit HDAC1 to the LTR, or does Tax inhibit the binding of HDAC1 to chromatin by squelching? A recent study reported that HDAC1 is present on the HTLV-1 LTR in HTLV-1-transformed cells and suggested that Tax recruits HDAC1 to the promoter (30). Given the fact that the HTLV-1-transformed cells used in the studies have multiple copies of the integrated LTR, however, it was not clear whether HDAC1 was associated with active or inactive chromatin templates.
To address this question, we utilized control and Tax-expressing pA-18G-BHK-21 cell lines, which contain a single copy of the HTLV-1 LTR, and compared the association of HDAC1 with the activated and nonactivated LTR in a ChIP assay. To generate the Tax-expressing cell line, pA-18G-BHK-21 cells were infected with Tax-expressing retrovirus, and stable clones that constitutively express Tax protein were obtained. The expression of Tax protein in these cells was examined by immunostaining with an anti-Tax monoclonal antibody (Tab172) (Fig. 4A). Tax was excluded from the nucleolus and localized to discrete nuclear sites, the Tax speckled structures, as reported previously (20, 40, 47). The control (Tax-negative) cells were all negative for the immunostaining (Fig. 4B).
FIG. 4.
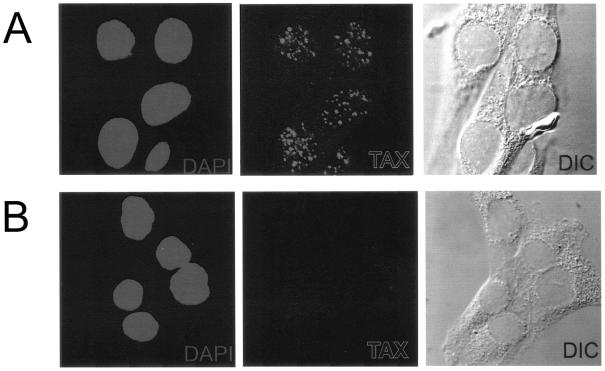
Tax localizes in discrete nuclear foci. Tax-expressing pA-18G-BHK-21 cells were generated through retroviral transduction. Tax-expressing cells (A) and control cells (B) were subjected to a direct immunofluorescence assay using an anti-Tax antibody. Each panel in a series is of the same field of view. DIC, differential interference contrast.
The activation of the HTLV-1 LTR was then further confirmed by β-Gal staining. In situ β-Gal staining showed that 100% of cells were positive, indicating the activation of the single-copy HTLV-1 LTR in Tax-expressing cells (Fig. 5A, right panel). We then performed ChIP assays utilizing Tax-negative or Tax-positive cells. Equivalent amounts of cross-linked chromatin were immunoprecipitated, and the precipitated DNA was then subjected to PCR amplification. Figure 4B shows that Tax binds to the integrated LTR (lane 6). The specificity of this assay is shown by the failure to detect the promoter when Tax-negative cells were used in the analysis (lane 5). In addition, Tax was not found associated with the control β-globin promoter (Fig. 4B, lane 6, bottom panel). We also examined the association of CREB, CBP, and RNAP II with the LTR. RNAP II and CBP were not observed on the nonactivated LTR (Fig. 4B, lanes 7 and 9) but were associated with the LTR from Tax-positive cells (Fig. 5B, lanes 8 and 10). A low but detectable level of CREB protein could be found on the inactive promoter (lane 11). Its binding was enhanced in the presence of Tax (lane 12). CBP, RNAP II, and CREB had minimal association with the β-globin promoter from both Tax− and Tax+ cells (Fig. 5B, bottom panel).
FIG. 5.


Presence of HDAC1 on the HTLV-1 LTR. (A) Tax-expressing pA-18G-BHK-21 cells were generated through retroviral transduction, and Tax-expressing cells (right panel) were positive for in situ β-Gal staining. Tax− cells (left panel) were all negative for the staining. The ChIP assay was performed using chromatin either from Tax+ or Tax− pA-18G-BHK-21 cells. (B) Tax, CREB, CBP, and RNAP II associated with the HTLV-1 LTR in Tax-expressing cells. ChIP assays were performed using anti-Tax, CREB, CBP, and RNAP II antibodies on extract from Tax+ or Tax− pA-18G-BHK-21 cells. (C) Enhanced histone H3 and H4 acetylation of the LTR in the Tax-expressing cells as shown in a ChIP assay of acetylated histone H3 and H4 in the LTR or control β-globin gene promoter from Tax− or Tax+ pA-18G-BHK-21 cells. (D) HDAC1 dissociated from activated HTLV-1 LTR. The ChIP assays were performed in Tax− or Tax+ pA-18G-BHK-21 cells using anti-HDAC1 (lanes 2 and 4) or a control IgG (lanes 1 and 3), as indicated. PCR products from the LTR template and β-globin promoter were separated on 2% agarose gel and visualized by ethidium bromide staining.
In addition, we examined histone acetylation. Low but reproducible levels of acetylated H3 and H4 were detected on the nonactivated LTR (Fig. 5C, lanes 5 and 7). In the presence of Tax, the levels of acetylated histone H3 and H4 on the LTR were enhanced (Fig. 5C, lanes 6 and 8), consistent with the association of CBP with the LTR (Fig. 5B, lane 10). The level of acetylated histone H3 and H4 associated with the β-globin promoter remained unchanged in Tax-negative and Tax-positive cells.
HDAC can be associated with the promoter either through targeted recruitment or constitutive binding. Interestingly, we found that while HDAC1 associates with nonactivated HTLV-1 LTR, the association was dramatically decreased on the Tax-transactivated LTR (Fig. 5D, lanes 3 and 6, top panel). In contrast, no Tax-regulated change in HDAC1 binding was observed on the β-globin promoter.
Tax competes with HDAC1 binding to the LTR.
Our studies indicate that Tax interacts with HDAC1 but that the interaction does not contribute to the recruitment of HDAC1 on the HTLV-1 promoter in vivo. In fact, our results suggest that the interaction between Tax and HDAC1 may inhibit HDAC1 binding to the LTR. To test this hypothesis, an in vitro chromatin pull-down assay was utilized. Two templates, the control −52 and the Tax-responsive 4xTRE (Fig. 6A), were reconstituted with Drosophila melanogaster embryo extract as described previously (33). Purified chromatin templates were incubated with nuclear extract in the presence or absence of purified Tax protein. As shown in Fig. 6B, HDAC1 associated with the 4xTRE chromatin template, but not the −52 chromatin template (Fig. 5B, lanes 1 and 4). The addition of increasing amounts of Tax to the reaction mixture reduced HDAC1 binding to the 4xTRE chromatin template (Fig. 5B, lanes 2 and 3, top panel). The decline in HDAC1 binding was accompanied by an increasing level of Tax bound to the template (Fig. 6B, lanes 2 and 3, bottom panel).
FIG. 6.
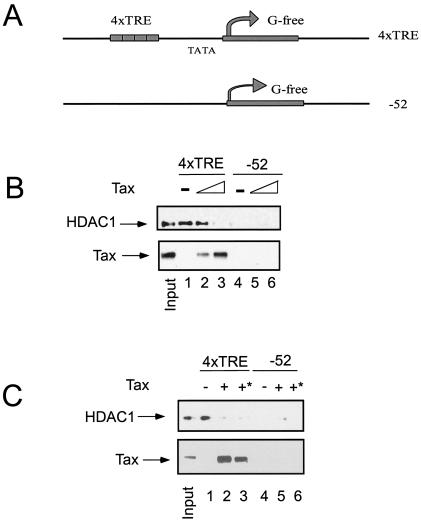
Tax competes with HDAC1 binding to the LTR. Biotinylated LTR chromatin templates were incubated with purified Tax protein and HeLa cell nuclear extract as described in Materials and Methods. After incubation and washing, proteins were eluted from beads and analyzed by SDS-PAGE and Western blotting using antibody against HDAC1. (A) Schematics of the 4xTRE and −52 chromatin templates. (B) Tax inhibits HDAC1 association with the 4xTRE chromatin template. Different levels of Tax protein were incubated with either 4xTRE chromatin or a control −52 chromatin. (C) Tax promotes HDAC1 dissociation from the 4xTRE chromatin template. The asterisk indicates that Tax protein was added after incubation of HeLa extract with chromatin template.
We also carried out experiments to determine if Tax could dissociate HDAC1 from the chromatin template. Consistent with the results presented above, HDAC1 bound to the 4xTRE template in the absence of Tax (Fig. 6C, lane 1, top panel). When Tax was added to the incubation mixture, inhibition of HDAC1 binding was observed (Fig. 6C, lane 2, top panel). Interestingly, when Tax was added to chromatin complexes containing bound HDAC1, Tax dissociated HDAC1 from the complex (Fig. 6C, lane 3, top panel). Together, these results provide direct evidence that Tax inhibits the interaction of HDAC1 with the HTLV-1 promoter.
DISCUSSION
We have demonstrated that HDACs play an important role in the regulation of HTLV-1 transcription. Inhibition of HDAC activity by TSA results in an increase in Tax transactivation in HTLV-1-transformed cells. Using ChIP assays, we have demonstrated that HDAC1 is associated with the inactive basal promoter but not the transcriptionally active template (Fig. 5D). Ego et al. (15) recently proposed that HDAC1 may be directly recruited to the HTLV-1 promoter by Tax. Further studies by Lemasson et al. (30) suggested that TSA inhibition of HDAC function on the template DNA resulted in increased histone H4 acetylation. The conclusions of these studies were complicated by the presence of active and inactive transcription templates present in the cells. It has been estimated that SLB-1, an HTLV-1-transformed T-cell line, has five copies of the HTLV-1 LTR, while MT-2, another HTLV-1 T-cell line, has two copies of the HTLV-1 LTR (30). At least one copy of the HTLV-1 LTR is inactive in these cell lines (30). Thus, it is difficult to distinguish between activated and nonactivated LTRs in HTLV-1-transformed cells in terms of coactivator and corepressor loading. Our analyses of inactive and active single-copy templates clarify these earlier studies by demonstrating that HDAC1 association with the promoter is not facilitated by Tax. In fact, our studies suggest that the ability of Tax to interact directly with HDAC1 inhibits the binding of HDAC1 to the promoter in the presence of Tax. It is interesting that a similar pattern in which HDAC1 is associated with inactive templates, but not activated templates, has been reported for some p53- and CREB-responsive genes (28).
Because our data indicate that HDAC1 associates with the nonactivated LTR, a question arises as to how HDAC1 is recruited. In general, HDACs associate with chromatin templates in two different manners (31). First, HDACs may bind to the promoter DNA in a nontargeted manner as components of the histone-binding Sin32 and NuRD complexes, which repress global chromatin transcription. Second, HDACs can be recruited to the promoter in a targeted manner by transcriptional factors, corepressor, DNA methylases, and methyl-CpG binding proteins (10, 41). Based on the chromatin pull-down assay (Fig. 6), in which HDAC1 associated with the 4xTRE chromatin but not the −52 control promoter chromatin, it is likely that HDAC1 associates with the HTLV-1 LTR through targeted recruitment. A recent study by Canettieri et al. indicated that CREB recruits HDAC1 to the CRE promoter and phosphorylation of CREB could dissociate HDAC1 from that promoter (6). ChIP assays demonstrated that a low level of CREB associated with the nonactivated LTR (Fig. 5B), leaving open the possibility that HDAC1 may be recruited to the LTR through CREB. It is also possible that other factors, such as Sp1, which has been shown to interact with HDAC1 and the LTR, may contribute to the recruitment of HDAC1 (3, 14). We also observed the loading of CBP and RNAP II on the LTR in the presence of Tax, consistent with the observation of Tax-dependent protein loading on the HTLV-1 LTR as assayed by genomic footprinting (11, 12). Based on our finding of HDAC1 association with the LTR and literature regarding the interaction between CREB, Tax, and HDAC1, we propose a working model of HTLV-1 transcriptional regulation (Fig. 7). On the nonactivated LTR, HDAC1 is recruited to the promoter, probably through its interaction with cellular proteins such as CREB or Sp1 bound to the LTR. In the presence of Tax, HDAC1 is released from the LTR, presumably through its interaction with Tax. Tax increases CREB association on the LTR, and the Tax/CREB/DNA complex recruits p300/CBP and RNAP II to the LTR to initiate transcription.
FIG. 7.
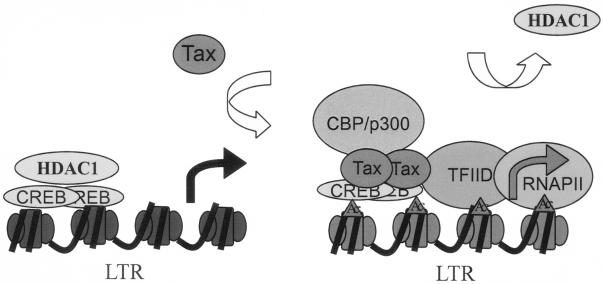
Model of HTLV-1 LTR activation by Tax. Tax relieves transcriptional repression. On the nonactivated LTR, HDAC1 is associated with the template, probably through its interaction with cellular proteins bound to the LTR. In the presence of Tax, HDAC1 is released from the LTR through its interaction with Tax. Tax increases CREB association on the LTR, and the Tax/CREB/DNA complex recruits p300/CBP and RNAP II to activate transcription.
Our data provide insight into the dynamic interaction between Tax and HDAC1 in Tax transactivation. Interaction of HDAC1 with a number of other transcription factors, including NF-κB, MyoD, p65, and p53, leads to transcriptional repression (1, 26, 36, 44, 56). For example, interaction between HDAC1 and p65 has been suggested to turn off transcription by recruitment of the deacetylase to the promoter (1). Our data suggest that the interaction between Tax and HDAC1 does not repress transcription but is part of the transcriptional activation pathway, since Tax can decrease HDAC1 binding to the template DNA by inhibiting the binding and/or by dissociating bound HDAC1. Future studies will be directed toward understanding the interplay between activator and repressor complexes that regulate the HTLV-1 LTR transcription.
REFERENCES
- 1.Ashburner, B. P., S. D. Westerheide, and A. S. J. Baldwin. 2001. The p65 (RelA) subunit of NF-κB interacts with the histone deacetylase (HDAC) corepressors HDAC1 and HDAC2 to negatively regulate gene expression. Mol. Cell. Biol. 21:7065-7077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Baranger, A. M., C. R. Palmer, M. K. Hamm, H. A. Giebler, A. Brauweiler, J. K. Nyborg, and A. Schepartz. 1995. Mechanism of DNA-binding enhancement by the human T-cell leukaemia virus transactivator Tax. Nature 376:606-608. [DOI] [PubMed] [Google Scholar]
- 3.Barnhart, M. K., L. M. Connor, and S. J. Marriott. 1997. Function of the human T-cell leukemia virus type 1 21-base-pair repeats in basal transcription. J. Virol. 71:337-344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bex, F., and R. B. Gaynor. 1998. Regulation of gene expression by HTLV-I Tax protein. Methods 16:83-94. [DOI] [PubMed] [Google Scholar]
- 5.Brady, J. N. 1996. Biology of HTLV-I: host cell interactions, p. 79-112. In P. Hollsberg and D. A. Hafl (ed.), Human T-cell lymphotropic virus type I. John Wiley and Sons Ltd., Chichester, England.
- 6.Canettieri, G., I. Morantte, E. Guzman, H. Asahara, S. Herzig, S. D. Anderson, J. R. Yates, and M. Montminy. 2003. Attenuation of a phosphorylation-dependent activator by an HDAC-PP1 complex. Nat. Struct. Biol. 10:175-181. [DOI] [PubMed] [Google Scholar]
- 7.Carruthers, L. M., C. Tse, K. P. Walker, and J. C. Hansen. 1999. Assembly of defined nucleosomal and chromatin arrays from pure components. Methods Enzymol. 304:19-35. [DOI] [PubMed] [Google Scholar]
- 8.Chan, H. M., and N. B. La Thangue. 2001. p300/CBP proteins: HATs for transcriptional bridges and scaffolds. J. Cell Sci. 114:2363-2373. [DOI] [PubMed] [Google Scholar]
- 9.Chen, L., W. Fischle, E. Verdin, and W. C. Greene. 2001. Duration of nuclear NF-κB action regulated by reversible acetylation. Science 293:1653-1657. [DOI] [PubMed] [Google Scholar]
- 10.Cress, W. D., and E. Seto. 2000. Histone deacetylases, transcriptional control, and cancer. J. Cell Physiol. 184:1-16. [DOI] [PubMed] [Google Scholar]
- 11.Datta, S., N. H. Kothari, and H. Fan. 2000. In vivo genomic footprinting of the human T-cell leukemia virus type 1 (HTLV-1) long terminal repeat enhancer sequences in HTLV-1-infected human T-cell lines with different levels of Tax I activity. J. Virol. 74:8277-8285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Datta, S., N. H. Kothari, and H. Fan. 2001. Induction of Tax in expression in MT-4 cells by 5-azacytidine leads to protein binding in the HTLV-1 LTR in vivo. Virology 283:207-214. [DOI] [PubMed] [Google Scholar]
- 13.de La Fuente, C., L. Deng, F. Santiago, L. Arce, L. Wang, and F. Kashanchi. 2000. Gene expression array of HTLV type 1-infected T cells: up-regulation of transcription factors and cell cycle genes. AIDS Res. Hum. Retrovir. 16:1695-1700. [DOI] [PubMed] [Google Scholar]
- 14.Doetzlhofer, A., H. Rotheneder, G. Lagger, M. Koranda, V. Kurtev, G. Brosch, E. Wintersberger, and C. Seiser. 1999. Histone deacetylase 1 can repress transcription by binding to Sp1. Mol. Cell. Biol. 19:5504-5511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ego, T., Y. Ariumi, and K. Shimotohno. 2002. The interaction of HTLV-1 Tax with HDAC1 negatively regulates the viral gene expression. Oncogene 21:7241-7246. [DOI] [PubMed] [Google Scholar]
- 16.Franklin, A. A., M. F. Kubik, M. N. Uittenbogaard, A. Brauweiler, P. Utaisincharoen, M. A. Matthews, W. S. Dynan, J. P. Hoeffler, and J. K. Nyborg. 1993. Transactivation by the human T-cell leukemia virus Tax protein is mediated through enhanced binding of activating transcription factor-2 (ATF-2): ATF-2 response and cAMP element-binding protein (CREB). J. Biol. Chem. 268:21225-21231. [PubMed] [Google Scholar]
- 17.Georges, S. A., H. A. Giebler, P. A. Cole, K. Luger, P. J. Laybourn, and J. K. Nyborg. 2003. Tax recruitment of CBP/p300, via the KIX domain, reveals a potent requirement for acetyltransferase activity that is chromatin dependent and histone tail independent. Mol. Cell. Biol. 23:3392-3404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Georges, S. A., W. L. Kraus, K. Luger, J. K. Nyborg, and P. J. Laybourn. 2002. p300-mediated Tax transactivation from recombinant chromatin: histone tail deletion mimics coactivator function. Mol. Cell. Biol. 22:127-137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Grozinger, C. M., and S. L. Schreiber. 2002. Deacetylase enzymes: biological functions and the use of small-molecule inhibitors. Chem. Biol. 9:3-16. [DOI] [PubMed] [Google Scholar]
- 20.Haoudi, A., R. C. Daniels, E. Wong, G. Kupfer, and O. J. Semmes. 2003. Human T-cell leukemia virus-I tax oncoprotein functionally targets a subnuclear complex involved in cellular DNA damage response. J. Biol. Chem. 278:37736-37744. [DOI] [PubMed] [Google Scholar]
- 21.Harrod, R., Y. L. Kuo, Y. Tang, Y. Yao, A. Vassilev, Y. Nakatani, and C. Z. Giam. 2000. p300 and p300/cAMP-responsive element-binding protein associated factor interact with human T-cell lymphotropic virus type-1 Tax in a multi-histone acetyltransferase/activator-enhancer complex. J. Biol. Chem. 275:11852-11857. [DOI] [PubMed] [Google Scholar]
- 22.Harrod, R., Y. Tang, C. Nicot, H. S. Lu, A. Vassilev, Y. Nakatani, and C. Z. Giam. 1998. An exposed KID-like domain in human T-cell lymphotropic virus type 1 Tax is responsible for the recruitment of coactivators CBP/p300. Mol. Cell. Biol. 18:5052-5061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hassig, C. A., J. K. Tong, T. C. Fleischer, T. Owa, P. G. Grable, D. E. Ayer, and S. L. Schreiber. 1998. A role for histone deacetylase activity in HDAC1-mediated transcriptional repression. Proc. Natl. Acad. Sci. USA 95:3519-3524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jiang, H., H. Lu, R. L. Schiltz, C. A. Pise-Masison, V. V. Ogryzko, Y. Nakatani, and J. N. Brady. 1999. PCAF interacts with tax and stimulates tax transactivation in a histone acetyltransferase-independent manner. Mol. Cell. Biol. 19:8136-8145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jones, P. L., and Y. B. Shi. 1903. N-CoR-HDAC corepressor complexes: roles in transcriptional regulation by nuclear hormone receptors. Curr. Top. Microbiol. Immunol. 274:237-268. [DOI] [PubMed] [Google Scholar]
- 26.Juan, L. J., W. J. Shia, M. H. Chen, W. M. Yang, E. Seto, Y. S. Lin, and C. W. Wu. 2000. Histone deacetylases specifically down-regulate p53-dependent gene activation. J. Biol. Chem. 275:20436-20443. [DOI] [PubMed] [Google Scholar]
- 27.Kwok, R. P., M. E. Laurance, J. R. Lundblad, P. S. Goldman, H. Shih, L. M. Connor, S. J. Marriott, and R. H. Goodman. 1996. Control of cAMP-regulated enhancers by the viral transactivator Tax through CREB and the co-activator CBP. Nature 380:642-646. [DOI] [PubMed] [Google Scholar]
- 28.Lagger, G., A. Doetzlhofer, B. Schuettengruber, E. Haidweger, E. Simboeck, J. Tischler, S. Chiocca, G. Suske, H. Rotheneder, E. Wintersberger, and C. Seiser. 2003. The tumor suppressor p53 and histone deacetylase 1 are antagonistic regulators of the cyclin-dependent kinase inhibitor p21/WAF1/CIP1 gene. Mol. Cell. Biol. 23:2669-2679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lee, D. K., B. C. Kim, J. N. Brady, K. T. Jeang, and S. J. Kim. 2002. Human T-cell lymphotropic virus type 1 tax inhibits transforming growth factor-β signaling by blocking the association of Smad proteins with Smad-binding element. J. Biol. Chem. 277:33766-33775. [DOI] [PubMed] [Google Scholar]
- 30.Lemasson, I., N. J. Polakowski, P. J. Laybourn, and J. K. Nyborg. 2002. Transcription factor binding and histone modifications on the integrated proviral promoter in human T-cell leukemia virus-I-infected T-cells. J. Biol. Chem. 277:49459-49465. [DOI] [PubMed] [Google Scholar]
- 31.Li, J., Q. Lin, W. Wang, P. Wade, and J. Wong. 2002. Specific targeting and constitutive association of histone deacetylase complexes during transcriptional repression. Genes Dev. 16:687-692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lin, R. J., L. Nagy, S. Inoue, W. Shao, W. H. J. Miller, and R. M. Evans. 1998. Role of the histone deacetylase complex in acute promyelocytic leukaemia. Nature 391:811-814. [DOI] [PubMed] [Google Scholar]
- 33.Lu, H., C. A. Pise-Masison, T. M. Fletcher, R. L. Schiltz, A. K. Nagaich, M. Radonovich, G. Hager, P. A. Cole, and J. N. Brady. 2002. Acetylation of nucleosomal histones by p300 facilitates transcription from tax-responsive human T-cell leukemia virus type 1 chromatin template. Mol. Cell. Biol. 22:4450-4462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Macaluso, M., C. Cinti, G. Russo, A. Russo, and A. Giordano. 2003. pRb2/p130-E2F4/5-HDAC1-SUV39H1-p300 and pRb2/p130-E2F4/5-HDAC1-SUV39H1-DNMT1 multimolecular complexes mediate the transcription of estrogen receptor-alpha in breast cancer. Oncogene 22:3511-3517. [DOI] [PubMed] [Google Scholar]
- 35.Mal, A., and M. L. Harter. 2003. MyoD is functionally linked to the silencing of a muscle-specific regulatory gene prior to skeletal myogenesis. Proc. Natl. Acad. Sci. USA 100:1735-1739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mal, A., M. Sturniolo, R. L. Schiltz, M. K. Ghosh, and M. L. Harter. 2001. A role for histone deacetylase HDAC1 in modulating the transcriptional activity of MyoD: inhibition of the myogenic program. EMBO J. 20:1739-1753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Michael, L. F., H. Asahara, A. I. Shulman, W. L. Kraus, and M. Montminy. 2000. The phosphorylation status of a cyclic AMP-responsive activator is modulated via a chromatin-dependent mechanism. Mol. Cell. Biol. 20:1596-1603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ng, P. W., H. Iha, Y. Iwanaga, M. Bittner, Y. Chen, Y. Jiang, G. Gooden, J. M. Trent, P. Meltzer, K. T. Jeang, and S. L. Zeichner. 2001. Genome-wide expression changes induced by HTLV-1 Tax: evidence for MLK-3 mixed lineage kinase involvement in Tax-mediated NF-κB activation. Oncogene 20:4484-4496. [DOI] [PubMed] [Google Scholar]
- 39.Nicot, C., R. Mahieux, C. Pise-Masison, J. Brady, A. Gessain, S. Yamaoka, and G. Franchini. 2001. Human T-cell lymphotropic virus type 1 Tax represses c-Myb-dependent transcription through activation of the NF-κB pathway and modulation of coactivator usage. Mol. Cell. Biol. 21:7391-7402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Park, H. U., J. H. Jeong, J. H. Chung, and J. N. Brady. Human T-cell leukemia virus type 1 Tax interacts with Chk1 and attenuates DNA-damage induced G2 arrest mediated by Chk1. Oncogene, in press. [DOI] [PubMed]
- 41.Peterson, C. L. 2002. HDAC's at work: everyone doing their part. Mol. Cell 9:921-922. [DOI] [PubMed] [Google Scholar]
- 42.Pise-Masison, C. A., R. Mahieux, M. Radonovich, H. Jiang, and J. N. Brady. 2001. Human T-lymphotropic virus type I Tax protein utilizes distinct pathways for p53 inhibition that are cell type-dependent. J. Biol. Chem. 276:200-205. [DOI] [PubMed] [Google Scholar]
- 43.Pise-Masison, C. A., M. Radonovich, R. Mahieux, P. Chatterjee, C. Whiteford, J. Duvall, C. Guillerm, A. Gessain, and J. N. Brady. 2002. Transcription profile of cells infected with human T-cell leukemia virus type I compared with activated lymphocytes. Cancer Res. 62:3562-3571. [PubMed] [Google Scholar]
- 44.Puri, P. L., S. Iezzi, P. Stiegler, T. T. Chen, R. L. Schiltz, G. E. Muscat, A. Giordano, L. Kedes, J. Y. Wang, and V. Sartorelli. 2001. Class I histone deacetylases sequentially interact with MyoD and pRb during skeletal myogenesis. Mol. Cell 8:885-897. [DOI] [PubMed] [Google Scholar]
- 45.Ressler, S., G. F. Morris, and S. J. Marriott. 1997. Human T-cell leukemia virus type 1 Tax transactivates the human proliferating cell nuclear antigen promoter. J. Virol. 71:1181-1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rivera-Walsh, I., M. E. Cvijic, G. Xiao, and S. C. Sun. 2000. The NF-kappa B signaling pathway is not required for Fas ligand gene induction but mediates protection from activation-induced cell death. J. Biol. Chem. 275:25222-25230. [DOI] [PubMed] [Google Scholar]
- 47.Semmes, O. J., and K. T. Jeang. 1996. Localization of human T-cell leukemia virus type 1 tax to subnuclear compartments that overlap with interchromatin speckles. J. Virol. 70:6347-6357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sterner, D. E., and S. L. Berger. 2000. Acetylation of histones and transcription-related factors. Microbiol. Mol. Biol. Rev. 64:435-459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sun, J. M., H. Y. Chen, and J. R. Davie. 2001. Effect of estradiol on histone acetylation dynamics in human breast cancer cells. J. Biol. Chem. 276:49435-49442. [DOI] [PubMed] [Google Scholar]
- 50.Utley, R. T., T. A. Owen-Hughes, L. J. Juan, J. Cote, C. C. Adams, and J. L. Workman. 1996. In vitro analysis of transcription factor binding to nucleosomes and nucleosome disruption/displacement. Methods Enzymol. 274:276-291. [DOI] [PubMed] [Google Scholar]
- 51.Van Lint, C., S. Emiliani, and E. Verdin. 1996. The expression of a small fraction of cellular genes is changed in response to histone hyperacetylation. Gene Expr. 5:245-253. [PMC free article] [PubMed] [Google Scholar]
- 52.Yang, X. J., and E. Seto. 2003. Collaborative spirit of histone deacetylases in regulating chromatin structure and gene expression. Curr. Opin. Genet. Dev. 13:143-153. [DOI] [PubMed] [Google Scholar]
- 53.Yoshida, M. 1995. HTLV-1 oncoprotein Tax deregulates transcription of cellular genes through multiple mechanisms. J. Cancer Res. Clin. Oncol. 121:521-528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yoshida, M., R. Furumai, M. Nishiyama, Y. Komatsu, N. Nishino, and S. Horinouchi. 2001. Histone deacetylase as a new target for cancer chemotherapy. Cancer Chemother. Pharmacol. 48(Suppl. 1):S20-S26. [DOI] [PubMed] [Google Scholar]
- 55.Yoshida, M., and M. Seiki. 1987. Recent advances in the molecular biology of HTLV-1: trans-activation of viral and cellular genes. Annu. Rev. Immunol. 5:541-559. [DOI] [PubMed] [Google Scholar]
- 56.Zhong, H., M. J. May, E. Jimi, and S. Ghosh. 2002. The phosphorylation status of nuclear NF-kappa B determines its association with CBP/p300 or HDAC-1. Mol. Cell 9:625-636. [DOI] [PubMed] [Google Scholar]