

Abstract
The steroid receptor coactivator 1 (SRC1) regulates key metabolic pathways, including glucose homeostasis. SRC1−/− mice have decreased hepatic expression of gluconeogenic enzymes and a reduction in the rate of endogenous glucose production (EGP). We sought to determine whether decreasing hepatic and adipose SRC1 expression in normal adult rats would alter glucose homeostasis and insulin action. Regular chow-fed and high-fat-fed male Sprage-Dawley rats were treated with an antisense oligonucleotide (ASO) against SRC1 or a control ASO for 4 wk, followed by metabolic assessments. SRC1 ASO did not alter basal EGP or expression of gluconeogenic enzymes. Instead, SRC1 ASO increased insulin-stimulated whole body glucose disposal by ∼30%, which was attributable largely to an increase in insulin-stimulated muscle glucose uptake. This was associated with an approximately sevenfold increase in adipose expression of lipocalin-type prostaglandin D2 synthase, a previously reported regulator of insulin sensitivity, and an approximately 70% increase in plasma PGD2 concentration. Muscle insulin signaling, AMPK activation, and tissue perfusion were unchanged. Although GLUT4 content was unchanged, SRC1 ASO increased the cleavage of tether-containing UBX domain for GLUT4, a regulator of GLUT4 translocation. These studies point to a novel role of adipose SRC1 as a regulator of insulin-stimulated muscle glucose uptake.
Keywords: insulin resistance, glucose transporter type 4, skeletal muscle, white adipose tissue
glucose homeostasis requires the complex coordination of cellular and molecular pathways in multiple tissues. In many instances, these pathways collate hormonal and metabolic cues to regulate transcription factors that alter the expression of genes encoding key metabolic enzymes. The activity of transcription factors is regulated further by transcriptional coactivators and corepressors. The three members of the p160 steroid coactivator receptor family, the steroid receptor coactivator 1 (SRC1 or NcoA-1), the transcriptional intermediary factor 2 (TIF2, SRC2, GRIP1, or NCOA2), and steroid receptor coactivator 3 (SRC3, p/CIP, AIB1, ACTR, RAC3, or TRAM-1) have all been shown to control energy balance through their interactions with many transcription factors, including nuclear receptors (14, 16, 18, 22). The SRC1 family has also been implicated in many other physiological processes, including reproduction, cancer, and immunity (22, 27, 42, 49, 50).
A significant body of work has shown that SRC1 is important for the tissue-specific activation of progesterone and estrogen receptors (1, 16, 42). Additionally, studies using the SRC1 global knockout mice have shown that SRC1 is required for uterine growth and function, prostate development, mammary gland growth and elongation, brain development and function, and metabolism (20, 23, 24, 28, 31, 49, 50, 52). SRC1−/− mice also have features of thyroid hormone resistance, with evidence that SRC1 is critical for proper activation of the thyroid hormone receptor-β (48). SRC1−/− mice have impairment in brown adipose tissue (BAT) function that makes them prone to weight gain potentially through decreased activation of PGC-1α, although this phenotype is not always apparent (30). And recently, studies investigating the mechanism of fasting hypoglycemia in SRC1−/− mice suggest that SRC1 regulates hepatic gluconeogenic gene expression in the fed and fasted states (23).
However, because SRC1 is involved in the regulation of cellular processes in many tissues, the tissue-specific function of SRC1 is difficult to interpret using a global SRC1-knockout mouse. Moreover, genetic models may trigger developmental changes that complicate phenotype interpretation. Antisense oligonucleotides (ASOs) allow us to selectively silence protein expression in key metabolic tissues in normal adult animals, circumventing the developmental concerns with knockout mice. ASOs decrease target expression primarily in white adipose tissue (WAT) and liver. ASOs do not decrease expression in other key tissues (e.g., skeletal or cardiac muscle, brown adipose tissue, pancreas, brain, etc.) (29). Thus, these compounds are useful tools to assess the role of key proteins in a more tissue-selective fashion. We treated regular chow-fed and high-fat-fed adult rats with SRC1 ASO and performed hyperinsulinemic euglycemic clamp studies to determine whether hepatic or adipose tissue SRC1 controls gluconeogenesis or peripheral insulin sensitivity.
MATERIALS AND METHODS
Animals.
Male Sprague-Dawley rats (200–225 g; Charles River Laboratories, Wilmington, MA) were housed on a 12:12-h light-dark cycle and received food and water ad libitum. They were given ≥7 days to acclimate before any studies. Rats were fed either regular rodent chow (60% carbohydrate, 10% fat, and 30% protein calories) and a high-fat diet (Dyets 112245: 26% carbohydrate, 59% fat, and 15% protein calories; Dyets, Bethlehem, PA). Body weight was monitored twice weekly. Rats were treated with either an ASO that lacks any homology to known sequences (Control ASO, CCTTCCCTGAAGGTTCCTCC) or an ASO against SRC1 (SRC1 ASO, AGATGTCGGACTTCTGCACG). ASOs were reconstituted in sterile saline, and rats were injected intraperitoneally at a dose of 50 mg·kg−1·wk−1 for 4–5 wk. Rats underwent the placement of jugular venous (for blood sampling) and carotid artery (for infusion) catheters ∼10 days before the infusion studies. All procedures were approved by the Institutional Animal Care and Use Committee of Yale University School of Medicine.
Hyperinsulinemic euglycemic clamp studies.
The hyperinsulemic euglycemic clamp studies were performed as described previously in overnight-fasted animals (19). Insulin was infused after a 2-h basal period with a primed continuous infusion of human insulin (40 mU·kg−1·10 min−1)/(4 mU·kg−1·min−1; Novo Nordisk, Princeton, NJ) and a variable infusion of 20% dextrose to maintain euglycemia. After 130 min, 2-deoxy-d-[1-14C]glucose (American Radiolabeled Chemicals, St. Louis, MO) was injected and glucose metabolism analyzed for 20 min.
Biochemical analysis and calculations.
Plasma glucose was measured using a YSI 2700D glucose analyzer. Plasma insulin and glucagon concentrations were measured by a RIA kit (Linco). Serum triglycerides, plasma nonesterified fatty acids, aspartate aminotransferase (AST), and alanine aminotransferase (ALT) were analyzed using COBASMira Plus (Roche). Plasma adiponectin was measured using a mouse multiplex assay kit (Meso Scale Discovery). Prostaglandin D2 (PGD2) was measured using the Prostaglandin D2-MOX EIA Kit (Cayman Chemical).
Total RNA preparation and RT-PCR analysis.
Total RNA and RT-PCR were performed as described previously (7).
Immunoblotting.
Protein preparation and immunoblotting were performed as described previously (7). Antibodies used were AMP-activated protein kinase-α (AMPKα), phospho-Thr172 AMPKα, GAPDH, and Akt2 (Cell Signaling Technology), phospho-Ser474 Akt2 (SAB Signalway), ACCβ, and phospho-Ser219 acetyl-CoA carboxylase-β (ACCβ; Santa Cruz Biotechnology). Sortilin (BD Biosciences), GLUT1 (Abcam), glucose transporter 4 (GLUT4), hexokinase II (HKII), and tether-containing UBX domain for GLUT4 (TUG) antibodies were a kind gift from Jonathan Bogan (Yale University, New Haven, CT).
Postmortem γ-counting and analysis of tissue perfusion.
Animals (n = 20) were given an in vivo intravenous injection of technetium-99m (99mTc)-tetrofosmin (251.6 ± 25.9 MBq) under basal or insulin-stimulated conditions. For insulin-stimulated conditions, rats were subjected to hyperinsulinemic euglycemic conditions until steady-state serum glucose values of 100–110 mg/dl were maintained for 20 min. Fifteen minutes after radiotracer injection, animals were euthanized, and the gastrocnemius, soleus, and whole heart were excised, weighed, and evaluated for 99mTc-tetrofosmin radioactivity with a γ-counter (Cobra 5003; Packard Instrument, Meriden, CT). All values for 99mTc-tetrofosmin retention were calculated as a percent of the injected dose per gram of tissue (after correction for background activity and radioactive decay), as described previously (43).
Statistical analysis.
Statistical significance has been calculated by Student's t-test or ANOVA, as stated in the figure legends. All data are displayed as means ± SE, unless stated otherwise.
RESULTS
SRC1 ASO treatment reduces fasting insulin concentration without changing fasting glucose concentration or weight gain.
Sprague-Dawley rats were injected with control ASO or SRC1 antisense ASO intraperitoneally for 4 wk prior to metabolic parameters being assessed. Control ASO treatment has previously been shown to be similar to saline injection (38, 39), and thus for these studies, SRC1 ASO-treated rats were compared with control ASO-treated rats. SRC1 ASO specifically reduced SRC1 expression in the liver and WAT but not skeletal muscle or brown adipose tissue (Fig. 1A). SRC1 ASO did not affect SRC2 or SRC3 expression in liver or WAT (Fig. 1, B and C). SRC1 ASO treatment also did not affect body weight, epididymal adipose tissue weight, plasma triglyceride concentration, tissue triglyceride concentration in the liver or gastrocnemius muscle, plasma nonesterified fatty acid concentration, or glucagon concentration in regular chow-fed rats (Tables 1 and 2). SRC1 ASO treatment did not alter plasma AST or ALT concentrations (Table 1). Surprisingly, unlike the SRC1−/− mice, SRC1 ASO treatment did not alter basal plasma glucose concentration (Table 2) or gluconeogenic gene mRNA expression (Fig. 1D). However, SRC1 ASO treatment decreased fasting plasma insulin concentration by ∼50% (Table 2). Taken together, these data suggested that SRC1 ASO treatment may increase insulin sensitivity in chow-fed rats.
Fig. 1.

Steroid receptor coactivator 1 (SRC1) antisense oligonucleotide (ASO) specifically decreases SRC1 expression in the liver and white adipose tissue (WAT). Sprague-Dawley rats were treated with control or SRC1 ASO for 4 wk, and mRNA expression was analyzed by real-time PCR in euglycemic hyperinsulinemic-clamped rats. A–C: SRC1 expression in the liver, WAT, brown adipose tissue (BAT), and muscle (A), SRC2 (B), and SRC3 expression (C) in the liver and WAT. D: real-time PCR analysis of hepatic gluconeogenic gene expression in overnight-fasted rats; n = 6–8/group. P value calculated by Student's t-test. *P < 0.05. GP6C, glucose 6-phosphatase catalytic; G6PT, glucose 6 phosphatase translocase; FBPase, fructose-1,6-bisphosphatase; PC, pyruvate carboxylase; PEPCK1, phosphoenolpyruvate carboxykinase 1.
Table 1.
Effect of SRC1 ASO on various metabolic parameters in regular chow-fed rats
| Control ASO | SRC1 ASO | P Value | |
|---|---|---|---|
| Epidydimal adipose tissue weight, g | 2.35 ± 0.22 | 2.15 ± 0.31 | 0.62 |
| Plasma triglyceride, mg/dl | 16.5 ± 2.14 | 16.2 ± 1.47 | 0.93 |
| Plasma NEFA, mmol/l | 0.76 ± 0.05 | 0.68 ± 0.04 | 0.21 |
| Plasma AST, U/l | 95.6 ± 15.04 | 72.7 ± 6.48 | 0.15 |
| Plasma ALT, U/l | 40.6 ± 4.62 | 31.3 ± 4.08 | 0.17 |
| Basal glucagon, pg/ml | 18.4 ± 5.7 | 23.5 ± 4.3 | 0.50 |
| Liver triglyceride, mg/g tissue | 6.98 ± 1.29 | 6.9 ± 1.06 | 0.96 |
| Gastrocnemius muscle triglyceride, mg/g tissue | 3.78 ± 0.99 | 1.85 ± 0.57 | 0.13 |
Values are means ± SE; n = 5–6/group. SRC1, steroid receptor coactivator 1; ASO, antisense oligonucleotide; NEFA, nonesterified fatty acid; AST, aspartate aminotransferase; ALT, alanine aminotransferase. Glucagon was measured in overnight-fasted rats treated with control or SRC1 ASO for 4 wk. All other parameters were determined in hyperinsulinemic euglycemic-clamped rats. P value calculated by Student's t-test.
Table 2.
SRC1 ASO treatment does not alter basal glucose concentration in regular chow- or high-fat-fed rats
| Regular Chow |
High-Fat Diet |
|||||
|---|---|---|---|---|---|---|
| Control ASO | SRC1 ASO | P Value | Control ASO | SRC1 ASO | P Value | |
| Body weight, g | 426 ± 62 | 394 ± 66 | 0.11 | 460 ± 16 | 424 ± 7 | 0.06 |
| Basal glucose, mg/dl | 117 ± 1.8 | 114 ± 2.4 | 0.34 | 114 ± 2 | 114 ± 2.2 | 0.62 |
| Clamped glucose, mg/dl | 108 ± 2.1 | 107 ± 1.9 | 0.67 | 110 ± 6.4 | 100 ± 1.7 | 0.99 |
| Basal insulin, μU/ml | 17.4 ± 3.8 | 8.17 ± 0.88* | 0.01 | 24.7 ± 3.97 | 15.91 ± 2.48 | 0.09 |
| Clamped insulin, μU/ml | 81 ± 6.98 | 81.5 ± 6.22 | 0.96 | 112.9 ± 3.91 | 98.3 ± 10.5 | 0.32 |
Values are means ± SE; n = 15–18/group. Sprague-Dawley rats were fed a regular chow or high-fat diet for 4 wk and treated with control or SRC1 ASO. Body weight, basal glucose, and basal insulin were analyzed in overnight-fasted rats. Clamped glucose is the average plasma glucose concentration during the last 40 min of the hyperinsulinemic euglycemic clamp studies. Clamped insulin is the average serum insulin concentration during the hyperinsulinemic euglycemic clamp studies. P value calculated by Student's t-test.
P < 0.05.
SRC1 ASO treatment increases peripheral insulin sensitivity and glucose disposal in gastrocnemius muscle.
To determine whether reduction in SRC1 expression affects insulin sensitivity, regular chow-fed SRC1 or control ASO-treated rats underwent hyperinsulinemic euglycemic clamp studies to quantify basal and insulin-stimulated changes in whole body and tissue-specific glucose metabolism. Plasma glucose values were maintained at euglycemia and matched during the steady-state portion (90–150 min; Fig. 2, A and B).
Fig. 2.

SRC1 ASO treatment increases peripheral insulin sensitivity and glucose disposal in gastrocnemius skeletal muscle. Sprague-Dawley rats were fed a regular chow diet and treated with control or SRC1 ASO for 4 wk. A: serum glucose levels during the hyperinsulinemic euglycemic clamp studies in rats treated with control or SRC1 ASO for 4 wk. B: glucose infusion rate in the clamp studies. C: average glucose infusion rate required to maintain euglycemia during steady state. D: insulin-stimulated peripheral glucose disposal during the hyperinsulinemic euglycemic clamp studies. E: endogenous glucose production during the hyperinsulinemic euglycemic clamp studies. F–I: glucose transport rate during the hyperinsulinemic euglycemic clamp studies into soleus muscle (F), WAT (G), gastrocnemius muscle (H), and BAT (I); n = 15–18/group. P value calculated by Student's t-test. *P < 0.05.
Unlike the SRC1−/− mice (23), decreasing SRC1 expression in normal rats did not alter endogenous glucose production in either the basal or insulin-stimulated state (Fig. 2E). Surprisingly, SRC1 ASO treatment increased the response to insulin, as reflected by a 40% increase in the glucose infusion rate required to maintain euglycemia (Fig. 2, B and C). This increase was largely attributable to a 28% increase in insulin-stimulated whole body peripheral glucose disposal (Fig. 2D).
We further quantified tissue-specific changes in glucose uptake by assessing 2-[14C]deoxyglucose uptake in key insulin responsive tissues. SRC1 ASO treatment increased glucose uptake by 74% in the gastrocnemius muscle (Fig. 2G) but did not increase uptake in the soleus muscle, WAT, or BAT (Fig. 2, F, H, and I). There was also no difference in the mRNA expression of gluconeogenic enzymes under hyperinsulinemic conditions (data not shown). Therefore, knockdown of SRC1 in the adult rat increases peripheral insulin sensitivity due to enhanced insulin-stimulated glucose uptake in specific skeletal muscle groups but does not affect the expression of gluconeogenic enzymes or basal and insulin-stimulated rates of endogenous glucose production.
SRC1 ASO treatment prevents insulin resistance in high-fat-fed rats without preventing weight gain.
Some reports have demonstrated that SRC1−/− mice are prone to diet-induced obesity (13, 26, 30). To determine whether SRC1 ASO treatment alters weight gain and diet-induced obesity and insulin resistance, rats were treated with control or SRC1 ASO and fed a high-fat diet for 4 wk. Insulin sensitivity was again assessed using hyperinsulinemic euglycemic clamp studies (Fig. 3, A and B). SRC1 ASO treatment did not alter body weight or basal glucose concentration, but basal insulin concentration did tend to be less (Table 2). However, as in the regular chow-fed rats, SRC1 ASO increased the glucose infusion rate by 66% (Fig. 3C) and glucose disposal by 46% (Fig. 3D) compared with control ASO-treated animals. This increase in glucose disposal was associated with insulin-stimulated increases in 2-deoxyglucose uptake in the whole gastrocnemius muscle (Fig. 3G) but not the soleus muscle or WAT (Fig. 3, F and H). Furthermore, there were no significant differences in endogenous glucose production in the basal or clamped state between the groups (Fig. 3E).
Fig. 3.

SRC1 ASO treatment increases peripheral insulin sensitivity and glucose disposal in gastrocnemius skeletal muscle in high-fat-fed rats. Sprague-Dawley rats were fed a high-fat diet and treated with control or SRC1 ASO for 4 wk. A: serum glucose levels during the hyperinsulinemic euglycemic clamp studies in rats treated with control or SRC1 ASO for 4 wk. B: glucose infusion rate in the clamp studies. C: average glucose infusion rate required to maintain euglycemia during steady state. D: insulin-stimulated peripheral glucose disposal during the hyperinsulinemic euglycemic clamp studies. E: endogenous glucose production during the hyperinsulinemic euglycemic clamp studies. F–H: glucose transport rate during the hyperinsulinemic euglycemic clamp studies into soleus muscle (F), gastrocnemius muscle (G), and WAT (H); n = 15–18/group. P value calculated by Student's t-test. *P < 0.05.
In summary, similar to rats fed a regular chow diet, SRC1 ASO improved insulin sensitivity in rats fed a high-fat diet by increasing glucose disposal in the gastrocnemius muscle but without altering hepatic insulin sensitivity. This improvement in insulin sensitivity in skeletal muscle is not due to protection from diet-induced obesity, since SRC1 ASO did not alter weight gain. It also cannot be explained by a direct effect of ASO treatment; ASOs decreased SRC1 expression primarily in the liver and WAT and not the skeletal muscle, where insulin sensitivity is enhanced. Instead, these data suggest that the improved insulin-stimulated glucose uptake in mixed-fiber-type skeletal muscle is an indirect effect, due possibly to alteration in a circulating factor that improves insulin-stimulated glucose uptake in muscle.
SRC1 ASO treatment increases the expression of the insulin-sensitizing factor PGD2 synthase.
One candidate for this circulating factor is PGD2. Lipocalin type PGD2 synthase (L-PGDS)-knockout mice develop glucose intolerance and insulin resistance (34), suggesting that L-PGDS positively regulates whole body insulin sensitivity. L-PGDS is a bifunctional enzyme; it synthesizes PGD2 and acts as a lipocalin, transporting various lipophilic small molecules extracellularly (45). It is currently unclear whether L-PGDS increases insulin sensitivity by acting as a lipocalin or through PGD2 production.
Interestingly, SRC1 ASO caused a striking 6.8-fold increase in the WAT expression of L-PGDS (Table 3). Expression of L-PGDS in the liver was actually decreased by ∼90% (P = 0.05). Another enzyme responsible for PGD2 production, hematopoietic type PGD2 synthase, has also been shown to promote insulin sensitivity (15), but the expression of this enzyme did not change with SRC1 knockdown in the WAT (Table 3) or liver (control ASO: 1.0 ± 0.25 vs. SRC1 ASO: 0.7 ± 0.15). The increased expression of white adipose L-PGDS was associated with an increase in plasma PGD2 concentration of 65% in regular chow-fed rats (Fig. 4A) and 70% in high-fat-fed rats (Fig. 4B), suggesting that L-PGDS activity was indeed increased. Interestingly, plasma PGD2 levels decreased within 15–30 min of insulin treatment during the hyperinsulinemic euglycemic clamp in both regular chow and high-fat-fed rats (Fig. 4, A and B).
Table 3.
SRC1 ASO treatment increases the expression of the insulin-sensitizing factor PGDS
| Control ASO | SRC1 ASO | P Value | |
|---|---|---|---|
| L-PGDS | 2.2 ± 1.03 | 17.28 ± 7.4 | 0.04 |
| H-PGDS | 1.07 ± 0.16 | 1.58 ± 0.42 | 0.24 |
| Leptin | 1.17 ± 0.24 | 0.69 ± 0.19 | 0.14 |
| RBP4 | 1.04 ± 0.11 | 0.82 ± 0.11 | 0.19 |
| Resistin | 1.04 ± 0.11 | 1.23 ± 0.18 | 0.38 |
| PPARγ | 1.1 ± 0.19 | 0.65 ± 0.15 | 0.10 |
| CIDEA | 1.05 ± 0.13 | 0.97 ± 0.11 | 0.65 |
| ACOX1 | 1.09 ± 0.18 | 0.80 ± 0.07 | 0.17 |
| PNPLA3 | 1.09 ± 0.18 | 0.33 ± 0.05 | 0.002 |
| PEPCK | 1.06 ± 0.14 | 0.39 ± 0.07 | 0.001 |
| LPL | 1.11 ± 0.2 | 0.92 ± 0.08 | 0.41 |
Values are mean fold change ± SE; n = 6–8/group. L-PGDS, lipocalin type prostaglandin D2 synthase; H-PGDS, hematopoietic type prostaglandin D2 synthase; RBP4, retinol-binding protein-4; PPARγ, peroxisome proliferator-activated receptor-α; CIDEA, cell death-inducing DNA fragmentation factor α-subunit-like effector A; ACOX1, acyl-CoA oxidase 1; PNPLA3, phospholipase domain-containing 3; PEPCK, phospoenolpyruvate carboxykinase; LPL, lipoprotein lipase. Sprague-Dawley rats were treated with control or SRC1 ASO and fed a regular chow diet for 4 wk. Real-time PCR analysis of mRNA expression in the white adipose tissue of hyperinsulinemic euglycemic-clamped rats. P value calculated by Student's t-test.
Fig. 4.

SRC1 ASO treatment increases the expression of the insulin-sensitizing factor prostaglandin D2 (PGD2) synthase. Sprague-Dawley rats were treated with control or SRC1 ASO and fed a regular chow diet for 4 wk. A and B: plasma PGD2 levels as determined by ELISA in chow-fed (A) and high-fat-fed rats (B) during a hyperinsulinemic euglycemic clamp. P value calculated by ANOVA. C: plasma adiponectin concentration in hyperinsulinemic euglycemic-clamped rats. P value calculated by Student's t-test; n = 6–8/group. *P < 0.05.
SRC1 ASO treatment did not alter WAT mRNA expression of other adipokines, such as leptin, retinol-binding protein 4 (RBP4; another lipocalin), and resistin (Table 3) (32, 37). Additionally, the concentration of adiponectin, an insulin-sensitizing adipokine, was reduced by 42% with SRC1 knockdown (Fig. 4C). Although activation of PPARγ in the WAT can increase whole body insulin sensitivity (25, 35, 51), this was not evident in this model. SRC1 ASO treatment did not change expression of the PPARγ target genes acyl-CoA oxidase 1 and liporotein lipase and was associated with a decrease in the expression of phospholipase domain-containing 3 and phosphoenolpyruvate carboxykinase (Table 3).
Insulin and L-PGDS do not increase glucose transport in muscle by increasing tissue perfusion.
Prostaglandins are well known to cause vasodilation and increase muscle blood flow (6, 36). There are also data to suggest that increases in insulin-stimulated glucose uptake are associated with alterations in muscle blood flow (9, 10, 46). Thus, we hypothesized that PGD2 may promote vasodilation and increased perfusion in the gastrocnemius muscle. The increase in muscle glucose delivery would then explain the increased glucose transport with SRC1 ASO treatment. To test this, we measured tissue perfusion in regular chow-fed control and SRC1 ASO-treated animals in both the basal and clamped state. Tissue-specific perfusion was quantified by postmortem γ-counting of 99mTc-tetrofosmin radioactivity in the soleus and gastrocnemius muscles. However, in both the basal and hyperinsulinemic states, SRC1 knockdown did not increase skeletal muscle perfusion (Fig. 5), suggesting that increases in insulin-stimulated glucose uptake in these tissues are not attributable to changes in muscle perfusion in this model (Fig. 5).
Fig. 5.
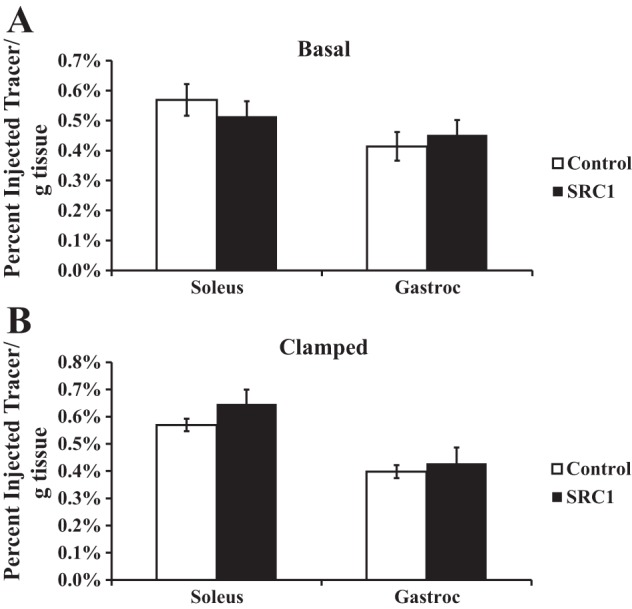
SRC1 ASO treatment and insulin do not change tissue perfusion. Regular chow-fed Sprague-Dawley rats were treated with control or SRC1 ASO for 4 wk. Tissue perfusion was determined in the basal (A) or hyperinsulinemic euglycemic-clamped state (B) by γ-counting of technetium 99 m-tetrofosmin radioactivity in the soleus and gastrocnemius (gastroc) muscles; n = 5–6/group.
SRC1 ASO treatment enhances gastrocnemius glucose transport via TUG cleavage but not Akt or AMPK signaling.
Glucose transport in muscle can be activated by both insulin-dependent and -independent pathways (3, 8, 47). The insulin-independent pathway increases glucose transport for exercising muscle via AMPK activation, leading to Akt substrate of 160 kDa (AS160) phosphorylation and GLUT4 translocation to the plasma membrane (3, 44). The insulin-dependent pathway allows for glucose uptake after a meal. Insulin action in skeletal muscle can be further divided into phosphatidylinositol 3-kinase (PI3K)-dependent or PI3K-independent arms. The canonical PI3K-dependent pathway involves phosphorylation and activation of Akt2 (17), which then phosphorylates many downstream targets, including AS160 (12), leading to GLUT4 translocation. Recent studies have suggested that there is an insulin-dependent but PI3K-independent pathway that also promotes GLUT4 translocation (2, 3). This arm of insulin signaling involves TC10α activation, which may act through PDZ domain-containing protein that interacts specifically with TC10α to promote cleavage of TUG, a protein that sequesters GLUT4 storage vesicles in a perinuclear storage compartment (3–5, 21). Upon insulin stimulation, cleavage of TUG enables the release of GLUT4 storage vesicles (GSVs) for GLUT4 translocation to the plasma membrane and glucose transport.
Analyses of these various pathways in gastrocnemius muscle indicate that SRC1 knockdown does not enhance Akt2 phosphorylation, suggesting the PI3K/Akt2 pathway is not affected (Fig. 6). Likewise, SRC1 ASO treatment does not cause AMPK activation, as assessed by AMPK phosphorylation or phosphorylation of its downstream target ACCβ (Fig. 6). Phosphorylation of these proteins was not different whether analyzed after 2 h (Fig. 6) or 20 min of insulin treatment (data not shown). Additionally, HKII expression was not changed after 20 min of insulin treatment, arguing against the possibility that increased glycolysis due to increased HKII expression may account for the increases in glucose disposal (Fig. 7, A and C).
Fig. 6.
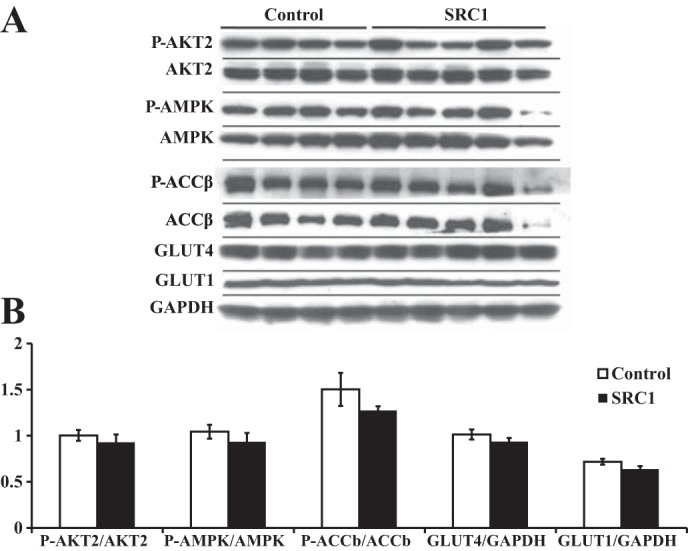
SRC1 ASO treatment does not enhance Akt2 or AMP-activated protein kinase (AMPK) signaling in the gastrocnemius muscle. A: Western blot analysis of phosphorylated (p)-Akt2, p-AMPK, and p-ACCβ (acetyl-CoA carboxylase-β) as well as total glucose transporter (GLUT)1, GLUT4, and GAPDH levels in the gastrocnemius muscle of hyperinsulinemic euglycemic-clamped rats treated with insulin for 2 h. B: densitometry of the Western blot analysis shown in A; n = 5–6/group.
Fig. 7.

SRC1 ASO treatment increases tether-containing UBX domain for GLUT4 (TUG) cleavage in the gastrocnemius muscle. A: Western blot analysis of hexokinase II (HKII), GLUT4, GLUT1, and GAPDH in the gastrocnemius muscle of hyperinsulinemic euglycemic-clamped rats treated with insulin for 20 min. B: Western blot analysis of sortilin, TUG, TUG cleavage products, and GAPDH in the same rats described in A. C: densitometry of the Western blot analysis shown in A and B; n = 5/group. P value determined by Student's t-test. *P < 0.05.
Glucose transport is mediated by the transporters GLUT1 and GLUT4 in muscle. SRC1 knockdown did not change muscle GLUT1 or GLUT4 expression acutely after either 20 min (Fig. 7, A and C) or 2 h (Fig. 6) of hyperinsulinemia. But the expression of GLUT4 in whole tissue extracts does not always correlate with the amount of GLUT4 at the plasma membrane. The recruitment of GLUT4 to the plasma membrane after insulin stimulation is regulated by an intricate network of proteins, including the key protein TUG (5, 21). After 20 min of insulin treatment, TUG cleavage was increased in rats treated with SRC1 ASO, as indicated by a 40% decrease in full-length 60-kDa TUG (Fig. 7, B and C), along with a 2.5-fold increase in the 42-kDa COOH-terminal cleavage product of TUG (Fig. 7, B and C) and a 50% increase in an 18-kDa cleavage product of TUG (Fig. 7, B and C). The decrease in full-length TUG is not due to a decrease in TUG transcription, since TUG mRNA is unchanged (Table 4). Therefore, our data suggest that SRC1 ASO treatment induces the PI3K-independent pathway, leading to TUG cleavage in the gastrocnemius muscle and increased glucose transport compared with control ASO-treated animals.
Table 4.
SRC1 ASO does not alter skeletal muscle mRNA expression of key genes regulating glucose and lipid metabolism
| Control ASO | SRC1 ASO | P Value | |
|---|---|---|---|
| L-PGDS | 2.13 ± 1.15 | 1.57 ± 0.82 | 0.72 |
| GLUT4 | 1.02 ± 0.07 | 1.19 ± 0.19 | 0.38 |
| ATGL | 1.04 ± 0.12 | 1.27 ± 0.13 | 0.22 |
| HSL | 1.08 ± 0.19 | 1.14 ± 0.23 | 0.84 |
| mtGPAT | 1.02 ± 0.08 | 0.97 ± 0.14 | 0.75 |
| DGAT1 | 1.03 ± 0.09 | 1.00 ± 0.13 | 0.87 |
| DGAT2 | 1.07 ± 0.14 | 1.07 ± 0.16 | 0.99 |
| AGPAT1 | 1.03 ± 0.10 | 1.39 ± 0.27 | 0.21 |
| AGPAT2 | 1.03 ± 0.10 | 1.05 ± 0.13 | 0.93 |
| AGPAT5 | 1.00 ± 0.04 | 1.19 ± 0.10 | 0.08 |
| PPARα | 1.02 ± 0.08 | 1.21 ± 0.22 | 0.40 |
| PPARδ | 1.06 ± 0.13 | 1.14 ± 0.19 | 0.72 |
| PGC-1α | 1.02 ± 0.07 | 1.26 ± 0.19 | 0.23 |
| SREBP-1c | 1.02 ± 0.07 | 1.22 ± 0.11 | 0.14 |
| FASN | 1.06 ± 0.14 | 1.33 ± 0.15 | 0.21 |
| SCD1 | 1.29 ± 0.32 | 1.13 ± 0.23 | 0.70 |
| TUG | 1.01 ± 0.06 | 0.96 ± 0.10 | 0.64 |
Values are mean fold change ± SE; n = 6–8/group. GLUT4, glucose transporter 4; ATGL, adipose triglyceride lipase; HSL, hormone-sensitive lipase; mtGPAT; mitochondrial glycerol-3-phosphate acyltransferase; DGAT1 and -2, diaclyglycerol acyltransferase 1 and 2, respectively; AGPAT1, -2, and -5, acylglycerolphosphate acyltransferase 1, 2, and 5, respectively; PPARα and -δ, peroxisome proliferator-activated receptor-α and -δ, respectively; PGC-1α, PPARγ coactivator-1α; SREBP-1c, sterol regulatory element-binding protein-1c; FASN, fatty acid synthase; SCD1, stearoyl-CoA desaturase-1; TUG, tether-containing UBX domain for GLUT4. Sprague-Dawley rats were treated with control or SRC1 ASO for 4 wk, and mRNA expression was analyzed by real-time PCR in the gastrocnemius muscle of hyperinsulinemic euglycemic-clamped rats. P value calculated by Student's t-test.
Interestingly, TUG cleavage products were not observed in the basal state (data not shown), suggesting that enhanced TUG proteolytic cleavage after SRC1 ASO treatment results from increased insulin signaling. Alternatively, enhanced TUG processing may result from increased basal GLUT4 trafficking into the insulin-responsive GSVs. Sortilin is a protein required for proper formation of the GSVs (41). However, SRC1 knockdown did not change sortilin abundance, suggesting that increased TUG processing is not due to changes in sortilin-dependent GLUT4 sorting (Fig. 7B).
Taken together, analyses of the signaling pathways controlling glucose transport in gastrocnemius muscle indicate that the increased insulin-mediated glucose transport with SRC1 ASO treatment is through an increase in TUG cleavage and increased GLUT4 translocation to the plasma membrane.
DISCUSSION
SRC1 is a transcriptional coactivator for many nuclear steroid receptors and transcription factors, including PPARγ, PPARα, and CEBPα (26, 30, 49). SRC1 has been implicated in regulating a variety of metabolic processes. Studies in SRC1−/− mice suggest that SRC1 regulates hepatic gluconeogenesis and glucose output in the fasting state. We undertook these studies to determine whether decreasing SRC1 expression in normal adult rats would similarly alter hepatic gluconeogenesis.
In contrast to the findings in SRC1−/− mice, decreasing hepatic and adipose SRC1 expression with a specific ASO did not alter the hepatic expression of gluconeogenic enzymes, hepatic glucose production, or hepatic insulin sensitivity. Instead, we observed that the main effect of decreasing SRC1 expression in adults rats was an improvement in skeletal muscle insulin sensitivity. This is likely an indirect effect of the SRC1 ASO. ASOs reduce target gene expression primarily in liver and WAT, but in this study they did not alter skeletal muscle SRC1 expression. Thus, these data suggest that SRC1 may regulate the expression of a circulating factor that augments glucose disposal in the skeletal muscle (Fig. 8). The data from these studies suggest a novel connection between adipose tissue and muscle metabolism in which increased adipose expression of L-PGDS and plasma PGD2 concentration may serve to potentiate insulin-stimulated GLUT4 trafficking.
Fig. 8.
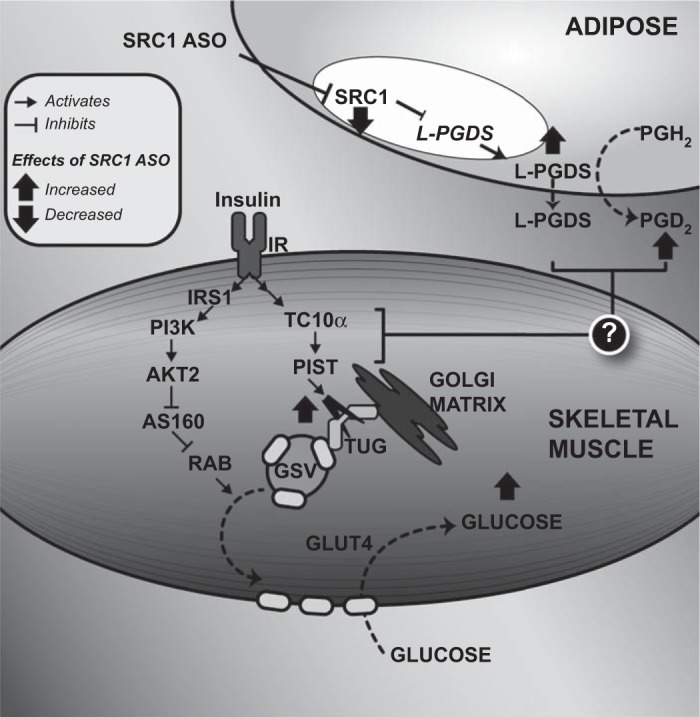
Schematic diagram showing the effect of SRC1 ASO on adipose tissue lipocalin type PGD2 synthase (L-PGDS) expression and muscle insulin-mediated glucose transport. SRC1 ASO decreases the expression of the transcription factor SRC1, leading to an increase in L-PGDS expression and secretion. L-PGDS then increases PGD2 synthesis, and L-PGDS, PGD2, or both lead to proteolysis of TUG proteins in insulin-stimulated gastrocnemius muscle. Since intact TUG prevents exocytic translocation of GLUT4 storage vesicles (GSV), the decrease in TUG abundance releases intracellular GLUT4 to fuse with the plasma membrane and to facilitate glucose transport in the presence of insulin (2). Therefore, SRC1 knockdown in the WAT enhances insulin-mediated glucose transport in the muscle. PGH2, prostaglandin H2; IRS-1, insulin receptor substrate-1; PI3K, phosphatidylinositol 3-kinase; AS160, Akt substrate of 160 kDa; RAB, Ras homologous from brain; PIST, PDZ domain-containing protein that interacts specifically with TC10α.
Our finding that SRC1 knockdown in the liver does not affect gluconeogenesis is contrary to previous studies of SRC1−/− mice (23). There are several possibilities that may explain these discrepancies. Clearly, there are the marked differences in experimental models. Louet et al. (23) studied male C57BL/J6 mice, whereas our study used male SD rats. And unlike a genetic model entirely devoid of SRC1, we decreased expression of SRC1 primarily in WAT and liver in normal adult rats using a synthetic ASO. Since SRC1 is expressed nearly ubiquitously, a global knockout may elicit developmental changes that impact the phenotype of the adult animal. Similarly, global knockout models are not ideal for assessing the effect of a gene on organ-specific physiology. Indeed, studies in SRC1−/− mice also have shown alterations in thyroid and sex hormone action (48, 50). The low expression of SRC1 with ASO treatment compared with complete absence in SRC1−/− mice may also allow for different tissue action, depending on the proteins interacting with SRC1 and threshold for activity of the complex. Finally, SRC1−/− mice gain more weight than WT mice when fed a high-fat diet (30). This may be due to the alterations in BAT or skeletal muscle energetics, where SRC1 competes with SRC2 to regulate genes controlling energy expenditure and uncoupling (13, 30). In comparison, the SRC1 ASOs did not alter BAT or muscle expression, and SRC1 ASO treatment did not impact weight gain in either chow-fed or high-fat-fed rats. Thus, the differences in model systems may account for the variance in the hepatic glucose production phenotype.
Surprisingly, the main physiological effect of decreasing hepatic and adipose SRC1 expression was augmenting insulin-stimulated muscle glucose uptake. SRC1 ASO increased the response to insulin even in the regular chow-fed rats, which were already insulin sensitive. Hepatic and muscle insulin action can be affected by intracellular diacylglycerol content, novel PKC activation (PKC-ϵ and -θ), and impairment in insulin signaling (40). However, intracellular lipid content is normally low in regular chow-fed animals and not affected by SRC1 ASO. Additionally, SRC1 knockdown did not affect insulin-stimulated Akt2 phosphorylation. Thus, the improvement in insulin sensitivity with SRC1 ASO treatment cannot be attributed to a decrease in muscle lipid content.
SRC1 ASO treatment also did not affect other proposed mediators of muscle insulin action, such as leptin, RBP4, or resistin (32, 37). Likewise, it did not increase adiponectin secretion or change PPARγ activity in the WAT, both of which can impact whole body insulin sensitivity (25, 35, 51). Although increases in muscle GLUT4 content or activation of AMPK have both been shown to increase muscle glucose transport (44), neither of these phenomena occurred with SRC1 ASO treatment. Thus, changes in previously described pathways that regulate muscle insulin responsiveness were unable to explain the improvement in insulin-stimulated muscle glucose uptake seen with SRC1 ASO treatment.
In seeking other possibilities, we observed an increase in WAT L-PGDS expression, suggesting that WAT L-PGDS expression and/or plasma PGD2 concentration may impact insulin-stimulated muscle glucose uptake. There is precedent for this model; L-PGDS−/− mice are insulin resistant (34), supporting a role for L-PGDS in controlling insulin sensitivity. L-PGDS and PGD2 have been shown to increase GLUT4 translocation and glucose transport in myotubes (33). We observed a nearly sevenfold rise in L-PGDS expression in the WAT, increased plasma concentration in its enzymatic product PGD2 in both chow-fed and high-fat-fed conditions, and increased glucose transport in the muscle. Taken together, these data suggest that SRC1 may suppress expression of L-PGDS and that L-PGDS or PGD2 may promote insulin-stimulated muscle glucose uptake.
Since PGD2 is a potent vasodilator, we tested the possibility that changes in muscle glucose uptake were primarily a function of increased muscle vasodilation and tissue perfusion. Interestingly, PGD2 concentrations fell with insulin stimulation in SRC1 ASO-treated animals. Thus, we measured muscle perfusion under basal and insulin-stimulated conditions with 99mTc-tetrofosmin, a diffusible radiotracer that is used in preclinical models and clinically for the evaluation of skeletal muscle perfusion (43). However, it was apparent that SRC1 ASO did not alter cardiac or skeletal muscle perfusion under either basal or insulin-stimulated conditions.
It is unlikely that PGD2 promotes TUG cleavage directly. Plasma PGD2 concentrations are high in the basal state, but the differences in TUG cleavage are evident only with insulin stimulation. If PGD2 was responsible, then the insulin-sensitizing effects would be more durable than the tonic stimulation from PGD2 action. From a more generalized view, prostaglandins may act to potentiate the action of growth factor signals. In an analogous model, PGE2 may augment the action of platelet-derived growth factor; PGE2 has low mitotic activity on its own but augments the platelet-derived growth factor signal to increase cell growth (11). Thus, some prostaglandins may act to modulate trophic signals, providing another level of regulation for key signaling pathways.
Alternatively, it is possible that the insulin-sensitizing effects may be due to L-PGDS. One prior study had suggested that L-PGDS could increase GLUT4 translocation and glucose transport in myotubes (33). Thus, we focused on mechanisms that may regulate GLUT4 vesicle transport. GLUT4 is stored in GSVs, which are retained intracellularly by TUG. Upon insulin stimulation, TUG is cleaved, releasing the GSVs to fuse with the plasma membrane (2, 3, 5). In SRC1 ASO-treated rats, we observed increased TUG cleavage in the gastrocnemius muscle in the presence of insulin, which could explain the increased glucose transport seen in the muscle and enhanced insulin sensitivity. The links between TUG cleavage and the increase in L-PGDS and/or PGD2 are not straightforward. Further studies are necessary to discern the roles of L-PGDS and PGD2 on insulin action and explore the connections to TUG cleavage and GLUT4 translocation. The present studies suggest that this may be an avenue worth pursuing.
Controlling insulin sensitivity and glucose homeostasis requires coordination of metabolic processes across several tissues, including the adipose tissue, muscle, and liver. These studies ascribe a new role to SRC1 as a regulator of a potentially novel adipose tissue-skeletal muscle axis. Specifically, SRC1 regulates adipose L-PGDS expression and plasma PGD2 concentration. L-PGDS or PGD2 may act in specific muscle types to fine-tune the GLUT4 response to insulin by potentiating cleavage of TUG (Fig. 8). This axis may hold potential for new therapies that could potentiate muscle glucose uptake in patients with type 2 diabetes.
GRANTS
This project was supported by grants from the US Public Health Service (R24-DK-085638, R01-DK-40936, R01-AG-23686, R01-DK-088231, R01-DK-34989, R01-DK-075772, R01-DK-092661, UL1-RR-0241395, P30-DK-034989, P30-DK-45735, and T32-HL-098069) and a Veterans Affairs Merit Grant (V. T. Samuel). This project was also funded by National Institutes of Health Grant DK-34989 and the Silvio O. Conte Digestive Diseases Research Core Centers (P30-DK-034989.5I01BX000901; V. T. Samuel).
DISCLOSURES
V. P. Manchem and S. Bhanot are employees of Isis Pharmaceuticals and may own stock in the company. The other authors have no competing interests to declare.
AUTHOR CONTRIBUTIONS
J.L.C., M.R.S., D.P.D., A.J.S., L.R., J.S.B., and V.T.S. conception and design of research; J.L.C., D.F.V., T.G., A.M., M.P., R.J.P., N.K., F.G.-E., A.K.G., M.R.S., D.P.D., A.J.S., C.E.H., and V.P.M. performed experiments; J.L.C., D.F.V., T.G., A.M., M.P., R.J.P., N.K., F.G.-E., A.K.G., M.R.S., D.P.D., A.J.S., V.P.M., S.B., and V.T.S. analyzed data; J.L.C., D.F.V., T.G., A.M., M.P., R.J.P., N.K., F.G.-E., A.K.G., M.R.S., D.P.D., A.J.S., L.R., C.E.H., V.P.M., S.B., J.S.B., and V.T.S. interpreted results of experiments; J.L.C. and V.T.S. prepared figures; J.L.C. and V.T.S. drafted manuscript; J.L.C., M.R.S., V.P.M., J.S.B., and V.T.S. edited and revised manuscript; J.L.C., A.M., M.P., R.J.P., N.K., F.G.-E., M.R.S., D.P.D., A.J.S., V.P.M., S.B., J.S.B., and V.T.S. approved final version of manuscript.
REFERENCES
- 1.Amazit L, Roseau A, Khan JA, Chauchereau A, Tyagi RK, Loosfelt H, Leclerc P, Lombes M, Guiochon-Mantel A. Ligand-dependent degradation of SRC-1 is pivotal for progesterone receptor transcriptional activity. Mol Endocrinol 25: 394–408, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Belman JP, Habtemichael EN, Bogan JS. A proteolytic pathway that controls glucose uptake in fat and muscle. Rev Endocr Metab Disord 15: 55–66, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bogan JS. Regulation of glucose transporter translocation in health and diabetes. Annu Rev Biochem 81: 507–532, 2012 [DOI] [PubMed] [Google Scholar]
- 4.Bogan JS, Hendon N, McKee AE, Tsao TS, Lodish HF. Functional cloning of TUG as a regulator of GLUT4 glucose transporter trafficking. Nature 425: 727–733, 2003 [DOI] [PubMed] [Google Scholar]
- 5.Bogan JS, Rubin BR, Yu C, Loffler MG, Orme CM, Belman JP, McNally LJ, Hao M, Cresswell JA. Endoproteolytic cleavage of TUG protein regulates GLUT4 glucose transporter translocation. J Biol Chem 287: 23932–23947, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Boushel R, Langberg H, Gemmer C, Olesen J, Crameri R, Scheede C, Sander M, Kjaer M. Combined inhibition of nitric oxide and prostaglandins reduces human skeletal muscle blood flow during exercise. J Physiol 543: 691–698, 2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cantley JL, Yoshimura T, Camporez JP, Zhang D, Jornayvaz FR, Kumashiro N, Guebre-Egziabher F, Jurczak MJ, Kahn M, Guigni BA, Serr J, Hankin J, Murphy RC, Cline GW, Bhanot S, Manchem VP, Brown JM, Samuel VT, Shulman GI. CGI-58 knockdown sequesters diacylglycerols in lipid droplets/ER-preventing diacylglycerol-mediated hepatic insulin resistance. Proc Natl Acad Sci USA 110: 1869–1874, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chang L, Chiang SH, Saltiel AR. Insulin signaling and the regulation of glucose transport. Mol Med 10: 65–71, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Clark MG, Wallis MG, Barrett EJ, Vincent MA, Richards SM, Clerk LH, Rattigan S. Blood flow and muscle metabolism: a focus on insulin action. Am J Physiol Endocrinol Metab 284: E241–E258, 2003 [DOI] [PubMed] [Google Scholar]
- 10.Clerk LH, Vincent MA, Lindner JR, Clark MG, Rattigan S, Barrett EJ. The vasodilatory actions of insulin on resistance and terminal arterioles and their impact on muscle glucose uptake. Diabetes Metab Res Rev 20: 3–12, 2004 [DOI] [PubMed] [Google Scholar]
- 11.Dajani OF, Meisdalen K, Guren TK, Aasrum M, Tveteraas IH, Lilleby P, Thoresen GH, Sandnes D, Christoffersen T. Prostaglandin E2 upregulates EGF-stimulated signaling in mitogenic pathways involving Akt and ERK in hepatocytes. J Cell Physiol 214: 371–380, 2008 [DOI] [PubMed] [Google Scholar]
- 12.Deshmukh A, Coffey VG, Zhong Z, Chibalin AV, Hawley JA, Zierath JR. Exercise-induced phosphorylation of the novel Akt substrates AS160 and filamin A in human skeletal muscle. Diabetes 55: 1776–1782, 2006 [DOI] [PubMed] [Google Scholar]
- 13.Duteil D, Chambon C, Ali F, Malivindi R, Zoll J, Kato S, Geny B, Chambon P, Metzger D. The transcriptional coregulators TIF2 and SRC-1 regulate energy homeostasis by modulating mitochondrial respiration in skeletal muscles. Cell Metab 12: 496–508, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Feige JN, Auwerx J. Transcriptional coregulators in the control of energy homeostasis. Trends Cell Biol 17: 292–301, 2007 [DOI] [PubMed] [Google Scholar]
- 15.Fujitani Y, Aritake K, Kanaoka Y, Goto T, Takahashi N, Fujimori K, Kawada T. Pronounced adipogenesis and increased insulin sensitivity caused by overproduction of prostaglandin D2 in vivo. FEBS J 277: 1410–1419, 2010 [DOI] [PubMed] [Google Scholar]
- 16.Han SJ, DeMayo FJ, Xu J, Tsai SY, Tsai MJ, O'Malley BW. Steroid receptor coactivator (SRC)-1 and SRC-3 differentially modulate tissue-specific activation functions of the progesterone receptor. Mol Endocrinol 20: 45–55, 2006 [DOI] [PubMed] [Google Scholar]
- 17.Jiang ZY, Zhou QL, Coleman KA, Chouinard M, Boese Q, Czech MP. Insulin signaling through Akt/protein kinase B analyzed by small interfering RNA-mediated gene silencing. Proc Natl Acad Sci USA 100: 7569–7574, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kino T, Ichijo T, Chrousos GP. FLASH interacts with p160 coactivator subtypes and differentially suppresses transcriptional activity of steroid hormone receptors. J Steroid Biochem Mol Biol 92: 357–363, 2004 [DOI] [PubMed] [Google Scholar]
- 19.Kumashiro N, Yoshimura T, Cantley JL, Majumdar SK, Guebre-Egziabher F, Kursawe R, Vatner DF, Fat I, Kahn M, Erion DM, Zhang XM, Zhang D, Manchem VP, Bhanot S, Gerhard GS, Petersen KF, Cline GW, Samuel VT, Shulman GI. Role of patatin-like phospholipase domain-containing 3 on lipid-induced hepatic steatosis and insulin resistance in rats. Hepatology 57: 1763–1772, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lachize S, Apostolakis EM, van der Laan S, Tijssen AM, Xu J, de Kloet ER, Meijer OC. Steroid receptor coactivator-1 is necessary for regulation of corticotropin-releasing hormone by chronic stress and glucocorticoids. Proc Natl Acad Sci USA 106: 8038–8042, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Loffler MG, Birkenfeld AL, Philbrick KM, Belman JP, Habtemichael EN, Booth CJ, Castorena CM, Choi CS, Jornayvaz FR, Gassaway BM, Lee HY, Cartee GD, Philbrick W, Shulman GI, Samuel VT, Bogan JS. Enhanced fasting glucose turnover in mice with disrupted action of TUG protein in skeletal muscle. J Biol Chem 288: 20135–20150, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lonard DM, Kumar R, O'Malley BW. Minireview: the SRC family of coactivators: an entree to understanding a subset of polygenic diseases? Mol Endocrinol 24: 279–285, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Louet JF, Chopra AR, Sagen JV, An J, York B, Tannour-Louet M, Saha PK, Stevens RD, Wenner BR, Ilkayeva OR, Bain JR, Zhou S, DeMayo F, Xu J, Newgard CB, O'Malley BW. The coactivator SRC-1 is an essential coordinator of hepatic glucose production. Cell Metab 12: 606–618, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Louet JF, O'Malley BW. Coregulators in adipogenesis: what could we learn from the SRC (p160) coactivator family? Cell Cycle 6: 2448–2452, 2007 [DOI] [PubMed] [Google Scholar]
- 25.Medina-Gomez G, Virtue S, Lelliott C, Boiani R, Campbell M, Christodoulides C, Perrin C, Jimenez-Linan M, Blount M, Dixon J, Zahn D, Thresher RR, Aparicio S, Carlton M, Colledge WH, Kettunen MI, Seppanen-Laakso T, Sethi JK, O'Rahilly S, Brindle K, Cinti S, Oresic M, Burcelin R, Vidal-Puig A. The link between nutritional status and insulin sensitivity is dependent on the adipocyte-specific peroxisome proliferator-activated receptor-gamma2 isoform. Diabetes 54: 1706–1716, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Miard S, Dombrowski L, Carter S, Boivin L, Picard F. Aging alters PPARgamma in rodent and human adipose tissue by modulating the balance in steroid receptor coactivator-1. Aging Cell 8: 449–459, 2009 [DOI] [PubMed] [Google Scholar]
- 27.Mukherjee A, Soyal SM, Fernandez-Valdivia R, Gehin M, Chambon P, Demayo FJ, Lydon JP, O'Malley BW. Steroid receptor coactivator 2 is critical for progesterone-dependent uterine function and mammary morphogenesis in the mouse. Mol Cell Biol 26: 6571–6583, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nishihara E, Yoshida-Komiya H, Chan CS, Liao L, Davis RL, O'Malley BW, Xu J. SRC-1 null mice exhibit moderate motor dysfunction and delayed development of cerebellar Purkinje cells. J Neurosci 23: 213–222, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Peng B, Andrews J, Nestorov I, Brennan B, Nicklin P, Rowland M. Tissue distribution and physiologically based pharmacokinetics of antisense phosphorothioate oligonucleotide ISIS 1082 in rat. Antisense Nucleic Acid Drug Dev 11: 15–27, 2001 [DOI] [PubMed] [Google Scholar]
- 30.Picard F, Gehin M, Annicotte J, Rocchi S, Champy MF, O'Malley BW, Chambon P, Auwerx J. SRC-1 and TIF2 control energy balance between white and brown adipose tissues. Cell 111: 931–941, 2002 [DOI] [PubMed] [Google Scholar]
- 31.Qin L, Liu Z, Chen H, Xu J. The steroid receptor coactivator-1 regulates twist expression and promotes breast cancer metastasis. Cancer Res 69: 3819–3827, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rabe K, Lehrke M, Parhofer KG, Broedl UC. Adipokines and insulin resistance. Mol Med 14: 741–751, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ragolia L, Hall CE, Palaia T. Lipocalin-type prostaglandin D(2) synthase stimulates glucose transport via enhanced GLUT4 translocation. Prostaglandins Other Lipid Mediat 87: 34–41, 2008 [DOI] [PubMed] [Google Scholar]
- 34.Ragolia L, Palaia T, Hall CE, Maesaka JK, Eguchi N, Urade Y. Accelerated glucose intolerance, nephropathy, and atherosclerosis in prostaglandin D2 synthase knock-out mice. J Biol Chem 280: 29946–29955, 2005 [DOI] [PubMed] [Google Scholar]
- 35.Rangwala SM, Lazar MA. Peroxisome proliferator-activated receptor gamma in diabetes and metabolism. Trends Pharmacol Sci 25: 331–336, 2004 [DOI] [PubMed] [Google Scholar]
- 36.Rao PS, Cavanagh D, O'Brien WF, Spaziani E. The effect of prostaglandin D2 on angiotensin converting enzyme. Prostaglandins Leukot Essent Fatty Acids 33: 111–114, 1988 [DOI] [PubMed] [Google Scholar]
- 37.Rasouli N, Kern PA. Adipocytokines and the metabolic complications of obesity. J Clin Endocrinol Metab 93: S64–S73, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Samuel VT, Choi CS, Phillips TG, Romanelli AJ, Geisler JG, Bhanot S, McKay R, Monia B, Shutter JR, Lindberg RA, Shulman GI, Veniant MM. Targeting foxo1 in mice using antisense oligonucleotide improves hepatic and peripheral insulin action. Diabetes 55: 2042–2050, 2006 [DOI] [PubMed] [Google Scholar]
- 39.Samuel VT, Liu ZX, Wang A, Beddow SA, Geisler JG, Kahn M, Zhang XM, Monia BP, Bhanot S, Shulman GI. Inhibition of protein kinase Cepsilon prevents hepatic insulin resistance in nonalcoholic fatty liver disease. J Clin Invest 117: 739–745, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Samuel VT, Shulman GI. Mechanisms for insulin resistance: common threads and missing links. Cell 148: 852–871, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shi J, Kandror KV. Sortilin is essential and sufficient for the formation of Glut4 storage vesicles in 3T3-L1 adipocytes. Dev Cell 9: 99–108, 2005 [DOI] [PubMed] [Google Scholar]
- 42.Spears M, Bartlett J. The potential role of estrogen receptors and the SRC family as targets for the treatment of breast cancer. Expert Opin Ther Targets 13: 665–674, 2009 [DOI] [PubMed] [Google Scholar]
- 43.Stacy MR, Zhou W, Sinusas AJ. Radiotracer imaging of peripheral vascular disease. J Nucl Med 54: 2104–2110, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Thong FS, Bilan PJ, Klip A. The Rab GTPase-activating protein AS160 integrates Akt, protein kinase C, and AMP-activated protein kinase signals regulating GLUT4 traffic. Diabetes 56: 414–423, 2007 [DOI] [PubMed] [Google Scholar]
- 45.Urade Y, Eguchi N. Lipocalin-type and hematopoietic prostaglandin D synthases as a novel example of functional convergence. Prostaglandins Other Lipid Mediat 68–69: 375–382, 2002 [DOI] [PubMed] [Google Scholar]
- 46.Vincent MA, Clerk LH, Lindner JR, Klibanov AL, Clark MG, Rattigan S, Barrett EJ. Microvascular recruitment is an early insulin effect that regulates skeletal muscle glucose uptake in vivo. Diabetes 53: 1418–1423, 2004 [DOI] [PubMed] [Google Scholar]
- 47.Watson RT, Kanzaki M, Pessin JE. Regulated membrane trafficking of the insulin-responsive glucose transporter 4 in adipocytes. Endocr Rev 25: 177–204, 2004 [DOI] [PubMed] [Google Scholar]
- 48.Weiss RE, Xu J, Ning G, Pohlenz J, O'Malley BW, Refetoff S. Mice deficient in the steroid receptor co-activator 1 (SRC-1) are resistant to thyroid hormone. EMBO J 18: 1900–1904, 1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Xu J, Li Q. Review of the in vivo functions of the p160 steroid receptor coactivator family. Mol Endocrinol 17: 1681–1692, 2003 [DOI] [PubMed] [Google Scholar]
- 50.Yamada T, Kawano H, Sekine K, Matsumoto T, Fukuda T, Azuma Y, Itaka K, Chung UI, Chambon P, Nakamura K, Kato S, Kawaguchi H. SRC-1 is necessary for skeletal responses to sex hormones in both males and females. J Bone Miner Res 19: 1452–1461, 2004 [DOI] [PubMed] [Google Scholar]
- 51.Zhang J, Fu M, Cui T, Xiong C, Xu K, Zhong W, Xiao Y, Floyd D, Liang J, Li E, Song Q, Chen YE. Selective disruption of PPARgamma 2 impairs the development of adipose tissue and insulin sensitivity. Proc Natl Acad Sci USA 101: 10703–10708, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhu L, Yang Y, Xu P, Zou F, Yan X, Liao L, Xu J, O'Malley BW, Xu Y. Steroid receptor coactivator-1 mediates estrogenic actions to prevent body weight gain in female mice. Endocrinology 154: 150–158, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]