

Abstract
Peptidyl arginine deiminase (PAD)4 is a nuclear enzyme that catalyzes the posttranslational conversion of arginine residues to citrulline. Posttranslational protein citrullination has been implicated in several inflammatory autoimmune diseases, including rheumatoid arthritis, colitis, and multiple sclerosis. Here, we tested the hypothesis that PAD4 contributes to ischemic acute kidney injury (AKI) by exacerbating the inflammatory response after renal ischemia-reperfusion (I/R). Renal I/R injury in mice increased PAD4 activity as well as PAD4 expression in the mouse kidney. After 30 min of renal I/R, vehicle-treated mice developed severe AKI with large increases in plasma creatinine. In contrast, mice pretreated with PAD4 inhibitors (2-chloroamidine or streptonigrin) had significantly reduced renal I/R injury. Further supporting a critical role for PAD4 in generating ischemic AKI, mice pretreated with recombinant human PAD4 (rPAD4) protein and subjected to mild (20 min) renal I/R developed exacerbated ischemic AKI. Consistent with the hypothesis that PAD4 regulates renal tubular inflammation after I/R, mice treated with a PAD4 inhibitor had significantly reduced renal neutrophil chemotactic cytokine (macrophage inflammatory protein-2 and keratinocyte-derived cytokine) expression and had decreased neutrophil infiltration. Furthermore, mice treated with rPAD4 had significantly increased renal tubular macrophage inflammatory protein-2 and keratinocyte-derived cytokine expression as well as increased neutrophil infiltration and necrosis. Finally, cultured mouse kidney proximal tubules treated with rPAD4 had significantly increased proinflammatory chemokine expression compared with vehicle-treated cells. Taken together, our results suggest that PAD4 plays a critical role in renal I/R injury by increasing renal tubular inflammatory responses and neutrophil infiltration after renal I/R.
Keywords: acute kidney injury, apoptosis, citrullination, histone, inflammation, necrosis
renal ischemia-reperfusion (I/R) injury is a major cause of perioperative acute kidney injury (AKI) for patients undergoing major vascular, abdominal organ transplantation or cardiac surgery and is directly associated with high mortality, morbidity, and cost during the perioperative period (3, 17). Furthermore, patients with AKI frequently develop extrarenal organ dysfunction, including hepatic dysfunction, intestinal barrier disruption, respiratory failure, and systemic inflammatory response syndrome, which frequently lead to the development of sepsis and multiorgan failure (31, 44). Unfortunately, the severity and incidence of AKI has been increasing without effective therapy or improvements in patient survival for the past 70 yr (16).
Renal I/R injury leading to AKI is characterized by renal tubular necrosis and apoptosis as well as by a severe inflammatory response (5, 18, 35). Indeed, renal inflammation is a major cause of ischemic AKI and is mediated by the renal tubular upregulation of proinflammatory cytokines followed by intrarenal leukocyte infiltration. Infiltrating leukocytes, including T lymphocytes, natural killer cells, and neutrophils, directly exacerbate kidney inflammation and injury (11, 35). Depletion or blockade of these proinflammatory leukocytes protects against I/R-induced AKI. Therefore, limiting the inflammatory response after renal I/R would attenuate the degree of renal tubular injury and protect against ischemic AKI.
Peptidyl arginine deiminases (PADs) are enzymes that catalyze the conversion of peptidyl arginine residues to peptidyl citrulline in target proteins in a Ca2+-dependent manner (2, 42). Posttranslational protein citrullination leads to drastic changes on protein charge, structure, and function (12, 15, 42). Of the five known subtypes of PAD (PAD1, PAD2, PAD3, PAD4, and PAD6), PAD4 is the only PAD enzyme localized in the nucleus and is the only PAD protein detected in the kidney (39, 41). PAD4-mediated posttranslational protein citrullination has been implicated in several inflammatory autoimmune diseases, including colitis, lupus, rheumatoid arthritis, and multiple sclerosis (2, 15, 27). Furthermore, PAD4-mediated citrullination of histones is critical for neutrophil extracellular trap formation after bacterial infection or the development of deep vein thrombosis (28, 29). Although the role of PAD4 in neutrophils, leukocytes, and autoimmune diseases has been well characterized, the role for PAD4-mediated protein citrullination in renal I/R injury has never been examined. Here, we tested the hypothesis that renal I/R induces PAD4 activity and expression. In addition, we tested whether kidney PAD4 activity contributes to ischemic AKI by exacerbating the inflammatory response after renal I/R.
MATERIALS AND METHODS
Murine model of renal I/R injury.
The Institutional Animal Care and Use Committee of Columbia University gave approval for the present study. Adult male C57BL/7 mice (∼20 g, Charles River, Wilmington, MA) were anesthetized with intraperioneal pentobarbital (Henry Schein Veterinary, Indianapolis, IN, 50 mg/kg body wt or to effect) and subjected to right nephrectomy and 20 or 30 min of left renal ischemia as previously described (10, 19). Sham-operated animals underwent the same surgical procedures (anesthesia, laparotomy, bowel manipulations, and wound closure) without renal ischemia. The body temperature was maintained between 36 and 38°C using a surgical heating pad during and after surgery. To test whether PAD4 inhibition protects against renal I/R injury, some mice were pretreated with 2-chloroamidine (100 mg/kg, a specific PAD inhibitor, EMD Millipore, Darmstadt, Germany) or with streptonigrin (0.4 mg/kg, a selective PAD4 inhibitor, Sigma Chemicals, St. Louis, MO) 15 min before the induction of 30 min of renal ischemia (6, 7). To test whether PAD4 supplementation exacerbates renal I/R injury, separate cohorts of mice were pretreated with 10 μg iv recombinant human PAD4 (rPAD4, Cayman Chemical, Ann Arbor, MI) 15 min before the induction of 20 min of renal ischemia.
Measurement of renal function.
Plasma creatinine was measured 24 h after renal I/R using an enzymatic creatinine reagent kit according to the manufacturer's protocol (Thermo Fisher Scientific, Waltham, MA). This method of creatinine measurement largely eliminates the interferences from mouse plasma chromagens well known to the Jaffe method (38).
Histological detection of kidney injury.
Kidney histology was examined 24 h after renal I/R or sham surgery by an experienced pathologist (V. D'Agati) who was unaware of the treatment that each animal had received. An established grading scale of necrotic injury (renal injury scores of 0–4) to the proximal tubules was used for the histopathological assessment of I/R-induced damage as outlined by Jablonski et al. (14) and as previously described in our studies (20, 21).
Histological detection of kidney apoptosis and neutrophil infiltration.
We detected apoptosis with TUNEL staining using a commercially available in situ cell death detection kit (Roche, Indianapolis, IN) 24 h after renal I/R or sham surgery as previously described (33, 34). Apoptotic TUNEL-positive cells were quantified in five to seven randomly chosen ×100 microscope image fields in the corticomedullary junction, and results were expressed as apoptotic cells counted per ×100 field. To corroborate our apoptosis quantification with TUNEL staining, an experienced pathologist (V. D'Agati) counted the number of apoptotic cells from 10 high-power fields in the corticomedullary junction (×400) per kidney.
Immunohistochemistry for neutrophils was performed using rat anti-mouse lymphocyte antigen (Ly)6B monoclonal antibody against neutrophils (AbD Serotec, Raleigh, NC), as previously described (33, 34). Primary antibody recognizing IgG2a (MCA1212, AbD Serotec) was used as a negative isotype control. Neutrophils infiltrating the kidney were quantified in five to seven randomly chosen ×200 microscope image fields in the corticomedullary junction, and results were expressed as neutrophils counted per ×200 field.
Quantitative RT-PCR.
Kidney inflammation after renal ischemia was also determined by measuring mRNA encoding markers of inflammation, including keratinocyte-derived cytokine (KC), ICAM-1, monocyte chemoattractant protein (MCP)-1, macrophage inflammatory protein (MIP)-2, and TNF-α 24 h after renal I/R. RT-PCR was performed as previously described (33, 34) with the primers shown in Table 1. Primer design was based on published GenBank sequences. Quantitative RT-PCR was performed using the MyiQ Real Time Detection System (Bio-Rad, Hercules, CA) using FastStart Universal SYBR Green Master Mix (Roche). To confirm equal RNA input, GAPDH mRNA expression and the relative expression of proinflammatory mRNA were calculated with the ΔΔCt method (where Ct is threshold cycle). Specificity of the amplification was checked by melting curve analysis.
Table 1.
Primers used in quantitative RT-PCRs to amplify mouse cDNAs based on published GenBank sequences for mice as well as annealing temperatures used for each primer
| Sense Primer | Antisense Primer | Annealing Temperature, °C | |
|---|---|---|---|
| TNF-α | 5′-TACTGAACTTCGGGGTGATTGGTCC-3′ | 5′-CAGCCTTGTCCCTTGAAGAGAACC-3′ | 65 |
| ICAM-1 | 5′-TGTTTCCTGCCTCTGAAGC-3′ | 5′-CTTCGTTTGTGATCCTCCG-3′ | 60 |
| Monocyte chemoattractant protein-1 | 5′-ACCTGCTGCTACTCATTCAC-3′ | 5′-TTGAGGTGGTTGTGGAAAAG-3′ | 60 |
| Macrophage inflammatory protein-2 | 5′-CCAAGGGTTGACTTCAAGAAC-3′ | 5′-AGCGAGGCACATCAGGTACG-3′ | 60 |
| Keratinocyte-derived cytokine | 5′-CAATGAGCTGCGCTGTCAGTG-3′ | 5′-CTTGGGGACACCTTTTAGCATC-3′ | 60 |
| GAPDH | 5′-ACCACAGTCCATGCCATCAC-3′ | 5′-CACCACCCTGTTGCTGTAGCC-3′ | 65 |
Isolation of mouse kidney proximal tubule cells and culture.
Mouse kidney proximal tubules were isolated using Percoll density gradient separation as previously described (32, 40). Mouse proximal tubules were grown in low-glucose DMEM-Ham's F-12 medium plus 10% FBS, 2 mM l-glutamine, and 10 mM HEPES.
PAD4 activity assay.
Mouse kidneys were collected 24 h after renal I/R injury or sham surgery. PAD activity was measured according to methods previously described by Wildeman and Pires (43) with slight modifications. This is a fluorescence-based assay and uses a small-molecule mimic of PAD4's natural substrate followed by trypsin cleavage subsequently unmasking the fluorophore. Increased PAD4 activity increases the citrulline product, which is not recognized by trypsin, leading to a reduced fluorescence reading. In brief, kidney tissue homogenates were incubated with the enzyme substrate (benzoyl-dl-arginine-7-amino-4-methylcoumarine, Santa Cruz Biotechnology, Santa Cruz, CA) plus 10 mM CaCl2 at 37°C for 10 min in the presence or absence of 200 μM 2-chloroamidine (a PAD4 inhibitor) in 50 mM Tris·HCl buffer (pH 7.6). After trypsin hydrolysis, the reaction was quenched in liquid nitrogen. The 14,000-g supernatant was analyzed with HPLC to measure the product of PAD4 enzyme (7-amino-4-methylcoumarine) on a C18 reversed-phase column with a binary low-pressure gradient elution system with a fluorescence detector set to 441 nm upon excitation with light at 342 nm.
PAD4 immunohistochemistry.
Immunohistochemistry was performed on mouse kidneys subjected to sham surgery or to 30 min of renal I/R injury 24 h postreperfusion. Kidneys were fixed with 4% paraformaldehyde, dehydrated with 30% sucrose, frozen in OCT (Tissue-Tek, Torrance, CA), and cryosectioned (5 μm thick). After being washed, sections were blocked with 2% BSA for 1 h at room temperature and stained with goat anti-PAD4 antibody (Santa Cruz Biotechnology) overnight followed by Alexa fluor 594-conjugated donkey anti-goat secondary antibody (Invitrogen, Carlsbad, CA) plus 4′,6-diamidino-2-phenylindole (DAPI; Invitrogen) nuclear staining. Cryosections were mounted in ProLong Gold antifade reagent-containing DAPI from Molecular Probes (Invitrogen) and imaged under a fluorescence microscope. PAD4 immunofluorescence intensities were quantified in ×400 images with Adobe Photoshop software. For each image, fluorescence intensities from four equal areas were averaged.
Citrullinated histone H3 immunohistochemistry.
Paraffin-embedded kidney tissues collected 24 h after renal I/R or sham surgery were cut at 5 μm, deparaffinized, and rehydrated in a graded ethanol series. Endogenous peroxidase was inhibited using 0.3% H2O2 in PBS for 30 min. Sections were then heated in 97°C sodium citrate buffer (10 mM, pH 6) for 40 min for antigen retrieval. Sections were incubated with 3% BSA in PBS for 60 min and then treated with the Vector Blocking kit (Vector Laboratories, Burlingame, CA) for endogenous biotin inhibition. Sections were then incubated overnight at 4°C with rabbit monoclonal citrulline R26 anti-citrullinated histone H3 antibody (1:100, 1% BSA in PBS-Tween, Abcam, Cambridge, MA). Sections were subsequently incubated with anti-rabbit secondary antibody (1:100, 1% BSA in PBS, Vector Laboratories) for 1 h, washed, and then incubated with avidin-biotin complex (ABC kit, Vector Laboratories). Sections were developed with 3,3′-diaminobenzidine and counterstained with hematoxylin. Some sections were stained without hematoxylin to confirm that nuclear 3,3′-diaminobenzidine stains citrullinated histone H3.
Statistical analysis.
Data were analyzed with Student's t-test when means between two groups were compared or with one-way ANOVA plus Tukey's post hoc multiple comparison test when multiple groups were compared. The ordinal values of renal and hepatic injury scores were analyzed by a Mann-Whitney nonparametric test. In all cases, P values of <0.05 were used to indicate significance. All data are expressed throughout the text as means ± SE.
RESULTS
Renal I/R induces PAD4 expression and activity.
We first determined whether renal I/R injury induces mouse kidney PAD4 expression. Figure 1A shows selective PAD4 mRNA induction (∼15-fold) in the kidneys of mice subjected to 24 h of renal I/R compared with sham-operated mice. PAD2 mRNA expression did not change after renal I/R compared with the sham-operated group. Consistent with the induction of PAD4 mRNA, we found increased PAD4 protein expression in mouse kidneys subjected to 24 h of renal I/R injury compared with sham-operated mice. Figure 1B shows representative images (from 4 experiments), and Fig. 1C shows the average fluorescence intensity quantification of PAD4 immunohistochemistry. Finally, there was a significant (∼5-fold) induction of PAD4 activity in the kidneys of mice subjected to 24 h of renal I/R compared with sham-operated mice (Fig. 1C).
Fig. 1.

Induction and activation of kidney peptidyl arginine deiminase (PAD)4 after ischemia-reperfusion (I/R) injury. A: PAD4 mRNA expression measured with quantitative RT-PCR increased in the mouse kidney 24 h after renal I/R injury (n = 5–6 experiments). Note the lack of increase in PAD2 mRNA after renal I/R. B: immunohistochemistry images (n = 4 representative experiments) showing increased renal tubular PAD4 protein expression (red fluorescence, ×400) in the mouse kidney 24 h after renal I/R injury. Blue fluorescence indicates 4′,6-diamidino-2-phenylindole (DAPI) nuclear staining. C: PAD4 immunofluorescence intensities quantified in ×400 image fields were significantly higher in mice subjected to renal I/R compared with sham-operated (sham) mice (n = 4). D: increased mouse kidney PAD4 activity 24 h after renal I/R injury (n = 5 experiments). *P < 0.05 vs. the sham group. Error bars represent 1 SE.
Renal I/R increases renal tubular nuclear histone H3 citrullination.
Considering that PAD4 converts peptidyl arginine residue to peptidyl citrulline on the histone tail and since PAD4 expression and activity were significantly increased after renal I/R, we performed immunohistochemistry to test whether renal I/R increases histone H3 citrullination. Twenty-four hours after renal I/R, we found increased citrullinated histone H3 immunoreactivity in the nucleus of renal proximal tubules compared with sham-operated mice (representative of 4 experiments; Fig. 2). Mice treated with a PAD4 inhibitor (2-chloroamidine) and subjected to renal I/R consequently showed markedly reduced citrullinated histone H3 expression, supporting the hypothesis that increased PAD4 activity after renal I/R increases histone H3 citrullination.
Fig. 2.

Renal I/R causes PAD4-mediated citrullination of nuclear histone H3. Images are representative of four immunohistochemistry experiments performed to stain for citrullinated histone H3. Kidneys subjected to renal I/R showed increased nuclear citrullinated histone H3 staining (dark brown, ×1,000) compared with those from sham mice. Mice treated with the selective PAD inhibitor 2-chloroamidine (2-Cl-amidine) showed markedly attenuated nuclear citrullinated histone H3 staining after renal I/R. Kidneys incubated with negative isotype control antibody showed no staining in the nucleus (n = 4 experiments).
PAD4 inhibition protects against renal I/R injury.
After demonstrating selective induction of PAD4 expression and increased PAD4 activity after renal I/R, we tested whether PAD4 inhibition would protect against ischemic AKI in mice. We used two distinct inhibitors of PAD: 2-chloroamidine and streptonigrin (6, 7). Plasma creatinine values were similar between vehicle-treated and 2-chloroamidine-treated sham-operated mice (anesthesia, laparotomy, right nephrectomy, and recovery; Fig. 3). As expected, plasma creatinine increased significantly in vehicle-treated mice 24 h after 30 min of renal I/R injury. However, PAD4 inhibition with 2-chloroamidine or streptonigrin treatment before renal ischemia protected against renal injury, as evidenced by the significantly lower plasma creatinine levels compared with vehicle-treated mice subjected to renal I/R (Fig. 3).
Fig. 3.
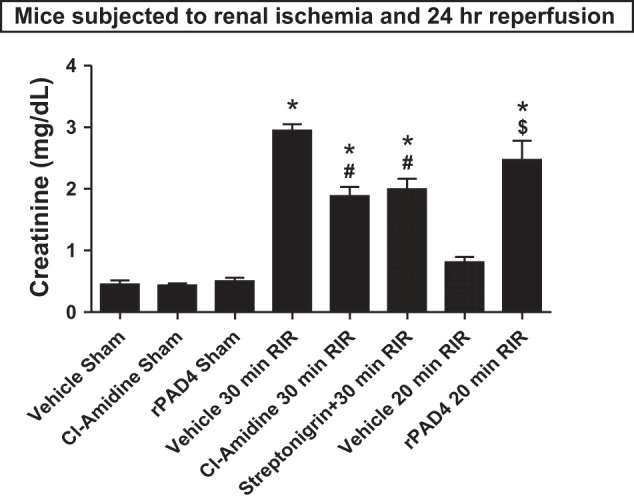
PAD4 plays a critical role in renal I/R (RIR) injury. C57/BL6 mice were subjected to sham operation or to 30 min of renal I/R after pretreatment with vehicle (1% DMSO) or with two distinct classes of PAD4 inhibitors (100 mg/kg 2-Cl-amidine or 0.4 mg/kg streptonigrin). Inhibition of PAD4 with 2-Cl-amidine or streptonigrin treatment protected against renal I/R injury. Separate cohorts of mice were subjected to mild (20 min) renal I/R injury after treatment with vehicle or with 10 μg recombinant human PAD4 (rPAD4). In contrast to the renal protection provided by PAD4 inhibitors, rPAD4 treatment exacerbated renal injury after I/R. *P < 0.001 vs. the vehicle-treated sham group; #P < 0.05 vs. the vehicle-treated group subjected to 30 min of renal I/R; $P < 0.001 vs. the vehicle-treated group subjected to 20 min of renal I/R. Error bars represent 1 SE; n = 5–6/group.
Exogenous recombinant PAD4 treatment exacerbates renal I/R injury.
We then tested the effects of rPAD4 administration on kidney injury induced with mild (20 min) renal I/R. Mice subjected to sham operation after rPAD4 treatment had similar baseline plasma creatinine compared with vehicle-treated sham-operated mice (Fig. 3). Vehicle-treated mice showed increased renal injury after mild (20 min) renal I/R. However, rPAD4-treated mice subjected to 20 min of renal I/R had significantly elevated (>3-fold) plasma creatinine, consistent with the hypothesis that exogenous PAD4 administration exacerbates ischemic AKI.
PAD4 inhibition protects and rPAD4 treatment exacerbates kidney necrosis, neutrophil infiltration and apoptosis after renal I/R.
Figure 4A shows representative hematoxylin and eosin-stained images (from 5 experiments) of mice subjected to 20 or 30 min of renal ischemia and 24 h of reperfusion (magnification: ×200). As expected, kidneys of vehicle-treated mice subjected to 30 min of renal I/R showed severe tubular necrosis and proteinaceous casts with increased tubular dilatation and congestion (Fig. 4A). Consistent with the plasma creatinine data, 2-chloroamidine-treated mice subjected to 30 min of renal I/R had decreased renal tubular necrosis, congestion, and cast formation. Mice subjected to 20 min of renal I/R had mild renal tubular necrosis. In contrast, rPAD4-treated mice subjected to renal I/R had markedly increased renal tubular necrosis, proteinaceous casts with increased tubular dilatation, and congestion.
Fig. 4.

PAD4 inhibition reduces and rPAD4 treatment exacerbates kidney necrosis after renal I/R injury. A: representative hematoxylin and eosin-stained images (from 5 experiments) of mice subjected to 20 or 30 min of renal ischemia and 24 h of reperfusion (magnification: ×200). As expected, kidneys of vehicle-treated mice subjected to 30 min of renal I/R showed severe tubular necrosis and proteinaceous casts with increased tubular dilatation and congestion. PAD4 inhibition with 2-Cl-amidine treatment decreased renal tubular necrosis, congestion, and cast formation after 30 min of renal I/R. Mice subjected to 20 min of renal I/R had mild renal tubular necrosis and proteinaceous casts. In contrast, rPAD4-treated mice subjected to renal I/R had markedly increased renal tubular necrosis with increased tubular dilatation and congestion. B: the renal injury score (scale: 0–4, n = 5) for histology grading was used to grade renal tubular necrosis 24 h after 20 or 30 min of renal I/R. Thirty minutes of renal I/R and 24 h of reperfusion resulted in severe renal tubular injury in vehicle-treated mice. 2-Cl-treated mice subjected to 30 min of renal I/R had significantly lower renal injury scores. Furthermore, 20 min of renal ischemia and 24 h of reperfusion resulted in mild to moderate acute tubular necrosis in vehicle-treated mice. In contrast, rPAD4-treated mice had significantly higher renal injury scores after 20 min renal of I/R. *P < 0.05 vs. the vehicle-treated group subjected to 30 min of renal I/R; #P < 0.001 vs. the vehicle-treated group subjected to 20 min of renal I/R. Error bars represent 1 SE.
The Jablonski scale (14) renal injury score (scale: 0–4) for histology grading was used to grade renal tubular necrosis 24 h after 20 or 30 min of renal I/R (Fig. 4B). Thirty minutes of renal I/R and 24 h of reperfusion resulted in severe renal tubular injury in vehicle-treated mice. 2-Chloroamidine-treated mice subjected to 30 min of renal I/R had significantly lower (by ∼50% less) renal injury scores. Furthermore, 20 min of renal ischemia and 24 h of reperfusion resulted in mild-to-moderate acute tubular necrosis in vehicle-treated mice. In contrast, rPAD4-treated mice had significantly higher (by ∼100%) renal injury scores after 20 min of renal I/R (Fig. 4B).
Figure 5A shows representative images (from 5 experiments) of immunohistochemistry for neutrophils (dark brown) in the kidneys of mice subjected to 20 or 30 min of renal ischemia and 24 h of reperfusion (magnification: ×200). Neutrophil infiltration markedly increased in vehicle-treated mice subjected to 30 min of renal I/R concentrated near the corticomedullary junction (Fig. 5, A and B). Again, 2-chloroamidine-treated mice subjected to 30 min of renal I/R had decreased neutrophil infiltration after 24 h of reperfusion. Consistent with the plasma creatinine and renal histology data, 20 min of renal ischemia and 24 h of reperfusion resulted in a scant number of neutrophils infiltrating the kidney. In contrast, rPAD4-treated mice had significantly higher (∼3-fold) numbers of neutrophils infiltrating the kidney after 20 min of renal I/R (Fig. 5, A and B).
Fig. 5.

PAD4 inhibition decreases and rPAD4 treatment exacerbates neutrophil infiltration after renal I/R. A and B: representative images (from 5 experiments, magnification: ×200; A) and quantification of infiltrated neutrophils per ×200 field (n = 5; B) of immunohistochemistry of neutrophil infiltration (dark brown) in the kidneys (corticomedullary junction) of mice subjected to 20 or 30 min of renal ischemia and 24 h of reperfusion. Neutrophil infiltration markedly increased in vehicle-treated mice subjected to 30 min of renal I/R concentrated near the corticomedullary junction Again, 2-Cl-amidine-treated mice subjected to 30 min of renal I/R had decreased neutrophil infiltration after 24 h of reperfusion. Twenty minutes of renal ischemia and 24 h of reperfusion resulted in a scant number of neutrophils infiltrating the kidney. In contrast, rPAD4-treated mice had significantly increased numbers of neutrophils infiltrating the kidney after 20 min of renal IR (A and B). *P < 0.05 vs. the vehicle-treated group subjected to 30 min of renal I/R; #P < 0.001 vs. the vehicle-treated group subjected to 20 min of renal I/R. Error bars represent 1 SE.
TUNEL staining detected fragmented DNA suggestive of apoptosis in renal tubular cells in the kidneys of mice (magnification: ×100, representative of 5 experiments; Fig. 6A). Increased renal tubule cell apoptosis occurred in vehicle-treated wild-type mice subjected to 30 min of renal I/R (Fig. 6, A and B). Again, 2-chloroamidine-treated mice subjected to 30 min of renal I/R had significantly decreased (∼40%) TUNEL-positive renal tubule cells 24 h after reperfusion. Twenty minutes of renal ischemia and 24 h of reperfusion resulted in a few number of TUNEL-positive kidney cells. In contrast, rPAD4-treated mice had significantly higher (>2-fold) numbers of TUNEL-positive renal tubule cells after 20 min of renal I/R and 24 h of reperfusion (Fig. 6, A and B). Since TUNEL stains fragmented DNA rather than specifically indicating apoptotic cells, we counted the number of apoptotic cells from 10 high-power fields in the corticomedullary junction (×400) per kidney. Consistent with the TUNEL data, PAD4 inhibition significantly decreased apoptotic renal tubular cells 24 h after 30 min of renal I/R (Fig. 6C). In contrast, rPAD4-treated mice had significantly increased apoptotic renal tubule cells after 20 min of renal I/R.
Fig. 6.

PAD4 inhibition decreases and rPAD4 treatment exacerbates renal tubular apoptosis after renal IR. A and B: representative images (from 5 experiments, magnification: ×200; A) and quantification of TUNEL staining to demonstrate renal tubular apoptosis per ×100 field (n = 5; B) in the kidneys of mice subjected to 20 or 30 min of renal ischemia and 24 h of reperfusion. TUNEL staining showed that renal tubule cell apoptosis increased in vehicle-treated wild-type mice subjected to 30 min of renal I/R (representative of 5 experiments; magnification: ×100). 2-Cl-amidine-treated mice subjected to 30 min of renal I/R had significantly decreased TUNEL-positive renal tubular cells 24 h after reperfusion (B). Twenty minutes of renal ischemia and 24 h of reperfusion resulted in a few TUNEL-positive cells. In contrast, rPAD4-treated mice had significantly increased numbers of TUNEL-positive cells after 20 min of renal I/R. C: since TUNEL stains fragmented DNA rather than specifically indicating apoptotic cells, we also counted the number of apoptotic cells from 10 high-power fields in the corticomedullary junction (×400) per kidney. PAD4 inhibition significantly decreased apoptotic renal tubular cells 24 h after 30 min of renal I/R. In contrast, rPAD4-treated mice had significantly increased apoptotic renal tubule cells after 20 min of renal I/R. *P < 0.05 vs. the vehicle-treated group subjected to 30 min of renal I/R; #P < 0.001 vs. the vehicle-treated group subjected to 20 min of renal I/R. Error bars represent 1 SE.
PAD4 modulates MIP-2 and KC expression after renal I/R injury.
We measured the expression of proinflammatory cytokine mRNA in the kidney (KC, TNF-α, ICAM-1, MCP-1, and MIP-2) 24 h after 30 min of renal I/R by quantitative RT-PCR (with the primer sequences shown in Table 1). Although all of the proinflammatory mRNA expression increased after renal I/R compared with sham-operated mice, we saw the greatest increases in the expression of KC, MCP-1, and MIP-2 (Fig. 7A). Of these, PAD4 inhibition with 2-chloroamidine treatment significantly attenuated the expression of neutrophil chemotactic cytokines KC and MIP-2. Mice subjected to mild (20 min) renal I/R had reduced upregulation of proinflammatory cytokines 24 h after injury compared with mice subjected to 30 min of renal I/R (Fig. 7B). Again, the induction of KC, MCP-1, and MIP-2 expression was most prominent after 20 min of renal I/R. Treatment of mice with recombinant human PAD4 selectively increased the expression of KC and MIP-2 after 20 min of renal I/R. These data support the hypothesis that PAD4 modulates the expression of neutrophil chemotactic cytokines KC and MIP-2 after renal I/R injury.
Fig. 7.
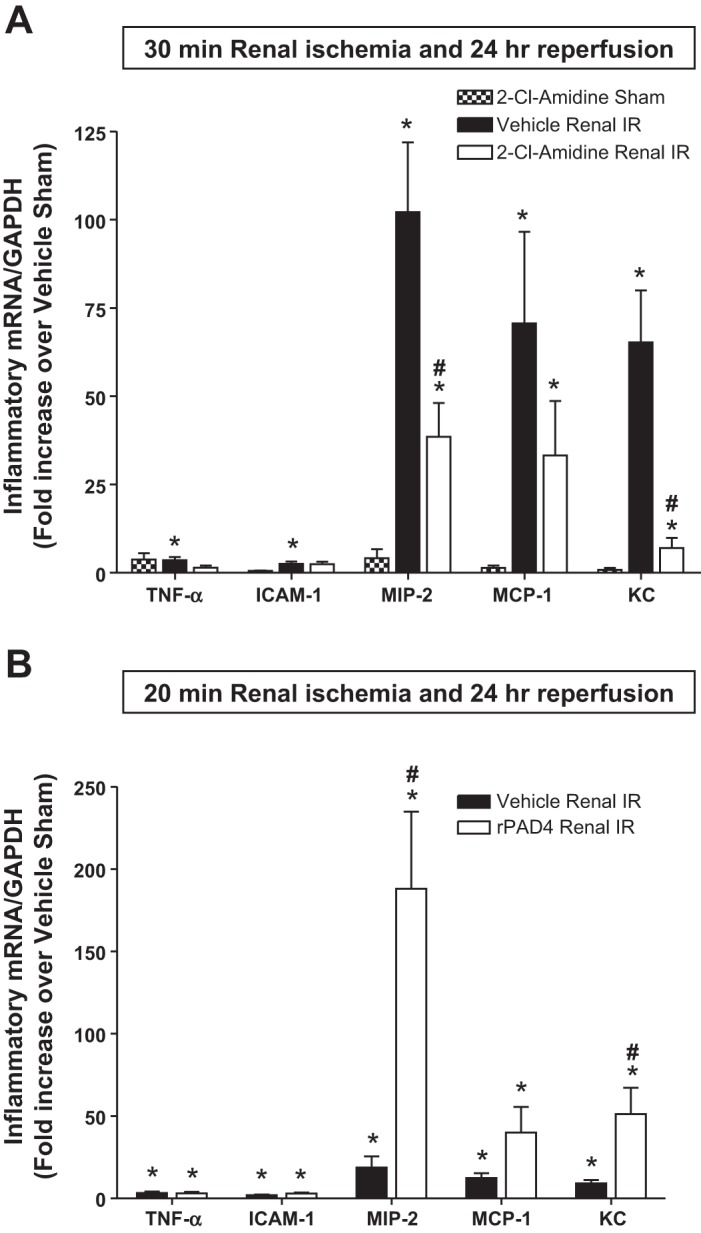
PAD4 modulates macrophage inflammatory protein (MIP)-2 and keratinocyte-derived cytokine (KC) expression after renal I/R injury. With quantitative RT-PCR, we measured the expression of proinflammatory cytokine and chemokine mRNA in the kidney [TNF-α, ICAM-1, MIP-2, monocyte chemoattractant protein (MCP)-1, and KC] 24 h after renal I/R. Fold increases in proinflammatory mRNAs normalized to GAPDH from quantitative RT-PCRs for each indicated mRNA (n = 6) are shown. A: PAD4 inhibition with 2-Cl-amidine treatment significantly attenuated the expression of neutrophil chemotactic cytokines MIP-2 and KC after 30 min of renal I/R injury. B: mice subjected to mild (20 min) renal I/R had reduced upregulation of proinflammatory cytokines 24 h after injury compared with mice subjected to 30 min of renal I/R. Treatment of mice subjected to 20 min of renal I/R with rPAD4 selectively increased the expression of MIP-2 and KC mRNA. *P < 0.05 vs. sham mice; #P < 0.05 vs. vehicle-treated mice subjected to renal I/R. Error bars represent 1 SE.
rPAD4 induces proinflammatory mRNA expression in primary cultures of mouse proximal tubules.
We next tested whether rPAD4 treatment directly induces proinflammatory gene expression in mouse proximal tubules in culture. Primary cultures of mouse proximal tubules were treated with vehicle (saline) or 5 μg/ml rPAD4 for 6 h. Quantitative RT-PCR revealed that rPAD4 treatment significantly induced the expression of the proinflammatory cytokine mRNAs measured (ICAM-1, KC, MCP-1, MIP-2, and TNF-α; Fig. 8). Therefore, these results support the hypothesis that PAD4 directly increases inflammatory cytokine expression in mouse proximal tubules.
Fig. 8.
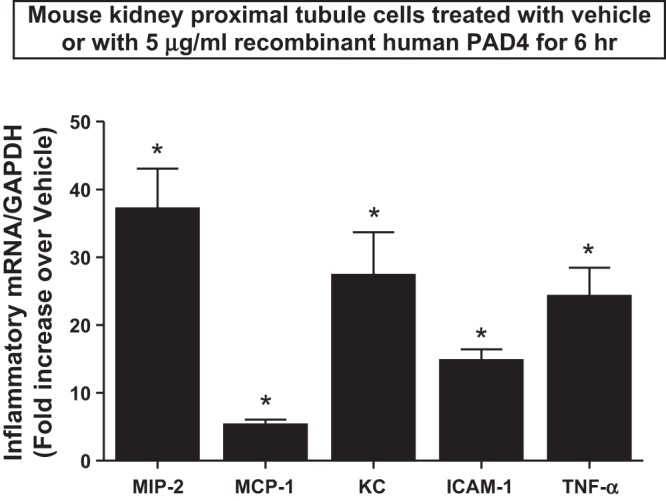
rPAD4 induces proinflammatory mRNA expression in primary cultures of mouse proximal tubules. Primary cultures of mouse proximal tubules were treated with vehicle or 5 μg/ml rPAD4 for 6 h. With quantitative RT-PCR, we measured the expression of proinflammatory cytokine and chemokine mRNA in these cells (MIP-2, MCP-1, KC, ICAM-1, and TNF-α). Fold increases in proinflammatory mRNAs normalized to GAPDH from quantitative RT-PCR reactions for each indicated mRNA (n = 6) are shown. Quantitative RT-PCR revealed that rPAD4 treatment significantly induced the expression of all proinflammatory cytokine mRNAs measured (MIP-2, MCP-1, KC, ICAM-1, and TNF-α). *P < 0.05 vs. vehicle-treated cells. Error bars represent 1 SE.
DISCUSSION
Citrullination or deamination is the posttranslational modification of peptidyl arginine to peptidyl citrulline residue catalyzed by the PAD family of enzymes (4, 15, 42). Of the five PAD subtypes in humans and rodents, PAD4 activity has received significant scientific and clinical interest due to its active role in innate immunity and autoimmune diseases, including rheumatoid arthritis, multiple sclerosis, ulcerative colitis, and lupus (2, 4). PAD4 was first identified in the nucleus of a neutrophil-like cell line (HL-60) (2), and PAD4-mediated citrullination of core histones (H3, H4, and H2A) is critical for the formation of neutrophil extracellular trap formation after bacterial infection and deep vein thrombosis (28). In the kidney, PAD2 and PAD4 mRNAs are present, although only PAD4 protein expression has been detected (39, 41). However, the role of PAD4 in renal inflammation and injury due to ischemic AKI has never been studied.
In the present study, we detected an induction of PAD4 expression concomitant with a robust increase in PAD4 activity after renal I/R. PAD4 requires dimerization and Ca2+-mediated conformational changes to promote the deamination process (4, 15, 42). PAD4 is known to be activated by the rise in intracellular Ca2+ as well as by ROS and proinflammatory cytokines (e.g., TNF-α), chemokines (e.g., IL-8), and Toll-like receptor ligands (e.g., lipopolysaccharides) (37). All of these stimuli that result in PAD4 activation occur during and after renal I/R injury (5, 18, 35).
The mechanisms by which PAD4-mediated peptidyl citrullination exacerbates ischemic AKI remain to be determined. The conversion of peptidyl arginine to peptidyl citrulline results in a <1-Da change in molecular mass and the loss of a positive charge of an arginine residue. The resulting conformational changes due to peptidyl citrullination may change binding properties and protein folding, leading to alterations of function and in the half-life of the protein (2, 4). Since PAD4 is specifically localized to the nucleus, it is most likely that PAD4 induction and activation increase the citrullination of nuclear proteins. Here, we show that histone H3 citrullination increases after renal I/R and is blocked by the specific PAD4 inhibitor 2-chloroamidine (Fig. 2). Interestingly, histones, including histone H3, have been recognized as one of the damage-associated molecular pattern ligands that are released after sepsis or ischemia (1, 13). PAD4 citrullinates arginine and methylarginine residues of histones and therefore prevents or even reverses methylation of these histones residues. It is possible that peptidyl citrullination potentiates the inflammatory effects of histone H3 after renal I/R. Furthermore, PAD4-mediated histone citrullination may also lead to chromatin decondensation followed by nuclear and cellular damage and death (2, 4).
The majority of renal cell death after I/R occurs due to necrosis (24). A recent elegant study (25) defined a caspase-independent receptor-interacting protein kinase 3-dependent regulated necrosis termed as necroptosis. Indeed, it is becoming increasingly clear that necroptosis is a well-defined and significant component of cell death after ischemic injury (23). We propose that PAD4 is released by necrotic renal tubular cells generated after renal I/R injury. The PAD4 enzymes that are released after renal I/R may act as damage-associated molecular pattern ligands to induce inflammatory responses in neighboring tubular or endothelial cells or in remote organs (e.g., the liver). It is possible that regulated necrosis (necroptosis) after renal I/R may promote the release of PAD4. Consistent with our hypothesis, we found significantly increased release of plasma IL-33 in mice subjected to renal I/R injury (data not shown). A recent study (36) has shown that IL-33 is released from cells that have undergone regulated necrosis.
Our experiments showed that two distinct classes of PAD4 inhibitors (2-chloroamidine and streptonigrin) protected against renal I/R injury with significantly reduced plasma creatinine, renal tubular necrosis, inflammation, and apoptosis. Streptonigrin is an aminoquinone with antibiotic and anticancer activity, and the 7-amino-quinoline-5,8-dione core of streptonigrin has been recently identified as a potent and selective PAD4 inhibitor (7). 2-Chloroamidine is also a potent inhibitor of PAD4 deimination, with an IC50 value of ∼6 μM (26). It irreversibly inactivates PAD4 by covalently modifying the catalytically active cysteine sites. These PAD4 inhibitors may be useful as therapeutic agents to attenuate the complication and severity of ischemic AKI. Consistent with our findings that PAD4 inhibition attenuated ischemic AKI and renal tubular inflammation, 2-chlroamidine treatment attenuates the progression and severity of several mouse models of inflammatory diseases, including colitis, lupus, and collagen-induced arthritis (4, 15, 22, 42). A study (6) in a mouse model of colitis showed that chronic treatment with 2-chloroamidine had no overall long-term toxicity or mortality.
As expected, all of the proinflammatory mRNAs (ICAM-1, KC, MCP-1, MIP-2, and TNF-α) were induced after renal I/R. Interestingly, we demonstrated reductions in KC and MIP-2 mRNA with selective PAD4 inhibition in mice after renal I/R. In contrast, rPAD4 treatment significantly and selectively induced the expression of KC and MIP-2 mRNA after renal I/R. The proinflammatory chemokines KC and MIP-2 play a major role in the pathogenesis of AKI by serving as neutrophil chemotactic factors (9, 35). Modulation of neutrophil chemotactic cytokines KC and MIP-2 is consistent with our finding that PAD4 inhibition significantly attenuated and rPAD4 treatment significantly increased neutrophil infiltration after renal I/R.
In the present study, we show that exogenous recombinant PAD4 protein directly increases cytokine mRNA expression in cultured proximal tubules. The exact mechanism of PAD4-mediated induction of proinflammatory mRNA expression is unclear. It is possible that PAD4-mediated citrullination may increase the potency of or even enable certain proteins to induce inflammatory cytokine expression. For example, vimentin citrullination stimulates TNF-α and IL-1 production in fibroblast-like synoviocytes (8). Furthermore, PAD4 may directly citrullinate several chemokine and cytokines [e.g., chemokine (C-X-C motif) ligands 5 and 8] to directly modulate the inflammatory potential of these cytokines (30).
In summary, we demonstrate, in this study, that renal I/R injury in mice upregulates renal tubular PAD4 expression and activity. We also show that PAD4 inhibition protected against ischemic AKI by reducing renal tubular necrosis, inflammation, and apoptosis. Taken together, our results allow us to hypothesize that PAD4 plays a critical role in generating AKI, most likely via inducing neutrophil chemotactic cytokine KC and MIP-2 synthesis, neutrophil infiltration, and the inflammatory response after renal I/R (Fig. 9). Selective inhibition of renal tubular PAD4 activity may reduce the morbidity and mortality arising from ischemic AKI.
Fig. 9.

Schematic of proposed role for PAD4 activation and induction in ischemic acute kidney injury (AKI). After renal ischemia and reperfusion, PAD4 activation and induction converts peptidyl arginine to peptidyl citrulline. Via mechanisms that are still unclear, peptidyl citrullination results in significant renal tubular inflammation, KC and MIP-2 mRNA induction, and increased kidney necrosis, inflammation, and apoptosis after renal I/R. Inhibition of PAD4 may be an effective therapeutic strategy to protect against ischemic AKI. Selective PAD4 inhibitors [2-Cl-amidine and streptonigrin (structures shown)] protected against kidney necrosis, apoptosis, and inflammation after renal I/R. PMNs, polymorphonuclear neutrophils.
GRANTS
This work was supported by Department of Anesthesiology, Columbia University, and in part by National Institutes of Health Grants R01-DK-058547 and R01-GM-067081 (to H. T. Lee).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
Author contributions: A.H., M.M.R., M.K., and H.T.L. conception and design of research; A.H., M.M.R., M.K., K.M.B., Z.M., and V.D. performed experiments; A.H., M.M.R., M.K., K.M.B., Z.M., V.D., and H.T.L. analyzed data; A.H., M.M.R., M.K., K.M.B., Z.M., and H.T.L. interpreted results of experiments; A.H., M.M.R., M.K., Z.M., and H.T.L. prepared figures; A.H., M.M.R., and H.T.L. drafted manuscript; A.H., M.M.R., M.K., and H.T.L. edited and revised manuscript; A.H., M.M.R., M.K., K.M.B., Z.M., V.D., and H.T.L. approved final version of manuscript.
ACKNOWLEDGMENTS
The authors appreciate the technical assistance provided by Dr. Joo Yun Kim and Dr. Kyota Fukazawa.
REFERENCES
- 1.Allam R, Scherbaum CR, Darisipudi MN, Mulay SR, Hagele H, Lichtnekert J, Hagemann JH, Rupanagudi KV, Ryu M, Schwarzenberger C, Hohenstein B, Hugo C, Uhl B, Reichel CA, Krombach F, Monestier M, Liapis H, Moreth K, Schaefer L, Anders HJ. Histones from dying renal cells aggravate kidney injury via TLR2 and TLR4. J Am Soc Nephrol 23: 1375–1388, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Anzilotti C, Pratesi F, Tommasi C, Migliorini P. Peptidylarginine deiminase 4 and citrullination in health and disease. Autoimmun Rev 9: 158–160, 2010 [DOI] [PubMed] [Google Scholar]
- 3.Aronson S, Blumenthal R. Perioperative renal dysfunction and cardiovascular anesthesia: concerns and controversies. J Cardiothorac Vasc Anesth 17: 117–130, 1998 [DOI] [PubMed] [Google Scholar]
- 4.Bicker KL, Thompson PR. The protein arginine deiminases: structure, function, inhibition, and disease. Biopolymers 99: 155–163, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bonventre JV, Weinberg JM. Recent advances in the pathophysiology of ischemic acute renal failure. J Am Soc Nephrol 14: 2199–2210, 2003 [DOI] [PubMed] [Google Scholar]
- 6.Chumanevich AA, Causey CP, Knuckley BA, Jones JE, Poudyal D, Chumanevich AP, Davis T, Matesic LE, Thompson PR, Hofseth LJ. Suppression of colitis in mice by Cl-amidine: a novel peptidylarginine deiminase inhibitor. Am J Physiol Gastrointest Liver Physiol 300: G929–G938, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dreyton CJ, Anderson ED, Subramanian V, Boger DL, Thompson PR. Insights into the mechanism of streptonigrin-induced protein arginine deiminase inactivation. Bioorg Med Chem 22: 1362–1369, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fan LY, He DY, Wang Q, Zong M, Zhang H, Yang L, Sun LS. Citrullinated vimentin stimulates proliferation, pro-inflammatory cytokine secretion, and PADI4 and RANKL expression of fibroblast-like synoviocytes in rheumatoid arthritis. Scand J Rheumatol 41: 354–358, 2012 [DOI] [PubMed] [Google Scholar]
- 9.Frangogiannis NG. Chemokines in ischemia and reperfusion. Thromb Haemost 97: 738–747, 2007 [PubMed] [Google Scholar]
- 10.Gallos G, Ruyle TD, Emala CW, Lee HT. A1 adenosine receptor knockout mice exhibit increased mortality, renal dysfunction, and hepatic injury in murine septic peritonitis. Am J Physiol Renal Physiol 289: F369–F376, 2005 [DOI] [PubMed] [Google Scholar]
- 11.Heinzelmann M, Mercer-Jones MA, Passmore JC. Neutrophils and renal failure. Am J Kidney Dis 34: 384–399, 1999 [DOI] [PubMed] [Google Scholar]
- 12.Horibata S, Coonrod SA, Cherrington BD. Role for peptidylarginine deiminase enzymes in disease and female reproduction. J Reprod Dev 58: 274–282, 2012 [DOI] [PubMed] [Google Scholar]
- 13.Huang H, Evankovich J, Yan W, Nace G, Zhang L, Ross M, Liao X, Billiar T, Xu J, Esmon CT, Tsung A. Endogenous histones function as alarmins in sterile inflammatory liver injury through Toll-like receptor 9 in mice. Hepatology 54: 999–1008, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jablonski P, Howden BO, Rae DA, Birrel CS, Marshall VC, Tange J. An experimental model for assessment of renal recovery from warm ischemia. Transplantation 35: 198–204, 1983 [DOI] [PubMed] [Google Scholar]
- 15.Jang B, Ishigami A, Maruyama N, Carp RI, Kim YS, Choi EK. Peptidylarginine deiminase and protein citrullination in prion diseases: strong evidence of neurodegeneration. Prion 7: 42–46, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jo SK, Rosner MH, Okusa MD. Pharmacologic treatment of acute kidney injury: why drugs haven't worked and what is on the horizon. Clin J Am Soc Nephrol 2: 356–365, 2007 [DOI] [PubMed] [Google Scholar]
- 17.Jones DR, Lee HT. Perioperative renal protection. Best Pract Res Clin Anaesthesiol 22: 193–208, 2008 [DOI] [PubMed] [Google Scholar]
- 18.Kaczorowski DJ, Tsung A, Billiar TR. Innate immune mechanisms in ischemia/reperfusion. Front Biosci (Elite Ed) 1: 91–98, 2009 [DOI] [PubMed] [Google Scholar]
- 19.Kim M, Kim M, Kim N, D'Agati VD, Emala CW, Sr, Lee HT. Isoflurane mediates protection from renal ischemia-reperfusion injury via sphingosine kinase and sphingosine-1-phosphate-dependent pathways. Am J Physiol Renal Physiol 293: F1827–F1835, 2007 [DOI] [PubMed] [Google Scholar]
- 20.Kim M, Kim M, Park SW, Pitson SM, Lee HT. Isoflurane protects human kidney proximal tubule cells against necrosis via sphingosine kinase and sphingosine-1-phosphate generation. Am J Nephrol 31: 353–362, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kim M, Park SW, Kim M, Chen SW, Gerthoffer WT, D'Agati VD, Lee HT. Selective renal over-expression of human heat shock protein 27 reduces renal ischemia-reperfusion injury in mice. Am J Physiol Renal Physiol 299: F347–F358, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Knight JS, Zhao W, Luo W, Subramanian V, O'Dell AA, Yalavarthi S, Hodgin JB, Eitzman DT, Thompson PR, Kaplan MJ. Peptidylarginine deiminase inhibition is immunomodulatory and vasculoprotective in murine lupus. J Clin Invest 123: 2981–2993, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Linkermann A, Brasen JH, Darding M, Jin MK, Sanz AB, Heller JO, De ZF, Weinlich R, Ortiz A, Walczak H, Weinberg JM, Green DR, Kunzendorf U, Krautwald S. Two independent pathways of regulated necrosis mediate ischemia-reperfusion injury. Proc Natl Acad Sci USA 110: 12024–12029, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Linkermann A, Brasen JH, Himmerkus N, Liu S, Huber TB, Kunzendorf U, Krautwald S. Rip1 (receptor-interacting protein kinase 1) mediates necroptosis and contributes to renal ischemia/reperfusion injury. Kidney Int 81: 751–761, 2012 [DOI] [PubMed] [Google Scholar]
- 25.Linkermann A, Green DR. Necroptosis. N Engl J Med 370: 455–465, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Luo Y, Arita K, Bhatia M, Knuckley B, Lee YH, Stallcup MR, Sato M, Thompson PR. Inhibitors and inactivators of protein arginine deiminase 4: functional and structural characterization. Biochemistry 45: 11727–11736, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mangat P, Wegner N, Venables PJ, Potempa J. Bacterial and human peptidylarginine deiminases: targets for inhibiting the autoimmune response in rheumatoid arthritis? Arthritis Res Ther 12: 209, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Martinod K, Demers M, Fuchs TA, Wong SL, Brill A, Gallant M, Hu J, Wang Y, Wagner DD. Neutrophil histone modification by peptidylarginine deiminase 4 is critical for deep vein thrombosis in mice. Proc Natl Acad Sci USA 110: 8674–8679, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Martinod K, Wagner DD. Thrombosis: tangled up in NETs. Blood 123: 2768–2776, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mortier A, Gouwy M, Van DJ, Proost P. Effect of posttranslational processing on the in vitro and in vivo activity of chemokines. Exp Cell Res 317: 642–654, 2011 [DOI] [PubMed] [Google Scholar]
- 31.Paladino JD, Hotchkiss JR, Rabb H. Acute kidney injury and lung dysfunction: a paradigm for remote organ effects of kidney disease? Microvasc Res 77: 8–12, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Park SW, Kim M, Brown KM, D'Agati VD, Lee HT. Inhibition of sphingosine 1-phosphate receptor 2 protects against renal ischemia-reperfusion injury. J Am Soc Nephrol 23: 266–280, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Park SW, Kim M, Chen SW, Brown KM, D'Agati VD, Lee HT. Sphinganine-1-phosphate protects kidney and liver after hepatic ischemia and reperfusion in mice through S1P(1) receptor activation. Lab Invest 90: 1209–1224, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Park SW, Kim M, Chen SW, D'Agati VD, Lee HT. Sphinganine-1-phosphate attenuates both hepatic and renal injury induced by hepatic ischemia and reperfusion in mice. Shock 33: 31–42, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ramesh G, Reeves WB. Inflammatory cytokines in acute renal failure. Kidney Int Suppl 66: S56–S61, 2004 [DOI] [PubMed] [Google Scholar]
- 36.Rickard JA, O'Donnell JA, Evans JM, Lalaoui N, Poh AR, Rogers T, Vince JE, Lawlor KE, Ninnis RL, Anderton H, Hall C, Spall SK, Phesse TJ, Abud HE, Cengia LH, Corbin J, Mifsud S, Di RL, Metcalf D, Ernst M, Dewson G, Roberts AW, Alexander WS, Murphy JM, Ekert PG, Masters SL, Vaux DL, Croker BA, Gerlic M, Silke J. RIPK1 regulates RIPK3-MLKL-driven systemic inflammation and emergency hematopoiesis. Cell 157: 1175–1188, 2014 [DOI] [PubMed] [Google Scholar]
- 37.Rohrbach AS, Slade DJ, Thompson PR, Mowen KA. Activation of PAD4 in NET formation. Front Immunol 3: 360, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Slot C. Plasma creatinine determination. A new and specific Jaffe reaction method. Scand J Clin Lab Invest 17: 381–387, 1965 [DOI] [PubMed] [Google Scholar]
- 39.van Beers JJ, Zendman AJ, Raijmakers R, Stammen-Vogelzangs J, Pruijn GJ. Peptidylarginine deiminase expression and activity in PAD2 knock-out and PAD4-low mice. Biochimie 95: 299–308, 2013 [DOI] [PubMed] [Google Scholar]
- 40.Vinay P, Gougoux A, Lemieux G. Isolation of a pure suspension of rat proximal tubules. Am J Physiol Renal Fluid Electrolyte Physiol 241: F403–F411, 1981 [DOI] [PubMed] [Google Scholar]
- 41.Vossenaar ER, Zendman AJ, van Venrooij WJ, Pruijn GJ. PAD, a growing family of citrullinating enzymes: genes, features and involvement in disease. Bioessays 25: 1106–1118, 2003 [DOI] [PubMed] [Google Scholar]
- 42.Wang S, Wang Y. Peptidylarginine deiminases in citrullination, gene regulation, health and pathogenesis. Biochim Biophys Acta 1829: 1126–1135, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wildeman E, Pires MM. Facile fluorescence-based detection of PAD4-mediated citrullination. Chembiochem 14: 963–967, 2013 [DOI] [PubMed] [Google Scholar]
- 44.Yap SC, Lee HT. Acute kidney injury and extrarenal organ dysfunction: new concepts and experimental evidence. Anesthesiology 116: 1139–1148, 2012 [DOI] [PubMed] [Google Scholar]