

Abstract
The Kaposi's sarcoma-associated herpesvirus (KSHV) gene product virally encoded G protein-coupled receptor (vGPCR) is a homolog of cellular GPCRs and has been proposed to play important roles in KSHV-induced angiogenesis. The most abundant vGPCR-containing transcripts are K14/vGPCR bicistronic RNAs that are strongly induced during lytic reactivation. Here we show that the promoter governing this transcript is strongly responsive to activation by the viral lytic switch protein RTA. By deletion mapping and scanning mutation analyses, we have identified three putative RTA response elements (A, B, and C) in this promoter. However, none of these sites appear to directly bind RTA in electrophoretic mobility shift assays (EMSA). Site C corresponds to a canonical binding site for RBP-J, a sequence-specific transcriptional repressor that is normally the target of Notch signaling. RBP-J can bind RTA and recruit it to its cognate recognition site; when this happens, the activation function of RTA can relieve RBP-J-mediated repression and upregulate expression of the targeted gene. EMSA studies reveal that both sites A and C can bind to RBP-J; sequence inspection reveals that site A is a novel functional variant of known RBP-J recognition sites. (Site B corresponds to an as-yet-unknown host DNA-binding protein.) The importance of sites A and C in vivo is underscored by the observation that K14/vGPCR promoter function is dramatically inhibited in cells genetically deficient in RBP-J. The regulation of K14/vGPCR transcripts by RBP-J raises the possibility that other modulators of Notch signaling might be able to induce expression of this RNA outside the context of lytic KSHV replication.
Kaposi's sarcoma-associated herpesvirus (KSHV) is a gammaherpesvirus associated with at least three human malignancies: Kaposi's sarcoma (KS) (13, 19, 27, 31, 33, 41, 42, 56, 69), primary effusion lymphoma (10), and multicentric Castleman's disease (MCD) (61). The tumorigenic and angiogenic mechanisms of KSHV still remain unclear. A striking feature of KSHV is that its genome carries many open reading frames resembling genes for cellular cytokines, chemokines, and chemokine receptors and genes involved in apoptosis and cell cycle regulation, indicating that these viral genes might play important roles in KSHV-induced proliferation and angiogenesis (7, 11, 15, 40, 45, 46, 51, 52). One outstanding example is the viral encoded G protein-coupled receptor (vGPCR), which is related to interleukin-8 (IL-8) receptor, a CXC chemokine receptor (11). vGPCR can signal in both ligand-dependent (20) and -independent (2) ways and through multiple signal transduction pathways to enhance the expression of NF-κB- and AP-1-dependent proinflammatory cytokines (IL-6, IL-1β, tumor necrosis factor alpha, RANTES, etc), basic fibroblast growth factor, and vascular endothelial growth factor (VEGF) (3, 9, 49, 53, 57). Overexpression of vGPCR in NIH 3T3 cells (3) has been shown to induce cell transformation and angiogenesis, and its expression in primary endothelial cells (4) can cause cellular immortalization and spindle shape conversion. Several transgenic mouse lines expressing vGPCR in either hematopoietic cells (mainly T and NK cells) (70), endothelial cells (39), or many other tissues (under simian virus 40 [SV40] early promoter-enhancer control) (23) have been established. These mice develop KS-like angioproliferative lesions composed of spindle-shaped endothelial cells, vascular structures, and infiltrating inflammatory cells in some models (23, 39, 70).
Extensive studies have focused on how vGPCR triggers signal transduction pathways to enhance the expression of genes that mediate these effects. However, regulation of vGPCR gene expression is still unclear. While a monocistronic transcript of vGPCR was detected in one study, its expression level is extremely low (44). A K14/vGPCR bicistronic transcript, however, has been identified by several groups and may be the predominant messenger for vGPCR (16, 32, 44). This transcript also encodes another viral signaling molecule, K14, at its 5′ end. In contrast to its cellular homolog OX2, K14 was found to activate myeloid-lineage cells to produce inflammatory cytokines; thus, it might be involved in recruiting lymphocytes for virus dissemination and in promoting the cytokine-mediated proliferation of KSHV-infected cells (17). This K14/vGPCR bicistronic transcript is strongly induced during early KSHV lytic reactivation (16, 32, 44), suggesting that it may be a target of the viral lytic switch protein RTA (replication and transcription activator).
Expression of RTA is necessary and sufficient to induce lytic reactivation of KSHV (21, 37, 38, 62). RTA is an effective transcription activator with DNA-binding and dimerization domains at the N terminus and a strong activation domain at the C terminus (37, 54). Although RTA is able to bind with high affinity to its own response element for activation of some target genes, such as kaposin and the noncoding polyadenylated nuclear (PAN) RNA (12, 58, 60), the complexity of its response elements in other target promoters suggests the existence of an additional mechanism(s) of activation (8, 12, 14, 36, 59, 60, 64). Indeed, we have previously shown that RTA can be directed to target promoters through interaction with a cellular transcription factor, RBP-J, that recognizes the specific sequence GTGGGAA (34). Other identified cellular DNA-binding protein partners of RTA include STAT3 and CCAAT/enhancer-binding protein α (24, 67). Given the large number of RTA target genes, additional cellular protein partners (66) or alternative DNA-binding sites for RTA might still exist. By exploring the transcriptional regulation of K14/vGPCR by RTA, we hope not only to understand the regulation of K14 and vGPCR gene expression but also to advance our knowledge of RTA activation mechanisms.
Using a transient-reporter assay, we first mapped three regions in the K14/vGPCR promoter that are important for RTA activation. By scanning mutagenesis, we then identified three potential RTA response elements (A, B, and C) and studied their roles in RTA activation through genetic and biochemical analyses. Two sites (A and C) were found to be binding sites for RBP-J, which was found to play a central role in mediating RTA-induced activation of the promoter. The third site binds an as-yet-unrecognized cellular DNA-binding protein that appears to play a demonstrable but subsidiary role in RTA-dependent activation of the locus.
MATERIALS AND METHODS
Cells.
293T, CV-1, and SLK cells were grown in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum. Mouse RBP-J−/− (OT11) and wild-type (WT) (OT13) fibroblast cell lines were kindly provided by T. Honjo and were grown in high-glucose Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum and 100 U of mouse gamma interferon (Life Technologies) per ml at 32°C in 5% CO2 incubator.
Plasmids.
To create the reporter construct of the K14/vGPCR promoter, pGL3-1257, a 1.3-kb DNA fragment upstream of the ATG codon of the K14 gene (corresponding to the 1,257 nucleotides [nt] upstream of the transcription start site) was PCR amplified from KSHV genomic DNA and cloned immediately upstream of the ATG codon of the luciferase gene in pGL3-basic at Bgl II and NcoI sites (the NcoI site on the vector was eliminated). Further deletions from the 5′ end of pGL3-1257 were made by either restriction enzyme digestion or PCR amplification (Fig. 1A). The large fragment recovered from SmaI-cleaved pGL3-1257 was recircularized to create pGL3-562. Likewise, pGL3-242 was constructed after XhoI and NcoI digestion of pGL3-1257. Constructs pGL3-222, pGL3-184, pGL3-130, and pGL3-98 were created by amplifying the corresponding fragments upstream of the ATG codon of the K14 gene and cloning the fragments into pGL3-basic by using XhoI and NcoI sites, respectively (the NcoI site on the vector was destroyed). The sequences of the oligonucleotides used in PCRs are available upon request. For luciferase reporter constructs containing tandem repeats, synthesized oligonucleotides of complementary sequence were annealed to form a 5′-end BglII overhang and a 3′-end BamHI overhang, followed by ligation and BglII digestion and finally ligation with a BglII-digested pGL3 promoter. The number of tandem repeats was detected by PCR amplification followed by separation on polyacrylamide gels. To make the pGST-RTA fusion construct, a 1.5-kb fragment encoding amino acids 1 to 530 of RTA was amplified from pcDNA3-ORF50 and inserted into pGEX-4T-1 at EcoRI and SalI sites. All constructs were verified by direct sequencing. pGST-RBPJ was described before (34).
FIG. 1.

Mapping of RTA response elements in the K14/vGPCR promoter. (A) Schematic representation of serial deletions of the putative K14/vGPCR promoter. A 1.3-kb fragment upstream of the K14 ATG codon (corresponding to nt −1257 upstream of the RNA initiation site [32]) was cloned in front of a reporter gene. Serial deletions were made on the 1.3-kb fragment from the 5′ end, with the number indicating the nucleotide position upstream of the RNA initiation site. The mapped mRNA initiation site is shown by the arrow. (B) RTA activation of K14/vGPCR promoters with serial deletions. Reporter constructs with the indicated promoter deletions (0.3 μg) were used to transfect 293T cells with either empty vector or RTA expression vector (0.6 μg). The fold induction of each construct by RTA was plotted, with the error bars representing standard deviations of the results from at least two independent experiments.
Luciferase assay.
Cells were seeded onto 12-well dishes and grown to 70 to 80% confluency. DNA transfection was performed by using Fugene 6 (Roche) according to the manufacturer's protocol. For each transfection, 0.3 μg of a reporter plasmid and 0 to 0.6 μg of pcDNA3-ORF50 plasmid (36) were used; 0.1 μg of pcDNA3.1-lacZ was also included in each transfection as an internal control. The amount of total DNAs was normalized to 1 μg with vector pcDNA3.1. Cell extracts were prepared and subjected to luciferase assay with a luciferase assay kit (Promega) and to β-galactosidase assay with a β-galactosidase assay kit (Promega). Each assay was done in duplicate in at least two independent experiments.
Purification of GST fusion proteins.
BL21 cells transformed with pGST, pGST-RTA, or pGST-RBPJ plasmid DNA were grown to exponential phase at 37°C before induction with 0.2 mM IPTG (isopropyl-β-d-thiogalactopyranoside) for 2 to 3 h. Cell pellets were harvested and lysed by repeated freeze-thawing and sonication. The cleared lysate was incubated at 4°C with glutathione-Sepharose beads (Amersham Pharmacia), which were then washed four times with NETN buffer (20 mM Tris [pH 8] 100 mM NaCl, 1 mM EDTA, and 0.5% NP-40) and resuspended in NETN buffer. Glutathione S-transferase (GST) proteins were eluted from the beads by incubation with glutathione at a concentration of 1.1 g/ml overnight at 4°C, and aliquots were stored at −80°C. The purified proteins were analyzed by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) followed by Coomassie blue staining.
EMSA.
Electrophoretic mobility shift assay (EMSA) was performed essentially as described previously (34). Purified GST fusion proteins were prepared as described above. Nuclear extracts were prepared according to the protocol of the manufacturer (Active Motif, Carlsbad, Calif.). The 32P-labeled oligonucleotides were incubated with either purified proteins or nuclear extracts for 30 min at room temperature, in the presence or absence of unlabeled competitor oligonucleotides, in a total volume of 20 μl of binding buffer (20 mM HEPES [pH 7.4], 50 mM KCl, 1 mM EDTA, 1 mM MgCl2, 10 mM dithiothreitol; 250 ng of dI-dC, 8.5% glycerol). Complexes were resolved on a 4% native polyacrylamide gel in 0.5× Tris-borate-EDTA. The gel was dried and exposed on autoradiographic film.
RESULTS
The K14/vGPCR promoter contains three major RTA response elements.
The K14/vGPCR bicistronic transcript is strongly activated during tetradecanoyl phorbol acetate-induced KSHV lytic replication (16, 32, 44) and therefore represents a potential direct target of RTA. We tested the ability of RTA to activate the K14/vGPCR promoter by use of a transient-reporter assay. The full-length construct pGL3-1257 includes about 1.3 kb of sequences upstream of the ATG codon of the K14 gene and drives the expression of a luciferase reporter gene (Fig. 1A). 293T cells were transfected with this reporter in the presence or absence of an RTA expression vector, and the fold induction of luciferase activity by RTA was calculated. RTA activation of this construct is robust (200- to 500-fold higher than for the control with empty vector) (Fig. 1B), suggesting the presence of a strong and possibly multiple RTA response element(s) in this 1.3-kb promoter region. To identify the potential element(s), we made serial deletions from the 5′ end of construct pGL3-1257 (i.e., pGL3-562, pLG3-242, pGL3-222, pGL3-184, pGL3-130, and pGL3-98) (Fig. 1A). RTA activation of these constructs in 293T cells revealed multiple RTA response elements (Fig. 1B). RTA activated the constructs pGL3-1257, pGL3-562, pGL3-242, and pGL3-222 at similar levels, varying from 350- to 500-fold. However, RTA activated the construct pGL3-184 by ∼110-fold, a significant decrease compared to the construct pGL3-222 (∼360-fold). Further deletion to the construct pGL3-130 led to even lower RTA activation (18-fold). Moreover, RTA activated the construct pGL3-98 by only threefold, a sixfold difference from the construct pGL3-130. The assays were repeated with SLK human endothelial cells, CV1 simian kidney cells, and mouse embryonic fibroblasts with similar results (data not shown). Taken together, these data suggest that the K14/vGPCR promoter contained three major RTA-responsive elements, putatively located between positions −222 and −184, between positions −184 and −130, and between positions −130 and −98.
To identify the exact sequences important for RTA activation, point substitution mutations were created across the region in the construct pGL3-222, pGL3-184, or pGL3-130 (Fig. 2A). The m6 mutation of the construct pGL3-222 was activated by RTA at the same level as the deletion mutant pGL3-184 (Fig. 2B), suggesting that the sequence GTGAGA is one of the RTA-responsive elements, which hereafter is called site A. Scanning mutations in the construct pGL3-184 revealed two consecutive mutants, m15 and m16, that showed a defect in RTA activation similar to that for the construct pGL3-130 (Fig. 2C), suggesting another RTA-responsive site (site B). As determined by sequence comparison analysis, a potential RBP-J-binding site (TTCCCAC) exists between nucleotides −130 and −98. We have previously shown that RTA can activate through an RBP-J-binding site via interaction with the RBP-J protein. Indeed, mutations (−130m) introduced into the construct pGL3-130 at the putative RBP-J-binding site significantly lowered the RTA activation level to one similar to that for the deletion mutant pGL3-98 (Fig. 2D), suggesting that this RBP-J-binding site is the third RTA-responsive element (site C).
FIG. 2.
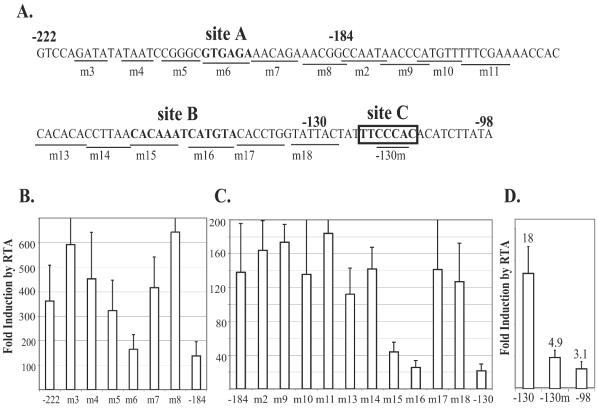
Scanning mutations reveal three major RTA response elements in the K14/vGPCR promoter. (A) Nucleotide sequences from nt −222 to −98 nt of the transcription initiation site. Sites A, B, and C, are in boldface. The position of each mutation is shown by underlining. The predicted RBP-J-binding site is boxed. (B) Scanning mutations between nt −222 and −184 revealed site A (defined by m6), which is important for RTA activation. Luciferase assay was performed with 293T cells as described for Fig. 1B. (C) Scanning mutations between nt −184 and −130 suggest that site B (defined by m15 and m16) is important for RTA activation. (D) The predicted RBP-J-binding site between nt −130 and −98 is the third RTA response element, site C (also defined by mutant −130m). Error bars indicate standard deviations.
The identified RTA-responsive elements (sites A, B, and C) do not form complexes with RTA.
Studies from several groups have demonstrated that RTA activates its target genes through multiple mechanisms: direct DNA binding, protein-protein interaction with other cellular DNA-binding factors, or both. For site C (a canonical RBP-J-binding site), by analogy with our previous work on RBP-J sites in other viral promoters (34), we propose that RTA can be tethered to this site by interaction with RBP-J. As for sites A and B, they could represent novel direct RTA-binding sites or sites for interaction with additional host proteins that can recruit RTA. We first examined the direct RTA binding in an EMSA system using GST-RTA protein and oligonucleotides spanning site A, B, or C. The sequences of the oligonucleotides used in this study are shown in Fig. 3A. The GST-RTA protein contained GST sequences at its N terminus fused with residues 1 to 530 of RTA at its C terminus. As determined by SDS-PAGE analysis (Fig. 3B) after purification on a glutathione-Sepharose column, the protein migrated as an 85-kDa band, which is the predicted size for a fusion protein of this composition. (The SDS-PAGE also showed the presence of some proteins of 26 kDa, which represent GST fragments presumably produced by cleavage by Escherichia coli proteases during the extraction process.) Using this material for EMSA with a 32P-labeled oligonucleotide for the known RTA-binding site from the PAN promoter, we observed strong complex formation with GST-RTA (Fig. 3C, lane 3) but not with the GST alone (lane 2). This interaction can be competed efficiently by WT PAN RTA response element (RRE) (lanes 4 and 5) and less efficiently by kaposin RRE (12, 59) (lanes 6 and 7). However, we observed no competition of this complex by oligonucleotides A, B, C, or BC (lanes 8 to 10). In accord with these results, when 32P-labeled oligonucleotide A or B was tested directly for binding to GST-RTA, neither site was observed to form complexes by EMSA under these conditions (Fig. 3D). As expected, oligonucleotide C, an RBP-J-binding site, also was not shifted by RTA protein in vitro (Fig. 3D).
FIG. 3.
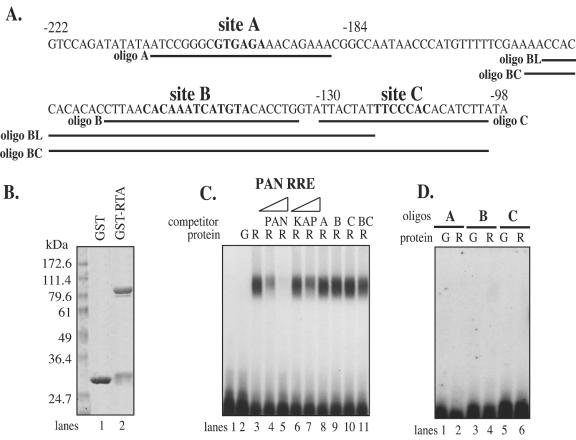
Sites A, B, and C do not bind to RTA efficiently in vitro. (A) Sequences from nt −222 to −98 upstream of the transcription site, with sites A, B, and C in boldface. Positions of all oligonucleotides (A, B, BL, C, and BC) used in this study are indicated underneath the corresponding sequences. (B) GST and GST-RTA proteins purified from E. coli lysates. A truncated RTA (amino acids 1 to 530) was fused to the N-terminal GST to create GST-RTA fusion protein. Both GST and GST-RTA fusion proteins were purified as described in Materials and Methods and analyzed in an SDS-polyacrylamide gel. The sizes of the molecular mass markers are indicated at the left. (C) In gel shift assay, a 32P-labeled PAN RRE (GATCCTTCCAAAAATGGGTGGCTAACCTGTCCAAAATATGGGAA) probe (lane 1) was incubated with either purified GST (lane 2) or GST-RTA fusion protein (lanes 3 to 11). Different competitor oligonucleotides were included in the probe-RTA incubation reaction: PAN RRE (20- and 400-fold excesses over probe) (lanes 4 and 5, respectively), kaposin RRE (GATCCCGGGAAATGGGTGGCTAACCCCTACATAAGCAGTTTGA) (20- and 400-fold) (lanes 6 and 7, respectively), oligonucleotide A (400-fold) (lane 8), oligonucleotide B (400-fold) (lane 9), oligonucleotide C (400-fold) (lane 10), or oligonucleotide BC (400-fold) (lane 11). G, GST protein; R, GST-RTA ΔSTAD protein. (D) RTA does not form a stable complex with oligonucleotide A, B, or C. Oligonucleotide A (lanes 1 and 2), B (lanes 3 and 4), or C (lanes 5 and 6) was incubated with either purified GST protein (lanes 1, 3, and 5) or GST-RTA fusion protein (lanes 2, 4 and 6) and separated on a native polyacrylamide gel.
We note that the RTA protein used in these in vitro DNA binding assays lacks the Ser/Thr-rich region and the C-terminal activation domain of WT RTA. As such, it lacks the heavy phosphorylation that characterizes the protein produced in mammalian cells. This raises the formal possibility that sites A and B could represent authentic direct binding sites that require these modifications. While we cannot fully exclude this possibility, we consider it unlikely, since both sites can be shown to bind host factors in a fashion that can be nullified by mutations that abolish RTA activation (see below).
Sites A and C contain functional RBP-J-binding sites and mediate RTA activation in a heterogeneous promoter.
To determine whether any cellular factor(s) binds to site A, we performed EMSA on oligonucleotide A with nuclear extracts of the 293T cells. We noticed that oligonucleotide A produced a strong and specific band of the same size as oligonucleotide C, which represents the conventional RBP-J-binding site (data not shown), suggesting that oligonucleotide A may contain an RBP-J-binding site. Using the purified GST-RBP-J fusion protein in a gel shift assay (Fig. 4A), we showed that oligonucleotide A indeed bound directly to RBP-J. The 32P-labeled oligonucleotide A (lane 1) formed a strong complex with RBP-J protein (lane 3) but not with GST alone (lane 2). This complex can be efficiently competed by WT (lanes 4 and 5) but not mutant (lanes 6 and 7) RBP-J oligonucleotide. Similar results were obtained for the control oligonucleotide C (lanes 8 to 12).
FIG. 4.

Oligonucleotide A or C can form a functional complex with RBP-J protein. (A) Gel shift assay of oligonucleotide A or C with purified GST-RBPJ protein. Labeled oligonucleotide A (lanes 1 to 7) or C (lanes 8 to 12) was incubated with either GST (lane 2) or GST-RBP-J (lanes 3 to 12) protein, in the absence of RBP-J oligonucleotide (lanes 3 and 8) or in the presence of unlabeled WT (10- and 40-fold) (lanes 4 and 5 and lanes 9 and 10) or mutant (10- and 40-fold) (lanes 6 and 7 and lanes 11 and 12) RBP-J oligonucleotide. G, GST protein; RP, GST-RBP-J protein. (B) Repeats of oligonucleotide A or C mediate RTA activation in a heterogeneous promoter. Oligonucleotide A or C (WT or mutant) was inserted in tandem repeats (the number of repeats is indicated) into the SV40 minimal promoter in the pGL3 promoter to drive the expression of reporter gene luciferase. The individual reporter construct was cotransfected into 293T cells with either empty vector or RTA expression vector. The fold induction by RTA was plotted, with the error bars representing standard deviations of the results from at least two independent experiments. A, WT oligonucleotide A; Am, mutant oligonucleotide A; C; WT oligonucleotide C; Cm, mutant oligonucleotide C.
To demonstrate that sites A and C are each sufficient for RTA activation, tandem repeats of either the WT or mutated sequences of site A or C were cloned into an SV40 minimal promoter driving the expression of a luciferase reporter gene. As positive controls, PAN and kaposin RREs were cloned in the same way. These constructs were tested by transient-reporter assay for RTA transactivation (Fig. 4B). As expected, RTA efficiently activated the promoter containing either PAN or KAP RRE at ∼10- to 20-fold. RTA activated the promoters containing WT oligonucleotide A as well, and the activation increased with increasing number of repeats. In contrast, RTA failed to activate those containing mutant oligonucleotide A. Likewise, WT but not mutant oligonucleotide C repeats efficiently mediated RTA activation in the SV40 minimal promoter in a repeat number-dependent manner (Fig. 4B).
These data clearly supported the idea that both A and C contain RBP-J-binding sites, which can mediate RTA activation through RTA-RBP-J protein interaction. While site C contains a predicted RBP-J-binding site, site A (GTGAGAA) contains a G→A change (underlined) in the conventional RBP-J-binding site (GTGGGAA). Although RBP-J has been reported to be able to bind to the sequence 5′-GTGGGAA specifically (63), alternative RBP-J-binding sequences (5′-TGGGAAA and 5′-TGGGAAAGAA) have also been reported (18, 30). The GTGAGAA sequence of site A reported here thus represents a novel alternative RBP-J-binding site.
Site B contains a binding site(s) for an additional cellular factor(s).
In a gel shift assay using nuclear extracts prepared from 293T cells, the 32P-labeled oligonucleotide B formed two specific complexes (Fig. 5A, lane 2), which can be efficiently competed by unlabeled WT oligonucleotide B (lanes 3 and 4). Mutant oligonucleotide 1 (mut-1), carrying the same mutation as m15 described above, served as a much weaker competitor than the WT oligonucleotide (compare lanes 5 and 3 and lanes 7 and 4). Mutant oligonucleotide 2 (mut-2), carrying the same mutation as m16, failed to compete the two specific bands even at a 200-fold molar excess (lanes 7 and 8). Thus, the interactions of this factor with site B are consistent with the above genetic analysis, in which m16 showed a more severe defect than m15 (Fig. 2C). These data strongly suggest that the site B-interacting host factor plays a role in facilitating RTA-mediated transactivation in vivo. However, it does not allow determination of the mechanism by which it may do so.
FIG. 5.

Oligonucleotide B is bound by a cellular factor(s) but functions poorly in heterologous contexts. (A) Oligonucleotide B forms specific complexes with 293T cell nuclear extracts. Labeled oligonucleotide B (lane 1) was incubated with 293T cell nuclear extracts (lanes 2 to 8) in the absence of competitor oligonucleotide (lane 2) or in the presence of the following unlabeled competitor oligonucleotides: WT oligonucleotide B (20- and 200-fold) (lanes 3 and 4, respectively), mutant oligonucleotide 1 (20- and 200-fold) (lanes 5 and 6, respectively), and mutant oligonucleotide 2 (20- and 200-fold) (lanes 7 and 8, respectively). The specific shifted bands are shown by arrows. WT-B, WT oligonucleotide B; mut-1, mutant oligonucleotide 1; mut-2, mutant oligonucleotide 2. (B) Repeats of oligonucleotide B mediate RTA activation poorly in a heterogeneous promoter. Oligonucleotide B or a long oligonucleotide spanning site B (oligonucleotide BL) (both illustrated in Fig. 3A) was inserted in tandem repeats (with the number of repeats as shown) into the SV40 minimal promoter in the pGL3 promoter, which drives the expression of luciferase. Individual reporter constructs were cotransfected into 293T cells with either empty vector or RTA expression vector, and the fold induction by RTA was calculated; the error bars represent standard deviations of the results from at least two independent experiments.
To further examine this issue, we asked whether site B, which is clearly necessary for full RTA activation (Fig. 2C), can be sufficient for such activation. To do this, we introduced tandem repeats of WT oligonucleotide B into an SV40 minimal promoter to drive the expression of luciferase. Surprisingly, we failed to observe significant activation by RTA in 293T cells (Fig. 5B). Similar results were obtained with SLK and MEF cells as well (data not shown). We next used a longer oligonucleotide, BL, which spans a larger region around site B (Fig. 3A). This oligonucleotide did mediate a modest activation (three- to fourfold) by RTA when oligomerized, although it was barely active as a solitary element (Fig. 5B). Taken together, these data suggest that whatever role site B plays in RTA activation of the K14/vGPCR promoter depends on being in its native promoter context and is poorly reproduced outside this context (see below).
RBP-J plays an essential role in RTA activation of the K14/vGPCR promoters.
In the experiments described above, we identified three sites (A, B, and C) that are important for RTA activation of the K14/vGPCR promoter and studied them individually in terms of RTA binding and activation. We next asked how each site contributes to the overall RTA activation of the intact K14/vGPCR promoter. To do this, we used construct pGL3-222, which contains all three sites in their native context and mediates efficient RTA activation. Single or double mutations were introduced into the A, B, or C site in construct pGL3-222, and their effects on RTA activation were examined by luciferase reporter assay (Fig. 6A). This revealed that the A and C elements are critical, since mutation of either site caused a 70% loss of RTA activation and mutation of both almost completely abolished RTA activation. These data strongly suggested that RBP-J binding plays a central role in RTA-mediated activation of the K14/vGPCR promoter. To further validate this, we examined the ability of this promoter to be activated in cells derived from mice in which the RBP-J gene has been inactivated by homologous recombination. We observed a near-total loss of RTA activation of the full-length promoter pGL3-222 in RBP-J−/− OT11cells (7.5-fold) compared to the isogenic RBP-J+/+ OT13 cells (283-fold) (Fig. 6B). In contrast, RTA activation of pGL3-222(Am+Cm), which contained mutations in both RBP-J sites, was significantly lower in OT13 cells (13.4-fold), which was a similarly low level as in OT11 cells (6.8-fold) (Fig. 6B). Moreover, introduction of RBP-J protein into OT11 cells by cotransfection of an RBP-J expression vector partially restored RTA activation of pGL3-222 (46-fold) but not that of the mutant promoter (5.3-fold) (Fig. 6B). Correspondingly, the construct pGL3-184 (with a deletion of site A) was activated only 25-fold by RTA in WT cells, considerably lower than the construct pGL3-222 (280-fold), and the construct pGL3-98 (with the deletion of both sites A and C) had a nearly no responsiveness (2.4-fold) to RTA (Fig. 6B), further confirming that RTA activation of the K14/vGPCR promoter is mainly RBP-J dependent and through sites A and C.
FIG. 6.
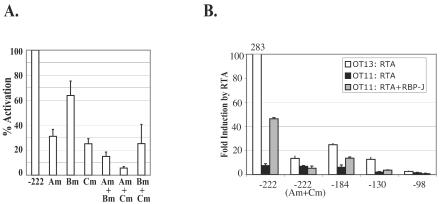
RBP-J plays a central role in RTA-mediated activation of the K14/vGPCR promoter. (A) Reporter construct pGL3-222 contains all three major RTA response elements, A, B, and C. Single mutation of site A, B, or C is indicated by Am, Bm, and Cm, respectively; corresponding double mutants are indicated by Am+Bm, Am+Cm, and Bm+Cm. The fold activation by RTA on each mutant was determined, normalized against that for the WT construct pGL3-222, and plotted as percent activation. The error bars represent standard deviations of the results from at least two independent experiments. (B) Comparison of RTA activation of the K14/vGPCR promoter constructs in OT13 (WT) and OT11 (RBP-J−/−) cells. OT13 or OT11 cells were transfected with 0.6 μg of individual K14/vGPCR reporter constructs [pGL3-222, pGL3-222(Am+Cm), pGL3-184, pGL3-130, or pGL3-98], 0 to 0.6 μg of RTA expression vector, and 0 to 0.6 μg of RBP-J expression vector (in OT11 cells only). pcDNA3.1-lacZ was included in each transfection as an internal control, and total DNA was normalized to 2 μg with vector pcDNA3.1. The fold induction by RTA was plotted, with the error bars representing standard deviations of the results from at least two independent experiments.
The role of site B in RTA activation of the K14/vGPCR promoter is more ambiguous. Single mutation of site B in the construct pGL3-222 had only a modest impact on activation of the native promoter, reducing it by only 35% (Fig. 6A). When site A was left intact and site C was removed by mutagenesis, mutation of B had little additional effect (25% for both Cm and Bm+Cm). By contrast, when site A was mutated and site C was left intact, mutation of B had a considerable impact on residual RTA activation (30 and 15% for Am and Am+Bm, respectively). A similar result was also obtained from the deletion mapping analysis (Fig. 2C): mutation of site B in the construct pGL3-198 (in which site A was deleted and site was C intact) significantly lowered the RTA activation, from 140- to 25-fold. Combined with the above observation that oligonucleotide B did not mediate RTA activation in a heterogeneous promoter (Fig. 5B), these data suggest an indirect role for site B. For example, it might function to enhance RTA interactions at site C, or it might augment the accessibility of site C to complex formation by RBP-J-RTA complexes.
DISCUSSION
We have previously identified RBP-J as an interaction partner of KSHV RTA and a critical component in mediating RTA activation of several KSHV target genes, including those for ORF57 and thymidine kinase (34). We have also shown that RBP-J plays an essential role in RTA-mediated lytic reactivation of KSHV, since such reactivation is completely defective in RBP-J−/− cells while being efficient in WT cells (35). Here we demonstrate that RBP-J also plays a role in the activation of genes (K14 and vGPCR) that play roles outside those known to be required for core lytic replication functions. Both of these are signaling molecules that play roles in modulation of inflammatory and angiogenic pathways (17), although it has not been excluded that they may also contribute more directly to lytic replication.
RBP-J is a downstream target of the Notch pathway, a conserved signal transduction pathway that is important in development and cell fate determination (43). The intracellular domain (ICD) of activated Notch is released from the membrane through proteolytic cleavage and is translocated to the nucleus, where it is directed to target promoters through interaction with RBP-J (47, 68). RBP-J is a repressor in the ground state; its interaction with Notch ICD relieves this repression and turns on target genes. Interestingly, KSHV is not the only virus that has parasitized this pathway. Several viral transcription factors, e.g., EBNA2 and EBNA3 of Epstein-Barr virus and the 13S isoform of adenovirus E1A, are known to bind and activate target genes via RBP-J interactions (1, 22, 25, 26, 29). In all cases, the viral proteins target the same (central repressive) domain of RBP-J that is targeted by Notch, although KSHV RTA is capable of interactions with an additional region of RBP-J in vitro (33).
The expression of vGPCR has been prominently linked to phenotypic changes that suggest important roles in KS pathogenesis. Not only does the protein trigger cell-autonomous proliferation, but it also can induce the expression of the proangiogenic molecule VEGF (3, 4). However, both of these activities are expected to be curtailed in the environment of lytic replication: cell proliferation is limited by virus-induced cytopathology, and VEGF expression is reduced by a virus-induced shutoff of host mRNA accumulation (B. Glaunsinger and D. Ganem, unpublished data). One way to resolve this conundrum is to imagine that vGPCR, which is not part of the standard latency program of KSHV, can be expressed outside the lytic program under certain conditions. Our finding of two functional RBP-J sites in the K14/vGPCR promoter suggests a potential role for Notch signaling in the induction of K14/vGPCR gene expression in the absence of RTA induction. That is, the activated Notch ICD molecule might also be able to utilize the same RBP-J-binding sites to activate vGPCR expression from latent viral genomes. Different Notch molecules (mammalian cells contain four Notch molecules, Notch-1 to -4) and their ligands (Jagged-1 and -2 and Delta-1, -3, and -4) are expressed in diverse cell types, including endothelial cells (55, 65) and B cells (6, 48), the two host cells for KSHV infection. Studies have suggested that Notch signals are involved in endothelial cell differentiation and B-cell development (5, 28, 48, 50). Therefore, it is possible that the Notch molecules in these cells can be activated by local signals and thus induce the expression of the K14/vGPCR transcript. Since the activated Notch-1 molecule has been shown not to induce the whole lytic cycle of KSHV (34), vGPCR would be free to function without the constraints of host shutoff and cell injury imposed by lytic growth. Further studies are now under way to explore this hypothesis.
Acknowledgments
We thank T. Honjo for providing OT13 and OT11 cells, members of the Ganem lab for advice and support, and Hinh Ly for reviewing the manuscript.
D.G. is an investigator of the Howard Hughes Medical Institute.
REFERENCES
- 1.Ansieau, S., L. J. Strobl, and A. Leutz. 2001. Activation of the Notch-regulated transcription factor CBF1/RBP-Jkappa through the 13SE1A oncoprotein. Genes Dev. 15:380-385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Arvanitakis, L., E. Geras-Raaka, A. Varma, M. C. Gershengorn, and E. Cesarman. 1997. Human herpesvirus KSHV encodes a constitutively active G-protein-coupled receptor linked to cell proliferation. Nature 385:347-350. [DOI] [PubMed] [Google Scholar]
- 3.Bais, C., B. Santomasso, O. Coso, L. Arvanitakis, E. G. Raaka, J. S. Gutkind, A. S. Asch, E. Cesarman, M. C. Gershengorn, E. A. Mesri, and M. C. Gerhengorn. 1998. G-protein-coupled receptor of Kaposi's sarcoma-associated herpesvirus is a viral oncogene and angiogenesis activator. Nature 391:86-89. [DOI] [PubMed] [Google Scholar]
- 4.Bais, C., A. Van Geelen, P. Eroles, A. Mutlu, C. Chiozzini, S. Dias, R. L. Silverstein, S. Rafii, and E. A. Mesri. 2003. Kaposi's sarcoma associated herpesvirus G protein-coupled receptor immortalizes human endothelial cells by activation of the VEGF receptor-2/ KDR. Cancer Cell 3:131-143. [DOI] [PubMed] [Google Scholar]
- 5.Bertrand, F. E., C. E. Eckfeldt, J. R. Fink, A. S. Lysholm, J. A. Pribyl, N. Shah, and T. W. LeBien. 2000. Microenvironmental influences on human B-cell development. Immunol. Rev. 175:175-186. [PubMed] [Google Scholar]
- 6.Bertrand, F. E., C. E. Eckfeldt, A. S. Lysholm, and T. W. LeBien. 2000. Notch-1 and Notch-2 exhibit unique patterns of expression in human B-lineage cells. Leukemia 14:2095-2102. [DOI] [PubMed] [Google Scholar]
- 7.Boshoff, C., Y. Endo, P. D. Collins, Y. Takeuchi, J. D. Reeves, V. L. Schweickart, M. A. Siani, T. Sasaki, T. J. Williams, P. W. Gray, P. S. Moore, Y. Chang, and R. A. Weiss. 1997. Angiogenic and HIV-inhibitory functions of KSHV-encoded chemokines. Science 278:290-294. [DOI] [PubMed] [Google Scholar]
- 8.Byun, H., Y. Gwack, S. Hwang, and J. Choe. 2002. Kaposi's sarcoma-associated herpesvirus open reading frame (ORF) 50 transactivates K8 and ORF57 promoters via heterogeneous response elements. Mol. Cell 14:185-191. [PubMed] [Google Scholar]
- 9.Cannon, M., N. J. Philpott, and E. Cesarman. 2003. The Kaposi's sarcoma-associated herpesvirus G protein-coupled receptor has broad signaling effects in primary effusion lymphoma cells. J. Virol. 77:57-67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cesarman, E., Y. Chang, P. S. Moore, J. W. Said, and D. M. Knowles. 1995. Kaposi's sarcoma-associated herpesvirus-like DNA sequences in AIDS-related body-cavity-based lymphomas. N. Engl. J. Med. 332:1186-1191. [DOI] [PubMed] [Google Scholar]
- 11.Cesarman, E., R. G. Nador, F. Bai, R. A. Bohenzky, J. J. Russo, P. S. Moore, Y. Chang, and D. M. Knowles. 1996. Kaposi's sarcoma-associated herpesvirus contains G protein-coupled receptor and cyclin D homologs which are expressed in Kaposi's sarcoma and malignant lymphoma. J. Virol. 70:8218-8223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chang, P. J., D. Shedd, L. Gradoville, M. S. Cho, L. W. Chen, J. Chang, and G. Miller. 2002. Open reading frame 50 protein of Kaposi's sarcoma-associated herpesvirus directly activates the viral PAN and K12 genes by binding to related response elements. J. Virol. 76:3168-3178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chang, Y., E. Cesarman, M. S. Pessin, F. Lee, J. Culpepper, D. M. Knowles, and P. S. Moore. 1994. Identification of herpesvirus-like DNA sequences in AIDS-associated Kaposi's sarcoma. Science 266:1865-1869. [DOI] [PubMed] [Google Scholar]
- 14.Chen, J., K. Ueda, S. Sakakibara, T. Okuno, and K. Yamanishi. 2000. Transcriptional regulation of the Kaposi's sarcoma-associated herpesvirus viral interferon regulatory factor gene. J. Virol. 74:8623-8634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cheng, E. H., J. Nicholas, D. S. Bellows, G. S. Hayward, H. G. Guo, M. S. Reitz, and J. M. Hardwick. 1997. A Bcl-2 homolog encoded by Kaposi sarcoma-associated virus, human herpesvirus 8, inhibits apoptosis but does not heterodimerize with Bax or Bak. Proc. Natl. Acad. Sci. USA 94:690-694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chiou, C. J., L. J. Poole, P. S. Kim, D. M. Ciufo, J. S. Cannon, C. M. ap Rhys, D. J. Alcendor, J. C. Zong, R. F. Ambinder, and G. S. Hayward. 2002. Patterns of gene expression and a transactivation function exhibited by the vGCR (ORF74) chemokine receptor protein of Kaposi's sarcoma-associated herpesvirus. J. Virol. 76:3421-3439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chung, Y. H., R. E. Means, J. K. Choi, B. S. Lee, and J. U. Jung. 2002. Kaposi's sarcoma-associated herpesvirus OX2 glycoprotein activates myeloid-lineage cells to induce inflammatory cytokine production. J. Virol. 76:4688-4698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dou, S., X. Zeng, P. Cortes, H. Erdjument-Bromage, P. Tempst, T. Honjo, and L. D. Vales. 1994. The recombination signal sequence-binding protein RBP-2N functions as a transcriptional repressor. Mol. Cell. Biol. 14:3310-3319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gao, S. J., L. Kingsley, D. R. Hoover, T. J. Spira, C. R. Rinaldo, A. Saah, J. Phair, R. Detels, P. Parry, Y. Chang, and P. S. Moore. 1996. Seroconversion to antibodies against Kaposi's sarcoma-associated herpesvirus-related latent nuclear antigens before the development of Kaposi's sarcoma. N. Engl. J. Med. 335:233-241. [DOI] [PubMed] [Google Scholar]
- 20.Gershengorn, M. C., E. Geras-Raaka, A. Varma, and I. Clark-Lewis. 1998. Chemokines activate Kaposi's sarcoma-associated herpesvirus G protein-coupled receptor in mammalian cells in culture. J. Clin. Investig. 102:1469-1472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gradoville, L., J. Gerlach, E. Grogan, D. Shedd, S. Nikiforow, C. Metroka, and G. Miller. 2000. Kaposi's sarcoma-associated herpesvirus open reading frame 50/Rta protein activates the entire viral lytic cycle in the HH-B2 primary effusion lymphoma cell line. J. Virol. 74:6207-6212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Grossman, S. R., E. Johannsen, X. Tong, R. Yalamanchili, and E. Kieff. 1994. The Epstein-Barr virus nuclear antigen 2 transactivator is directed to response elements by the J kappa recombination signal binding protein. Proc. Natl. Acad. Sci. USA 91:7568-7572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Guo, H. G., M. Sadowska, W. Reid, E. Tschachler, G. Hayward, and M. Reitz. 2003. Kaposi's sarcoma-like tumors in a human herpesvirus 8 ORF74 transgenic mouse. J. Virol. 77:2631-2639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gwack, Y., S. Hwang, C. Lim, Y. S. Won, C. H. Lee, and J. Choe. 2002. Kaposi's sarcoma-associated herpesvirus open reading frame 50 stimulates the transcriptional activity of STAT3. J. Biol. Chem. 277:6438-6442. [DOI] [PubMed] [Google Scholar]
- 25.Henkel, T., P. D. Ling, S. D. Hayward, and M. G. Peterson. 1994. Mediation of Epstein-Barr virus EBNA2 transactivation by recombination signal-binding protein J kappa. Science 265:92-95. [DOI] [PubMed] [Google Scholar]
- 26.Hsieh, J. J., and S. D. Hayward. 1995. Masking of the CBF1/RBPJ kappa transcriptional repression domain by Epstein-Barr virus EBNA2. Science 268:560-563. [DOI] [PubMed] [Google Scholar]
- 27.Huang, Y. Q., J. J. Li, M. H. Kaplan, B. Poiesz, E. Katabira, W. C. Zhang, D. Feiner, and A. E. Friedman-Kien. 1995. Human herpesvirus-like nucleic acid in various forms of Kaposi's sarcoma. Lancet 345:759-761. [DOI] [PubMed] [Google Scholar]
- 28.Izon, D. J., J. A. Punt, and W. S. Pear. 2002. Deciphering the role of Notch signaling in lymphopoiesis. Curr. Opin. Immunol. 14:192-199. [DOI] [PubMed] [Google Scholar]
- 29.Johannsen, E., C. L. Miller, S. R. Grossman, and E. Kieff. 1996. EBNA-2 and EBNA-3C extensively and mutually exclusively associate with RBPJkappa inEpstein-Barr virus-transformed B lymphocytes. J. Virol. 70:4179-4183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kannabiran, C., X. Zeng, and L. D. Vales. 1997. The mammalian transcriptional repressor RBP (CBF1) regulates interleukin-6 gene expression. Mol. Cell. Biol. 17:1-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kedes, D. H., E. Operskalski, M. Busch, R. Kohn, J. Flood, and D. Ganem. 1996. The seroepidemiology of human herpesvirus 8 (Kaposi's sarcoma-associated herpesvirus): distribution of infection in KS risk groups and evidence for sexual transmission. Nat. Med. 2:918-924. [DOI] [PubMed] [Google Scholar]
- 32.Kirshner, J. R., K. Staskus, A. Haase, M. Lagunoff, and D. Ganem. 1999. Expression of the open reading frame 74 (G-protein-coupled receptor) gene of Kaposi's sarcoma (KS)-associated herpesvirus: implications for KS pathogenesis. J. Virol. 73:6006-6014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lefrere, J. J., M. C. Meyohas, M. Mariotti, J. L. Meynard, M. Thauvin, and J. Frottier. 1996. Detection of human herpesvirus 8 DNA sequences before the appearance of Kaposi's sarcoma in human immunodeficiency virus (HIV)-positive subjects with a known date of HIV seroconversion. J. Infect. Dis. 174:283-287. [DOI] [PubMed] [Google Scholar]
- 34.Liang, Y., J. Chang, S. J. Lynch, D. M. Lukac, and D. Ganem. 2002. The lytic switch protein of KSHV activates gene expression via functional interaction with RBP-Jkappa (CSL), the target of the Notch signaling pathway. Genes Dev. 16:1977-1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Liang, Y., and D. Ganem. 2003. Lytic but not latent infection by Kaposi's sarcoma-associated herpesvirus requires host CSL protein, the mediator of Notch signaling. Proc. Natl. Acad. Sci. USA 100:8490-8495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lukac, D. M., L. Garibyan, J. R. Kirshner, D. Palmeri, and D. Ganem. 2001. DNA binding by Kaposi's sarcoma-associated herpesvirus lytic switch protein is necessary for transcriptional activation of two viral delayed early promoters. J. Virol. 75:6786-6799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lukac, D. M., J. R. Kirshner, and D. Ganem. 1999. Transcriptional activation by the product of open reading frame 50 of Kaposi's sarcoma-associated herpesvirus is required for lytic viral reactivation in B cells. J. Virol. 73:9348-9361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lukac, D. M., R. Renne, J. R. Kirshner, and D. Ganem. 1998. Reactivation of Kaposi's sarcoma-associated herpesvirus infection from latency by expression of the ORF 50 transactivator, a homolog of the EBV R protein. Virology 252:304-312. [DOI] [PubMed] [Google Scholar]
- 39.Montaner, S., A. Sodhi, A. Molinolo, T. H. Bugge, E. T. Sawai, Y. He, Y. Li, P. E. Ray, and J. S. Gutkind. 2003. Endothelial infection with KSHV genes in vivo reveals that vGPCR initiates Kaposi's sarcomagenesis and can promote the tumorigenic potential of viral latent genes. Cancer Cell 3:23-36. [DOI] [PubMed] [Google Scholar]
- 40.Moore, P. S., C. Boshoff, R. A. Weiss, and Y. Chang. 1996. Molecular mimicry of human cytokine and cytokine response pathway genes by KSHV. Science 274:1739-1744. [DOI] [PubMed] [Google Scholar]
- 41.Moore, P. S., and Y. Chang. 1995. Detection of herpesvirus-like DNA sequences in Kaposi's sarcoma in patients with and without HIV infection. N. Engl. J. Med. 332:1181-1185. [DOI] [PubMed] [Google Scholar]
- 42.Moore, P. S., L. A. Kingsley, S. D. Holmberg, T. Spira, P. Gupta, D. R. Hoover, J. P. Parry, L. J. Conley, H. W. Jaffe, and Y. Chang. 1996. Kaposi's sarcoma-associated herpesvirus infection prior to onset of Kaposi's sarcoma. AIDS 10:175-180. [DOI] [PubMed] [Google Scholar]
- 43.Mumm, J. S., and R. Kopan. 2000. Notch signaling: from the outside in. Dev. Biol. 228:151-165. [DOI] [PubMed] [Google Scholar]
- 44.Nador, R. G., L. L. Milligan, O. Flore, X. Wang, L. Arvanitakis, D. M. Knowles, and E. Cesarman. 2001. Expression of Kaposi's sarcoma-associated herpesvirus G protein-coupled receptor monocistronic and bicistronic transcripts in primary effusion lymphomas. Virology 287:62-70. [DOI] [PubMed] [Google Scholar]
- 45.Neipel, F., J. C. Albrecht, A. Ensser, Y. Q. Huang, J. J. Li, A. E. Friedman-Kien, and B. Fleckenstein. 1997. Human herpesvirus 8 encodes a homolog of interleukin-6. J. Virol. 71:839-842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nicholas, J., V. R. Ruvolo, W. H. Burns, G. Sandford, X. Wan, D. Ciufo, S. B. Hendrickson, H. G. Guo, G. S. Hayward, and M. S. Reitz. 1997. Kaposi's sarcoma-associated human herpesvirus-8 encodes homologues of macrophage inflammatory protein-1 and interleukin-6. Nat. Med. 3:287-292. [DOI] [PubMed] [Google Scholar]
- 47.Panin, V. M., and K. D. Irvine. 1998. Modulators of Notch signaling. Semin. Cell. Dev. Biol. 9:609-617. [DOI] [PubMed] [Google Scholar]
- 48.Parreira, L., H. Neves, and S. Simoes. 2003. Notch and lymphopoiesis: a view from the microenvironment. Semin. Immunol. 15:81-89. [DOI] [PubMed] [Google Scholar]
- 49.Pati, S., M. Cavrois, H. G. Guo, J. S. Foulke, Jr., J. Kim, R. A. Feldman, and M. Reitz. 2001. Activation of NF-κB by the human herpesvirus 8 chemokine receptor ORF74: evidence for a paracrine model of Kaposi's sarcoma pathogenesis. J. Virol. 75:8660-8673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rossant, J., and M. Hirashima. 2003. Vascular development and patterning: making the right choices. Curr. Opin. Genet. Dev. 13:408-412. [DOI] [PubMed] [Google Scholar]
- 51.Russo, J. J., R. A. Bohenzky, M. C. Chien, J. Chen, M. Yan, D. Maddalena, J. P. Parry, D. Peruzzi, I. S. Edelman, Y. Chang, and P. S. Moore. 1996. Nucleotide sequence of the Kaposi sarcoma-associated herpesvirus (HHV8). Proc. Natl. Acad. Sci. USA 93:14862-14867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sarid, R., T. Sato, R. A. Bohenzky, J. J. Russo, and Y. Chang. 1997. Kaposi's sarcoma-associated herpesvirus encodes a functional bcl-2 homologue. Nat. Med. 3:293-298. [DOI] [PubMed] [Google Scholar]
- 53.Schwarz, M., and P. M. Murphy. 2001. Kaposi's sarcoma-associated herpesvirus G protein-coupled receptor constitutively activates NF-kappa B and induces proinflammatory cytokine and chemokine production via a C-terminal signaling determinant. J. Immunol. 167:505-513. [DOI] [PubMed] [Google Scholar]
- 54.Seaman, W. T., D. Ye, R. X. Wang, E. E. Hale, M. Weisse, and E. B. Quinlivan. 1999. Gene expression from the ORF50/K8 region of Kaposi's sarcoma-associated herpesvirus. Virology 263:436-449. [DOI] [PubMed] [Google Scholar]
- 55.Shutter, J. R., S. Scully, W. Fan, W. G. Richards, J. Kitajewski, G. A. Deblandre, C. R. Kintner, and K. L. Stark. 2000. Dll4, a novel Notch ligand expressed in arterial endothelium. Genes Dev. 14:1313-1318. [PMC free article] [PubMed] [Google Scholar]
- 56.Simpson, G. R., T. F. Schulz, D. Whitby, P. M. Cook, C. Boshoff, L. Rainbow, M. R. Howard, S. J. Gao, R. A. Bohenzky, P. Simmonds, C. Lee, A. de Ruiter, A. Hatzakis, R. S. Tedder, I. V. Weller, R. A. Weiss, and P. S. Moore. 1996. Prevalence of Kaposi's sarcoma associated herpesvirus infection measured by antibodies to recombinant capsid protein and latent immunofluorescence antigen. Lancet 348:1133-1138. [DOI] [PubMed] [Google Scholar]
- 57.Sodhi, A., S. Montaner, V. Patel, M. Zohar, C. Bais, E. A. Mesri, and J. S. Gutkind. 2000. The Kaposi's sarcoma-associated herpes virus G protein-coupled receptor up-regulates vascular endothelial growth factor expression and secretion through mitogen-activated protein kinase and p38 pathways acting on hypoxia-inducible factor 1alpha. Cancer Res. 60:4873-4880. [PubMed] [Google Scholar]
- 58.Song, M. J., H. J. Brown, T. T. Wu, and R. Sun. 2001. Transcription activation of polyadenylated nuclear RNA by RTA in human herpesvirus 8/Kaposi's sarcoma-associated herpesvirus. J. Virol. 75:3129-3140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Song, M. J., H. Deng, and R. Sun. 2003. Comparative study of regulation of RTA-responsive genes in Kaposi's sarcoma-associated herpesvirus/human herpesvirus 8. J. Virol. 77:9451-9462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Song, M. J., X. Li, H. J. Brown, and R. Sun. 2002. Characterization of interactions between RTA and the promoter of polyadenylated nuclear RNA in Kaposi's sarcoma-associated herpesvirus/human herpesvirus 8. J. Virol. 76:5000-5013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Soulier, J., L. Grollet, E. Oksenhendler, P. Cacoub, D. Cazals-Hatem, P. Babinet, M. F. d'Agay, J. P. Clauvel, M. Raphael, L. Degos, et al. 1995. Kaposi's sarcoma-associated herpesvirus-like DNA sequences in multicentric Castleman's disease. Blood 86:1276-1280. [PubMed] [Google Scholar]
- 62.Sun, R., S. F. Lin, L. Gradoville, Y. Yuan, F. Zhu, and G. Miller. 1998. A viral gene that activates lytic cycle expression of Kaposi's sarcoma-associated herpesvirus. Proc. Natl. Acad. Sci. USA 95:10866-10871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Tun, T., Y. Hamaguchi, N. Matsunami, T. Furukawa, T. Honjo, and M. Kawaichi. 1994. Recognition sequence of a highly conserved DNA binding protein RBP-J kappa. Nucleic Acids Res. 22:965-971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ueda, K., K. Ishikawa, K. Nishimura, S. Sakakibara, E. Do, and K. Yamanishi. 2002. Kaposi's sarcoma-associated herpesvirus (human herpesvirus 8) replication and transcription factor activates the K9 (vIRF) gene through two distinct cis elements by a non-DNA-binding mechanism. J. Virol. 76:12044-12054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Uyttendaele, H., G. Marazzi, G. Wu, Q. Yan, D. Sassoon, and J. Kitajewski. 1996. Notch4/int-3, a mammary proto-oncogene, is an endothelial cell-specific mammalian Notch gene. Development 122:2251-2259. [DOI] [PubMed] [Google Scholar]
- 66.Wang, S., S. Liu, M. H. Wu, Y. Geng, and C. Wood. 2001. Identification of a cellular protein that interacts and synergizes with the RTA (ORF50) protein of Kaposi's sarcoma-associated herpesvirus in transcriptional activation. J. Virol. 75:11961-11973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wang, S. E., F. Y. Wu, M. Fujimuro, J. Zong, S. D. Hayward, and G. S. Hayward. 2003. Role of CCAAT/enhancer-binding protein alpha (C/EBPα) in activation of the Kaposi's sarcoma-associated herpesvirus (KSHV) lytic-cycle replication-associated protein (RAP) promoter in cooperation with the KSHV replication and transcription activator (RTA) and RAP. J. Virol. 77:600-623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Weinmaster, G. 2000. Notch signal transduction: a real rip and more. Curr. Opin. Genet. Dev. 10:363-369. [DOI] [PubMed] [Google Scholar]
- 69.Whitby, D., M. R. Howard, M. Tenant-Flowers, N. S. Brink, A. Copas, C. Boshoff, T. Hatzioannou, F. E. Suggett, D. M. Aldam, A. S. Denton, et al. 1995. Detection of Kaposi sarcoma associated herpesvirus in peripheral blood of HIV-infected individuals and progression to Kaposi's sarcoma. Lancet 346:799-802. [DOI] [PubMed] [Google Scholar]
- 70.Yang, T. Y., S. C. Chen, M. W. Leach, D. Manfra, B. Homey, M. Wiekowski, L. Sullivan, C. H. Jenh, S. K. Narula, S. W. Chensue, and S. A. Lira. 2000. Transgenic expression of the chemokine receptor encoded by human herpesvirus 8 induces an angioproliferative disease resembling Kaposi's sarcoma. J. Exp Med. 191:445-454. [DOI] [PMC free article] [PubMed] [Google Scholar]