

Abstract
Microvascular stability and regulation of capillary tonus are regulated by pericytes and their interactions with endothelial cells (EC). While the RhoA/Rho kinase (ROCK) pathway has been implicated in modulation of pericyte contractility, in part via regulation of the myosin light chain phosphatase (MLCP), the mechanisms linking Rho GTPase activity with actomyosin-based contraction and the cytoskeleton are equivocal. Recently, the myosin phosphatase-RhoA-interacting protein (MRIP) was shown to mediate the RhoA/ROCK-directed MLCP inactivation in vascular smooth muscle. Here we report that MRIP directly interacts with the β-actin-specific capping protein βcap73. Furthermore, manipulation of MRIP expression influences pericyte contractility, with MRIP silencing inducing cytoskeletal remodeling and cellular hypertrophy. MRIP knockdown induces a repositioning of βcap73 from the leading edge to stress fibers; thus MRIP-silenced pericytes increase F-actin-driven cell spreading twofold. These hypertrophied and cytoskeleton-enriched pericytes demonstrate a 2.2-fold increase in contractility upon MRIP knockdown when cells are plated on a deformable substrate. In turn, silencing pericyte MRIP significantly affects EC cycle progression and angiogenic activation. When MRIP-silenced pericytes are cocultured with capillary EC, there is a 2.0-fold increase in EC cycle entry. Furthermore, in three-dimensional models of injury and repair, silencing pericyte MRIP results in a 1.6-fold elevation of total tube area due to EC network formation and increased angiogenic sprouting. The pivotal role of MRIP expression in governing pericyte contractile phenotype and endothelial growth should lend important new insights into how chemomechanical signaling pathways control the “angiogenic switch” and pathological angiogenic induction.
Keywords: cytoskeleton, Rho kinase, myosin light chain phosphatase, myosin light chain, capillary, mural cell, angiogenesis
in arteries and veins, vascular tone is largely regulated via vascular smooth muscle. In capillaries and venules, however, tonus is coregulated by endothelial cells (EC) and pericytes. In the microcirculation, pericyte dysfunction contributes to several debilitating vascular pathologies, including diabetic retinopathy (DR), hypertension, atherosclerosis, ischemia, stroke, asthma, and Alzheimer's disease (10, 13, 30, 53).
A growing body of evidence in pericyte pathophysiology indicates that pericytes play a critical role in the maintenance of capillary EC (CEC) physiology and angiogenic induction (2, 12, 16, 38). For example, in the retina, perturbations in pericyte-EC interactions can result in pathological angiogenesis accompanying proliferative DR (2, 13, 21). The notion has been widely held that, early in DR, vascular cell death manifests, first by pericyte dropout and then by EC apoptosis and an excessive retinal neovascular response (2, 13, 40). However, recent work strongly suggests that pericyte dysfunction, not death or dropout, precedes pathological angiogenic induction and misregulation of microvascular growth (36, 39, 41).
Recent genetic and pharmacological studies have demonstrated that pericyte contractile phenotype is dependent on the RhoA/Rho kinase (ROCK) pathway. Overexpression and selective inhibition studies have revealed that RhoA GTPase activity affects specifically the smooth muscle actin (SMA)-based cytoskeletal networks (35). Furthermore, alteration of pericyte Rho GTPase status via viral transduction of dominant-active or dominant-negative RhoA constructs leads to altered force generation on deformable substrates and hyper- or hypocontractile cells, respectively (39). Interestingly, alteration of normal pericyte tonus in either direction was shown to equally culminate in a loss of contact-dependent EC quiescence (39). Thus a more complete understanding of the RhoA/ROCK pathway will lend insights into the physiological/pathological regulation of contractile phenotype and how pericyte-EC interactions govern angiogenesis.
As the Rho effector pathway is central to the control of cytoskeletal dynamics and contractile phenotype, further studies may reveal promising new therapeutic targets in treating microvascular pathologies. Recently, myosin phosphatase-RhoA interacting protein (MRIP) was identified as a critical molecular scaffold that coordinates RhoA and myosin light chain (MLC) phosphatase (MLCP) localization to stress fibers in smooth muscle cells (55, 66, 67). MRIP binds 1) MLCP through its COOH-terminal leucine zipper domain (66), 2) RhoA via its central, Rho-binding domain (66), and 3) F-actin through the NH2 terminus; importantly, while ROCK is associated with the MRIP-RhoA-MLCP complex, MRIP and ROCK do not directly interact (47). The novel complex, containing MRIP, RhoA, MLCP, and ROCK, is localized on actomyosin-containing filament bundles, and silencing of MRIP leads to an uncoupling of stress fiber-associated MLCP, an accumulation of phosphorylated MLC, and a stress fiber-enriched phenotype in smooth muscle cells (55, 67). While this MRIP regulatory complex coordinates the localization of RhoA and MLCP to microfilaments, the specific mechanisms governing stress fiber assembly/remodeling and the targeting of ROCK to the cytoskeleton remain incompletely understood.
As increasing evidence supports a critical role for the RhoA/ROCK effector pathway in microvascular (dys)function, we performed genetic screens linked to cell- and molecule-based assays to reveal the complex molecular interplay governing vascular cell contractile phenotype. Interestingly, we discovered that ROCK is a direct interactor of the β-actin-specific, barbed-end capping protein βcap73 and that MRIP directly binds βcap73, thereby linking MRIP and the Rho effector pathway with the isoactin and cytoskeletal effector network. Furthermore, MRIP silencing leads to a marked pericyte hypertrophy, mirrored by an extensively remodeled isoactin network, without significant changes in overall cytoskeletal protein expression. Silencing MRIP culminates in a relocalization of βcap73 from the plasma membrane and increased cell spreading rates. These hypertrophic, MRIP-silenced pericytes are unable to maintain contact-dependent EC growth arrest in coculture studies. Using a three-dimensional (3D) model of angiogenesis, MRIP-silenced pericytes elevate CEC tube network formation and EC sprouting. Together, these findings reveal a novel cytoskeletal protein-protein interaction between MRIP-βcap73 and βcap73-ROCK, while they point to the pivotal role of MRIP in modulation of pericyte cytoskeletal dynamics, EC growth, and angiogenic induction. Perturbations in this pathway may contribute to the abnormal pericyte contractility that underlies essential hypertension and/or proliferative DR.
MATERIALS AND METHODS
Cell Culture
Microvascular pericytes, isolated from bovine retina as previously described (25), were maintained in 10% bovine calf serum (BCS)-containing DMEM with 1% l-glutamine and penicillin-streptomycin-amphotericin B [Atlanta Biological (Flowery Branch, GA) and Life Technologies (Grand Island, NY)] and used at passages 2–6. Bovine retinal CEC, isolated as previously reported (18), were grown in 5% BCS-containing DMEM and used at passages 10–15.
MRIP Yeast-Two-Hybrid Screen
Yeast-two-hybrid screens were performed as previously described (66). Briefly, the COOH-terminal domain of MRIP was cloned into the yeast-two-hybrid vector pGBT9 expressing the Gal4 DNA-binding domain. The yeast strain Y190 was systematically transformed using the lithium acetate method with a human aorta cDNA library fused with the yeast activation domain vector (Clontech, Mountain View, CA). After growth on selective medium, colonies were confirmed by colony lift filter β-galactosidase assay. The cDNA library plasmid from positive colonies was harvested and sequenced at the Tufts University Core Sequencing Facility.
βcap73 Yeast-Two-Hybrid Screen
The Matchmaker two-hybrid system was employed according to the manufacturer's instructions (Clontech). Briefly, the yeast strain PJ629-2A was transformed with the pAS2 bait vector containing the COOH terminus of βcap73 and selected on plates coated with synthetic dropout (SD) medium lacking Trp. Concentrated cultures transformed with pAS2-βcap73-C were incubated overnight with the mouse Matchmaker cDNA library, which was subcloned into the αACT2 activator vector and pretransformed into the Y187 strain containing LacZ and MEL1 reporters (Clontech) in yeast extract-peptone-dextrose medium (+0.005% adenine hemisulfate and kanamycin). Approximately 5 × 106 diploid colonies, selected on TDO (SD medium lacking Leu, Trp, and His) plates for 14 days at 30°C, were screened. Positive colonies, subcultured in 1 ml of SD medium lacking Leu and Trp and containing 0.003% adenine hemisulfate, were tested for α-galactosidase activity using plates coated with TDO lacking Ade and containing 20 μg/μl X-galactosidase. PCR sorting was employed, and DNA from unique clones was purified and transformed into the Escherichia coli DH5α strain. Purified plasmids were confirmed using restriction digest and sequenced using the GAL4-AD sequencing primer 5′-TACCACTACAATGGATG-3′.
MRIP:βcap73 GST-Binding Assay
Fusion proteins were expressed and binding assays were performed as previously described (66). Briefly, COS1 cells transfected with a myc-tagged βcap73 plasmid were washed twice with cold PBS and lysed in a buffer containing 50 mM Tris, pH 7.6, 7 mM MgCl2, 2 mM EDTA, 2 mg/ml n-dodecyl-β-maltoside, 0.4 mg/ml cholesteryl hemisuccinate, 0.6 M NaCl, 10 mM sodium molybdate, 2 mM PMSF, and protease inhibitor cocktail (Sigma, St. Louis, MO). After 1 h of incubation, lysates were clarified and mixed with GST-fusion proteins prebound to glutathione-agarose beads. After incubation overnight at 4°C, beads were washed, and proteins were eluted in SDS-containing sample buffer. Bound proteins were detected using Western blot analysis for c-myc and GST epitopes.
MRIP-βcap73 Coimmunoprecipitation
Coimmunoprecipitation assays were performed in pericytes as described previously (66). Briefly, cells were lysed in a buffer containing 40 mM Tris, pH 7.6, 0.275 M MgCl2, 4 mM EDTA, 2% Triton X-100, 20% glycerol, 50 mM β-glycerol phosphate, 2 mM PMSF, and protease inhibitor cocktail (Sigma) for 1 h at room temperature. After high-speed centrifugation for 20 min at 4°C, the supernatant was precleared with Protein A-Sepharose beads (Pierce, Rockford, IL) and incubated overnight with anti-MRIP antibodies. Protein A beads were added, and after 2 h of incubation the beads were washed with 50 mM Tris, pH 7.6, 7 mM MgCl2, and 2 mM EDTA. Proteins were eluted with sample buffer and analyzed using Western blotting.
βcap73-ROCK Coimmunoprecipitation
NIH 3T3 cells, transfected with 0.2 μg of pEF BOS-ROCK wild-type vector (a generous gift from Dr. Kozo Kaibuchi, Nagoya University, Japan) with Effectene (Qiagen, Valencia, CA) according to the manufacturer's instructions, were lysed for 10 min in RIPA buffer containing 150 mM NaCl, 30 mM Tris·HCl, pH 8.0, 0.1% SDS, 0.5% sodium deoxycholate, 1% NP-40, and protease inhibitor cocktail. Approximately 10 μg of primary antibody (ROCK K18, Sigma) were incubated with 10 μl of Protein A-Sepharose beads for 1 h at room temperature with gentle rotation. Then 250 μl of cell lysate (∼400 μg) were precleared with 10 μl of beads for 1 h at room temperature and incubated with antibody-bound Protein A-Sepharose overnight at 4°C with gentle rotation. The immunoprecipitate was washed with RIPA buffer before elution in sample buffer, run using SDS-PAGE, and immunoblotted for βcap73 (mouse monoclonal) (63).
Transfection of Pericytes With siRNAs
Pericytes, seeded at 20–30% confluence, were transfected for 96 h with 100 nM control [scrambled (SCR)] or MRIP-specific oligonucleotides (67) using Lipofectamine 2000 according to the manufacturer's instructions for siRNA (Life Technologies).
Western Blotting
Pericytes were lysed in sample buffer and subjected to SDS-PAGE. Proteins were transferred overnight to Protran nitrocellulose (GE, Piscataway, NJ). Membranes, blocked in 5% milk-Tris-buffered saline containing Tween 20 for 1 h at room temperature, were incubated overnight at 4°C with primary antibodies, washed, and then incubated for 2 h at room temperature with horseradish peroxidase-labeled secondary antibodies (1:1,000–1:20,000 dilution; Chemicon/Millipore, Billerica, MA). Membranes were treated with Western C reagent (Bio-Rad, Waltham, MA), and bands were visualized using a UVP imager (Vision Works, Upland, CA). Primary antibodies include β-actin (1:1,000 dilution) (28), nonmuscle myosin IIB (1:2,000 dilution) (24), βcap73 (1:1,000 dilution; Covance custom serum) (14), and lamin A/C (1:10,000 dilution; a generous gift from Dr. L. Gerace, Scripps Research Institute, La Jolla, CA).
Immunofluorescence Microscopy
Cells were plated on coverslips and then fixed and stained as previously reported (14). Primary antibodies included MRIP (Becton-Dickinson, Franklin Lakes, NJ), βcap73 (Covance custom research serum), β-actin (28), and SMA (Biogenex, Fremont, CA). Alexa Fluor 488-phalloidin (0.25 unit/coverslip) was used to stain actin stress fibers (Life Technologies). Cells were imaged as previously reported: wide-field images were captured using a Zeiss Axiovert 200M inverted microscope (14). ImageJ (National Institutes of Health) and the freehand measurement tool were used to measure area occupied by individual cells; for staining intensity measurements, cells were individually outlined, and the MetaMorph threshold function was used to determine the percentage of thresholded area.
Pericyte Spreading Assays
Pericytes were freshly trypsinized, and a suspension of 2–4 × 103 cells/ml was added to a customized chamber slide fitted for a 12-mm cover glass (56). Temperature-controlled, time-lapse microscopy was used to capture an image once every 2 min for 60 min using differential interference contrast. Using the caliper function in MetaMorph software, cell diameter was measured at time 0 and at 60 min postplating.
Pericyte Contractility Assays
Pericyte contractility was assayed as previously described (36, 39, 41), with the following modifications (22). Briefly, silicone (DMPS-12M, Sigma) was spread on coverslips at 42°C for 1 h. Silicone-coated coverslips were passed briefly over a flame to polymerize the substrate and then coated with plasma (36, 39, 41). Rat tail collagen type I (Becton-Dickinson), diluted to 3 μg/ml in PBS, was added to the coverslip and sterilized under UV light for 5 min. Trypsisinzed pericytes, treated with SCR or MRIP siRNA as described above, were plated on silicone-coated coverslips at 5–10% confluence to avoid confounding the analysis by neighboring pericytes. Wrinkles were manually counted and assigned a value of 0.25, 0.5, 0.75, or 1, depending on the approximate degree at which that the wrinkle crossed the cell diameter.
EdU Assays: Pericyte-EC Coculture
Pericytes, silenced as described above, were seeded onto coverslips in 10% BCS-containing DMEM at 1,500 cells/well. At 5 days after siRNA treatment, bovine retinal CEC were added at a 1:1 pericyte-to-EC coculture ratio. On day 6 (i.e., 24 h after addition of CEC), the Click-iT EdU Alexa Fluor 594 imaging kit (Life Technologies) was used according to the manufacturer's instructions with a 4-h pulse with 10 μM 5-ethynyl-2′-deoxyuridine (EdU). Nuclei were counterstained with Hoechst 33342 (1:1,000 dilution; Life Technologies). Pericytes were detected using SMA antibodies. Images were quantified by manual identification of EC that physically touch pericytes (contact-dependent) followed by determination of the percentage of S phase-positive EC nuclei (EdU-positive, SMA-negative cells).
3D Angiogenesis: Pericyte-EC Coculture
The 3D model of angiogenic sprouting was performed as previously reported (8, 26) with the following modifications. Control and MRIP-silenced pericytes were trypsinized 4 days after transfection with siRNA and labeled with DiO (Life Technologies) according to the manufacturer's instructions. The silenced pericytes were mixed into a single-cell suspension with bovine retinal CEC at a ratio of 1:25 (6,000 pericytes/1.5 × 105 EC per well) in 5% BCS-containing DMEM. The coculture was embedded between two layers of the matrix solution (2:1:1 growth factor-reduced Matrigel-collagen I-5% BCS DMEM) in eight-well chamber slides (Becton-Dickinson). Tube networks were allowed to form overnight; on the following day (day 0), a punch wound was generated through the entire thickness of the gel and immediately filled with fresh matrix solution, and live fluorescent and phase-contrast images were captured immediately postinjury and at 24 h after injury. Total endothelial tube formation was determined using ImageJ by subtraction of any cells/structures inside the wound on day 0 from the day 1 totals to normalize each wound and ensure that only new sprouts/tubes were measured. In this way, average tube length, total tube length, and total number of sprouts were calculated.
Statistical Analysis
For all experiments, two-tailed Student's t-tests were performed, and error is plotted as SE. All experiments were performed in triplicate.
RESULTS
βcap73 Directly Interacts With MRIP and ROCK
Because of the critical role of MRIP in regulating mural cell contractile function, in part by localizing RhoA and MLCP to actomyosin-containing stress fibers, we performed a yeast-two-hybrid screen, employing the COOH-terminal domain of MRIP as bait, to investigate how the RhoA pathway and actin effectors might regulate microvascular pericyte cytoskeletal function. Through this genetic analysis, we found that MRIP directly interacts with a previously identified and well-characterized cytoskeletal effector and β-actin-specific capping protein, βcap73 (Fig. 1, A and B). We confirmed this interaction biochemically with in vitro binding assays using various GST-purified MRIP constructs; furthermore, we determined that the Rho-binding domain of MRIP is sufficient to bind βcap73 (Fig. 1C). Additionally, immunoprecipitation and Western blot experiments reveal that the endogenous proteins interact, since antibodies against βcap73 or MRIP immunoprecipitate their respective binding partner from pericyte lysates (Fig. 1D and data not shown).
Fig. 1.

A novel interaction between myosin phosphatase-RhoA-interacting protein (MRIP) and the β-actin-specific capping protein βcap73. A: domain structures of MRIP and βcap73. MRIP has 2 NH2-terminal pleckstrin homology domains (PH), central proline-rich domains (PPP), a central Rho-binding domain (RBD, red-hatched box), and 3 COOH-terminal coiled-coil (CC1, CC2, and CC3) domains. The myosin-binding subunit (MBS) of myosin light chain phosphatase (MLCP) binds to MRIP via the tail portion of the CC2 domain. βcap73 has 6 NH2-terminal ankryin repeats, a central α-helical domain, and a COOH-terminal β-actin-binding domain. B: a yeast-2-hybrid assay reveals an association between MRIP and βcap73. C: in vitro binding assays reveal a direct association of βcap73 specifically with the RBD of MRIP. Top: Coomassie-stained GST expression constructs; bottom: corresponding Western blot for the myc-tagged βcap73 after incubation with various MRIP domains. D: immunoprecipitation with polyclonal antibodies against βcap73 pulls down βcap73 and MRIP from pericyte lysates. E: fluorescence microscopy reveals that MRIP (green, arrow) and βcap73 (red, arrowheads) overlap in a punctate pattern (insets show higher magnification of area in white boxes) along actomyosin stress fibers in pericytes. Scale bars = 25 μm.
With confirmation of the novel MRIP-βcap73 interaction, we used wide-field immunofluorescence microscopy to examine the subcellular localization of these proteins. An overlap of MRIP and βcap73 staining along the length of stress fibers occurs in a punctate pattern, as indicated by the yellow merge in Fig. 1E. There are stress fiber domains that contain solely MRIP; additionally, we noted enhanced MRIP staining in retraction fibers (Fig. 1E, arrow; see Fig. 3B). As previously reported (14, 63, 71), we observe a leading-edge enriched pool of βcap73; moreover, antibodies against βcap73 reveal a strongly positive, reticulated staining pattern in stress fibers (Fig. 1E, arrowheads and inset). Thus, in pericytes, MRIP and βcap73 staining appears in distinct subcellular areas as well as coinciding along actomyosin-containing fibers.
Fig. 3.

MRIP silencing alters pericyte phenotype. A: pericyte cultures treated with scrambled (SCR) or MRIP-specific siRNAs (MRIP) were fixed after 4 days of silencing and stained for MRIP and myosin. MRIP-silenced pericytes did not stain for MRIP but did retain myosin-positive fibers (*). Scale bar = 25 μm. B: quantification of percent thresholded area of MRIP and myosin localization in silenced pericytes. *P < 0.0001. C: Western blot analysis of pericytes treated with SCR or MRIP siRNA. D: pericyte cultures treated with SCR or MRIP siRNAs were fixed after 4 days of silencing and triple-labeled with DAPI (blue), phalloidin (green), and β-actin (red). Scale bar = 100 μm.
Interestingly, in a parallel set of experiments, we employed the β-actin-binding domain contained within βcap73 in an independent yeast-two-hybrid screen and identified p160-ROCK1 as a direct interactor of this isoactin capping protein (Fig. 2). To confirm these genetic data via biochemical and cell biology-based approaches, we performed coimmunoprecipitation and colocalization studies. Indeed, anti-ROCK1 IgG is able to coimmunoprecipitate endogenous βcap73 from 3T3 cells (Fig. 2B). Furthermore, βcap73 colocalizes with ROCK at the leading edges of migrating 3T3 cells (Fig. 2C). As previous data demonstrated an indirect interaction between MRIP and ROCK, our novel data demonstrate that βcap73 interacts directly with both MRIP and ROCK and potentially links MRIP and MLCP regulation/mural cell contractility with the Rho effector pathway.
Fig. 2.

A novel interaction between βcap73 and Rho kinase 1 (ROCK1). A: yeast-2-hybrid assay using the COOH-terminal domain of βcap73 as bait interacted positively with the B2/3.95.2 clone, which is 100% identical to residues 521–767 of Mus musculus ROCK1. B: antibodies against ROCK1 were used to immunoprecipitate βcap73 from 3T3 cell lysates. Lane 1, total cell lysate; lane 2, ROCK1 (immunoprecipitation) and βcap73 (immunoblotting). C: prominent colocalization of βcap73 and ROCK1 in migrating, ROCK1-overexpressing 3T3 cells.
MRIP Silencing Induces Cellular Hypertrophy and Cytoskeletal Reorganization
To determine the effects of MRIP expression on pericyte contractile phenotype, we employed well-characterized siRNAs targeting MRIP (55, 67). Also, to confirm that MRIP is silenced in pericytes, we employed high-resolution light microscopy, as we found that commercially available MRIP antibodies produced strong signals in this assay but were inefficient in Western blot studies. In general, we observe that many silenced cells display a robust reduction in MRIP staining, although they retain prominent myosin localization on stress fibers (Fig. 3A). Indeed, digital analysis of fluorescence intensity reveals MRIP staining in 39.9 ± 7.3% of the total cell area in control cells but only in 16.3 ± 7.3% of the total area in silenced pericytes; thus MRIP localization decreases by 59.1% upon siRNA treatment (Fig. 3B). Additionally, we found myosin intensities to be unchanged between treatments (68.3 ± 7.9% in SCR vs. 62.2 ± 9.5% in MRIP); thus MRIP silencing does not affect the localization of this particular cytoskeletal component (Fig. 3B). Together, our results suggest a reduction of MRIP without an effect on another cytoskeletal component, myosin. Furthermore, MRIP silencing does not alter steady-state levels of the cytoskeletal proteins β-actin, βcap73, and myosin (Fig. 3C); furthermore, preliminary analysis reveals that the levels of smooth muscle myosin and ROCK1/2 are equivalent between populations (data not shown).
MRIP silencing in pericytes also elicits a dramatic phenotypic change accompanied by a markedly reorganized F-actin cytoskeleton, as revealed by imaging using phalloidin staining and anti-β-actin antibodies (Fig. 3D). Consistently, stress fibers appear enlarged and arranged in a parallel, bundled-like orientation in MRIP-silenced pericytes; in SCR-treated cells, fibers appear more heterogeneous and oriented in orthogonal or branched arrays. Quantitative analysis reveals that MRIP-silenced pericytes are 100% larger than their control, SCR siRNA-treated counterparts: 30,902.7 ± 7,946.5 vs. 15,657.1 ± 4,155.5 pixels (data not shown). Together, MRIP silencing increases pericyte size, with an underlying reorganization of cytoskeletal architecture, without altering total cytoskeletal protein levels.
βcap73 Localization is Altered Upon MRIP Silencing
Because MRIP silencing induces a dramatic reorganization of the actin cytoskeleton without perturbing myosin localization, we were interested to learn whether localization of βcap73 would be affected. Under control conditions, MRIP and βcap73 are present along stress fibers in a reticulated pattern (Fig. 1E and Fig. 4A, top and inset). However, upon MRIP silencing, βcap73 localization appears transformed from a punctate to a smooth pattern on the hypertrophied stress fibers (Fig. 4, A and insets). Interestingly, in MRIP-silenced pericytes, not only is lamellipodial formation reduced by 2.4-fold compared with controls (data not shown), but also the few remaining leading-edge structures are largely devoid of βcap73 (Fig. 4B, white box and asterisk). Since βcap73 binds both β-actin and MRIP and a major role of βcap73 is to control β-actin polymerization at the leading edges of migratory or remodeling cells (14, 62, 63, 71), we were interested to learn whether silencing MRIP would similarly impair β-actin recruitment to the leading edge. However, MRIP silencing has no effect on β-actin localization to lamellipodia (Fig. 4C, white boxes and insets). Together, these results suggest that MRIP silencing causes βcap73 relocalization without affecting β-actin positioning at the leading edge in pericytes.
Fig. 4.

MRIP silencing alters βcap73 localization. A: pericytes were treated with siRNA as previously described. Scale bars = 25 μm. B: leading edge localization of βcap73 is prominent in SCR pericytes but is lacking upon MRIP silencing (*). C: β-actin localization is similarly observed in lamellae in SCR and MRIP pericytes (insets). Scale bars = 25 μm.
MRIP Silencing Increases Spreading in Pericytes
Since βcap73 regulates β-actin filament extension at the cytoskeleton-plasma membrane interface and MRIP silencing removes βcap73 from leading edges without a concomitant loss of β-actin, we reasoned that cell spreading in MRIP-silenced pericytes may be altered. Immediately after plating, both SCR and MRIP-silenced pericytes appear rounded; however, after 1 h, MRIP-silenced cells appeared larger and thinner than controls (Fig. 5A). Quantitative analysis reveals that the average cell diameters are not statistically different for both treatments at time 0 (30.3 ± 1.5 and 35.3 ± 1.4 μm for SCR and MRIP, respectively); however, at 60 min, the MRIP-silenced cells are twofold larger than their control counterparts (83.8 ± 5.4 vs. 54.9 ± 3.7 μm; Fig. 5B). Indeed, the cell spreading rate (0.41 ± 0.05 and 0.81 ± 0.09 μm/min for SCR and MRIP, respectively) is elevated by approximately twofold in MRIP-silenced pericytes (Fig. 5C). These data reveal that, despite a robust cytoskeletal architecture under static conditions, if subjected to kinetic assays, MRIP-silenced pericytes display increased β-actin-driven cell spreading.
Fig. 5.
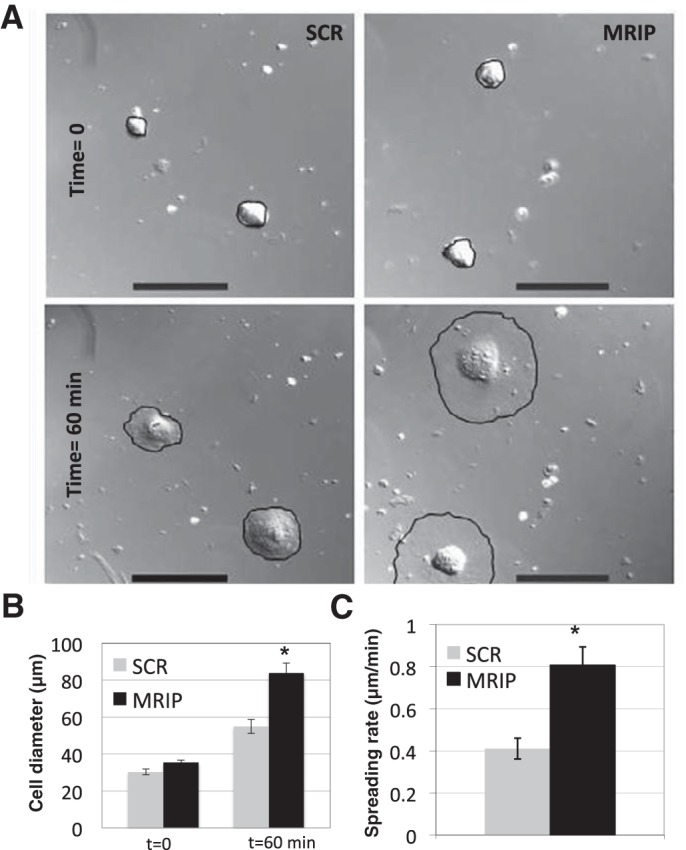
MRIP silencing enhances pericyte spreading. A: differential interference contrast images of SCR and MRIP-silenced pericytes at time 0 (top) and after 60 min of spreading (bottom). Cell edges are outlined in black to demarcate extent of spreading. Scale bars = 100 μm. B: cell diameters at time 0 and after 60 min of spreading. *P < 0.01. C: cell spreading rates. *P < 0.01.
MRIP Silencing Alters Pericyte Cytoskeletal Organization
As previous data demonstrated cytoskeletal hypertrophy and a repositioning of βcap73, we sought to more closely analyze cytoskeletal organization in MRIP-silenced pericytes. In control pericytes, β-actin is mainly situated at the leading edges, with a diffuse cytoplasmic localization and a punctate staining pattern, perhaps reflecting pools of β-actin monomers (Fig. 6A, inset); additionally, phalloidin-stained stress fibers appear short and thin (Fig. 6A, top). However, in MRIP-silenced pericytes, β-actin appears concentrated in the stress fiber-associated cytoskeleton (Fig. 6A, bottom). Quantitative image analysis reveals an increase in β-actin intensity, as the percentage of stained cell area is increased by 2.6-fold (from 21.3 ± 14.5% in SCR to 54.8 ± 13.4% in MRIP; data not shown), suggesting a shift toward increased β-actin incorporation into stress fibers in MRIP-silenced pericytes. While a qualitative reorganization of phalloidin-stained stress fibers is observed in MRIP-silenced cells, we do not observe a significant difference in phalloidin-stained area between treatments (data not shown). Normally, within pericyte populations, within each cell, there are isoactin-enriched domains possessing SMA or β-actin (9). In control pericytes, SMA staining is largely confined to the cell center, while β-actin is primarily localized at the cell periphery (Fig. 6B, top). However, in MRIP-silenced pericytes, SMA and β-actin isoforms are prominently localized on central stress fibers (Fig. 6B, bottom). Phalloidin and SMA costaining reveals a greater incorporation of SMA into stress fibers in MRIP-silenced cells (Fig. 6, A–C, merge) than in control cells, in which there are phalloidin-positive cells without a concomitant SMA colocalization (Fig. 6C). In addition, preliminary observations support an increase in SMA-positive cells in MRIP-silenced pericytes (1.25-fold; data not shown). Together, these data suggest that reduction of MRIP expression leads to a highly reorganized, actin-enriched cytoskeleton.
Fig. 6.

MRIP silencing alters pericyte cytoskeletal arrangement. A: pericytes transfected with SCR or MRIP siRNA were stained with phalloidin (green) and β-actin (red). Insets: pools of monomeric actin or stress fiber incorporation of β-actin in SCR and MRIP pericytes, respectively. Scale bars = 25 μm. B: SCR and MRIP-silenced pericytes were stained for smooth muscle actin (SMA, green) and β-actin (red). Scale bars = 25 μm. C: silenced pericytes were stained with phalloidin (green) and SMA (red). Scale bars = 25 μm.
MRIP Silencing Increases Pericyte Contraction on Deformable Substrates
With the observation that MRIP silencing results in a dramatically reorganized and hypertrophied cytoskeleton, we hypothesized that these cells might possess altered contractile properties. To test this hypothesis, control and MRIP-silenced pericytes were plated onto collagen-coated silicone substrates, and contractility was explored. Control, SCR siRNA-treated cells generate force capable of deforming underlying silicone substrata and possess differential wrinkling patterns, typically aligned in the direction of migration or retraction (Fig. 7A, left). In contrast, the extent to which MRIP-silenced pericytes deform substrates is 2.2 times that in control cells, and wrinkles appear more prominent, typically bisecting the entire cell diameter (Fig. 7A, right). A degree of wrinkling was assigned to each cell: SCR cells displayed an average of 6.7 ± 4.3 wrinkles per cell, while MRIP-silenced cells displayed an average of 13.6 ± 5.2, a twofold increase (Fig. 7B). Also, we find that a greater percentage of control than MRIP cells are unable to deform the substrate (11.5 ± 4.6% vs. 1 ± 0.66%, P < 0.05; data not shown). Thus the reorganized and hypertrophied cytoskeleton in MRIP-silenced pericytes leads to an increase in force production.
Fig. 7.
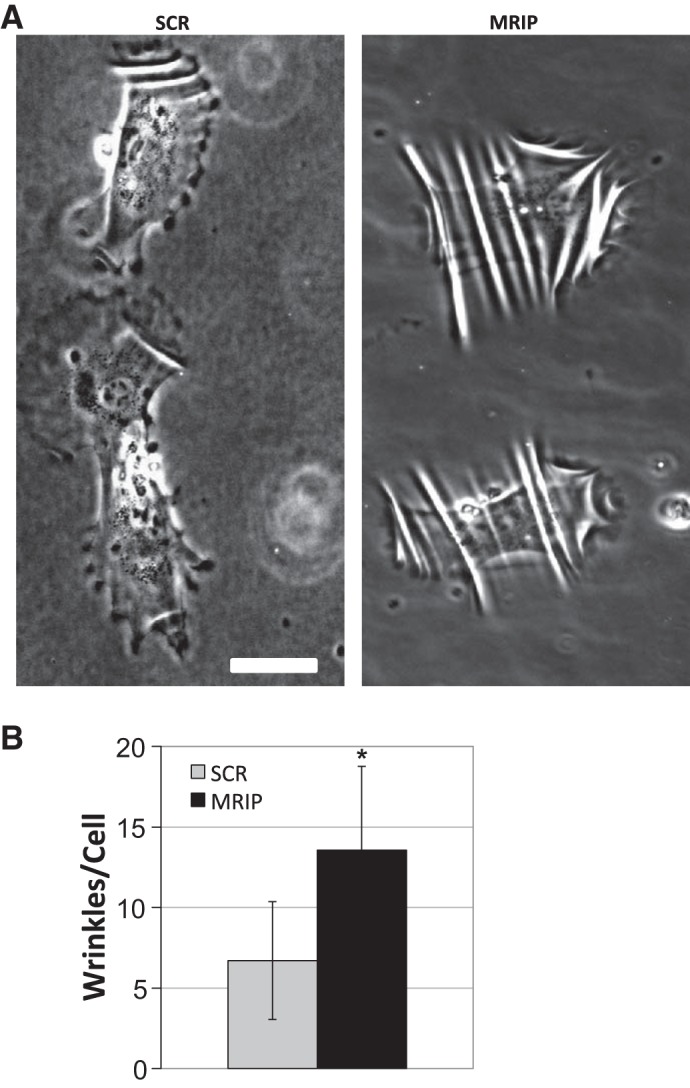
MRIP silencing increases pericyte contractility. A: pericytes treated with siRNA were trypsinized and plated on a collagen-coated silicone substrate. Live cells generating wrinkles on the deformable substrate were imaged 24 h after plating. Scale bar = 50 μm. B: average number of wrinkles per cell revealed an increase in contraction of MRIP-silenced pericytes. *P < 0.05.
MRIP-Deficient Pericytes Are Unable to Maintain EC Growth Arrest
Recently published results reveal that alterations in pericyte RhoA GTP level and activity affect contractile phenotype and impair the ability of pericytes to maintain contact-mediated EC growth arrest in coculture (36, 39, 41). Since MRIP silencing influences pericyte contractility, we tested whether these hypertrophic pericytes would alter EC cycle dynamics. After we silenced MRIP specifically in pericytes, we added CEC to cultures in an equal cell ratio, as this reflects the EC-to-pericyte ratio in vivo in the retina, for a 24-h coculture on coverslips. To determine S phase entry in CEC, live cells were pulsed with EdU, then they were fixed and immunolocalized. In control cells, prominent contact-dependent EC quiescence is observed; however, MRIP-silenced cells are unable to maintain CEC growth arrest (Fig. 8). CEC that are in direct contact with MRIP-silenced pericytes exhibit a significant 2.1-fold increase in the percentage of endothelial S phase entry from 35.5 ± 2.4% in SCR cocultures to 72.6 ± 2.1% in MRIP-silenced conditions (Fig. 8B). These data imply that altered MRIP expression may contribute to abnormal pericyte function and may have pathological implications, as these hypertrophic pericytes are unable to mediate EC growth arrest in a contact-dependent manner.
Fig. 8.
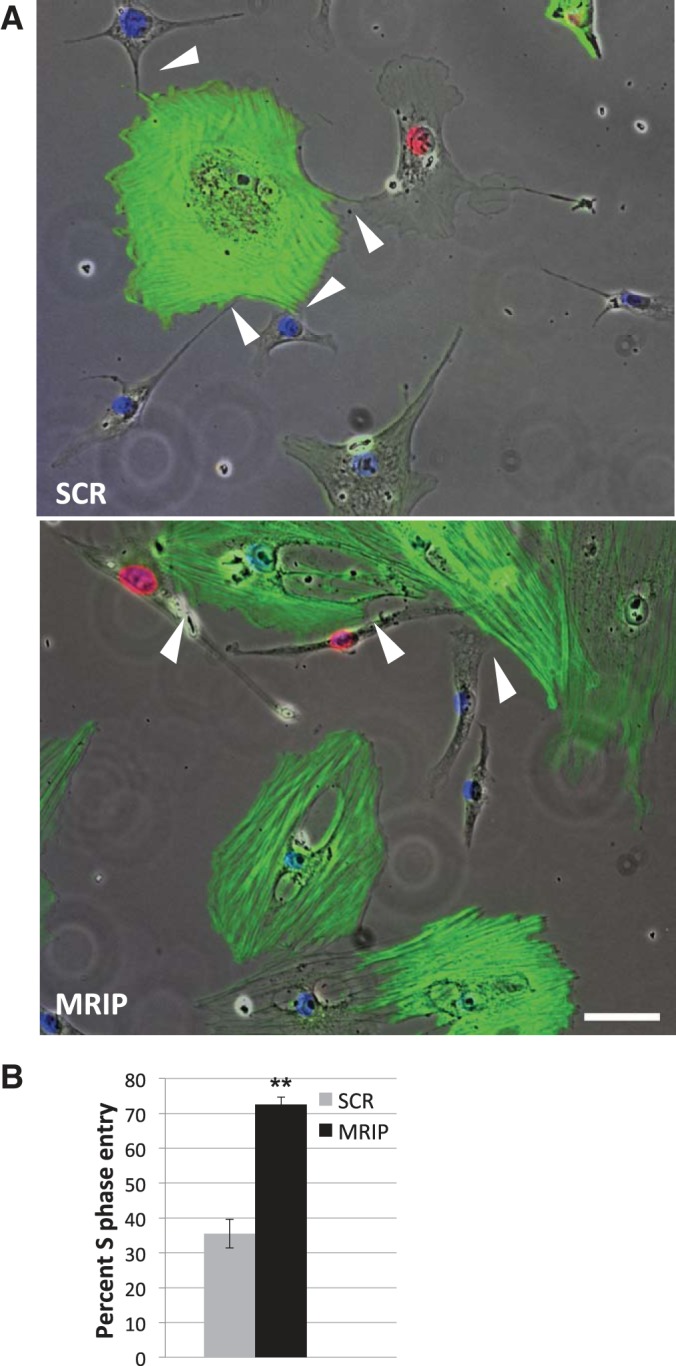
MRIP silencing abrogates pericyte-mediated endothelial cell (EC) growth arrest. A: pericytes treated with SCR or MRIP siRNA were plated in coculture with EC and stained with SMA (green), DAPI (blue), and 5-ethynyl-2′-deoxyuridine (red). All 3 fluorescent fields were merged against phase images, and EC physically contacting pericytes are evident (arrowheads). Scale bar = 25 μm. B: percentage of S phase-positive nuclei for capillary EC in direct contact with pericytes. **P < 0.001.
Pericyte MRIP Silencing Elevates Angiogenesis in a 3D Model of Microvascular Injury
We recently developed a 3D model enabling a quantitative analysis of the angiogenic response to injury. In this model system, CEC are able to form a microvascular network possessing capillary-like structures that generate bona fide lumens (8, 26). Using this method, we were able to test whether angiogenic responses to injury would be altered when we silenced MRIP specifically in pericytes and cultured these cells with normal CEC. After morphogenesis was established and a punch wound was generated, we imaged cocultures immediately after injury to compare and correct for any cell extension into the “wound” on day 0 compared with day 1 of angiogenic sprouting (Fig. 9, A and insets). Interestingly, the total tube length is significantly increased in MRIP-silenced pericyte cocultures compared with controls (to 1,597.9 ± 362.3 from 979.1 ± 271.9; Fig. 9B), an effect that was due to an increase in total number of angiogenic sprouts (to 63 ± 9.5 from 41.5 ± 8.8; Fig. 9B), rather than an increase in average tube length (to 24.1 ± 1.1 from 23.6 ± 2.1; data not shown). Thus, in a 3D model of angiogenesis, MRIP-silenced pericytes dramatically increase endothelial tube formation via an increase in sprouting. Together, alteration of pericyte mechanochemistry and contractile phenotype affects microvascular cell growth and angiogenic potential.
Fig. 9.

MRIP silencing in pericytes increases EC sprouting in a 3-dimensional coculture angiogenesis assay. A: DiO-labeled pericytes were treated with SCR or MRIP siRNA, and EC were encased between 2 layers of a gelatinous substrate. After an initial, 24-h phase of morphogenesis, a punch wound was generated through the gel, and the resulting defect was filled with fresh gel (left). Live cell image capture was used to observe angiogenesis (white arrows) into the “wound” on day 1 postinjury (right). B: quantification using ImageJ reveals a significant increase in total tube length in MRIP-silenced cocultures. This effect was due to a significant increase in angiogenic sprouts, rather than an increase in average tube length (data not shown). *P < 0.05.
DISCUSSION
We report novel cytoskeletal protein-protein interactions among key regulators of pericyte contractility: MRIP with βcap73 and βcap73 with ROCK. Results conclusively demonstrate that alteration of pericyte MRIP expression plays a pivotal role in control of pericyte contractility and force production, which, in turn, modulates CEC growth dynamics and angiogenic status. These findings provide important new insights into the essential role(s) of cytoskeletal protein-protein interactions and pericyte chemomechanics as “angiogenic activators” or “switches.”
MRIP Directly Associates With βcap73
It has been reported that MRIP associates with stress fibers together with MLCP and RhoA and, indirectly, with ROCK (55, 66, 67). Here we report novel βcap73 interactions with MRIP and ROCK. As MRIP indirectly binds ROCK, a key regulator of MLCP activity (55, 66, 67), these findings might provide insight into the RhoA/ROCK-mediated inactivation of MLCP. Future work will explore the functional consequence of these novel interactions. We hypothesize that multiprotein interactions would be possible, since 1) MRIP is able to simultaneously bind both the myosin-binding subunit of MLCP and RhoA (66) and 2) a direct interaction exists between βcap73 and ROCK. In future studies we aim to test whether MRIP control of pericyte contractile phenotype involves 1) stress fiber localization of MLCP and RhoA via its actin-binding property, 2) incorporation of ROCK into the MLCP regulatory complex indirectly through the ROCK-βcap73 interaction, and 3) MLCP activity via ROCK-mediated inhibition (Fig. 10).
Fig. 10.
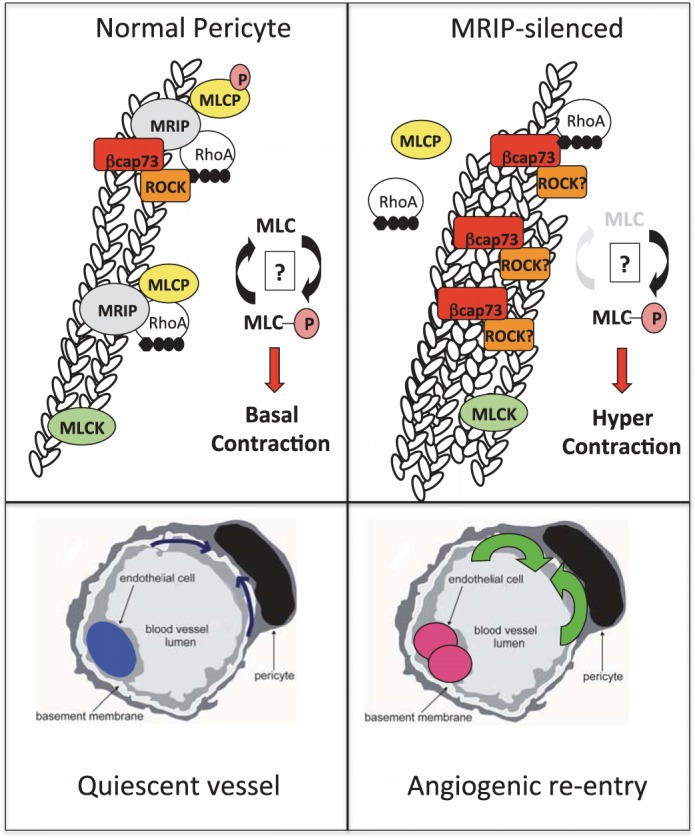
The MRIP-βcap73 complex governs pericyte contractile phenotype. Left: in normal pericytes, MRIP directly binds MLCP and RhoA and indirectly interacts with ROCK. Through the novel MRIP-βcap73 interaction and the newly described βcap73-ROCK interaction, we propose a novel signaling complex, with MRIP and βcap73 mediating the RhoA/ROCK signaling pathway, which ultimately regulates MLCP activity and pericyte contractility. It is likely that there are subdomains containing the 1) novel MRIP-βcap73 complex, 2) MRIP and MLCP, and 3) myosin light chain (MLC) kinase (MLCK), all working toward maintaining a balance of MLC-phosphorylated (P) MLC levels, which results in basal pericyte tonus. The pericyte, with normal mechanical stiffness, then relays signals to the underlying EC to maintain quiescence (growth arrest, blue nucleus). Right: upon MRIP silencing, MLCP is no longer stress fiber-associated. These events give rise to a diminution of MLC dephosphorylation; MLCK and ROCK phosphorylate MLC without MLCP opposition, which gives rise to the hypertrophic state; MRIP-silenced and hypercontractile pericytes cannot sustain contact-dependent EC growth arrest. In turn, EC enter the S phase (pink nuclei), “priming” microvascular growth and angiogenic activation.
MRIP Silencing in Pericytes
As in vascular smooth muscle cells (55, 67), MRIP silencing in pericytes increases cell size and induces marked cytoskeletal reorganization without perturbing other cytoskeletal proteins. Increased β-actin incorporation into phalloidin-positive pericyte stress fibers aligns with previous work demonstrating an augmented stress fiber-associated pool of actin in smooth muscle (67). Taken together, our results point to conservation of MRIP function across the mural cell spectrum.
MRIP silencing alters βcap73 localization and isoactin organization.
Interestingly, while silencing MRIP does not affect βcap73 expression levels, there is a marked βcap73 reorganization, as this isoactin-specific regulator becomes repositioned from the leading edge into the stress fiber pool. Similarly, βcap73 mislocalization occurs upon overexpression of a mutant Arf6, which causes βcap73 entrapment within the recycling endosomes, leading to dysregulated β-actin filament assembly and resultant alterations in cell motility, spreading, and projection formation (56). Moreover, ROCK-mediated phosphorylation of MLCP/MLC (1, 15, 33, 34) and ezrin (50, 20, 29) leads to alterations in contractility and motility, respectively; furthermore, with our observations that βcap73 and ROCK directly bind, we predict that βcap73 localization is central to modulation of ROCK signaling. We envision that regulation of pericyte contractile phenotype is achieved via balancing leading edge-associated βcap73-ROCK with stress fiber-localized βcap73 signaling complex, which consists of MRIP, RhoA, ROCK, and MLCP.
It has been well documented that microvascular pericytes express multiple cytoskeletal and contractile protein isoforms that possess unique subcellular roles (23, 25). In control pericytes, we find β-actin localized in plasma membrane regions of moving cytoplasm, reminiscent of structures observed in the leading edges of cells including fibroblasts, neurons, epithelial cells, and EC (4, 9, 19, 23, 28). In MRIP-silenced pericytes, we observe an increased localization of β-actin to stress fibers and a dramatic hypertrophy of the cytoskeleton. Altogether, we observe a marked shift in cytoskeletal dynamics, with hypertrophied stress fibers containing increased amounts of βcap73, β-actin, and SMA in MRIP-silenced pericytes.
MRIP silencing increases pericyte spreading.
We observe that silencing pericyte MRIP expression leads to a repositioning of βcap73 without a corresponding diminution of β-actin in lamellae, leading to elevated spreading rates. Cells released from mitotic growth arrest accumulate phosphorylated MLC, which decreases as cells complete spreading (72); furthermore, spreading cells display elevated ROCK activity and phosphorylated MLC (5, 61). In MRIP-silenced pericytes, MLCP is no longer targeted to the cytoskeleton and MLC kinase (MLCK)/ROCK kinase activity would be largely unopposed, potentially leading to increased levels of phosphorylated MLC to foster the enhanced cell spreading response. Future experiments will be central in determination of the precise role of each kinase in modulating pericyte physiology. Interestingly, in neuronal cells, silencing the murine homolog of MRIP, p116RIP, prevents cell spreading and the RhoA/ROCK-regulated neuronal outgrowth (46). These results suggest that while MRIP may similarly converge on the RhoA/ROCK pathway, the resultant signaling may be differentially regulated in a cell type-specific manner to affect cytoskeletal outcomes.
MRIP silencing increases pericyte contractility.
Silencing pericyte MRIP induces a marked hypertrophy and a forcible deformation of underlying substrata compared with controls, which is consistent with earlier studies where cytoskeletal reorganization and expression of SMA enhanced mural cell contractility (23, 27, 64). Furthermore, specific cytoskeleton-disrupting agents/pharmacological inhibitors similarly reverse the enhanced cytoskeletal stiffening and contractile activity (41). Reorganization/expression of smooth muscle proteins underlies contractile force generation and has been shown to be dependent on Rho signaling in a variety of cell types (32, 35, 39, 44, 48, 68). Moreover, ROCK can directly induce smooth muscle contraction via phosphorylation of MLC (37) and has been implicated in the generation of central stress fibers via increased phosphorylated MLC levels, while MLCK plays a role in assembly of peripheral stress fibers (31, 69). Thus ongoing experiments are aimed at exploring the role of each kinase in controlling pericyte contractile phenotype.
Coculture of CEC and MRIP-Silenced Pericytes
MRIP silencing prevents contact-mediated EC growth arrest.
Similar to MRIP-silenced, hypertrophic pericytes that are unable to sustain CEC growth arrest in a contact-dependent manner, pericytes with altered Rho GTP status or ROCK activity influence both pericyte contractile phenotype and abrogate maintenance of EC growth arrest (35, 36, 39, 41). While S phase entry is not always indicative of cytokinesis, preliminary data reveal an increase in the ratio of CEC to pericytes and in the total number of EC in MRIP-silenced cocultures (1.7- and 1.4-fold, respectively; data not shown) on day 7. Interestingly, we observe an increase in EC cycle entry when MRIP-silenced pericytes and EC are not in direct contact (52.3 ± 3.8% vs. 30.6 ± 4.1% for MRIP- and SCR-treated cocultures, respectively; data not shown), which has also been reported for cocultures in which pericyte Rho GTPase levels are manipulated (39). In preliminary studies using pericyte-conditioned medium from SCR-treated or MRIP-silenced pericytes, we were unable to observe a marked difference in CEC proliferation or S phase entry between treatments (data not shown). Importantly, Lee et al. (41) report that pericytes can deform their underlying substrates in a manner that may be capable of transmitting forces to adjacent CEC. Interestingly, our preliminary data reveal that mechanical strain, similar to that produced by pericytes, is sufficient to induce CEC S phase entry (unpublished observations). A rigorous scrutiny of the extracellular matrices/tension produced by SCR-treated and MRIP-silenced pericytes may offer important new insights linked to a pericyte-driven, angiogenically active endothelium.
Silencing pericyte MRIP alters angiogenic potential.
Endothelial morphogenesis is achieved through a balance between cell-cell interactions, cell-matrix binding, cytoskeletal reorganization, and alterations in intracellular tension (7, 11, 65). The RhoA/ROCK pathway has been implicated in the mechanical-dependent regulation of angiogenesis (43), and RhoA-dependent “mechanosensing” can be pathologically misregulated in tumors (17). Also, there is a growing body of evidence of pericyte-mediated mechanochemical regulation of microvascular physiology (36, 38, 39, 41). As MRIP plays an important role in regulating the RhoA/ROCK-mediated regulation of contractile phenotype, we used a 3D coculture model of injury and repair to test whether silencing pericyte MRIP would affect angiogenesis. Indeed, when MRIP-silenced pericytes are cocultured with CEC, we observe a marked increase in angiogenic activation and sprouting postinjury. Our data are consistent with the growing body of evidence of the pivotal roles of pericytes and pericyte-like cells in modulating microvascular physiology and function (2, 6, 10, 12, 13, 38, 49, 70). For example, when adipose-derived stem cells, which can be shifted to a pericyte-like phenotype with TGF-β pretreatment, are delivered intravitreally, prevention of vascular dropout or microvascular stabilization can be achieved (45). Ultimately, we predict a physiological-pathological spectrum of pericyte contractility, which either maintains vascular stability and CEC quiescence or fosters an angiogenic activation response downstream of mechanical alterations.
Conclusion
It is generally accepted that pericyte loss, via apoptosis and/or migration, represents a hallmark of DR (40, 42, 52, 58). Moreover, pericytes maintain EC growth arrest and vascular stability through direct contact (51) and soluble mediators (54). However, recently published work and our present findings support a paradigm shift wherein the frank loss of pericytes may not be required to reverse EC quiescence or induce angiogenic “switching.” Indeed, our findings suggest that subtle alterations in microenvironment or pericyte chemomechanics play key roles in “tipping” EC into a growth-promoted, angiogenically activated state (36, 39, 41). In this vein, pericyte loss may not be a prerequisite for angiogenic activation and diabetic vasculopathy. Perhaps dysregulation of microvascular tonus, initiated by diabetes-induced perturbations in pericyte MRIP and/or Rho GTPase signaling, alters pericyte contractility, causing retinal microvascular pathology and disease inception.
GRANTS
This work was supported by National Institute of Diabetes and Digestive and Kidney Diseases Grant T32 DK-07542, National Heart, Lung, and Blood Institute Training Program in Cardiovascular Research/Molecular Cardiology Research Grant HL-069770 (J. T. Durham), and National Institutes of Health Grants HL-074069 (H. K. Surks) and EY-15125, EY-19533, and EY-022063 (I. M. Herman).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
J.T.D., H.K.S., and I.M.H. are responsible for conception and design of the research; J.T.D., H.K.S., and B.M.D. performed the experiments; J.T.D., H.K.S., B.M.D., and I.M.H. analyzed the data; J.T.D., H.K.S., B.M.D., and I.M.H. interpreted the results of the experiments; J.T.D. prepared the figures; J.T.D. drafted the manuscript; J.T.D. and I.M.H. edited and revised the manuscript; J.T.D. and I.M.H. approved the final version of the manuscript.
ACKNOWLEDGMENTS
We are grateful to Patrice Tellier for experimental support in the βcap73-ROCK interaction project and to Dr. Tatiana Demidova-Rice for critical reading of the manuscript.
REFERENCES
- 1.Amano M, Ito M, Kimura K, Fukata Y, Chihara K, Nakano T, Matsuura Y, Kaibuchi K. Phosphorylation and activation of myosin by Rho-associated kinase (Rho-kinase). J Biol Chem 271: 20246–20249, 1996 [DOI] [PubMed] [Google Scholar]
- 2.Armulik A, Genove G, Betsholtz C. Pericytes: developmental, physiological, and pathological perspectives, problems, and promises. Dev Cell 21: 193–215, 2011 [DOI] [PubMed] [Google Scholar]
- 3.Arnaoutova I, George J, Kleinman HK, Benton G. The endothelial cell tube formation assay on basement membrane turns 20: state of the science and the art. Angiogenesis 12: 267–274, 2009 [DOI] [PubMed] [Google Scholar]
- 4.Bassell GJ, Zhang H, Byrd AL, Femino AM, Singer RH, Taneja KL, Lifshitz LM, Herman IM, Kosik KS. Sorting of β-actin mRNA and protein to neurites and growth cones in culture. J Neurosci 18: 251–265, 1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bhadriraju K, Yang M, Alom Ruiz S, Pirone D, Tan J, Chen CS. Activation of ROCK by RhoA is regulated by cell adhesion, shape, and cytoskeletal tension. Exp Cell Res 313: 3616–3623, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Blocki A, Wang Y, Koch M, Peh P, Beyer S, Law P, Hui J, Raghunath M. Not all MSCs can act as pericytes: functional in vitro assays to distinguish pericytes from other mesenchymal stem cells in angiogenesis. Stem Cells Dev 22: 2347–2355, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chen CS, Mrksich M, Huang S, Whitesides GM, Ingber DE. Micropatterned surfaces for control of cell shape, position, and function. Biotechnol Prog 14: 356–363, 1998 [DOI] [PubMed] [Google Scholar]
- 8.Demidova-Rice TN, Geevarghese A, Herman IM. Bioactive peptides derived from vascular endothelial cell extracellular matrices promote microvascular morphogenesis and wound healing in vitro. Wound Repair Regen 19: 59–70, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.DeNofrio D, Hoock TC, Herman IM. Functional sorting of actin isoforms in microvascular pericytes. J Cell Biol 109: 191–202, 1989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Diaz-Flores L, Gutierrez R, Madrid JF, Varela H, Valladares F, Acosta E, Martin-Vasallo P, Diaz-Flores L, Jr. Pericytes: morphofunction, interactions and pathology in a quiescent and activated mesenchymal cell niche. Histol Histopathol 24: 909–969, 2009 [DOI] [PubMed] [Google Scholar]
- 11.Dike LE, Chen CS, Mrksich M, Tien J, Whitesides GM, Ingber DE. Geometric control of switching between growth, apoptosis, and differentiation during angiogenesis using micropatterned substrates. In Vitro Cell Dev Biol Anim 35: 441–448, 1999 [DOI] [PubMed] [Google Scholar]
- 12.Dulmovits BM, Herman IM. Microvascular remodeling and wound healing: a role for pericytes. Int J Biochem Cell Biol 44: 1800–1812, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Durham JT, Herman IM. Microvascular modifications in diabetic retinopathy. Curr Diab Rep 11: 253–264, 2011 [DOI] [PubMed] [Google Scholar]
- 14.Durham JT, Herman IM. Inhibition of angiogenesis in vitro: a central role for β-actin dependent cytoskeletal remodeling. Microvasc Res 77: 281–288, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Feng J, Ito M, Ichikawa K, Isaka N, Nishikawa M, Hartshorne DJ, Nakano T. Inhibitory phosphorylation site for Rho-associated kinase on smooth muscle myosin phosphatase. J Biol Chem 274: 37385–37390, 1999 [DOI] [PubMed] [Google Scholar]
- 16.Fruttiger M. Development of the retinal vasculature. Angiogenesis 10: 77–88, 2007 [DOI] [PubMed] [Google Scholar]
- 17.Ghosh K, Thodeti CK, Dudley AC, Mammoto A, Klagsbrun M, Ingber DE. Tumor-derived endothelial cells exhibit aberrant Rho-mediated mechanosensing and abnormal angiogenesis in vitro. Proc Natl Acad Sci USA 105: 11305–11310, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gitlin JD, D'Amore PA. Culture of retinal capillary cells using selective growth media. Microvasc Res 26: 74–80, 1983 [DOI] [PubMed] [Google Scholar]
- 19.Gunning P, Weinberger R, Jeffrey P. Actin and tropomyosin isoforms in morphogenesis. Anat Embryol (Berl) 195: 311–315, 1997 [DOI] [PubMed] [Google Scholar]
- 20.Haas MA, Vickers JC, Dickson TC. Rho kinase activates ezrin-radixin-moesin (ERM) proteins and mediates their function in cortical neuron growth, morphology and motility in vitro. J Neurosci Res 85: 34–46, 2007 [DOI] [PubMed] [Google Scholar]
- 21.Hammes HP. Pericytes and the pathogenesis of diabetic retinopathy. Horm Metab Res 37 Suppl 1: 39–43, 2005 [DOI] [PubMed] [Google Scholar]
- 22.Harris AK, Wild P, Stopak D. Silicone rubber substrata: a new wrinkle in the study of cell locomotion. Science 208: 177–179, 1980 [DOI] [PubMed] [Google Scholar]
- 23.Herman IM. Actin isoforms. Curr Opin Cell Biol 5: 48–55, 1993 [DOI] [PubMed] [Google Scholar]
- 24.Herman IM, Crisona NJ, Pollard TD. Relation between cell activity and the distribution of cytoplasmic actin and myosin. J Cell Biol 90: 84–91, 1981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Herman IM, D'Amore PA. Microvascular pericytes contain muscle and nonmuscle actins. J Cell Biol 101: 43–52, 1985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Herman IM, Leung A. Creation of human skin equivalents for the in vitro study of angiogenesis in wound healing. Methods Mol Biol 467: 241–248, 2009 [DOI] [PubMed] [Google Scholar]
- 27.Herrera AM, McParland BE, Bienkowska A, Tait R, Pare PD, Seow CY. “Sarcomeres” of smooth muscle: functional characteristics and ultrastructural evidence. J Cell Sci 118: 2381–2392, 2005 [DOI] [PubMed] [Google Scholar]
- 28.Hoock TC, Newcomb PM, Herman IM. β-Actin and its mRNA are localized at the plasma membrane and the regions of moving cytoplasm during the cellular response to injury. J Cell Biol 112: 653–664, 1991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hopkins AM, Pineda AA, Winfree LM, Brown GT, Laukoetter MG, Nusrat A. Organized migration of epithelial cells requires control of adhesion and protrusion through Rho kinase effectors. Am J Physiol Gastrointest Liver Physiol 292: G806–G817, 2007 [DOI] [PubMed] [Google Scholar]
- 30.Kamouchi M, Ago T, Kuroda J, Kitazono T. The possible roles of brain pericytes in brain ischemia and stroke. Cell Mol Neurobiol 32: 159–165, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Katoh K, Kano Y, Amano M, Kaibuchi K, Fujiwara K. Stress fiber organization regulated by MLCK and Rho-kinase in cultured human fibroblasts. Am J Physiol Cell Physiol 280: C1669–C1679, 2001 [DOI] [PubMed] [Google Scholar]
- 32.Kawada N, Seki S, Kuroki T, Kaneda K. ROCK inhibitor Y-27632 attenuates stellate cell contraction and portal pressure increase induced by endothelin-1. Biochem Biophys Res Commun 266: 296–300, 1999 [DOI] [PubMed] [Google Scholar]
- 33.Kawano Y, Fukata Y, Oshiro N, Amano M, Nakamura T, Ito M, Matsumura F, Inagaki M, Kaibuchi K. Phosphorylation of myosin-binding subunit (MBS) of myosin phosphatase by Rho-kinase in vivo. J Cell Biol 147: 1023–1038, 1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kimura K, Ito M, Amano M, Chihara K, Fukata Y, Nakafuku M, Yamamori B, Feng J, Nakano T, Okawa K, Iwamatsu A, Kaibuchi K. Regulation of myosin phosphatase by Rho and Rho-associated kinase (Rho-kinase). Science 273: 245–248, 1996 [DOI] [PubMed] [Google Scholar]
- 35.Kolyada AY, Riley KN, Herman IM. Rho GTPase signaling modulates cell shape and contractile phenotype in an isoactin-specific manner. Am J Physiol Cell Physiol 285: C1116–C1121, 2003 [DOI] [PubMed] [Google Scholar]
- 36.Kotecki M, Zeiger AS, Van Vliet KJ, Herman IM. Calpain- and talin-dependent control of microvascular pericyte contractility and cellular stiffness. Microvasc Res 80: 339–348, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kureishi Y, Kobayashi S, Amano M, Kimura K, Kanaide H, Nakano T, Kaibuchi K, Ito M. Rho-associated kinase directly induces smooth muscle contraction through myosin light chain phosphorylation. J Biol Chem 272: 12257–12260, 1997 [DOI] [PubMed] [Google Scholar]
- 38.Kutcher ME, Herman IM. The pericyte: cellular regulator of microvascular blood flow. Microvasc Res 77: 235–246, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kutcher ME, Kolyada AY, Surks HK, Herman IM. Pericyte Rho GTPase mediates both pericyte contractile phenotype and capillary endothelial growth state. Am J Pathol 171: 693–701, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kuwabara T, Cogan DG. Retinal vascular patterns. VI. Mural cells of the retinal capillaries. Arch Ophthalmol 69: 492–502, 1963 [DOI] [PubMed] [Google Scholar]
- 41.Lee S, Zeiger A, Maloney J, Maciej K, Van Vliet K, Herman IM. Pericyte actomyosin-mediated contraction at the cell-material interface can modulate the microvascular niche. J Phys Condens Matter 22: 1–11, 2010 [DOI] [PubMed] [Google Scholar]
- 42.Li W, Yanoff M, Liu X, Ye X. Retinal capillary pericyte apoptosis in early human diabetic retinopathy. Chin Med J (Engl) 110: 659–663, 1997 [PubMed] [Google Scholar]
- 43.Mammoto A, Connor KM, Mammoto T, Yung CW, Huh D, Aderman CM, Mostoslavsky G, Smith LE, Ingber DE. A mechanosensitive transcriptional mechanism that controls angiogenesis. Nature 457: 1103–1108, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Masamune A, Kikuta K, Satoh M, Satoh K, Shimosegawa T. Rho kinase inhibitors block activation of pancreatic stellate cells. Br J Pharmacol 140: 1292–1302, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mendel TA, Clabough EB, Kao DS, Demidova-Rice TN, Durham JT, Zotter BC, Seaman SA, Cronk SM, Rakoczy EP, Katz AJ, Herman IM, Peirce SM, Yates PA. Pericytes derived from adipose-derived stem cells protect against retinal vasculopathy. PLos One 8: e65691, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mulder J, Ariaens A, van den Boomen D, Moolenaar WH. p116Rip targets myosin phosphatase to the actin cytoskeleton and is essential for RhoA/ROCK-regulated neuritogenesis. Mol Biol Cell 15: 5516–5527, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mulder J, Poland M, Gebbink MF, Calafat J, Moolenaar WH, Kranenburg O. p116Rip is a novel filamentous actin-binding protein. J Biol Chem 278: 27216–27223, 2003 [DOI] [PubMed] [Google Scholar]
- 48.Murata T, Arii S, Nakamura T, Mori A, Kaido T, Furuyama H, Furumoto K, Nakao T, Isobe N, Imamura M. Inhibitory effect of Y-27632, a ROCK inhibitor, on progression of rat liver fibrosis in association with inactivation of hepatic stellate cells. J Hepatol 35: 474–481, 2001 [DOI] [PubMed] [Google Scholar]
- 49.Nassiri SM, Rahbarghazi R. Interactions of mesenchymal stem cells with endothelial cells. Stem Cells Dev 23: 319–332, 2014 [DOI] [PubMed] [Google Scholar]
- 50.Onoue N, Nawata J, Tada T, Zhulanqiqige D, Wang H, Sugimura K, Fukumoto Y, Shirato K, Shimokawa H. Increased static pressure promotes migration of vascular smooth muscle cells: involvement of the Rho-kinase pathway. J Cardiovasc Pharmacol 51: 55–61, 2008 [DOI] [PubMed] [Google Scholar]
- 51.Orlidge A, D'Amore PA. Inhibition of capillary endothelial cell growth by pericytes and smooth muscle cells. J Cell Biol 105: 1455–1462, 1987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Pfister F, Feng Y, vom Hagen F, Hoffmann S, Molema G, Hillebrands JL, Shani M, Deutsch U, Hammes HP. Pericyte migration: a novel mechanism of pericyte loss in experimental diabetic retinopathy. Diabetes 57: 2495–2502, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Pimentel-Coelho PM, Rivest S. The early contribution of cerebrovascular factors to the pathogenesis of Alzheimer's disease. Eur J Neurosci 35: 1917–1937, 2012 [DOI] [PubMed] [Google Scholar]
- 54.RayChaudhury A, D'Amore PA. Endothelial cell regulation by transforming growth factor-β. J Cell Biochem 47: 224–229, 1991 [DOI] [PubMed] [Google Scholar]
- 55.Riddick N, Ohtani K, Surks HK. Targeting by myosin phosphatase-RhoA interacting protein mediates RhoA/ROCK regulation of myosin phosphatase. J Cell Biochem 103: 1158–1170, 2008 [DOI] [PubMed] [Google Scholar]
- 56.Riley KN, Maldonado AE, Tellier P, D'Souza-Schorey C, Herman IM. βcap73-ARF6 interactions modulate cell shape and motility after injury in vitro. Mol Biol Cell 14: 4155–4161, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Robison WG, Jr., McCaleb ML, Feld LG, Michaelis OE, Laver N, Mercandetti M. Degenerated intramural pericytes (“ghost cells”) in the retinal capillaries of diabetic rats. Curr Eye Res 10: 339–350, 1991 [DOI] [PubMed] [Google Scholar]
- 59.Schevzov G, Lloyd C, Gunning P. High level expression of transfected β- and γ-actin genes differentially impacts on myoblast cytoarchitecture. J Cell Biol 117: 775–785, 1992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Schultz GS, Davidson JM, Kirsner RS, Bornstein P, Herman IM. Dynamic reciprocity in the wound microenvironment. Wound Repair Regen 19: 134–148, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Senju Y, Miyata H. The role of actomyosin contractility in the formation and dynamics of actin bundles during fibroblast spreading. J Biochem 145: 137–150, 2009 [DOI] [PubMed] [Google Scholar]
- 62.Shuster CB, Herman IM. Indirect association of ezrin with F-actin: isoform specificity and calcium sensitivity. J Cell Biol 128: 837–848, 1995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Shuster CB, Lin AY, Nayak R, Herman IM. βcap73: a novel β-actin-specific binding protein. Cell Motil Cytoskeleton 35: 175–187, 1996 [DOI] [PubMed] [Google Scholar]
- 64.Sobue K, Hayashi K, Nishida W. Expressional regulation of smooth muscle cell-specific genes in association with phenotypic modulation. Mol Cell Biochem 190: 105–118, 1999 [PubMed] [Google Scholar]
- 65.Stroka KM, Aranda-Espinoza H. Effects of morphology vs. cell-cell interactions on endothelial cell stiffness. Cell Mol Bioeng 4: 9–27, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Surks HK, Richards CT, Mendelsohn ME. Myosin phosphatase-Rho interacting protein. A new member of the myosin phosphatase complex that directly binds RhoA. J Biol Chem 278: 51484–51493, 2003 [DOI] [PubMed] [Google Scholar]
- 67.Surks HK, Riddick N, Ohtani K. M-RIP targets myosin phosphatase to stress fibers to regulate myosin light chain phosphorylation in vascular smooth muscle cells. J Biol Chem 280: 42543–42551, 2005 [DOI] [PubMed] [Google Scholar]
- 68.Takaishi K, Matozaki T, Nakano K, Takai Y. Multiple downstream signalling pathways from ROCK, a target molecule of Rho small G protein, in reorganization of the actin cytoskeleton in Madin-Darby canine kidney cells. Genes Cells 5: 929–936, 2000 [DOI] [PubMed] [Google Scholar]
- 69.Totsukawa G, Yamakita Y, Yamashiro S, Hartshorne DJ, Sasaki Y, Matsumura F. Distinct roles of ROCK (Rho-kinase) and MLCK in spatial regulation of MLC phosphorylation for assembly of stress fibers and focal adhesions in 3T3 fibroblasts. J Cell Biol 150: 797–806, 2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Traktuev DO, Merfeld-Clauss S, Li J, Kolonin M, Arap W, Pasqualini R, Johnstone BH, March KL. A population of multipotent CD34-positive adipose stromal cells share pericyte and mesenchymal surface markers, reside in a periendothelial location, and stabilize endothelial networks. Circ Res 102: 77–85, 2008 [DOI] [PubMed] [Google Scholar]
- 71.Welch AY, Herman IM. Cloning and characterization of βCAP73, a novel regulator of β-actin assembly. Int J Biochem Cell Biol 34: 864–881, 2002 [DOI] [PubMed] [Google Scholar]
- 72.Yamakita Y, Yamashiro S, Matsumura F. In vivo phosphorylation of regulatory light chain of myosin II during mitosis of cultured cells. J Cell Biol 124: 129–137, 1994 [DOI] [PMC free article] [PubMed] [Google Scholar]