

Abstract
E-cadherin is essential for the integrity of adherens junctions between lung epithelial cells, and the loss of E-cadherin allows cell motility and is thought to promote lung cancer metastasis. While the downregulation of E-cadherin expression has been well characterized and is seen with transforming growth factor-β1 (TGF-β1) exposure, few studies have focused on E-cadherin upregulation. Here, we show that serum starvation causes increased E-cadherin expression via the activation of c-Src kinase in non-small-cell lung cancer A549 cells. Serum starvation increased E-cadherin protein levels in a time- and dose-dependent manner. E-cadherin mRNA transcripts were unchanged with starvation, while protein translation inhibition with cycloheximide attenuated E-cadherin protein induction by starvation, suggesting that E-cadherin is regulated at the translational level by serum starvation. c-Src is a nonreceptor tyrosine kinase known to regulate protein translation machinery; serum starvation caused early and sustained activation of c-Src in A549 cells followed by E-cadherin upregulation. Furthermore, overexpression of a dominant negative c-Src attenuated the induction of E-cadherin by serum deprivation. Finally, we observed that TGF-β1 treatment attenuated the serum activation of c-Src as well as E-cadherin expression when cells were deprived of serum. In conclusion, our data demonstrate that the c-Src kinase is activated by serum starvation to increase E-cadherin expression in A549 cells, and these phenomena are antagonized by TGF-β1. These novel observations implicate the c-Src kinase as an upstream inducer of E-cadherin protein translation with serum starvation and TGF-β1 diametrically regulating c-Src kinase activity and thus E-cadherin abundance in A549 cells.
Keywords: c-Src, E-cadherin, TGF-β1, non-small-cell lung cancer, starvation
tumorigenesis is a complex process, whereby accumulated genetic, epigenetic, and protein activity changes cause transformation of normal lung epithelial cells into hyperproliferative cancer cells (39), which can mobilize to cause distant metastases and escape surveillance by the immune system (14, 39, 40). Lung cancer is the leading cause of cancer death in the world (17, 18) and the most prevalent subtypes are the non-small-cell lung cancers (NSCLC). NSCLCs are highly invasive, less sensitive to chemotherapy and radiation therapy, and up to 36% of NSCLCs develop brain metastases, which confers a poor prognosis (3, 32). Reduction of epithelial adherens junctions can permit NSCLC invasion and an invasive cellular phenotype (33). Furthermore, variability within the tumor microenvironment can significantly alter cancer cell behaviors, with cell signaling molecules, hypoxia, and nutrient depletion all impacting cell metabolism and proliferation (10).
The formation and maintenance of adherens junctions is important in the structural integrity of epithelial tissues (4). E-cadherin is a major component of an adhesion complex that binds epithelia together at these junctions (35, 45, 49). Loss of E-cadherin can facilitate the invasive phenotype of epithelial-derived cancer cells and is associated with poor outcome in several tumor states, including NSCLC, invasive ductal breast carcinoma, and gastric adenocarcinoma (2, 6, 13, 25). Besides its role in cell-to-cell adhesion, E-cadherin is also critical in signal transduction via interactions with the actin cytoskeleton and signaling molecules like β-catenin (35, 49). Interestingly, expression of truncated α- or β-catenin abrogates E-cadherin function, and downregulation of β-catenin has been associated with malignant transformation (52–54). Since E-cadherin plays an important role in tissue morphogenesis, cell polarity, and tumor invasiveness (29, 45), understanding the regulation of E-cadherin at the molecular level may provide insights into metastatic cancer biology and could reveal therapeutic targets.
Transforming growth factor-β1 (TGF-β1) is a pleiotropic cytokine that plays a complex role in tumorigenesis (31, 46, 51). During early tumor development, TGF-β1 suppresses cell proliferation by regulating cytostasis, cell differentiation, and apoptosis. Later in tumor progression, TGF-β1 signaling shows pro-oncogenic effects, provoking epithelial cell migration, invasion, and metastasis (15, 24, 38). Importantly, TGF-β1 signaling leads to downregulation of E-cadherin in lung cancer cells (26, 33). However, the upregulation of E-cadherin expression has not been well studied.
The c-Src tyrosine kinase has been implicated in the development and progression of various human cancers of the colon, breast, pancreas, and brain (7, 28, 50). c-Src is known to regulate translation of several proteins (21, 43, 48) and possesses two major tyrosine phosphorylation sites: Y416 and Y530. Y416 phosphorylation activates c-Src, while Y530 phosphorylation inhibits c-Src kinase activity (9, 16, 55). c-Src activation is associated with cytoproliferation and transformation in tumorigenesis, and it regulates adhesion and invasion in the later stages of tumor progression (30, 44, 55). During epithelial morphogenesis, Src kinase plays an important role in maintenance of epithelial integrity by upregulating E-cadherin (42).
In this study, we demonstrate that nutrient depletion by serum starvation increases E-cadherin protein, but not its mRNA transcript. Serum starvation phosphorylates and activates c-Src, which then mediates E-cadherin expression in A549 cells. Further, we show that TGF-β1 prevents this starvation-induced c-Src activation and blunts E-cadherin upregulation by starvation. This is the first report to reveal an effect of nutrient depletion on E-cadherin expression in a cancer cell line and may improve our understanding of the biology of E-cadherin expression in cancer development and metastasis.
MATERIALS AND METHODS
Cells and reagents.
A549 cells (ATCC CCL-185) were cultured with RPMI 1640 medium containing 2 mM glutamine with 10% fetal bovine serum (FBS). Beas2B cells (ATCC CCL-9609) were cultured with HITES medium with 10% FBS. Human bronchial epithelial primary cells (HBEpCs, Lonza) were cultured in BEGM medium containing growth factors (Lonza). V5 antibody, E-cadherin antibody, the mammalian expression plasmid pcDNA3.1/V5-His TOPO, Escherichia coli Top 10 competent cells, lipofectamine transfection reagent, and recombinant TGF-β1 were purchased from Invitrogen (Carlsbad, CA). Phospho-Src Y416 (pY416-Src) antibody was obtained from Cell Signaling Technology (Danvers, MA). Cycloheximide, c-Src, and β-actin antibodies were from Sigma Aldrich (St. Louis, MO). All materials used in the experiments are commercially available.
Construction of a dominant negative kinase dead c-Src plasmid.
The dominant negative cDNA of human c-Src has two point mutations, a Lys-to-Arg substitution at residue 298 and a Tyr-to-Phe substitution at residue 530. The dominant negative c-Src cDNA was inserted into a pcDNA3.1D/V5-His vector (Invitrogen).
Immunoblotting.
A549 cells were washed with cold PBS and collected in cell lysis buffer containing 20 mM Tris HCl (pH 7.4), 150 mM NaCl, 2 mM EGTA, 5 mM β glycerophosphate, 1 mM MgCl2, 1% Triton X-100, 1 mM sodium orthovanadate, 10 μg/ml protease inhibitors, 1 μg/ml leupeptin, and 1 μg/ml pepstatin. An equal amount of cell lysates (20 μg) was subjected to SDS-PAGE gels, electrotransferred to membranes, and immunoblotted as described previously.
Immunostaining.
A549 cells were plated on 35 mm glass-bottom culture dishes. After treatment, cells were fixed in 3.7% formaldehyde for 20 min, followed by permeabilization with 0.1% Triton X-100 for 2 min. Cells were incubated with a 1:200 dilution of antibody against E-cadherin or pY416-Src, followed by a 1:200 dilution of fluorescence-conjugated secondary antibody sequentially for immunostaining. Immunofluorescent cell imaging was performed on a Nikon confocal microscope.
Plasmid transfection.
A549 cells were subcultured on six-well plates or 35-mm plates for 24 h. Lipofectamine transfection reagent was added to the mixture of 2 μg of plasmid and 200 μl of FBS free medium and incubated for 10 min to allow transfection reagent/DNA complexes to form. The mixture was added directly to the cells with complete medium. The transfected cells were cultured for 48 h.
TGF-β1 treatment.
A549 cells were cultured in six-well plates or 35-mm dishes in complete medium. After 48 h of transfection, the medium was replaced with 1 ml of FBS-free medium. TGF-β1 was added into the medium with the concentration range of 0, 1, 2, or 5 ng/ml. After incubation at 37°C, the cells were collected and analyzed by immunoblotting with E-cadherin and pY416-Src antibodies.
Relative quantitation of gene expression by real-time PCR and qRT-PCR.
Total RNA was isolated using Trizol reagent (Life Technologies, Grand Island, NY) following manufacturer's instructions. cDNA was obtained by reverse transcription followed by amplification with an SosoFast Evagreen supermix and detection by a CFX96 real-time PCR detection system (Bio-Rad, Hercules, CA). Levels of transcripts were normalized to GAPDH. Low-amplification cycle PCR was performed for 28 cycles using primers for full-length E-cadherin and GAPDH mRNAs. PCR products were run on an agarose gel containing ethidium bromide and visualized with UV light.
Statistics.
All results were subjected to statistical analysis using two-way analysis of variance and, wherever appropriate, analyzed by Student-Newman-Keuls test. Data are expressed as means ± SD of triplicate samples from at least three independent experiments, and values that were P < 0.05 were considered statistically significant.
RESULTS
Serum starvation upregulates E-cadherin expression at the translational level.
FBS is the most widely used growth supplement in cell culture medium, which provides nutrients and growth factors for cell proliferation. Ten percent FBS is recommended in culture media for most immortalized lung epithelial cell lines. Researchers often use serum starvation as a means to decrease the unpredictable effects of nutrients on protein expression and cellular responses of cell lines in molecular investigations, and nutrient depletion may emulate the tumor microenvironment of some cancers. Since many experiments on E-cadherin are conducted in serum-free conditions, we evaluated E-cadherin expression in A549 (NSCLC line) and Beas2B (bronchial epithelial cell line) cells after changing the culture media to FBS-free media for 0 h, 6 h, 1 day, and 2 days; E-cadherin was upregulated by starvation over this time course (Fig. 1A). Further, we observed that depletion of growth factors from culture media for human bronchial epithelial cells likewise increased immunoreactive E-cadherin levels after 24 h (Fig. 1A). We next examined if this effect was FBS dose dependent. A549 cells were cultured with media containing different concentrations of FBS for 24 h, and E-cadherin levels were determined by immunoblotting. E-cadherin expression decreased as FBS supplementation was increased in the media (Fig. 1B). To determine if these changes of E-cadherin are due to increases in transcription, we examined E-cadherin mRNA by qRT-PCR and “lower-amplification cycle” PCR, and found no difference between E-cadherin mRNA levels from cells cultured in serum-starved or -rich conditions (Fig. 1, C and D). Pretreatment with the protein synthesis inhibitor cycloheximide (CHX) attenuated the starvation-induced E-cadherin expression (Fig. 1E), suggesting that the starvation upregulates E-cadherin expression at a translational level rather than through transcriptional activation.
Fig. 1.

Serum starvation upregulates E-cadherin expression at the translational level. A: A549 or Beas2B cells were cultured in serum-free basal RPMI-1640 medium for 0, 6 h, 1 day (1d), and 2 days (2d). Human bronchial epithelial primary cells (HBEpCs) were cultured in growth factor-free medium for 1 day. E-cadherin and β-actin were detected by immunoblotting. B: A549 cells cultured with a range of serum concentrations (0, 0.5%, 1%, 5%, and 10%) for 24 h were analyzed for E-cadherin and β-actin by immunoblotting. Bottom: E-cadherin densitometries were normalized to β-actin intensities. *P < 0.05 compared with 0% of FBS. C: E-cadherin mRNA expression levels in A549 cells in serum-free media (0 h, 6 h, 1 day, 2 day) analyzed by qRT-PCR relative to GAPDH mRNA as internal control; fold changes were calculated relative to 0-h data. D: agarose gel image of E-cadherin and GAPDH full-length cDNA amplified by PCR with 28 cycles. E: cell lysates from serum-starved A549 cells treated with vehicle or 20 μg/ml cycloheximide (CHX) were analyzed by E-cadherin and β-actin immunoblotting. Blots shown are representative from 3 independent experiments except the bottom panel in A (2 experiments).
c-Src regulates serum starvation-induced E-cadherin expression.
c-Src is a multifunctional tyrosine kinase involved in the regulation of protein translation processes and has been heavily implicated in cancer pathophysiology (7, 21, 28, 50). We next examined if c-Src modulates the E-cadherin expression that we observed in the setting of serum starvation. In A549 cells, changing to serum-free culture media led to activation of c-Src over time as determined by Y416 phosphorylation (Fig. 2A). As seen for E-cadherin expression, c-Src Y416 phosphorylation correlated inversely with FBS concentration in the culture medium (Fig. 2B). This activation of c-Src was visualized by confocal microscopy in both cytosolic and membrane fractions of A549 cells (Fig. 2C). These results suggest that c-Src can be activated by nutrient starvation.
Fig. 2.

Serum starvation induces c-Src activity. A: c-Src activation was analyzed in A549 cells in serum-free media (0, 6 h, 24 h, 48 h) by immunoblotting for tyrosine 416 phosphorylation of c-Src (PY416-c-Src). Bottom panel: PY416-c-Src intensities were analyzed and normalized to β-actin intensities. *P < 0.05 compared with 0-h starvation data. B: A549 Phospho-c-Src Y416 analysis 24 h after exposure to a range of serum concentrations (0%, 0.5%, 1%, 5%, and 10 %) with PY416-c-Src intensities relative to β-actin intensities. *P < 0.05 compared with 0% FBS. C: immunocytochemistry of A549 cells cultured in media with or without serum for 24 h; IgG (green, top panel), PY416-c-Src (green, bottom panels), and DAPI (red) were visualized by fluorescence under equal exposure conditions. Representative immunoblots and images from 3 independent experiments are shown.
We next examined if our findings on c-Src and E-cadherin expression may be related. The V5 epitope-tagged dominant negative kinase dead c-Src (Dn-c-Src-V5) or control plasmid was transiently transfected into A549 cells which were later incubated for 24 h in FBS-free conditions. Dn-c-Src overexpression reduces the E-cadherin upregulation seen in response to serum starvation in A549 cells (Fig. 3A), and overexpression of this Dn-c-Src decreased E-cadherin levels in a dose-dependent manner (Fig. 3B). To investigate if c-Src directly phosphorylates E-cadherin we tried to detect E-cadherin by immunoblot after immunoprecipitation of phosphotyrosine-containing proteins, and as shown in Fig. 3C, tyrosine phosphorylation of E-cadherin was not detected in both basal and starvation conditions. Further, coimmunoprecipitation of c-Src with E-cadherin did not reveal any detectable association between these two proteins (Fig. 3D), suggesting that c-Src activity lies further upstream in E-cadherin induction rather than acting through direct phosphorylation.
Fig. 3.

c-Src mediates serum starvation-induced E-cadherin expression. A: A549 cells were transfected with V5 tagged dominant negative c-Src plasmid (Dn-c-Src-V5) for 48 h, and media was then replaced with serum-free medium for 24 h. E-cadherin and β-actin levels were analyzed by immunoblotting. B: A549 cells transfected with increasing Dn-c-Src-V5 plasmid (0, 0.5, 1, 2 μg) for 48 h were then cultured in serum-free medium for 24 h followed by immunoblotting. C: A549 cells were cultured in serum-free conditions for 0–24 h. Immunoprecipitation of tyrosine phosphorylated proteins with an antibody to phosphotyrosine (p-Tyr), followed by E-cadherin and p-Tyr immunoblotting. Whole cell lysates (WCL) were analyzed with E-cadherin and p-Tyr immunoblotting. D: coimmunoprecipitation of c-Src with E-cadherin was performed after starvation (24 h) by c-Src antibody immunoprecipitation of c-Src binding complex followed by E-cadherin immunoblotting. Shown are representative blots from 3 independent experiments except C (2 experiments).
TGF-β1 attenuates serum starvation-mediated E-cadherin upregulation.
Because TGF-β1 has been shown to cause downregulation of E-cadherin in cells (26, 33), we examined if TGF-β1 could modulate the E-cadherin induction observed with serum starvation. A549 cells were incubated with or without TGF-β1 in FBS-free media over time, and TGF-β1 attenuated the serum starvation-mediated E-cadherin upregulation (Fig. 4A) in a dose-dependent manner (Fig. 4B). These data were corroborated by immunofluorescence staining and confocal imaging (Fig. 4C), where we observed a robust increase in membrane E-cadherin expression with serum starvation that was less intense in the presence of TGF-β1, suggesting that TGF-β1 can blunt serum starvation-induced E-cadherin upregulation. Further, we found that the mRNA levels of E-cadherin did not change with TGF-β1 exposure for 24 h (Fig. 4D), indicating this inhibitory effect of TGF-β1 on E-cadherin is likely due to reductions in translation rather than transcription in 24 h.
Fig. 4.

TGF-β1 attenuates serum starvation-mediated E-cadherin upregulation. A: A549 cells were cultured in serum-free basal RPMI-1640 medium for 0, 3 h, 6 h, 1 day, 2 days, and 3 days in the presence or absence of TGF-β1 (5 ng/ml) and lysates analyzed for E-cadherin. B: A549 cells treated with a dose range of TGF-β1 (0, 1, 2, 5, 10 ng/ml) in serum-free conditions for 24 h were analyzed for E-cadherin. C: A549 cells were serum-starved for 24 h in the presence of TGF-β1 (5 ng/ml), and E-cadherin (green) was visualized by immunocytochemistry under identical exposure conditions. Fluorescence intensity profiles shown at right. Immunoblots and images shown are representative from 3 independent experiments. D: E-cadherin mRNA level in TGF-β1 (5 ng/ml, 24 h)-treated A549 cells by qRT-PCR shown as fold changes relative to GAPDH control. ns, not significant.
TGF-β1 attenuates serum starvation-mediated c-Src activity.
We have shown that serum starvation induced E-cadherin expression through the activation of c-Src in A549 cells. To investigate if TGF-β1 modulation of E-cadherin requires c-Src activity, we treated the cells with TGF-β1 (5 ng/ml) in FBS-free media. TGF-β1 reduced the serum starvation-mediated c-Src Y416 phosphorylation at 24 h (Fig. 5A), and this occurs in a dose-dependent manner (Fig. 5B). Immunostaining confirmed that serum starvation increased Y416 phosphorylation of c-Src in the cytoplasm and the plasma membrane, and this effect was suppressed by TGF-β1 in A549 cells (Fig. 5C).
Fig. 5.

TGF-β1 attenuates serum starvation-mediated c-Src activity. A: A549 cells were starved for 0, 6 h, and 24 h in the presence of TGF-β1 (5 ng/ml) followed by PY416-c-Src immunoblotting. B: PY416-c-Src analysis of 24-h serum-starved A549 cells in the presence of TGF-β1 (0–5 ng/ml). C: PY416-c-Src (green) and DAPI (red) immunocytochemical analysis of A549 cells in serum-rich media (left), or serum-starved A549 cells in the absence (middle) or presence of TGF-β1 (5 ng/ml, right). Immunoblots and images shown are representative from 3 independent experiments.
DISCUSSION
E-cadherin is implicated in the pathobiology of several types of cancer (2, 52, 53), as loss or dysfunction of E-cadherin is associated with increased lung cancer cell proliferation and invasiveness (2, 13, 52–54). Cancer nutrient depletion response pathways cause major shifts in stress resistance by reducing growth factors, such as insulin-like growth factor-1 (IGF-1), and modulating cellular energetics to protect cells against multiple stresses, including heat shock and oxidative damage (27, 37). Recent studies have revealed that starvation could reduce tumor growth and sensitize cancer cells to chemotherapeutic agents by reducing the level of IGF-1 and increasing p53 activation (22, 41). The effects of starvation on E-cadherin expression have not been studied previously. In this study we demonstrate that serum starvation increases E-cadherin expression in the A549 non-small-cell lung cancer cell line as well as in the bronchial epithelial cell line Beas-2B and in human bronchial epithelial cells, and this effect seems to modulate E-cadherin translation. We also show that the kinase activity of c-Src is required for the induction of E-cadherin translation. Furthermore, we demonstrate that TGF-β1 opposes these effects of serum starvation, as c-Src activation and E-cadherin are both decreased in the presence of TGF-β1. It has been previously reported that TGF-β1 reduces c-Src activities by degradation of activated c-Src in rat fibroblasts (12). Our findings are consistent with that report, and this study provides a novel molecular perspective on the regulation of E-cadherin expression by linking its induction to c-Src activity.
Studies on E-cadherin regulation in cancer pathobiology have largely been focused on its downregulation and dysfunction. TGF-β1 signaling reduces E-cadherin expression and promotes cell motility through regulation of Snail expression (1, 26, 33). Snail is an E-cadherin transcriptional suppressor that requires histone deacetylase 1 (HDAC1) modulation of histone 3 and histone 4 acetylation at the E-cadherin promoter (34). Other studies have shown that IGF-1 and the steroid hormone progesterone downregulated E-cadherin and promoted tumor invasiveness (20, 36). Also, in a study consistent with this report, inhibition of IGF-1 signaling attenuated IGF-1 induced β-catenin activity and increased levels of E-cadherin (23). Tamoxifen, a nonsteroidal anti-estrogen, is reported to restore E-cadherin function and suppresses breast cancer cells' invasive phenotype (8). Since E-cadherin enhances adhesion complexes and decreases cell motility, it has been identified as a metastasis suppression molecule, and restoration of E-cadherin function has been considered as a potential therapeutic strategy. This study provides a new molecular mechanism by which nutrient and growth factor starvation might inhibit tumor cell proliferation and mobilization via the upregulation of E-cadherin. Since FBS contains IGF-1, depletion of IGF-1 and other hormones may contribute to the E-cadherin upregulation we observed here (8, 23). Furthermore, we report that TGF-β1 antagonizes the starvation-induced E-cadherin upregulation, but that there is no change in E-cadherin transcript levels under either condition. It will be interesting to characterize how this effect of TGF-β1 in starvation conditions differs from the Snail-dependent E-cadherin downregulation previously reported.
In A549 cells, serum starvation-induced E-cadherin expression seems to be modulated by translational, not transcriptional, activation. The regulation of E-cadherin translation has not been well demonstrated, although one report has shown that Drosophila HnRNP A1 regulated E-cadherin translation by binding to the 5′-untranslated region (5′-UTR) of E-cadherin mRNA (19). Active c-Src has been shown to increased translation of some proteins, such as β-catenin, an E-cadherin binding partner (21). We show that c-Src activation correlates with the upregulation of E-cadherin in starvation conditions, but that dominant negative c-Src suppresses E-cadherin levels. We also report that c-Src is neither associated with the E-cadherin protein nor does c-Src cause E-cadherin tyrosine phosphorylation. Taken together, these data suggest that c-Src activity may modulate E-cadherin translation. c-Src is involved in a variety of normal and oncogenic processes, including proliferation and cell-cell adhesion (44, 47). Several studies indicate that c-Src negatively regulates E-cadherin function (5), and one report suggests that v-Src phosphorylates E-cadherin to cause ubiquitination and internalization of adhesion complexes in a canine kidney epithelial cell line (11). Shindo et al. (42) have demonstrated that activation of c-Src enhanced E-cadherin-mediated lung epithelial barrier integrity, and we have found that treatment of human bronchial epithelial cells with the Src kinase inhibitor (PP1) reduced epithelial barrier integrity (Zhao Y, unpublished observations). This is consistent with our present report that active c-Src promotes E-cadherin function through regulation of its expression. It is also possible that c-Src interacts with or phosphorylates E-cadherin-associated proteins such as β-catenin for the effects we report. Future studies are necessary to determine if any c-Src substrate(s) bind to E-cadherin mRNA or otherwise regulate E-cadherin translation.
In summary, the current study reveals the novel observation that E-cadherin is upregulated by serum starvation in the non-small-cell lung cancer A549 cell line without an increase in E-cadherin transcription (Fig. 6). Further, we demonstrate that c-Src activation mediates this upregulation and that TGF-β1 attenuates both the c-Src activation and E-cadherin upregulation in starved conditions. We propose translational upregulation downstream of c-Src as the molecular mechanism of this phenomenon. Extensive understanding of E-cadherin biology may be informative for the development of new therapeutic targets to inhibit tumor growth and metastasis.
Fig. 6.
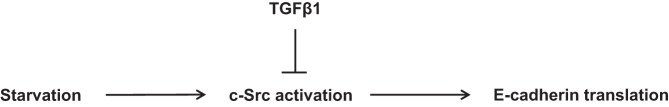
Serum starvation induces E-cadherin expression through activation of c-Src. Serum starvation increases c-Src activity as well as E-cadherin expression, which is attenuated by TGF-β1 in A549 cells.
GRANTS
This study was supported by National Heart, Lung, and Blood Institute Grants R01-HL-01916 and R01-HL-112791 to Y. Zhao and by American Heart Association Award 12SDG9050005 to J. Zhao.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
Author contributions: S.D., A.K., J.W., R.K.B., S.X., L.Z., and J.Z. performed experiments; S.D., Y.Z., and J.Z. analyzed data; S.D., A.K., N.M.W., S.X., L.Z., Y.Z., and J.Z. interpreted results of experiments; S.D., Y.Z., and J.Z. prepared figures; S.D., Y.Z., and J.Z. drafted manuscript; S.D., A.K., R.K.B., N.M.W., Y.Z., and J.Z. edited and revised manuscript; S.D., A.K., J.W., R.K.B., N.M.W., S.X., L.Z., H.M., Y.Z., and J.Z. approved final version of manuscript; H.M., Y.Z., and J.Z. conception and design of research.
ACKNOWLEDGMENTS
We thank M. Myerburg for the discussion.
REFERENCES
- 1.Attisano L, Wrana JL. Signal transduction by the TGF-beta superfamily. Science 296: 1646–1647, 2002 [DOI] [PubMed] [Google Scholar]
- 2.Bae GY, Choi SJ, Lee JS, Jo J, Lee J, Kim J, Cha HJ. Loss of E-cadherin activates EGFR-MEK/ERK signaling, which promotes invasion via the ZEB1/MMP2 axis in non-small cell lung cancer. Oncotarget 4: 2512–2522, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Barnholtz-Sloan JS, Maldonado JL, Williams VL, Curry WT, Rodkey EA, Barker FG, 2nd, Sloan AE. Racial/ethnic differences in survival among elderly patients with a primary glioblastoma. J Neuro-oncology 85: 171–180, 2007 [DOI] [PubMed] [Google Scholar]
- 4.Baum B, Georgiou M. Dynamics of adherens junctions in epithelial establishment, maintenance, and remodeling. J Cell Biol 192: 907–917, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Behrens J, Vakaet L, Friis R, Winterhager E, Van Roy F, Mareel MM, Birchmeier W. Loss of epithelial differentiation and gain of invasiveness correlates with tyrosine phosphorylation of the E-cadherin/beta-catenin complex in cells transformed with a temperature-sensitive v-SRC gene. J Cell Biol 120: 757–766, 1993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Berx G, Cleton-Jansen AM, Strumane K, de Leeuw WJ, Nollet F, van Roy F, Cornelisse C. E-cadherin is inactivated in a majority of invasive human lobular breast cancers by truncation mutations throughout its extracellular domain. Oncogene 13: 1919–1925, 1996 [PubMed] [Google Scholar]
- 7.Biscardi JS, Belsches AP, Parsons SJ. Characterization of human epidermal growth factor receptor and c-Src interactions in human breast tumor cells. Mol Carcinogenesis 21: 261–272, 1998 [DOI] [PubMed] [Google Scholar]
- 8.Bracke ME, Charlier C, Bruyneel EA, Labit C, Mareel MM, Castronovo V. Tamoxifen restores the E-cadherin function in human breast cancer MCF-7/6 cells and suppresses their invasive phenotype. Cancer Res 54: 4607–4609, 1994 [PubMed] [Google Scholar]
- 9.Fincham VJ, Unlu M, Brunton VG, Pitts JD, Wyke JA, Frame MC. Translocation of Src kinase to the cell periphery is mediated by the actin cytoskeleton under the control of the Rho family of small G proteins. J Cell Biol 135: 1551–1564, 1996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fritz V, Fajas L. Metabolism and proliferation share common regulatory pathways in cancer cells. Oncogene 29: 4369–4377, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fujita Y, Krause G, Scheffner M, Zechner D, Leddy HE, Behrens J, Sommer T, Birchmeier W. Hakai, a c-Cbl-like protein, ubiquitinates and induces endocytosis of the E-cadherin complex. Nat Cell Biol 4: 222–231, 2002 [DOI] [PubMed] [Google Scholar]
- 12.Fukuda K, Kawata S, Tamura S, Matsuda Y, Inui Y, Igura T, Inoue S, Kudara T, Matsuzawa Y. Transforming growth factor-beta1-induced degradation of activated Src tyrosine kinase in rat fibroblasts. Oncogene 16: 3349–3356, 1998 [DOI] [PubMed] [Google Scholar]
- 13.Gabbert HE, Mueller W, Schneiders A, Meier S, Moll R, Birchmeier W, Hommel G. Prognostic value of E-cadherin expression in 413 gastric carcinomas. Int J Cancer 69: 184–189, 1996 [DOI] [PubMed] [Google Scholar]
- 14.Herceg Z, Hainaut P. Genetic and epigenetic alterations as biomarkers for cancer detection, diagnosis and prognosis. Mol Oncol 1: 26–41, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ikushima H, Miyazono K. TGFbeta signalling: a complex web in cancer progression. Nat Rev Cancer 10: 415–424, 2010 [DOI] [PubMed] [Google Scholar]
- 16.Ingley E. Src family kinases: regulation of their activities, levels and identification of new pathways. Biochim Biophys Acta 1784: 56–65, 2008 [DOI] [PubMed] [Google Scholar]
- 17.Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA Cancer J Clin 61: 69–90, 2011 [DOI] [PubMed] [Google Scholar]
- 18.Jemal A, Siegel R, Ward E, Hao Y, Xu J, Thun MJ. Cancer statistics, 20009. CA Cancer J Clin 59: 225–249, 2009 [DOI] [PubMed] [Google Scholar]
- 19.Ji Y, Tulin AV. Poly(ADP-ribose) controls DE-cadherin-dependent stem cell maintenance and oocyte localization. Nat Commun 3: 760, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kariagina A, Xie J, Langohr IM, Opreanu RC, Basson MD, Haslam SZ. Progesterone decreases levels of the adhesion protein E-cadherin and promotes invasiveness of steroid receptor positive breast cancers. Horm Cancer 4: 371–380, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Karni R, Gus Y, Dor Y, Meyuhas O, Levitzki A. Active Src elevates the expression of beta-catenin by enhancement of cap-dependent translation. Mol Cell Biol 25: 5031–5039, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lee C, Raffaghello L, Brandhorst S, Safdie FM, Bianchi G, Martin-Montalvo A, Pistoia V, Wei M, Hwang S, Merlino A, Emionite L, de Cabo R, Longo VD. Fasting cycles retard growth of tumors and sensitize a range of cancer cell types to chemotherapy. Sci Transl Med 4: 124ra127, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lee J, Ju J, Park S, Hong SJ, Yoon S. Inhibition of IGF-1 signaling by genistein: modulation of E-cadherin expression and downregulation of beta-catenin signaling in hormone refractory PC-3 prostate cancer cells. Nutr Cancer 64: 153–162, 2012 [DOI] [PubMed] [Google Scholar]
- 24.Lee JM, Dedhar S, Kalluri R, Thompson EW. The epithelial-mesenchymal transition: new insights in signaling, development, and disease. J Cell Biol 172: 973–981, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lipponen P, Saarelainen E, Ji H, Aaltomaa S, Syrjanen K. Expression of E-cadherin (E-CD) as related to other prognostic factors and survival in breast cancer. J Pathol 174: 101–109, 1994 [DOI] [PubMed] [Google Scholar]
- 26.Liu J, Hu G, Chen D, Gong AY, Soori GS, Dobleman TJ, Chen XM. Suppression of SCARA5 by Snail1 is essential for EMT-associated cell migration of A549 cells. Oncogenesis 2: e73, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Longo VD, Finch CE. Evolutionary medicine: from dwarf model systems to healthy centenarians? Science 299: 1342–1346, 2003 [DOI] [PubMed] [Google Scholar]
- 28.Mao W, Irby R, Coppola D, Fu L, Wloch M, Turner J, Yu H, Garcia R, Jove R, Yeatman TJ. Activation of c-Src by receptor tyrosine kinases in human colon cancer cells with high metastatic potential. Oncogene 15: 3083–3090, 1997 [DOI] [PubMed] [Google Scholar]
- 29.Mareel M, Boterberg T, Noe V, Van Hoorde L, Vermeulen S, Bruyneel E, Bracke M. E-cadherin/catenin/cytoskeleton complex: a regulator of cancer invasion. J Cell Physiol 173: 271–274, 1997 [DOI] [PubMed] [Google Scholar]
- 30.Martin GS. The hunting of the Src. Nat Rev Mol Cell Biol 2: 467–475, 2001 [DOI] [PubMed] [Google Scholar]
- 31.Massague J. TGFbeta in Cancer. Cell 134: 215–230, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mujoomdar A, Austin JH, Malhotra R, Powell CA, Pearson GD, Shiau MC, Raftopoulos H. Clinical predictors of metastatic disease to the brain from non-small cell lung carcinoma: primary tumor size, cell type, and lymph node metastases. Radiology 242: 882–888, 2007 [DOI] [PubMed] [Google Scholar]
- 33.Onder TT, Gupta PB, Mani SA, Yang J, Lander ES, Weinberg RA. Loss of E-cadherin promotes metastasis via multiple downstream transcriptional pathways. Cancer Res 68: 3645–3654, 2008 [DOI] [PubMed] [Google Scholar]
- 34.Peinado H, Ballestar E, Esteller M, Cano A. Snail mediates E-cadherin repression by the recruitment of the Sin3A/histone deacetylase 1 (HDAC1)/HDAC2 complex. Mol Cell Biol 24: 306–319, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Perez-Moreno M, Jamora C, Fuchs E. Sticky business: orchestrating cellular signals at adherens junctions. Cell 112: 535–548, 2003 [DOI] [PubMed] [Google Scholar]
- 36.Playford MP, Bicknell D, Bodmer WF, Macaulay VM. Insulin-like growth factor 1 regulates the location, stability, and transcriptional activity of beta-catenin. Proc Natl Acad Sci USA 97: 12103–12108, 2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Raffaghello L, Lee C, Safdie FM, Wei M, Madia F, Bianchi G, Longo VD. Starvation-dependent differential stress resistance protects normal but not cancer cells against high-dose chemotherapy. Proc Natl Acad Sci USA 105: 8215–8220, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rahimi RA, Leof EB. TGF-beta signaling: a tale of two responses. J Cell Biochem 102: 593–608, 2007 [DOI] [PubMed] [Google Scholar]
- 39.Sato M, Shames DS, Gazdar AF, Minna JD. A translational view of the molecular pathogenesis of lung cancer. J Thoracic Oncol 2: 327–343, 2007 [DOI] [PubMed] [Google Scholar]
- 40.Sekido Y, Fong KM, Minna JD. Molecular genetics of lung cancer. Annu Rev Med 54: 73–87, 2003 [DOI] [PubMed] [Google Scholar]
- 41.Shi Y, Felley-Bosco E, Marti TM, Orlowski K, Pruschy M, Stahel RA. Starvation-induced activation of ATM/Chk2/p53 signaling sensitizes cancer cells to cisplatin. BMC Cancer 12: 571, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Shindo M, Wada H, Kaido M, Tateno M, Aigaki T, Tsuda L, Hayashi S. Dual function of Src in the maintenance of adherens junctions during tracheal epithelial morphogenesis. Development 135: 1355–1364, 2008 [DOI] [PubMed] [Google Scholar]
- 43.Soung YH, Korneeva N, Kim TH, Chung J. The role of c-Src in integrin (alpha6beta4) dependent translational control. BMC Cell Biol 14: 49, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Summy JM, Gallick GE. Src family kinases in tumor progression and metastasis. Cancer Metastasis Rev 22: 337–358, 2003 [DOI] [PubMed] [Google Scholar]
- 45.Takeichi M. Cadherin cell adhesion receptors as a morphogenetic regulator. Science 251: 1451–1455, 1991 [DOI] [PubMed] [Google Scholar]
- 46.Tang B, Vu M, Booker T, Santner SJ, Miller FR, Anver MR, Wakefield LM. TGF-beta switches from tumor suppressor to prometastatic factor in a model of breast cancer progression. J Clin Invest 112: 1116–1124, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Thomas SM, Brugge JS. Cellular functions regulated by Src family kinases. Annu Rev Cell Dev Biol 13: 513–609, 1997 [DOI] [PubMed] [Google Scholar]
- 48.Touyz RM, He G, Wu XH, Park JB, Mabrouk ME, Schiffrin EL. Src is an important mediator of extracellular signal-regulated kinase 1/2-dependent growth signaling by angiotensin II in smooth muscle cells from resistance arteries of hypertensive patients. Hypertension 38: 56–64, 2001 [DOI] [PubMed] [Google Scholar]
- 49.van Roy F, Berx G. The cell-cell adhesion molecule E-cadherin. Cell Mol Life Sci 65: 3756–3788, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Verbeek BS, Vroom TM, Adriaansen-Slot SS, Ottenhoff-Kalff AE, Geertzema JG, Hennipman A, Rijksen G. c-Src protein expression is increased in human breast cancer. An immunohistochemical and biochemical analysis. J Pathol 180: 383–388, 1996 [DOI] [PubMed] [Google Scholar]
- 51.Wakefield LM, Roberts AB. TGF-beta signaling: positive and negative effects on tumorigenesis. Curr Opin Genet Dev 12: 22–29, 2002 [DOI] [PubMed] [Google Scholar]
- 52.Wijnhoven BP, Dinjens WN, Pignatelli M. E-cadherin-catenin cell-cell adhesion complex and human cancer. Br J Surg 87: 992–1005, 2000 [DOI] [PubMed] [Google Scholar]
- 53.Wong AS, Gumbiner BM. Adhesion-independent mechanism for suppression of tumor cell invasion by E-cadherin. J Cell Biol 161: 1191–1203, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yanagisawa M, Anastasiadis PZ. p120 catenin is essential for mesenchymal cadherin-mediated regulation of cell motility and invasiveness. J Cell Biol 174: 1087–1096, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yeatman TJ. A renaissance for SRC. Nat Rev Cancer 4: 470–480, 2004 [DOI] [PubMed] [Google Scholar]