

Abstract
The growth properties and antigenic relatedness of the CAN98-75 (CAN75) and the CAN97-83 (CAN83) human metapneumovirus (HMPV) strains, which represent the two distinct HMPV genetic lineages and exhibit 5 and 63% amino acid divergence in the fusion (F) and attachment (G) proteins, respectively, were investigated in vitro and in rodents and nonhuman primates. Both strains replicated to high titers (≥6.0 log10) in the upper respiratory tract of hamsters and to moderate titers (≥3.6 log10) in the lower respiratory tract. The two lineages exhibited 48% antigenic relatedness based on reciprocal cross-neutralization assay with postinfection hamster sera, and infection with each strain provided a high level of resistance to reinfection with the homologous or heterologous strain. Hamsters immunized with a recombinant human parainfluenza virus type 1 expressing the fusion F protein of the CAN83 strain developed a serum antibody response that efficiently neutralized virus from both lineages and were protected from challenge with either HMPV strain. This result indicates that the HMPV F protein is a major antigenic determinant that mediates extensive cross-lineage neutralization and protection. Both HMPV strains replicated to low titers in the upper and lower respiratory tracts of rhesus macaques but induced high levels of HMPV-neutralizing antibodies in serum effective against both lineages. The level of HMPV replication in chimpanzees was moderately higher, and infected animals developed mild colds. HMPV replicated the most efficiently in the respiratory tracts of African green monkeys, and the infected animals developed a high level of HMPV serum-neutralizing antibodies (1:500 to 1:1,000) effective against both lineages. Reciprocal cross-neutralization assays in which postinfection sera from all three primate species were used indicated that CAN75 and CAN83 are 64 to 99% related antigenically. HMPV-infected chimpanzees and African green monkeys were highly protected from challenge with the heterologous HMPV strain. Taken together, the results from hamsters and nonhuman primates support the conclusion that the two HMPV genetic lineages are highly related antigenically and are not distinct antigenic subtypes or subgroups as defined by reciprocal cross-neutralization in vitro.
Human metapneumovirus (HMPV), which was first identified several years ago in The Netherlands (39), is a member of the Paramyxoviridae family and has been tentatively assigned to the Metapneumovirus genus of the Pneumovirus subfamily. This subfamily also includes the Pneumovirus genus, containing the respiratory syncytial virus (RSV). Since its initial discovery, HMPV has been found to infect humans worldwide (7, 29, 31). Extensive sequence analysis of isolates from around the world indicates that there are two major genetic lineages of HMPV (3). It has been suggested that the different HMPV lineages represent different HMPV serotypes (29, 40). However, the biology and epidemiology of the newly identified HMPVs are not well characterized. For example, the overall impact of HMPV disease on hospitalization for respiratory tract disease is not well established, and it is not known if infection with one HMPV genetic lineage can induce protective immunity against reinfection with homologous or heterologous HMPV strains. Moreover, the lack of established animal models of HMPV replication and the reportedly poor growth characteristics of this virus have impeded its characterization.
Like all members of the Paramyxoviridae family, HMPV is an enveloped single-stranded negative-sense RNA virus. The genome is approximately 13 kb in length, and the HMPV 3′-to-5′ gene order is N-P-M-F-M2-1/M2-2-SH-G-L, which is the same as that of the avian members of the Metapneumovirus genus (5, 38). The CAN98-75 (CAN75) and CAN97-83 (CAN83) strains of HMPV were isolated in Canada from clinical samples and represent members of each of the two major genetic lineages (31). The amino acid identity of the predicted encoded proteins between these two strains is 96% (N protein), 85% (P protein), 97% (M protein), 95% (F protein), 96% (M2-1 protein), 89% (M2-2 protein), 59% (SH protein), 37% (G protein), and 94% (L protein) (5). Thus, with the notable exception of the SH and G proteins, the polypeptides encoded by each of these genetic lineages are highly related at the amino acid level.
The major question addressed in this article is that of the antigenic relatedness of the two strains of HMPV. The answer to this question will have implications for HMPV epidemiology and for the formulation of a strategy to develop vaccines effective against this important respiratory pathogen. We first identified suitable cell culture growth conditions to generate HMPV suspensions of sufficient titer for administration to experimental animals. It was also necessary to identify suitable small-animal and nonhuman primate models for HMPV replication. The present study demonstrates that the two HMPV lineages exhibited a high level of antigenic relatedness in rodents and nonhuman primates including chimpanzees and a high level of cross-protective efficacy, indicating that these two strains do not represent distinct antigenic subtypes. This study also establishes hamsters and African green monkeys as suitable animal models for HMPV replication and immunogenicity.
MATERIALS AND METHODS
Cell lines and viruses.
Rhesus monkey kidney cells (LLC-MK2; ATCC CCL 7.1) cells were maintained in OptiMEM (Invitrogen, Grand Island, N.Y.) supplemented with 5% fetal bovine serum and gentamicin sulfate (50 μg/ml). HMPV strains CAN98-75 and CAN97-83, kindly provided by Guy Boivin (31), are clinical isolates that were propagated in LLC-MK2 cells. The recombinant human parainfluenza virus type 1 (PIV1) (rHPIV1) strain Washington/64, HPIV3, and HPIV2 were grown in LLC-MK2 cells as described previously (28, 34, 36). Virus pools were generated, and virus titer was determined at 32°C in LLC-MK2 cells grown in OptiMEM medium and supplemented with porcine-derived trypsin (5 μg/ml; BioWhittaker, Walkersville, Md.).
Quantification of HMPV and HPIV preparations.
Viral titer was quantified by serial 10-fold dilution of virus applied to LLC-MK2 monolayer cultures on 96-well plates. Before infection, LLC-MK2 monolayers were washed twice with OptiMEM to remove residual serum proteins that can inhibit trypsin activity. Infected cultures were incubated for 8 days at 32°C in OptiMEM medium with 5 μg of added trypsin/ml. HMPV-infected monolayers were detected by incubation with hamster polyclonal HMPV antiserum (see below) used at a 1:500 dilution followed by a second antibody (1:500 dilution) consisting of peroxidase-conjugated rabbit anti-Syrian hamster immunoglobulin G (Jackson Immunochemicals, West Grove, Pa.). Bound antibody-antigen complexes were detected by immunoperoxidase staining achieved with ECL chromogenic substrate (KPL, Gaithersburg, Md.). Cultures determined to be infected with HPIV were detected by hemadsorption with guinea pig erythrocytes, as described previously (28). The virus titer is reported as log10 50% tissue culture infectious dose per milliliter (log10 TCID50/ml).
Generation of rHPIV1 expressing the HMPV CAN83 fusion F protein.
The previously described full-length antigenomic HPIV1 cDNA (28) (GenBank accession number AF457102) was modified at nucleotides (nt) 113 to 118 (TTTTAT) to contain a unique cloning site (MluI; ACGCGT) immediately upstream of the N protein open reading frame (ORF). The CAN83 strain F protein ORF (GenBank accession number AY297749) was amplified from purified viral RNA and was modified to contain the flanking HPIV1 cis-acting transcription control elements indicated in Fig. 2. Thus, the HMPV F protein is expressed as a supernumerary ORF in the pre-N position of the rHPIV1. This rHPIV1 expressing the fusion F protein of the CAN83 strain (rHPIV1-F83) was recovered from transfected HEp-2 cells as described previously (28). rHPIV1-F83 was biologically cloned and amplified in LLC-MK2 cells as described previously (28).
FIG. 2.
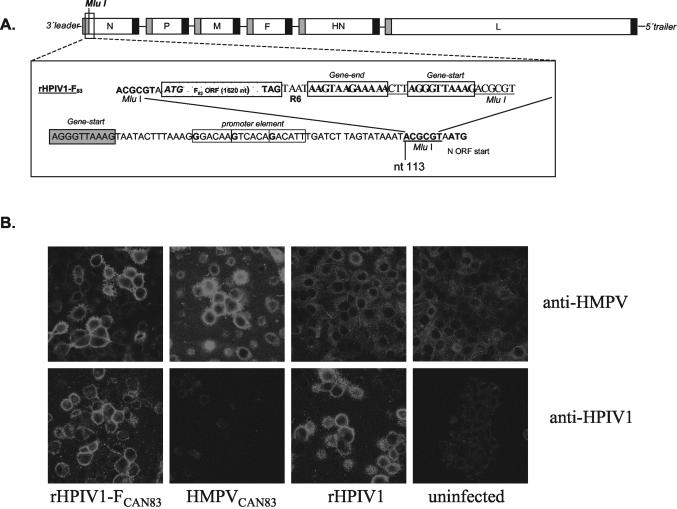
Construction of an rHPIV1 vector expressing CAN83 F. (A) Diagram of the construction of rHPIV1-F83. The HPIV1 genome was modified by the creation of an MluI restriction site (HPIV1 nt 113 to 118) 1 nt prior to the translational start codon of the N ORF (HPIV1 nt 119 to 121). The F ORF of HMPV strain CAN83 (1,620 nt in length and encoding a 539-amino-acid polypeptide; GenBank accession number AY297749) was engineered by PCR to be followed by the tetranucleotide TAAT (R6) and an HPIV1 gene junction consisting of a gene end signal, a CTT intergenic region, and a gene start signal. The length of the entire cassette was 1,656 nt and was designed, upon insertion into the MluI site, to conform to the rule of six and to maintain the HPIV1 gene start signal sequence phasing (23). Gene start and gene end signals are indicated. A portion of the predicted viral promoter that lies within the N gene is shown. (B) Indirect immunofluorescence of LLC-MK2 cells that were infected with rHPIV1-F83, HMPV CAN83, or rHPIV1 or that were mock infected (uninfected), as indicated, incubated for 72 h, and analyzed by indirect immunofluorescence by using anti-HMPV (αHMPV) polyclonal hamster serum or a mixture of two mouse monoclonal antibodies to the HPIV1 HN protein (αHPIV1), as indicated, as the first antibody.
Generation of hamster polyclonal antibodies to HMPV.
Eight-week-old Golden Syrian hamsters (Mesocricetus auratus; Charles River Laboratories, Stoneridge, N.Y.) were infected intranasally (IN) with 104 TCID50 of a mixture of HMPV strains CAN75 and CAN83. Animals were boosted 4 weeks later by the IN and intraperitoneal administration of 106 TCID50 of a mixture of HMPV strains CAN75 and CAN83. Serum that was collected at 4 weeks and again at 5 months after the first immunization was used routinely for immunostaining and immunofluorescence. In order to determine the extent of antigenic relatedness between the HMPV strains, immunologically naïve hamsters were subjected to a single IN inoculation of 104 TCID50 of either HMPV strain, and sera were collected 44 days later and analyzed in neutralization assays against each strain.
Indirect immunofluorescence of HMPV- and HPIV1-infected cells.
The expression of HMPV F protein by rHPIV1-F83 was detected by indirect immunofluorescence. LLC-MK2 cells grown on glass slides were infected with rHPIV1-F83, HPIV1, or HMPV, and 72 h postinfection, the cells were fixed with 3% formaldehyde and permeabilized with 0.1% Triton X-100, as described previously (33). To detect HMPV antigen expression, pooled hamster polyclonal anti-HMPV serum antibodies obtained from infected hamsters were applied to infected cells at a dilution of 1:500. HPIV1 infection was detected by indirect immunofluorescence by using a mixture of two mouse monoclonal antibodies to the HPIV1 HN protein (8.2.2A and 4.5; kindly provided by Yasuhiko Ito, Mie University School of Medicine, Tsu Shi, Japan). Fluorescein isothiocyanate-conjugated goat anti-hamster or goat anti-mouse immunoglobulin G antibody (Jackson Immunochemicals) was used for indirect immunofluorescence at a dilution of 1:500. Immunofluorescence was detected and images were collected on a TCS-SP2 laser scanning microscope (Leica Microsystems, Mannheim, Germany). Images were processed with TCS-NT/SP software (version 1.6.587; Leica), Imaris 3.1.1 (Bitplane AG, Zurich, Switzerland), and Adobe (San Jose, Calif.) Photoshop version 7.0.
Growth kinetics of HMPV in vitro.
The kinetics of replication of the CAN75 and CAN83 HMPV lineages was determined by inoculation of HMPV onto LLC-MK2 monolayers on 6-well plates (Costar; Corning, N.Y.) at a multiplicity of infection of 0.01, and the cultures were incubated at 32°C. A total of 0.5 ml of medium from each well was harvested and replaced with 0.5 ml of fresh OptiMEM medium supplemented with trypsin (5 μg/ml) at 0 h and at 24-h intervals for up to 12 days postinfection. Also, HMPV was grown under similar conditions but without any trypsin added to the medium. HPIV1, HPIV2, and HPIV3 control viruses were grown under identical conditions. HPIV1 was grown with trypsin, and HPIV2 and HPIV3 were grown in medium without added trypsin. The amount of virus present in the daily harvest was quantified by titration on LLC-MK2 monolayers in 96-well plates (Costar) as described above.
HMPV serum neutralization assays.
The susceptibility of HMPV strain CAN75 or CAN83 to neutralization by serum antibodies was determined by using an endpoint dilution neutralization assay. A total of 75 μl of OptiMEM containing approximately 200 TCID50 of CAN75 or CAN83 was mixed with an equal volume of OptiMEM containing serial twofold dilutions of hamster or primate serum that had been heat inactivated at 56°C for 30 min. The virus-antibody mixture was incubated at 37°C for 1 h. A portion (50 μl) of the virus-antibody mixture was then transferred to each of 2 wells of a 96-well plate containing LLC-MK2 cells and was incubated for approximately 1 h at 32°C. The virus-antibody mixture was removed, and the monolayers were washed twice with OptiMEM. OptiMEM (180 μl) supplemented with trypsin was added to the cultures. Cultures were incubated at 32°C, supplemented with the additional trypsin (250 ng in 50 μl) on day 6, and stained for the presence of HMPV antigen on day 8 by using polyclonal hamster anti-HMPV serum as described above. The neutralization titer is the highest dilution of antibody at which half of the cultures were negative for infection.
Determination of replication and protective efficacy of HMPV in hamsters.
Two groups of 4-week-old Golden Syrian hamsters were inoculated IN with 0.1 ml of L15 medium containing 106 TCID50 of CAN75 or CAN83. On day 3, 4, 5, 6, or 7 postinfection, the lungs and nasal turbinates were harvested from six animals from each group, and the virus titer of individual specimens was quantified by serial dilution of tissue homogenates on LLC-MK2 monolayers, as described above. The mean virus titer was calculated for each group of hamsters and is expressed as log10 TCID50 per gram of tissue (log10 TCID50/g). To determine the level of protective efficacy afforded by infection with either HMPV strain, hamsters in groups of six were infected with 105.5 TCID50 of CAN75 or CAN83 in a 0.1-ml inoculum or with L15 medium diluent. On day 42 postinfection, hamsters were challenged by IN administration of 105.5 TCID50 of CAN75 or CAN83 in a 0.1-ml inoculum. Nasal turbinates and lungs were harvested 4 days later, and the titer of virus in each tissue homogenate was determined as described above.
Nonhuman primate studies.
Rhesus macaques (Macaca mulatta) and African green monkeys (Cercopithecus aethiops) that were negative for serum-neutralizing antibodies to HMPV were inoculated simultaneously by the IN and intratracheal (IT) routes by using a 1-ml inoculum per site containing 105.2 TCID50 of virus in L15 medium. Seronegative or seropositive chimpanzees (Pan troglodytes) were similarly infected. The virus was administered by both the IN and IT routes to ensure the opportunity for it to replicate at each site (13, 17). Animals were housed and specimens were collected as previously described (15). The animals were observed clinically for 16 days postinoculation. Nasopharyngeal (NP) swab samples were collected on days 1 through 10 and on day 12 postinfection, and tracheal lavage (TL) samples were collected on days 2, 4, 6, 8, 10, and 12 postinfection. NP and TL samples from infected chimpanzees were also collected on days 14 and 16. The amount of virus present in NP and TL specimens was quantified by titration on LLC-MK2 cell monolayers as described above, and the mean peak virus titer obtained was expressed as log10 TCID50/ml. Chimpanzees and African green monkeys were challenged IN and IT with 1 ml per site containing 105.2 TCID50 of the CAN75 or CAN83 strain 35 or 30 days, respectively, after first infection, and NP and TL samples were collected on days 2, 4, 6, 8, and 10 postchallenge. HMPV present in the samples was quantified as described above. Serum samples were collected to determine the titer of neutralization antibodies before immunization (on day 0), postimmunization (on day 35 or 30), and 28 days postchallenge.
RESULTS
Growth properties of HMPV CAN75 and CAN83 strains in cell culture.
It was necessary to identify growth conditions for HMPV CAN75 and CAN83 that would give the high titers of both viruses that are needed for evaluation in vivo. The growth characteristics of the HMPV CAN75 and CAN83 strains, each representing one of the two identified HMPV lineages, were compared in a permissive rhesus monkey kidney cell line (LLC-MK2) since this line efficiently supports replication of a variety of paramyxoviruses (7, 28, 36). Previously, it was found that CAN83 replicated to approximately 106.0 TCID50/ml in LLC-MK2 cells (6) when grown in the presence of trypsin, indicating that this strain requires trypsin for efficient growth in vitro, as was previously reported for the original Netherlands 00-1 strain (39). The present study extended the previous findings with CAN83 to determine if CAN75 could also replicate to high titer in LLC-MK2 cells. We also compared the growth kinetics of HMPV CAN75 and CAN83 with those of HPIV1, HPIV2, and HPIV3. HPIV1 is a member of the Respirovirus genus that grows efficiently in LLC-MK2 cells in the presence of added trypsin, while HPIV2 and HPIV3 do not require added trypsin for growth in LLC-MK2 cells.
We found that CAN75 and CAN83 both require trypsin for efficient growth in LLC-MK2 cells (Fig. 1). CAN83 grew moderately faster and to a higher peak titer than CAN75. Both HMPV strains achieved peak titers (∼107 TCID50/ml) by 11 days postinfection. In contrast, HPIV1 achieved a titer of 108 TCID50/ml by day 3 and a peak titer of 109 TCID50/ml by day 5 postinfection. HPIV2 and HPIV3 achieve peak titers comparable to that of HPIV1 when grown in the absence of added trypsin under otherwise similar experimental conditions (Fig. 1) (35, 36). Thus, HMPV grows to an approximately 100-fold lower titer than members of the Respirovirus and Rubulavirus genera. The peak HMPV titers reported here (107.2 to 107.7 TCID50/ml) are substantially higher than previously reported for HMPV strains (39) grown in other tissue culture systems (39), which likely is due to the daily addition of trypsin-containing medium to the infected cell cultures in the present experiment and to the use of LLC-MK2 cells. Trypsin was replenished daily in LLC-MK2 cells (Fig. 1), since another monkey kidney cell line (Vero cells) has been previously found to rapidly inactivate trypsin added to the cell culture medium, and therefore, the yield of slow-growing, trypsin-requiring viruses can reach suboptimal levels unless trypsin is replenished (6, 22).
FIG. 1.
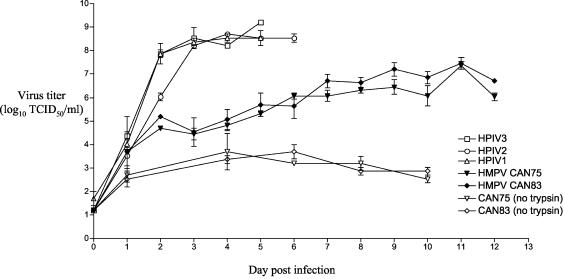
Growth kinetics of HMPV strains CAN75 and CAN83 in the presence or absence of trypsin. LLC-MK2 monolayers were infected with the indicated HMPV strain at a multiplicity of infection of 0.01 and were incubated at 32°C with or without added trypsin. Aliquots of the medium supernatants were harvested at 24-h intervals until the monolayer became detached from the plate. The virus titer was quantified on LLC-MK2 monolayers at 32°C. The level of replication of a biologically derived HPIV2, or rHPIV1 or rHPIV3, was determined in separate experiments under similar conditions (HPIV2 and HPIV3 were grown in the absence of added trypsin, and HPIV1 was grown with added trypsin). The virus titer is expressed as log10 TCID50/ml.
HMPV replicates to high titers in the respiratory tract of hamsters.
The evaluation of HMPV and potential vaccine candidates and recombinant HMPV derivatives would be facilitated by the availability of a small-animal model for evaluating replication, immunogenicity, and protective efficacy in vivo. Recently, it was reported that the original Netherlands HMPV isolate (strain 00-1) replicates in hamsters (37). In the present study, Golden Syrian hamsters were infected IN with 106 TCID50 of HMPV strain CAN75 or CAN83, and the level of replication in the nasal turbinates and lungs was determined on days 3 to 7 (Table 1). Each strain replicated to a peak titer greater than 106 TCID50 in the nasal turbinates and 3.6 to 4.4 log10 in the lower respiratory tract. Peak titers were achieved on days 3 and 4. Thus, hamsters are a permissive small-animal model for replication for members of both HMPV lineages. IN-administered HMPV was also found to be immunogenic in hamsters (see below).
TABLE 1.
Level of replication of HMPV strains CAN75 and CAN83 in the upper and lower respiratory tracts of hamsters
| Virusa | Tissue | Mean virus titer (log10 TCID50/g ± SE) on dayb:
|
||||
|---|---|---|---|---|---|---|
| 3 | 4 | 5 | 6 | 7 | ||
| HMPV CAN75 | Nasal turbinates | 6.0 ± 0.5 | 5.7 ± 0.3 | 5.1 ± 0.3 | 3.7 ± 1.2 | 1.7 ± 0.5 |
| Lungs | 3.6 ± 1.0 | 3.1 ± 0.3 | 3.0 ± 0.3 | ≤1.5 ± 0.0 | ≤1.5 ± 0.0 | |
| HMPV CAN83 | Nasal turbinates | 6.6 ± 0.4 | 6.4 ± 0.4 | 6.4 ± 0.6 | 5.2 ± 0.5 | 4.2 ± 0.7 |
| Lungs | 4.4 ± 0.6 | 4.3 ± 0.7 | 3.3 ± 0.2 | ≤1.5 ± 0.0 | ≤1.5 ± 0.0 | |
Hamsters in groups of 30 were inoculated intranasally with 106 TCID50 of the indicated virus.
Six animals from each group were sacrificed per day on days 3 through 7, nasal turbinates and lung tissues were harvested, and the virus present in tissue homogenates was quantified by serial dilution on LLC-MK2 monolayers at 32°C. The mean virus titer for each group of hamsters is expressed as log10 TCID50/g ± standard error (SE).
Evaluation of the antigenic relatedness of strains CAN83 and CAN75 by reciprocal cross-neutralization with hamster postinfection sera.
Previous studies have illustrated in detail the level of genetic diversity between the two proposed genetic lineages (3, 5, 30, 31), but the level of antigenic relatedness had not been described, due in part to the lack of defined animal models for HMPV replication and immunogenicity. The extent of antigenic diversity among circulating HMPV strains is of considerable importance to epidemiology and vaccine development. Hamsters that were seronegative for HMPV were infected IN with 104 TCID50 of strain CAN75 or CAN83, and postinfection sera were collected 44 days later. These individual sera were assayed for their neutralization titer to both HMPV strains, and the degree of antigenic relatedness was determined with the formula described by Archetti and Horsfall (1), which measures the percent antigenic relatedness as a function of the effectiveness of antibodies raised against each strain in neutralizing the homologous and heterologous strains in vitro (Table 2). The in vitro neutralization assay showed that hamsters infected with either strain developed a high titer of neutralizing antibody effective against either strain. The 10 animals infected with strain CAN83 yielded sera that neutralized the homologous CAN83 strain somewhat more efficiently than the heterologous CAN75 strain, but sera from animals infected with the CAN75 strain exhibited animal-to-animal variability with regard to whether the homologous or heterologous strain was neutralized more efficiently. Based on these neutralization titers, the level of antigenic relatedness between CAN75 and CAN83 was calculated to be approximately 48%, indicating that the two HMPV genetic lineages are highly related antigenically.
TABLE 2.
Homologous and heterologous HMPV serum neutralization titers in hamsters following primary infection with HMPV strain CAN75 or CAN83
| Animal no. and meanc | Immunizing virusa | Serum neutralizing antibody titerb against:
|
|
|---|---|---|---|
| CAN75 | CAN83 | ||
| 1 | CAN75 | 8.9 | 6.9 |
| 2 | CAN75 | 8.3 | 5.3 |
| 3 | CAN75 | 7.9 | 5.9 |
| 4 | CAN75 | 9.9 | 10.4 |
| 5 | CAN75 | 8.4 | 7.3 |
| 6 | CAN75 | 8.4 | 8.9 |
| 7 | CAN75 | 8.9 | 9.9 |
| 8 | CAN75 | 8.4 | 8.4 |
| 9 | CAN75 | 9.9 | 12.7 |
| 10 | CAN75 | 9.4 | 12.4 |
| 11 | CAN75 | 9.9 | 11.9 |
| Mean log2 | 8.9 ± 0.7 | 8.7 ± 2.6 | |
| 12 | CAN83 | 7.3 | 10.7 |
| 13 | CAN83 | 8.4 | 11.3 |
| 14 | CAN83 | 10.9 | 11.3 |
| 15 | CAN83 | 7.9 | 10.3 |
| 16 | CAN83 | 7.9 | 10.3 |
| 17 | CAN83 | 8.9 | 11.7 |
| 18 | CAN83 | 7.9 | 10.3 |
| 19 | CAN83 | 8.9 | 11.7 |
| 20 | CAN83 | 9.9 | 12.3 |
| 21 | CAN83 | 8.9 | 10.3 |
| Mean log2 | 8.6 ± 1.1 | 10.9 ± 0.7 | |
Hamsters were inoculated intranasally with 104 TCID50 of the indicated strain of HMPV.
Sera were collected 44 days postimmunization. The HMPV-neutralizing titer against each strain was determined and is expressed as the reciprocal log2. The mean log2 ± standard deviation is indicated. All animals had preimmunization neutralizing antibody titers of ≤1.0.
The antigenic relatedness between HMPV strains CAN75 and CAN83 was determined to be 48%, based on the Archetti-Horsfall formula [percent relatedness = (r1 × r2)1/2 × 100], where r1 is the geometric mean neutralization titer of antibody 1 to heterologous strain 2/geometric mean neutralization titer of antibody 1 to homologous strain 1, and r2 is the geometric mean neutralization titer of antibody 2 to heterologous strain 1/geometric mean neutralization titer of antibody 2 to homologous strain 2.
Infection of hamsters with either HMPV strain protects against homologous and heterologous challenge.
Groups of hamsters were infected with either strain of HMPV or with L15 diluent as a negative control. After 6 weeks, the hamsters were challenged with either strain of HMPV, and the level of pulmonary challenge virus replication in the respiratory tract on day 4 was determined, as described above. As shown in Table 3, previous infection by either HMPV strain induced a high level of protection against the homologous and heterologous HMPV strain in the upper respiratory tract. The lower respiratory tract was completely protected from challenge with either strain (data not shown). In addition, sera were collected 31 days following the first infection and assayed to determine the neutralizing titer against each strain (Table 3). The results were very similar to those shown in Table 2; infection with CAN75 induced antibodies that were modestly more effective in neutralizing the homologous strain than the heterologous strain, but antibodies induced by infection with CAN83 neutralized the homologous and heterologous strains with similar efficiencies. These cross-protection data and reciprocal neutralization assays indicated that CAN83 and CAN75 are antigenically highly related.
TABLE 3.
Hamsters infected with HMPV strain CAN83 or CAN75 are protected against challenge with the homologous and heterologous HMPV strains
| Immunizing agenta | Mean serum-neutralizing antibody titerb (reciprocal log2) against:
|
Mean virus titer (log10 TCID50/g) in nasal turbinates of hamsters administeredc:
|
||
|---|---|---|---|---|
| CAN75 | CAN83 | CAN75 | CAN83 | |
| CAN83 | 9.2 ± 0.2 | 10.4 ± 0.3 | 2.0 ± 0.2 | 1.7 ± 0.2 |
| CAN75 | 10.9 ± 0.2 | 7.7 ± 0.2 | ≤1.5 ± 0.0 | 2.4 ± 0.5 |
| L15 medium | ≤1.0 ± 0.0 | ≤1.0 ± 0.0 | 5.3 ± 0.1 | 5.6 ± 0.2 |
Hamsters in groups of 12 were immunized by IN infection with 105.5 TCID50 of the indicated virus or mock infected with L15 medium.
Sera were collected 2 to 3 days before and 31 days following the first infection, and the neutralizing titer against each of the two strains was determined. The preinfection anti-HMPV serum titers were ≤1.0 (reciprocal log2) for all animals in the study.
On day 42, six hamsters from each group were challenged IN with 105.5 TCID50 of CAN83, and the remaining six were challenged with CAN75. Nasal turbinates and lungs were harvested 4 days later, and the virus titer was determined on LLC- MK2 cells. The mean virus titer for each group of hamsters is expressed as log10 TCID50/g of nasal turbinate ± the standard error.
Infection of hamsters with HPIV1 expressing the CAN83 strain F protein protected hamsters from challenge with either HMPV strain.
The level of sequence identity for the F and G proteins of the CAN75 and CAN83 strains is 95 and 37%, respectively (3, 5). Thus, of these two major surface proteins, F is more likely to be the major contributor to the high level of cross-neutralization and cross-protection described above. To examine the ability of the HMPV F protein alone to induce serum-neutralizing antibodies and to confer protection against HMPV challenge, rHPIV1-F83 was recovered in vitro from an HPIV1 antigenomic cDNA containing the CAN83 F protein ORF inserted upstream of the HPIV1 N gene and flanked by HPIV1 gene start and gene end transcription signals (Fig. 2A). HPIV1 was selected as the vector since it and HMPV each replicate efficiently in the respiratory tract of hamsters, and the ability of the HMPV F expressed in the absence of other HMPV proteins could be readily assessed in hamsters. Expression of the F protein was confirmed by indirect immunofluorescence of infected cells (Fig. 2B). Thus, rHPIV1-F83 would express all of the proteins of HPIV1 as well as the F protein of HMPV.
Groups of hamsters were immunized with CAN83, wild-type rHPIV1, rHPIV1-F83 or L15 medium, and 33 days later, sera were collected and tested for the ability to neutralize the CAN75 and CAN83 strains. As shown in Table 4, sera from animals immunized with CAN83 or rHPIV1-F83 efficiently neutralized both strains of the virus. In each case, neutralization of the homologous CAN83 strain was somewhat more efficient than that of the heterologous CAN75 strain, which follows the pattern noted above for sera from CAN83-infected animals. Fifty days following immunization, the animals were challenged by IN infection with rHPIV1, CAN75, or CAN83. As shown in Table 4, animals previously infected with rHPIV1-F83 had a 125- and 158-fold reduction in the upper respiratory tract in the level of replication of the CAN75 and CAN83 challenge virus, respectively. Animals were completely protected in the lower respiratory tract (data not shown). The rHPIV1-F83 virus also protected against HPIV1 challenge, indicating that this single recombinant virus was able to protect against the three respiratory tract pathogens, namely CAN75, CAN83, and HPIV1. These findings indicate that the F protein of HMPV is a major protective antigen. Furthermore, the F protein of the CAN83 strain induced neutralizing antibodies and protection that were effective against both CAN83 and CAN75.
TABLE 4.
rHPIV1 expressing CAN83 F equally protects hamsters against challenge with HMPV subgroup CAN75 and CAN83 as well as against HPIV1
| Virus inoculuma | Mean serum neutralization titer (log2) to indicated virusb
|
Mean level of replication of indicated challenge virus in nasal turbinatesc
|
||||
|---|---|---|---|---|---|---|
| rHPIV1 | CAN75 | CAN83 | rHPIV1 | CAN75 | CAN83 | |
| L15 medium | ≤1.0 ± 0.0 | ≤1.0 ± 0.0 | ≤1.0 ± 0.0 | 4.9 ± 0.3 | 5.0 ± 0.2 | 6.1 ± 0.3 |
| CAN83 | ≤1.0 ± 0.0 | 8.3 ± 0.2 | 9.8 ± 0.2 | 4.6 ± 0.1 | 1.5 ± 0.0 | 1.5 ± 0.0 |
| rHPIV1wt | 6.1 ± 0.3 | ≤1.0 ± 0.0 | ≤1.0 ± 0.0 | 2.0 ± 0.2 | 4.4 ± 0.2d | 5.8 ± 0.2e |
| rHPIV1-F83 | 4.7 ± 0.2 | 6.0 ± 0.3 | 8.6 ± 0.2 | 1.9 ± 0.2 | 2.9 ± 0.3d | 3.9 ± 0.1e |
Groups of 18 hamsters were infected IN with 105 TCID50 of the indicated virus or with L15 medium as a negative control. rHPIV1 wt, wild-type rHPIV1.
Thirty-three days following the first infection, sera were collected and assayed to determine the neutralization titers against HPIV1, CAN75, and CAN83. Fifty days following the first infection, six hamsters from each group were challenged IN with 105 TCID50 of the indicated virus, and the nasal turbinates were harvested after 4 days.
Virus present in the tissue homogenates was quantified and is expressed as the mean log10 TCID50/g for each group ± the standard error.
Statistically significant difference between indicated values (P < 0.01 [unpaired t test]).
Statistically significant difference between indicated values (P < 0.0001 [unpaired t test]).
Natural HMPV infection of captive chimpanzees: prevalence of HMPV serum antibodies in chimpanzees.
Thirty-one captive-bred chimpanzees (Pan troglodytes), ranging in age from 1.5 to 3 years, were screened for the presence of serum-neutralizing antibodies to HMPV strain CAN75 or CAN83. A large number of animals (19 animals, or 61%) were found to be seropositive for either one or both HMPV strains, while the remaining 12 animals (39%) were seronegative for both strains (Fig. 3). In the seropositive animals, serum-neutralizing antibody titers to CAN75 ranged from low (2.6 log2) to ≥9.6 log2, and titers to CAN83 ranged from undetectable (≤1.0 log2) to 8.6 log2. Four of the animals that were seropositive for strain CAN75 and seronegative for strain CAN83 had very low neutralizing antibody titers (2.6 to 4.0 log2), and it is unclear if these values represent low antibody levels, as might occur for animals infected at an early age in the presence of maternal antibodies (27), or if they represent nonspecific neutralizing activity. The majority of seropositive animals had higher antibody titers to CAN75 than to the CAN83 strain. The significance of this finding is not known. The prevalence of HMPV antibodies in chimpanzees indicates that these animals readily become infected with HMPV in captivity, most likely from their human handlers. This also is characteristic of RSV (14) and HPIV3.
FIG. 3.
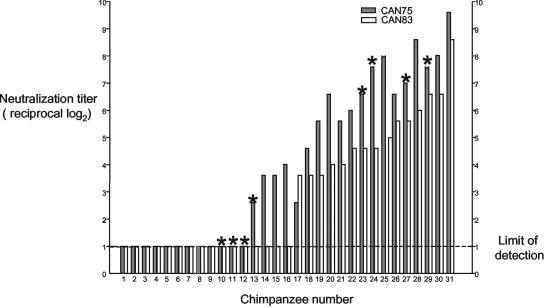
Seroprevalence of HMPV in captive chimpanzees. Sera obtained from 31 captive-bred chimpanzees were screened for the presence of neutralizing antibodies to HMPV strains CAN75 and CAN83. The serum neutralization titer (reciprocal log2) to CAN75 or CAN83 is indicated for each sample screened. The values obtained are arranged in order of increasing neutralization titer, and the animals are numbered accordingly. The animals indicated with asterisks were chosen for an infection and challenge study (Table 5).
Experimentally administered HMPV replicates and is immunogenic in chimpanzees.
We next evaluated the ability of strains CAN75 and CAN83 to infect chimpanzees that were seropositive for both strains or seronegative for the infecting strain (Table 5). Eight animals (indicated with asterisks in Fig. 4) were organized into four groups (two animals per group). Group 1 was seronegative for strain CAN75 and CAN83 and was administered CAN75. Group 2 was seronegative for CAN83, had low or no detectable neutralization titers to strain CAN75, and was administered CAN83. Group 3 and group 4 were seropositive for both strains and were administered CAN75 and CAN83, respectively. Each of the four seronegative animals (groups 1 and 2) had nasal discharge on days 7, 8, 9, and 10 postinfection. One of the animals in group 1 had thick mucus in the TL sample on day 8 and had decreased appetite. The seropositive animals (groups 3 and 4) showed no signs of illness throughout the course of the study. Thus, each of the HMPV seronegative chimpanzees developed illness following infection with the CAN75 or CAN83 HMPV strain, demonstrating that these viruses are infectious and virulent in chimpanzees. Furthermore, these findings from experimental infection and the findings that 61% of the chimpanzees were seropositive to HMPV indicate that chimpanzees are susceptible to natural infection and that this infection induces resistance to disease upon reinfection.
TABLE 5.
Levels of replication, immunogenicity, and cross-protection of HMPV strains CAN75 and CAN83 in chimpanzees
| Group no.a | Virus adminis- tered | Serostatus at time of inoculation | Preimmunization serum neutralization titerd to:
|
Level of virus replicatione for:
|
Postimmunization (day 28) serum neutral-ization titerd to:
|
Level of challenge virus replicationf for:
|
||||
|---|---|---|---|---|---|---|---|---|---|---|
| CAN75 | CAN83 | NP swab | TL sample | CAN75 | CAN83 | NP swab | TL sample | |||
| 1 | CAN75 | Negativeb | ≤1.0 ± 0.0 | ≤1.0 ± 0.0 | 1.9 ± 0.4 | 2.0 ± 0.8 | 8.7 ± 0.3 | 10.2 ± 0.3 | ≤0.5 ± 0.0 | ≤0.5 ± 0.0 |
| 2 | CAN83 | Negativeb | 3.8 ± 1.5 | ≤1.0 ± 0.0 | 3.2 ± 0.5 | 1.8 ± 0.3 | 8.9 ± 1.0 | 10.2 ± 0.3 | ≤0.5 ± 0.0 | ≤0.5 ± 0.0 |
| 3 | CAN75 | Positivec | 6.8 ± 0.2 | 5.1 ± 0.5 | 1.5 ± 0.0 | ≤0.5 ± 0.0 | 7.4 ± 0.5 | 9.2 ± 0.2 | ND | ND |
| 4 | CAN83 | Positivec | 7.6 ± 0.0 | 5.6 ± 1.0 | ≤0.5 ± 0.0 | 1.5 ± 0.3 | 8.4 ± 1.5 | 10.2 ± 0.2 | ND | ND |
Animals in groups of two were inoculated IN and IT with 105.2 TCID50 of the indicated virus in a 1-ml inoculum at each site.
Chimpanzees in this group were seronegative (mean antibody titer of <1:4) for the homologous infecting HMPV strain at the start of the study.
Chimpanzees in this group were seropositive (mean antibody titer of >1:20) for the homologous infecting HMPV strain at the start of the study.
Serum neutralization antibody titer to the indicated virus is expressed as the mean reciprocal log2 ± the standard error.
Level of virus replication is expressed as the mean of the peak virus titers (log10 TCID50/ml ± standard error) for the animals in each group irrespective of sampling day. The NP titers on day 0 were ≤0.5 log10 TCID50/ml. The lower limit of detection is 1.0 log10 TCID50/ml. A value of ≤0.5 log10 TCID50/ml is assigned to samples with no detectable virus.
On day 35, animals were challenged IN and IT with 105.2 TCID50 of the heterologous strain of HMPV in a 1-ml inoculum at each site. Level of virus replication is expressed as the mean of the peak virus titers (log10 TCID50/ml ± standard error) for the animals in each group irrespective of sampling day. The NP titers on day 0 were ≤0.5 log10 TCID50/ml. The lower limit of detection is 1.0 log10 TCID50/ml. ND, not done.
FIG. 4.
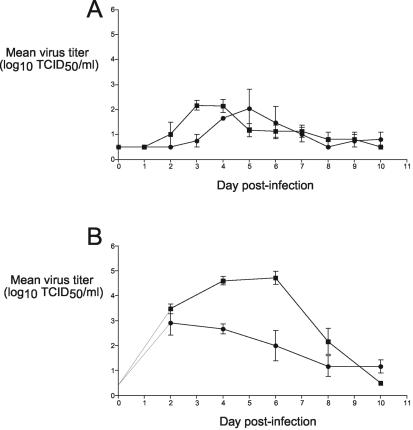
Level of replication of HMPV strains CAN75 and CAN83 in the upper and lower respiratory tract of African green monkeys. Four animals per group were infected IN and IT with 105.2 TCID50 of the indicated virus, CAN75 (•) or CAN83 (▪), and NP swab (A) and TL (B) specimens were taken on the indicated days, and the titer of shed virus was quantified.
Since the seronegative chimpanzees developed illness upon challenge with HMPV, we had expected virus to be shed in moderate to high titer from the upper and lower respiratory tract, as is the case with chimpanzees infected with RSV (4). Surprisingly, however, the HMPV-infected chimpanzees shed relatively low levels of virus. Even though all of the animals in the seronegative groups exhibited clinical disease signs in response to infection, the mean peak titer of virus recovered from the NP swabs ranged from 1.9 to 3.2 log10, and the titer from the TL ranged from 1.8 to 2.0 log10. The swabs and lavage samples from animals in groups 3 and 4, which were seropositive to HMPV at the start of the study, either lacked detectable HMPV or had a very low titer (1.5 log10). Thus, the lack of disease signs correlated with a reduced or undetectable level of virus shedding.
In this small sample, there was no evidence of antigenic differences between strains CAN75 and CAN83. Specifically, seronegative animals that were immunized with either strain (groups 1 and 2) developed antibodies with titers that were somewhat higher against strain CAN83 than against strain CAN75, irrespective of the immunizing virus.
The animals that were seronegative for the strain used in the primary infection were next challenged with the heterologous HMPV strain. As shown in Table 5, both groups of animals were completely protected from cross-challenge, indicating that previous infection by one HMPV lineage protects chimpanzees from subsequent infection by the heterologous lineage.
Replication of HMPV in monkeys.
The levels of replication and immunogenicity of HMPV CAN75 and CAN83 strains were determined in two species of monkeys. Twenty-two rhesus macaques and 20 African green monkeys were screened for serum-neutralizing antibodies to HMPV, and each animal was seronegative for HMPV (≤1.0 log2). Four animals of each species were infected IT with 105.2 TCID50 of either HMPV strain CAN75 or strain CAN83. Illness did not develop in any of the 16 monkeys. Samples from rhesus monkeys had very low titers of virus, ranging from 0.9 to 2.6 log10 TCID50 (Table 6). Furthermore, the postimmunization titers of serum-neutralizing antibodies were also relatively low, ranging from 6.3 to 8.4 log2. Thus, rhesus monkeys support the replication of HMPV, but the level of replication is low.
TABLE 6.
Level of replication, immunogenicity, and cross-protection of HMPV strains CAN75 and CAN83 in two species of monkeys
| Primate species | No. of animals | Virus inoculuma | Level of virus replicationb for:
|
Postimmunization (day 28) serum neutralization titerc to:
|
% Antigenic relatednessd | Level of challenge virus replicatione for:
|
|||
|---|---|---|---|---|---|---|---|---|---|
| NP swab | TL sample | CAN75 | CAN83 | NP swab | TL sample | ||||
| Rhesus | 4 | CAN75 | 1.3 ± 0.1 | 0.9 ± 0.2 | 6.4 ± 0.6 | 7.2 ± 0.3 | 64 | ND | ND |
| Rhesus | 4 | CAN83 | 1.6 ± 0.1 | 2.6 ± 0.3 | 6.3 ± 0.1 | 8.4 ± 0.3 | ND | ND | |
| African green | 4 | CAN75 | 2.6 ± 0.5 | 3.2 ± 0.4 | 9.4 ± 0.3 | 10.8 ± 0.4 | 99 | 1.4 ± 0.1 | 0.8 ± 0.1 |
| African green | 4 | CAN83 | 2.2 ± 0.2 | 4.9 ± 0.1 | 9.5 ± 0.5 | 10.9 ± 0.4 | 0.6 ± 0.1 | 0.9 ± 0.2 | |
Animals were inoculated IN and IT with 105.2 TCID50 of the indicated virus in a 1-ml inoculum at each site.
The level of virus replication is expressed as the mean of the peak virus titers (log10 TCID50/ml ± standard error) for the animals in each group irrespective of sampling day. Virus titrations were performed on LLC-MK2 cells. The lower limit of detection is 1.0 log10 TCID50/ml. A value of ≤0.5 log10 TCID50/ml is assigned to samples with no detectable virus.
Mean serum neutralization antibody titer to the indicated HMPV strain is expressed as the mean reciprocal log2 ± the standard error. All animals were seronegative for both strains of HMPV at the start of the study (i.e., day 0 reciprocal neutralization titer of ≤1.0 log2).
Percent antigenic relatedness was calculated with the formula of Archetti and Horsfall (see Table 2, footnote c).
On day 30, animals were challenged IN and IT with 105.2 TCID50 of the heterologous virus in a 1-ml inoculum at each site. Level of virus replication is expressed as the mean of the peak virus titers (log10 TCID50/ml ± standard error) for the animals in each group irrespective of sampling day. The lower limit of detection is 1.0 log10 TCID50/ml. ND, not done.
The highest level of HMPV replication and immunogenicity was found to occur in infected African green monkeys (Table 6), which shed titers of HMPV ranging from 2.2 to 4.9 log10 TCID50 and which developed serum-neutralizing antibody titers that ranged from 9.4 to 10.9 log2. The time course of HMPV replication in samples collected from the nasopharynx and the trachea of the African green monkeys is shown in Fig. 4 and indicates that virus shedding occurred over a broad window of more than 6 days.
Thirty days following the first infection, the African green monkeys were challenged with the heterologous HMPV strain (Table 6). Both groups of animals were highly protected from cross-challenge, indicating that previous infection by one HMPV lineage confers resistance to subsequent infection by the heterologous strain. These data indicate that African green monkeys are permissive for HMPV replication and, in particular, represent a suitable primate host with which to evaluate the replication and immunogenicity of HMPV.
We next evaluated the ability of the antibodies induced by the primary infection in the two species of monkeys to neutralize the homologous versus heterologous subgroup virus to determine if the high level of antigenic relatedness seen in hamsters was also seen in nonhuman primates. Each species was calculated separately, showing that the percent relatedness of the two HMPV lineages was 64 and 99%, based on the data from rhesus macaques and African green monkeys, respectively (Table 6).
DISCUSSION
Characterization of HMPV has been hampered by its relatively poor growth in vitro (39) and by the lack of established animal models of infection, immunogenicity, and protective efficacy. Although two strains of HMPV representing each of the major genetic lineages grew less well in vitro than HPIV1, which shares a requirement for added trypsin, it was found that moderately high titers could be achieved in vitro by the regular replenishment of the trypsin in the growth medium. It was also found that hamsters are relatively permissive for replication of members of both HMPV lineages and that the infected animals developed a strong immune response that protected the animals from challenge. While this paper was in preparation, another group showed that the original HMPV Netherlands isolate (00-1), which belongs to the same lineage as the CAN83 strain, was immunogenic and replicated in the respiratory tract of hamsters (37). HMPV infection in other small animal models such as ferrets and rabbits has been reported to induce a strong immune response (39), but the level of virus replication in these animals has not been reported. The present study extended these observations to show that members of both HMPV lineages replicated efficiently in hamsters and that infection induced a high level of neutralizing antibodies and resistance to challenge that was effective against both homologous and heterologous strains. In addition to identifying the hamster as a useful rodent model for HMPV infection and vaccine development, two species of nonhuman primates were also identified as useful models for the development of respiratory tract disease (chimpanzees) and for replication (African green monkeys).
Chimpanzees, but not rhesus macaques or African green monkeys, had a high seroprevalence for HMPV, indicating that chimpanzees can readily become infected by their human handlers and possibly by animal-to-animal transmission. Chimpanzees that were experimentally infected with either strain developed mild upper respiratory tract disease symptoms and cough, but HMPV was shed only in low titers from the respiratory tract. RSV, which also is a member of the Pneumovirus subfamily, has also been found to naturally infect chimpanzees in captivity (14), but in contrast to HMPV, RSV replicates to a high titer in the respiratory tract of chimpanzees (4). The apparent incongruity between the low level of HMPV shedding in the presence of disease symptoms might reflect a difficulty in retrieving infectious virus from secretions, possibly due to the inactivation by host immune factors or a high level of cell association of HMPV in the epithelial cells lining the respiratory tract of the chimpanzees. Since the chimpanzee is so closely related to humans, this raises the possibility that detection of HMPV in human secretions might also be inefficient. The inability to detect substantial HMPV shedding was unexpected, since RSV is known to be labile and cell associated in vitro but is nonetheless shed from RSV-infected chimpanzees presenting with RSV-associated illness at a level that is 100- to 1,000-fold greater than was the case for HMPV-infected chimpanzees (4, 41). Alternatively, the level of HMPV replication in chimpanzees may indeed be low, but infection may trigger immune- or chemokine-mediated pathology, resulting in the development of disease symptoms (16). Although the level of HMPV shedding was low, chimpanzees developed a robust immune response. Animals that were seropositive for both strains by either natural or experimental infection were protected from disease following challenge with either strain. This is the first demonstration of the feasibility of immunizing against HMPV in a primate animal model achieved in response to either natural exposure or IN immunization. Thus, chimpanzees may provide a useful nonhuman primate model for HMPV disease but are less than ideal models of virus replication. Moreover, their lack of availability and seropositivity will restrict their further use.
HMPV replicated to a high titer in African green monkeys, especially in the lower respiratory tract, and infected animals developed a high titer of serum-neutralizing antibodies. Resistance to replication of a heterologous challenge HMPV strain was also observed. We were unable to detect a substantial level of HMPV replication in rhesus macaques, although this species developed serum-neutralizing antibodies following HMPV infection, indicating that these macaques are not ideal animal models for the quantitation of HMPV replication. Thus, African green monkeys appear to be the best nonhuman primate model examined thus far for evaluating HMPV replication and immunogenicity. Previously, HMPV viral RNA was detected by reverse transcription-PCR in throat swabs of experimentally infected cynomolgus macaques (39); however, infectious virus was not quantified in that study.
It has been suggested that there are be multiple serotypes of HMPV (29, 40). In the present study, the antigenic relatedness between CAN75 and CAN83, representing the two known genetic lineages of HMPV, was examined by using reciprocal cross-neutralization assays based on primary infection sera from hamsters, two species of monkeys, and chimpanzees. The two HMPV genetic lineages were found to be highly related antigenically in each animal model. The level of relatedness was 48% in hamsters and from 64 to 99% in nonhuman primates. This high level of relatedness would be expected to confer a high level of cross-protection in challenge experiments, and this indeed was observed. The demonstration of a high level of cross-neutralization and cross-protection following a primary infection indicates that these two strains do not represent different serotypes or even antigenic subgroups of HMPV. In particular, the level of antigenic relatedness between the two lineages of HMPV is higher than that observed between the A and B antigenic subgroups of RSV (10, 21). Based on an analysis of rodent and human sera, the two subgroups of RSV were 25 and 31% related, respectively (19, 21).
The F proteins of the two RSV subgroups were 89% identical at the amino acid level and 50 to 100% antigenically related. The RSV subgroup A and B G proteins are 53% identical at the amino acid level but are only 5 to 7% related in their level of antigenic cross-reactivity (19-21). Thus, the F protein is responsible for the relatively high degree of cross-subgroup neutralization observed in vitro for RSV (21). It was reasonable to suggest that the F protein also played the major role in the antigenic relatedness between members of the two HMPV lineages since it is 95% identical between lineages at the amino acid level whereas the G protein is only 37% identical, which was found to be the case. Animals immunized with an rHPIV1 vector expressing the CAN83 F protein developed neutralizing antibodies against both strains and were protected from challenge from members of both lineages of HMPV, demonstrating that the HMPV F protein is a major contributor to the high level of antigenic relatedness observed between lineages. Recently, it was shown that a chimeric human-bovine PIV3 expressing the HMPV strain 00-1 F protein protected hamsters from infection with the HMPV strain 00-1 (37), although challenge with a heterologous strain was not performed. Thus, the HMPV F protein is a major neutralization antigen that confers substantial neutralization and protection across lineages. These findings suggest that the HMPV F protein would be a preferred HMPV antigen to express from a live-vaccine expression vector. The HPIV1 virus bearing the CAN83 F protein protected against both HPIV1 and HMPV, indicating that it, like the HPIV3 vector described (37), also has promise for use as a vaccine to induce protective immunity to multiple human viral pathogens.
The two HMPV lineages examined in this study are also more closely related antigenically than are the human and bovine PIV3 strains. These two members of the Respirovirus genus share 79 and 75% sequence identity in their F and HN glycoproteins, respectively, and are 25% related antigenically (11). Infection of hamsters or nonhuman primates with bovine PIV3 protects animals from challenge with HPIV3 (11, 18, 32). In contrast to RSV, PIV3, and HMPV, the HPIV4 subgroups A and B are highly distinct immunologically (8, 26), despite a comparatively high level of amino acid identity in the F and HN antigenic glycoproteins (95 and 84%, respectively) (2, 24). Studies suggest that this antigenic diversity may be caused by the creation or deletion of N-glycosylation sites on the HN glycoproteins (25). For the HPIV4 subgroups A and B, the F protein appears to be contributing less to the antigenic relatedness estimated with the neutralization test than for the RSV and HMPV viruses.
The present study did not address the relative contributions of the other two surface proteins of HMPV, namely, the G and SH proteins, to the observed antigenic relatedness or to heterologous protection. In the case of RSV, the F and G surface glycoproteins are the only significant neutralization antigens and are the major protective antigens (12), and in the case of HPIV1, HPIV2, HPIV3, and HPIV4, the two surface proteins F and HN also are the neutralization and major protective antigens (9). The SH protein of RSV is not a significant neutralization or protective antigen, but this might be because it has a very small extracellular domain. In contrast, the HMPV SH and G proteins have large extracellular domains, and it may be that each protein will be found to be a neutralization and protective antigen. Genetic analysis has shown that the SH and G glycoproteins of the two HMPV lineages were highly variable and that, in particular, the G protein did not have more than three contiguous amino acids in the extracellular domain conserved between the two lineages (5). This finding suggests that conserved antigenic sites would be rare or nonexistent and that these proteins likely contribute little to the cross-protection observed between members of the two lineages of HMPV. However, we do not rule out a role for G or SH in an HMPV vaccine. Because antigenic relatedness is defined by an in vitro neutralization assay, it can be dominated by a major conserved neutralization antigen such as the F protein. It may be that, in humans, G and SH play roles in protection that cannot be predicted by these studies. If so, the high degree of genetic diversity between the genetic lineages for these proteins would become important for immunoprophylaxis, such that both lineages would need to be represented in a vaccine. In sum, the present study identified hamsters and African green monkeys as useful animal models for the two lineages of HMPV and demonstrated a high degree of antigenic relatedness and cross protection that is mediated by immunity to the highly conserved F protein. An HPIV1 vector bearing the HMPV F protein provided protection against HPIV1 as well as both lineages of HMPV, indicating that such vectors might be useful as vaccines to protect against disease caused by both HPIV1 and HMPV.
Acknowledgments
We thank Guy Boivin (Laval University, Quebec, Canada) and Teresa Peret, Dean Erdman, and Larry Anderson (CDC, Atlanta, Georgia) for providing virus stocks of the CAN75 and CAN83 strains. We also thank Yasuhiko Ito (Mie University School of Medicine, Japan) for providing HPIV1 HN monoclonal hybridoma cell lines and Tammy Tobery and Josh Moore for excellent technical assistance and care of primates.
REFERENCES
- 1.Archetti, I., and F. Horsfall. 1950. Persistent antigenic variation of influenza A viruses after incomplete neutralization in ovo with heterologous immune serum. J. Exp. Med. 92:441-462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bando, H., K. Kondo, M. Kawano, H. Komada, M. Tsurudome, M. Nishio, and Y. Ito. 1990. Molecular cloning and sequence analysis of human parainfluenza type 4A virus HN gene: its irregularities on structure and activities. Virology 175:307-312. [DOI] [PubMed] [Google Scholar]
- 3.Bastien, N., S. Normand, T. Taylor, D. Ward, T. C. Peret, G. Boivin, L. J. Anderson, and Y. Li. 2003. Sequence analysis of the N, P, M and F genes of Canadian human metapneumovirus strains. Virus Res. 93:51-62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Belshe, R. B., L. S. Richardson, W. T. London, D. L. Sly, J. H. Lorfeld, E. Camargo, D. A. Prevar, and R. M. Chanock. 1977. Experimental respiratory syncytial virus infection of four species of primates. J. Med. Virol. 1:157-162. [DOI] [PubMed] [Google Scholar]
- 5.Biacchesi, S., M. H. Skiadopoulos, G. Boivin, C. T. Hanson, B. R. Murphy, P. L. Collins, and U. J. Buchholz. 2003. Genetic diversity between human metapneumovirus subgroups. Virology 315:1-9. [DOI] [PubMed] [Google Scholar]
- 6.Biacchesi, S., M. H. Skiadopoulos, K.-C. Tran, B. R. Murphy, P. L. Collins, and U. Buchholz. 2004.. Recovery of human metapneumovirus from cDNA: optimization of growth in vitro and expression of additional genes. Virology 247-259 321:. [DOI] [PubMed]
- 7.Boivin, G., G. De Serres, S. Cote, R. Gilca, Y. Abed, L. Rochette, M. G. Bergeron, and P. Dery. 2003. Human metapneumovirus infections in hospitalized children. Emerg. Infect. Dis. 9:634-640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Canchola, J., A. J. Vargosko, H. W. Kim, R. H. Parrott, E. E. Christmas, B. C. Jeffries, and R. M. Chanock. 1964. Antigenic variation among newly isolated strains of parainfluenza type 4 virus. Am. J. Hyg. 79:357-364. [DOI] [PubMed] [Google Scholar]
- 9.Chanock, R. M., B. R. Murphy, and P. L. Collins. 2001. Parainfluenza viruses, p. 1341-1379. In D. M. Knipe, P. M. Howley, D. E. Griffin, R. A. Lamb, M. A. Martin, B. Roizman, and S. E. Strauss (ed.), Fields virology, 4th ed., vol. 1. Lippincott Williams & Wilkins, Philadelphia, Pa.
- 10.Coates, H. V., D. W. Alling, and R. M. Chanock. 1966. An antigenic analysis of respiratory syncytial virus isolates by a plaque reduction neutralization test. Am. J. Epidemiol. 83:299-313. [DOI] [PubMed] [Google Scholar]
- 11.Coelingh, K., C. C. Winter, E. L. Tierney, W. T. London, and B. R. Murphy. 1988. Attenuation of bovine parainfluenza virus type 3 in nonhuman primates and its ability to confer immunity to human parainfluenza virus type 3 challenge. J. Infect. Dis. 157:655-662. [DOI] [PubMed] [Google Scholar]
- 12.Connors, M., P. L. Collins, C. Y. Firestone, and B. R. Murphy. 1991. Respiratory syncytial virus (RSV) F, G, M2 (22K), and N proteins each induce resistance to RSV challenge, but resistance induced by M2 and N proteins is relatively short-lived. J. Virol. 65:1634-1637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Crowe, J. E., Jr., P. T. Bui, W. T. London, A. R. Davis, P. P. Hung, R. M. Chanock, and B. R. Murphy. 1994. Satisfactorily attenuated and protective mutants derived from a partially attenuated cold-passaged respiratory syncytial virus mutant by introduction of additional attenuating mutations during chemical mutagenesis. Vaccine 12:691-699. [DOI] [PubMed] [Google Scholar]
- 14.Crowe, J. E., Jr., P. L. Collins, W. T. London, R. M. Chanock, and B. R. Murphy. 1993. A comparison in chimpanzees of the immunogenicity and efficacy of live attenuated respiratory syncytial virus (RSV) temperature-sensitive mutant vaccines and vaccinia virus recombinants that express the surface glycoproteins of RSV. Vaccine 11:1395-1404. [DOI] [PubMed] [Google Scholar]
- 15.Durbin, A. P., C. J. Cho, W. R. Elkins, L. S. Wyatt, B. Moss, and B. R. Murphy. 1999. Comparison of the immunogenicity and efficacy of a replication-defective vaccinia virus expressing antigens of human parainfluenza virus type 3 (HPIV3) with those of a live attenuated HPIV3 vaccine candidate in rhesus monkeys passively immunized with PIV3 antibodies. J. Infect. Dis. 179:1345-1351. [DOI] [PubMed] [Google Scholar]
- 16.Glass, W. G., H. F. Rosenberg, and P. M. Murphy. 2003. Chemokine regulation of inflammation during acute viral infection. Curr. Opin. Allergy Clin. Immunol. 3:467-473. [DOI] [PubMed] [Google Scholar]
- 17.Hall, S. L., A. Stokes, E. L. Tierney, W. T. London, R. B. Belshe, F. C. Newman, and B. R. Murphy. 1992. Cold-passaged human parainfluenza type 3 viruses contain ts and non-ts mutations leading to attenuation in rhesus monkeys. Virus Res. 22:173-184. [DOI] [PubMed] [Google Scholar]
- 18.Haller, A. A., M. MacPhail, M. Mitiku, and R. S. Tang. 2001. A single amino acid substitution in the viral polymerase creates a temperature-sensitive and attenuated recombinant bovine parainfluenza virus type 3. Virology 288:342-350. [DOI] [PubMed] [Google Scholar]
- 19.Hendry, R. M., J. C. Burns, E. E. Walsh, B. S. Graham, P. F. Wright, V. G. Hemming, W. J. Rodriguez, H. W. Kim, G. A. Prince, K. McIntosh, et al. 1988. Strain-specific serum antibody responses in infants undergoing primary infection with respiratory syncytial virus. J. Infect. Dis. 157:640-647. [DOI] [PubMed] [Google Scholar]
- 20.Johnson, P. R., M. K. Spriggs, R. A. Olmsted, and P. L. Collins. 1987. The G glycoprotein of human respiratory syncytial viruses of subgroups A and B: extensive sequence divergence between antigenically related proteins. Proc. Natl. Acad. Sci. USA 84:5625-5629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Johnson, P. R., Jr., R. A. Olmsted, G. A. Prince, B. R. Murphy, D. W. Alling, E. E. Walsh, and P. L. Collins. 1987. Antigenic relatedness between glycoproteins of human respiratory syncytial virus subgroups A and B: evaluation of the contributions of F and G glycoproteins to immunity. J. Virol. 61:3163-3166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kaverin, N. V., and R. G. Webster. 1995. Impairment of multicycle influenza virus growth in Vero (WHO) cells by loss of trypsin activity. J. Virol. 69:2700-2703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kolakofsky, D., T. Pelet, D. Garcin, S. Hausmann, J. Curran, and L. Roux. 1998. Paramyxovirus RNA synthesis and the requirement for hexamer genome length: the rule of six revisited. J. Virol. 72:891-899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Komada, H., H. Bando, M. Ito, H. Ohta, M. Kawano, M. Nishio, M. Tsurudome, N. Watanabe, N. Ikemura, S. Kusagawa, et al. 1995. Sequence analyses of human parainfluenza virus type 4A and type 4B fusion proteins. J. Gen. Virol. 76:3205-3210. [DOI] [PubMed] [Google Scholar]
- 25.Komada, H., M. Ito, M. Nishio, M. Kawano, H. Ohta, M. Tsurudome, S. Kusagawa, M. O'Brien, H. Bando, and Y. Ito. 2000. N-glycosylation contributes to the limited cross-reactivity between hemagglutinin neuraminidase proteins of human parainfluenza virus type 4A and 4B. Med. Microbiol. Immunol. (Berlin) 189:1-6. [DOI] [PubMed] [Google Scholar]
- 26.Komada, H., M. Tsurudome, M. Ueda, M. Nishio, H. Bando, and Y. Ito. 1989. Isolation and characterization of monoclonal antibodies to human parainfluenza virus type 4 and their use in revealing antigenic relation between subtypes 4A and 4B. Virology 171:28-37. [DOI] [PubMed] [Google Scholar]
- 27.Murphy, B. R., P. L. Collins, L. Lawrence, J. Zubak, R. M. Chanock, and G. A. Prince. 1989. Immunosuppression of the antibody response to respiratory syncytial virus (RSV) by pre-existing serum antibodies: partial prevention by topical infection of the respiratory tract with vaccinia virus-RSV recombinants. J. Gen. Virol. 70:2185-2190. [DOI] [PubMed] [Google Scholar]
- 28.Newman, J. T., S. R. Surman, J. M. Riggs, C. T. Hansen, P. L. Collins, B. R. Murphy, and M. H. Skiadopoulos. 2002. Sequence analysis of the Washington/1964 strain of human parainfluenza virus type 1 (HPIV1) and recovery and characterization of wild-type recombinant HPIV1 produced by reverse genetics. Virus Genes 24:77-92. [DOI] [PubMed] [Google Scholar]
- 29.Osterhaus, A., and R. Fouchier. 2003. Human metapneumovirus in the community. Lancet 361:890-891. [DOI] [PubMed] [Google Scholar]
- 30.Peiris, J. S., W. H. Tang, K. H. Chan, P. L. Khong, Y. Guan, Y. L. Lau, and S. S. Chiu. 2003. Children with respiratory disease associated with metapneumovirus in Hong Kong. Emerg. Infect. Dis. 9:628-633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Peret, T. C., G. Boivin, Y. Li, M. Couillard, C. Humphrey, A. D. Osterhaus, D. D. Erdman, and L. J. Anderson. 2002. Characterization of human metapneumoviruses isolated from patients in North America. J. Infect. Dis. 185:1660-1663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schmidt, A. C., J. M. McAuliffe, A. Huang, S. R. Surman, J. E. Bailly, W. R. Elkins, P. L. Collins, B. R. Murphy, and M. H. Skiadopoulos. 2000. Bovine parainfluenza virus type 3 (BPIV3) fusion and hemagglutinin-neuraminidase glycoproteins make an important contribution to the restricted replication of BPIV3 in primates. J. Virol. 74:8922-8929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Skiadopoulos, M. H., and A. A. McBride. 1996. The bovine papillomavirus type 1 E2 transactivator and repressor proteins use different nuclear localization signals. J. Virol. 70:1117-1124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Skiadopoulos, M. H., S. Surman, J. M. Tatem, M. Paschalis, S. L. Wu, S. A. Udem, A. P. Durbin, P. L. Collins, and B. R. Murphy. 1999. Identification of mutations contributing to the temperature-sensitive, cold-adapted, and attenuation phenotypes of the live-attenuated cold-passage 45 (cp45) human parainfluenza virus 3 candidate vaccine. J. Virol. 73:1374-1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Skiadopoulos, M. H., S. R. Surman, A. P. Durbin, P. L. Collins, and B. R. Murphy. 2000. Long nucleotide insertions between the HN and L protein coding regions of human parainfluenza virus type 3 yield viruses with temperature-sensitive and attenuation phenotypes. Virology 272:225-234. [DOI] [PubMed] [Google Scholar]
- 36.Skiadopoulos, M. H., L. Vogel, J. M. Riggs, S. R. Surman, P. L. Collins, and B. R. Murphy. 2003. The genome length of human parainfluenza virus type 2 follows the rule of six, and recombinant viruses recovered from non-polyhexameric-length antigenomic cDNAs contain a biased distribution of correcting mutations. J. Virol. 77:270-279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tang, R. S., J. H. Schickli, M. MacPhail, F. Fernandes, L. Bicha, J. Spaete, R. A. Fouchier, A. D. Osterhaus, R. Spaete, and A. A. Haller. 2003. Effects of human metapneumovirus and respiratory syncytial virus antigen insertion in two 3′ proximal genome positions of bovine/human parainfluenza virus type 3 on virus replication and immunogenicity. J. Virol. 77:10819-10828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.van den Hoogen, B. G., T. M. Bestebroer, A. D. Osterhaus, and R. A. Fouchier. 2002. Analysis of the genomic sequence of a human metapneumovirus. Virology 295:119-132. [DOI] [PubMed] [Google Scholar]
- 39.van den Hoogen, B. G., J. C. de Jong, J. Groen, T. Kuiken, R. de Groot, R. A. Fouchier, and A. D. Osterhaus. 2001. A newly discovered human pneumovirus isolated from young children with respiratory tract disease. Nat. Med. 7:719-724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.van den Hoogen, B. G., D. M. Osterhaus, and R. A. Fouchier. 2004. Clinical impact and diagnosis of human metapneumovirus infection. Pediatr. Infect. Dis. J. 23:S25-S32. [DOI] [PubMed] [Google Scholar]
- 41.Wechsler, S. L., D. M. Lambert, M. S. Galinski, B. E. Heineke, A. L. Lambert, M. Mink, O. M. Rochovansky, and M. W. Pons. 1985. A simple method for increased recovery of purified paramyxovirus virions. J. Virol. Methods 12:179-182. [DOI] [PubMed] [Google Scholar]