

Abstract
Vitamin D deficiency (VDD) or insufficiency is recognized for its association with nonalcoholic steatohepatitis (NASH), whereas the underlying mechanism remains unknown. Using animal models, we found that vitamin D deficiency promoted the high-fat diet (HFD)-initiated simple steatosis into typical NASH, characterized by elevated hepatic inflammation and fat degeneration. The NASH derived from VDD + HFD was related to poor retention of bile acids in the liver and biliary tree, in line with downregulation of the ileal apical sodium-dependent bile acid cotransporter (iASBT). The impediment of hepatic bile acids by the VDD + HFD mice was related to increased expression of hepatic SREBP-1c and fatty acid synthase, suggesting that VDD may upregulate endogenous fatty acid synthesis into NASH through impaired enterohepatic circulation. Administration of 1,25(OH)2VD3 (calcitriol) corrected the NASH phenotypes in line with restoration of iASBT, promotion of bile filling in the biliary tree, suppression of hepatic lipogenesis, and inflammation. Moreover, administration of a bile acid-sequestering agent suppressed ileal fibroblast growth factor 15 expression, leading to increased iASBT expression to restore bile filling in the liver and biliary tree, which ameliorates steatosis and inflammation in the liver. These results suggest a novel mechanism for NASH development, by which VDD downregulates iASBT expression, resulting in a poor bile acid pool and elevation of hepatic lipogenesis and inflammation. In conclusion, vitamin D and bile acid sequestration may be explored as new strategies to treat or prevent NASH.
Keywords: nonalcoholic steatohepatitis; vitamin D deficiency; vitamin D; 1,25-dihydroxyl vitamin D3; calcitriol; bile acid sequestration; enterohepatic circulation
nonalcoholic fatty liver diseases (NAFLD), in its various manifestations ranging from simple steatosis to nonalcoholic steatohepatitis (NASH) and even cirrhosis and hepatocellular carcinoma, are increasingly recognized worldwide (20). NASH was first described in 1980 by Ludwig et al. (18) for the liver biopsy findings resembling those of alcoholic steatohepatitis in patients without significant alcohol consumption (18). The prevalence of NAFLD is estimated at 15–20% in adult population, whereas NASH is about 3–5% in the general population for many countries (2). To explain how simple steatosis is transformed into NASH, Day et al. (6) proposed a “two-hit theory,” suggesting that a second hit is critical to drive fatty degeneration into inflammation-featured NASH.
Bile acids are discharged from the gallbladder into the intestinal lumen upon ingestion of food, which is mediated by cholecystokinin. After emulsification with dietary lipids, about 95% of the bile acids are reabsorbed through the intestinal bile acid transporter, including the ileal apical sodium-dependent bile acid cotransporter (iASBT), and returned to the liver in a process called enterohepatic circulation (16). Bile acids in the liver play critical roles in maintaining metabolic homeostasis of triglycerides (TG) and glucose by serving as ligands for nuclear receptor, farnesoid X receptor (FXR). The TG-lowering effect of bile acids was first described 35 years ago when the patients with hypertriglyceridemia were given chenodeoxycholic acid, which had a beneficial therapeutic effect (4). In animal models, evidence showed that bile acids such as cholic acid could lower the plasma TG levels in the plasma and the liver via activation of FXR to induce small heterodimer partner (SHP), which, in turn, suppresses SREBP-1c, a master transcription factor for lipogenic genes such as fatty acid synthase (FAS) and stearoyl-CoA desaturases-1 (28).
Vitamin D, a group of secosteroid hormones, has pleiotropic physiological functions (1). In addition to its classical roles in maintaining calcium/phosphate homeostasis, vitamin D is increasingly found to have other functions in cell proliferation, differentiation, suppression of hepatic virus proliferation, and immune regulation (29). Importantly, vitamin D deficiency (VDD) has been increasingly recognized for its association with various chronic liver diseases (14, 17). Low vitamin D serum level is related to severe fibrosis and impaired responsiveness in interferon-based therapy for genotype 1 chronic hepatitis C, whereas the latter is often associated with NASH despite the unknown mechanism (22). There is emerging although controversial evidence showing the association of VDD with NAFLD (3, 7).
There are two major unsolved questions in the field of NASH pathology. First, it is unknown as to how lipogenesis in the liver is persistently active even under high-fat feeding. Second, it is unknown how simple steatosis is transformed into NASH, marked by an inflammatory process. In this report, we showed that VDD exacerbated hepatic inflammation and steatosis, in part through alterations in bile acid transport. Moreover, evidence presented here links NASH formation with impaired expression of iASBT and reduced bile acids in the liver and gallbladder. Indeed, administration of calcitriol resolved NASH in association with restoration of iASBT expression, increased bile acid transporting, and suppression of hepatic steatosis and inflammation. In addition, because previous work showed that fibroblast growth factor 15 (FGF15, the equivalent of human FGF19) was able to suppress iASBT (26), we investigated and found that the suppressed expression of iASBT in the NASH model was associated with the upregulation of ileal FGF15. Conversely, treatment of the NASH mice with 1,25-dihydroxyl VD3 suppressed ileal FGF15 expression, restored iASBT expression, promoted bile acid filling, and suppressed hepatic lipogenesis and inflammation. Finally, manipulation of the enterohepatic circulation by bile acid sequestration suppressed ileal FGF15, resulting in elevation of iASBT expression together with bile filling in the gallbladder and liver, which suppressed the NASH phenotypes. These results suggest that VDD might serve as a “second hit” promoting simple steatosis into NASH, in part, through suppression of iASBT and impaired enterohepatic circulation of bile acids, which consequently results in hepatic lipogenesis and inflammation. Therefore, vitamin D therapy and bile acid sequestration should be further explored as approaches to prevent or treat NASH in patients.
MATERIALS AND METHODS
Animals and treatments.
All procedures in the present study followed the principles outlined in the Guide for the Care and Use of Laboratory Animals published by the National Institutes of Health (NIH). The protocols were examined and approved by the Institutional Animal Care and Use Committee of Beijing Capital Medical University. Balb/C mice, at 6–7 wk of age, were maintained in a temperature-controlled (23°C) facility with free access to food and water. Body weight and food consumption were recorded twice a week. Balb/C male mice (n = 9 for each conditions) were fed for 8 wk in the following conditions: 1) control (CONT), by which mice were fed with AIN-93-based chemical-defined diets containing sufficient vitamin supplements (3.8 kcal/g, 17% calories from fat, vitamin D3 at 1,000 IU/kg), 2) high-fat diet feeding (HFD) (5.1 kcal/g, 60% calories from fat, vitamin D3 at 1,000 IU/kg), 3) HFD plus 1,25-dihydroxyl VD3 treatment (HFD + calcitriol), by which the high-fat-fed mice were given 1,25-dihydroxyl VD3 (calcitriol from Roche) treatment at a dose of 5 ng/g body wt by intramuscular injection twice per week in the last 6 wk with the last injection at 48 h before death, 4) VDD diet, by which vitamin D was omitted in the standard AIN-93 diet, and the mice were devoid of light exposure, 5) VDD plus HFD feeding (VDD + HFD), by which the mice were fed with HFD without vitamin D additive, and the mice were devoid of light exposure, and 6) VDD + HFD plus 1,25-dihydroxyl VD3 treatment [VDD + HFD + 1,25(OH)2VD3], by which the mice were fed with HFD without vitamin D3 additive and devoid of light exposure, and given 1,25-dihydroxyl VD3 at a dose of 5 ng/g body wt by intramuscular injection twice per week during the last 6 wk with the last injection at 48 h before death. For bile acid sequestration treatment (n = 8), the mice were fed with HFD for 2 wk, and then cholestyramine was added to the same diet (at 3%, wt/wt) for the following 6 wk.
Three days before death, the circadian rhythm and metabolic oscillation for all the mice were subjected to synchronization by feeding at night for 12 h followed by fasting with free access to water for another 12 h in daytime. The distal region of ileum, liver tissue, whole gallbladder, and serum were collected for analysis. To measure the acute response to 1,25-dihydroxyl VD3 in the physiological condition, the healthy mice (n = 8) after being fasted for 10 h were given a single dose of 1,25-dihydroxyl VD3 (10 ng/g body wt) by intramuscular injection, and the mice were killed after 12 h.
Biochemical measurements.
Serum 25(OH)VD3 was measured by high-performance liquid chromatography-mass spectrometry, calibrated with isotope-labeled standard (Sigma Aldrich). Serum levels of alanine aminotransferase, aspartate aminotransferase, TG, total cholesterol, and nonesterified fatty acids (NEFA) were measured by automated instruments in the Beijing YouAn Hospital.
NAFLD Activity Scores.
Histological staining (hematoxylin and eosin, H and E) and Oil-Red-O staining were scored by two experienced clinical pathologists for levels of ballooning cells, Mallory bodies, and necro-inflammatory lesions. A score of 0 was assigned when ballooning cells were <5% of the parenchymal cells. Scores of 1–2 were used when ballooning cells accounted for 5–20% of the parenchymal cells and referred to as simple steatosis. Scores of 3–4 were used when fatty degenerated cells accounted for 20–50% of the parenchymal cells, and infiltrated granulocytes were scattered in the parenchyma. A score of 5 was given when fibrosis was evident, and fatty degeneration was >60% of the parenchymal cells.
Extraction of hepatic and gallbladder bile acids.
Liver tissues (90–110 mg) from three lobules were collected, weighed, and homogenized in cold anhydrous 1 ml ethanol, followed by heating at 50°C for 2 h. Gallbladder was cautiously collected and disrupted in the ethanol in 1 ml. After centrifugation at 6,000 g for 10 min, the supernatant was collected, and total bile acids were measured by automated devices in the Clinical Laboratory in the Beijing YouAn Hospital. The levels of total bile acids in the liver and gallbladder were presented as micromoles per gram of liver, and micromoles per gram of body weight, respectively.
qRT-PCR analysis.
Total RNA of liver tissues was extracted by TRIZOL (Invitrogen). First-strand cDNA was produced by SuperScript II Reverse Transcriptase with random primers. Two micrograms of total RNA were used for reverse transcription in 20 μl. The products were diluted 10 times with pure water. Ten-microliter reactions were set up in a 384-well PCR plate using the following final concentrations: 1 μM each of forward and reverse primers, SYBR Green master mix (Eurogentec), and 5 ng of cDNA. Cycling conditions were as follows: hot activation (95°C for 10 min), amplification (95°C for 15 s, 60°C for 1 min) repeated 40 times, and quantification with a single fluorescence measurement. Expression levels were analyzed in cDNA synthesized from total mRNA using real-time PCR as described in our previous report (23). The sequence information is given in Table 1.
Table 1.
Primers used for real-time qPCR analysis
| Primer | GeneBank | Sequence Forward (5′-3′) | Sequence Reverse (5′-3′) |
|---|---|---|---|
| GAPDH | NM_008084.2 | aac ttt ggc attgtg gaa gg | aca cat t gg ggg t ag gaa ca |
| CYP7A1 | NM_007824.2 | ccg tct acg cat gtt tct ca | ttg gcc agc act ctg taa tg |
| CYP27A1 | NM_024264.4 | gcc tca cct atg gga tct tca | tca aag cct gac gca gat g |
| SHP | NM_011850.2 | agc tgg gtc cca agg agt at | ctt gag ggt aga ggc cat ga |
| FXR | NM_009108.2 | tcc gga cat tca acc atc ac | tca ctg cac atc cca gat ctc |
| FGF15 | NM_008003.2 | aga cga ttg cca tca agg ac | ctg gtc ctg gag ctg ttc tc |
| ASBT | NM_011388.2 | gac t cg gga acg at t gt g at | ggt t ca at g at c cag gca ct |
| CYP3A11 | NM_007818.3 | aaa ctg cag gat gag atc gat ga | tcc agg tat tcc atc tcc atc ac |
| TNF-α | NM_013693.2 | gcc tct tct cat tcc tgc ttg t | ttg aga tcc atg ccg ttg |
| IL-6 | NM_031168.1 | agt tgc ctt ctt ggg act ga | tcc acg att tcc cag aga ac |
| FAS | NM_007988.3 | act ggg tga cct tca tgg ac | acc acc aga gac cgt t at gc |
| PPAR-α | NM_001113418.1 | ggg cag agc aag tca tct tc | ggt cac cta cga gtg gca tt |
| CPT-1α | NM_013495.2 | agc agc agg tgg aac tgt tt | ggc ttg tct caa gtg ctt cc |
| SREBP-1C | NM_011480.3 | gat cgc agt ctg agg agg ag | gat cgc caa gct tct cta cg |
SHP, small heterodimer partner; FXR, farnesoid X receptor; FGF15, fibroblast growth factor 15; ASBT, apical sodium-dependent bile acid cotransporter; FAS, fatty acid synthase; PPAR, peroxisome proliferator-activated receptor; CPT, carnitine palmitoyltransferase.
Statistical analysis.
Microsoft Excel 2003 (Microsoft) and GraphPad Prism 5.0 (GraphPad Software) were used for data recording, collection, processing, and calculation. Results were reported as means ± SE of the values recorded. Statistical differences were determined using SPSS 11.5 for Windows. Statistical significance is displayed as *P < 0.05 or **P < 0.01 by t-test for paired comparison.
RESULTS
VDD drives simple steatosis into NASH, which can be resolved by 1,25-dihydroxyl VD3.
Balb/C male mice (n = 9) at the age of 6 wk were fed for an additional 8 wk in the following conditions: 1) control diet, 2) HFD, 3) HFD with administration of 1,25-dihydroxyl VD3 (calcitriol) in the last 6 wk, HFD + calcitriol, 4) VDD, by which VD3 was depleted in the control diet, 5) VDD in HFD feeding, by which vitamin D3 as additive in the diet was omitted in the HFD feeding, VDD + HFD, and 6) VDD in HFD feeding by which the mice were treated with 1,25-dihydroxyl VD3 in the last 6 wk, VDD + HFD + VD (calcitriol). As shown in Fig. 1A, simple steatosis was evident by the mice after the HFD feeding. Fatty degeneration was indicated by large ballooning cells that were scattered around portal vein. However, under VDD + HFD feeding, fatty degeneration was significantly exacerbated, shown by ballooning cells overspreading in >50% of the parenchymal area. In contrast, VDD alone for 8 wk generated a low degree but histologically recognized initial steatosis. TG, as shown in Fig. 1B, were elevated by HFD feeding as expected but were additionally increased under VDD. The exacerbated fatty degeneration was further illustrated by the Oil-Red-O staining, shown in Fig. 1A. The vitamin D levels in the serum, in the format of 25-hydroxyl VD3, were measured by LC/MS for the mice under the experimental conditions (Table 2).
Fig. 1.

Vitamin D deficiency (VDD) accelerates the high fat diet (HFD)-induced simple steatosis into nonalcoholic steatohepatitis (NASH), which can be resolved by calcitriol treatment. Adult Balb/C male mice (n = 9 for each condition) were fed for 8 wk in the following conditions: 1) control, in AIN-93-purified diets with sufficient vitamin V3, 2) HFD with sufficient VD3, 3) HFD feeding plus calcitriol treatment (HFD + VD), by which mice in HFD feeding for 2 wk were administered with calcitriol treatment for last 6 wk, 4) VDD diet, by which vitamin D was omitted in the standard AIN-93 diet, and the mice were devoid of light exposure, 5) HFD in VDD (VDD + HFD), by which the mice were devoid of light exposure as well, and 6) mice in VDD + HFD for 2 wk, followed by calcitriol treatment for additional 6 wk in the same diet (VDD + HFD + VD). 3 days before death, the rhythm of metabolic oscillation for all the mice was synchronized by feeding in the night for 12 h followed by fasting with free access to water for 12 h in the daytime. A: hematoxylin and eosin (H&E) and Oil-Red-O staining. B: triglyceride (TG) levels in the liver. C: body weight. D: mRNA levels of proinflammatory cytokines were measured by qRT-PCR analysis. E: nonalcoholic fatty liver disease (NAFLD) Activity Score (NAS) scores. The data are presented as means ± SE. Comparison was conducted by t-test between the experimental groups, and *P < 0.05, **P < 0.01.
Table 2.
Metabolic parameters
| Group | CONT | HFD | HFD + Calcitriol | VDD + HFD | VDD + HFD + Calcitriol | HFD + BAS |
|---|---|---|---|---|---|---|
| Body weight gain, g, 50 days | 4.80 ± 2.77 | 7.40 ± 1.14 | 5.00 ± 1.50* | 6.80 ± 1.79 | 3.70 ± 3.43† | 2.00 ± 0.81* |
| Body weight gain, % | 20.08% | 29.92% | 20.62% | 29.32% | 16.41% | 8% |
| Total caloric intake, kcal/day | 10.72 | 13.27 | 14.88 | 11.40 | 11.99 | 13.55 |
| Liver/body weight, kg | 0.042 ± 0.001 | 0.045 ± 0.001 | 0.053 ± 0.003 | 0.047 ± 0.002 | 0.051 ± 0.002 | 0.039 ± 0.002 |
| Serum 25(OH) D3, ng/ml | 3.08 ± 2.11 | 2.21 ± 0.86 | 1.42 ± 0.40 | 0.27 ± 0.03 | 0.24 ± 0.06 | 2.21 ± 0.86 |
| Serum TG, μmol/l | 1.72 ± 0.22 | 2.13 ± 0.19 | 1.60 ± 0.18* | 2.04 ± 0.10 | 0.95 ± 0.06† | 0.89 ± 0.45† |
| Serum NEFA, μmol/l | 2.45 ± 0.21 | 3.00 ± 0.40* | 1.97 ± 0.25 | 2.58 ± 0.22 | 1.23 ± 0.06† | 1.45 ± 0.46* |
| Serum CHOL, μmol/l | 3.15 ± 0.21 | 4.74 ± 0.15** | 3.86 ± 0.31* | 4.70 ± 0.15 | 4.76 ± 0.10 | 6.29 ± 1.23* |
Applicable values are means ± SE. High-fat diet (HFD) vs. control (CONT); HFD + calcitriol vs. HFD; vitamin D deficiency (VDD) + HFD + calcitriol vs. VDD + HFD; HFD + bile acid sequestration (BAS) vs. HFD;
P < 0.05 vs. HFD,
P < 0.01. TG, triglycerides; NEFA, nonesterified fatty acid; CHOL, cholesterol.
Typical features of metabolic parameters were accessed. As expected, body weight, central obesity as indicated by visceral fat tissues, serum TG, total cholesterol, and NEFA were significantly elevated by HFD feeding, which were additionally exacerbated by VDD (VDD + HFD) (Table 2 and unpublished data). The mice under the HFD or VDD + HFD feeding were given calcitriol treatment. As the results, the serum makers for fatty liver including TG and NEFA were improved significantly. Importantly, as shown in Table 2, feeding under VDD did not increase calorie intake, whereas the increased TG deposition and steatosis were increased, strongly suggesting that the VDD-induced additional fat deposition in the liver may result from elevated de novo lipogenesis or/and impaired energy expenditure. Indeed, administration of calcitriol did not reduce the calorie intake for the mice under HFD feeding, which supports a notion that the calcitriol-exerted effect for NASH resolution is to control lipogenesis and/or energy expenditure. Such speculation was addressed by other experimental results in this study.
Another key finding is that inflammation, initiated by HFD feeding, was exacerbated by additional VDD (VDD + HFD). Histological staining showed granulocytes present as clustered lesions (in a green lined box, Fig. 1A). Inflammation, as measured by the mRNA levels of TNF-α and IL-6, was increased by HFD and further significantly exacerbated by VDD + HFD (Fig. 1D). Conversely, administration of calcitriol revoked the steatosis, resolved parenchymal ballooning cells, and suppressed the elevation of hepatic TG. Administration of calcitriol thoroughly suppressed the inflammatory lesions, reduced infiltration of granulocytes, and suppressed TNF-α and IL-6 transcription. Body weight, central obesity, and serum parameters such as TG and NEFA for metabolic disorders were significantly improved by calcitriol treatment (Table 2 and data not shown). Severity of fatty degeneration, assessed by NAFLD Activity Score (NAS), was increased to grade 1–2 by HFD, and it was additionally escalated to grade 4–5 by VDD + HFD but suppressed by calcitriol treatment (Fig. 1E). Thus the experimental results provided two important observations. First, VDD exacerbates the HFD-initiated steatosis, suggesting that vitamin D may have regulatory roles in controlling lipid homeostasis. Second, VDD promotes the transition from simple steatosis to NASH, indicating that vitamin D signal may suppress inflammation.
NASH, induced by VDD + HFD, is associated with impaired enterohepatic circulation of bile acids.
Although direct evidence for the linkage of enterohepatic circulation to the development of NAFLD is seemingly unknown, the knowledge about bile acids in regulating HFD-induced hyperglycemia, obesity, and fatty acid synthesis pathway in the liver is available (11, 28). We reasoned that impaired enterohepatic circulation of bile acids might underlie the effects we observed with VDD. Total bile acids in the liver and gallbladder were measured. As shown in Fig. 2A, the levels of bile acids in the liver and gallbladder were not significantly altered by HFD feeding but were significantly decreased by additional VDD (VDD + HFD). In contrast, administration of calcitriol restored bile acid levels in the liver and gallbladder. These findings suggested that vitamin D signal might upregulate bile acids in the liver, which consequently suppresses NASH.
Fig. 2.

The NASH derived from VDD in HFD feeding (VDD + HFD) is associated with impaired enterohepatic circulation, which can be resolved by calcitriol treatment. Balb/C mice (n = 9) were treated as described in Fig. 1. A: total bile aids in the liver and gallbladder were measured and presented as micromoles per gram of liver tissue and micromoles per gram of body weight for the gallbladder, respectively. The data are presented as means ± SE. B: expression of ileal apical sodium-dependent bile acid cotransporter (ASBT) and fibroblast growth factor 15 (FGF15) was determined by qRT-PCR analysis. C: expression of hepatic Cyp7A1 and Cyp27A1 was determined by qRT-PCR analysis. *P < 0.05, **P < 0.01.
Impaired enterohepatic circulation in NASH is related to downregulation of intestinal ASBT.
Given the evidence of decreased retention of bile acids in the liver and gallbladder by the VDD + HFD mice in the NASH phenotype (Fig. 2A), we considered the possibility that VDD might inhibit reabsorption of bile acids and decrease the bile acid pool. Therefore, we measured iASBT, a key component in the enterohepatic circulation. Our results showed that iASBT in its mRNA format was upregulated by HFD feeding, which was in agreement with the elevated bile acids in the liver and gallbladder (Fig. 2A). Importantly, in the condition of VDD + HFD, the iASBT levels were drastically decreased, which was in line with the decreased bile acid pool in the liver and gallbladder. Administration of calcitriol restored the expression of iASBT significantly in the gut, which is also related to the increased bile acids in the liver and biliary system.
A previous study showed that ileal FGF15 could suppress iASBT expression presumably through a paracrine mechanism (26). As shown in Fig. 2, the expression of ileal FGF15 was initially induced by HFD feeding and additionally upregulated by VDD + HFD feeding. Importantly, administration of calcitriol significantly downregulated ileal FGF15 expression and restored iASBT expression at the same pace. These results suggest that the effect of vitamin D on iASBT and the enterohepatic circulation is likely mediated through ileal FGF15.
Whereas 90% bile acids in the enterohepatic circulation are contributed by ileal-based readsorption, about 10% are lost and replenished through de novo synthesis by Cyp7A1 and Cyp27A1 in the liver, through the classic and acidic pathways, respectively. As shown in Fig. 2C, whereas the expression of Cyp7A1 was downregulated by HFD feeding, it was increased under VDD + HFD, suggesting that vitamin D signaling may suppress its expression in the liver. Indeed, administration of calcitriol suppressed the Cyp7A1 expression significantly. On the other hand, Cyp27A1 was slightly increased by HFD feeding, and it was also suppressed by calcitriol. These results implicate that, in the context of VDD + HFD, the impaired bile acid retention in the liver is related to impaired iASBT for the uptake of bile acids, rather than the endogenous synthesis of bile acids.
Upregulation of lipogenesis machinery in the VDD + HFD feeding is attenuated by calcitriol.
The fact that VDD + HFD feeding escalated accumulation of TG but did not affect the calorie uptake (Table 2) indicates that de novo synthesis of fatty acid may contribute significantly to the NASH. SREBP-1c, a key transcription factor for lipogenesis, was substantially increased under VDD + HFD feeding (Fig. 3). Conversely, treating the mice with calcitriol (VDD + HFD + VD) suppressed the expression of SREBP-1c in the liver. It was found that FAS, a major enzyme controlling fatty acid synthesis and TG accumulation, is highly expressed even under HFD feeding, leading to NASH (19). Such a seemingly paradoxical fact was confirmed in our study, shown in Fig. 3. Importantly, administration of calcitriol simultaneously suppressed hepatic SREBP-1c and its target gene FAS, suggesting a mechanism for vitamin D signaling in inhibiting hepatic lipogenesis in the NASH.
Fig. 3.

Altered expression of the genes related to hepatic lipogenesis and energy expenditure is related to the NASH derived from VDD and HFD feeding. The experimental conditions are described in Fig. 1. The expression of hepatic SREBP-1c, fatty acid synthase (FAS), carnitine palmitoyltransferase 1α (CPT-1α), peroxisome proliferator-activated receptor-α (PPAR-α), farnesoid X receptor (FXR), and small heterodimer partner (SHP) was determined by qRT-PCR analysis. The data are presented as means ± SE. *P < 0.05, **P < 0.01.
Carnitine palmitoyltransferase 1α (CPT-1α) is a mitochondrial enzyme to transport long-chain fatty acids for β-oxidation and energy expenditure. Hepatic CPT-1α expression was downregulated under the VDD + HFD feeding, indicating that impaired β-oxidation might contribute to the NASH formation as well (Fig. 3). Actually, administration of calcitriol restored the expression of CPT-1α in the liver under VDD + HFD, suggesting increased expenditure of fatty acids. Peroxisome proliferator-activated receptor-α (PPAR-α), which serves as an upstream transcription factor controlling the expression of CPT-1α, was also decreased by HFD feeding and additionally downregulated in the VDD condition (VDD + HFD). Conversely, administration of calcitriol fully restored hepatic PPAR-α expression, supporting a notion that vitamin D signaling may facilitate fatty acid oxidation to minimize steatosis.
Calcitriol initiates the ileal FGF15/hepatic FXR/SHP/SREBP-1c pathway to suppress lipogenic enzymes and enhance gallbladder filling at physiological conditions.
We next measured vitamin D for its roles in enterohepatic circulation and lipogenesis in the liver under physiological conditions. Expression of lipogenic enzymes regulated by SREBP-1c is known to be suppressed by SHP, a master nuclear suppressor (28). On the other hand, SHP is also a hierarchy suppressor controlling the expression of Cyp7A1 to restrain bile acid synthesis. According to current concepts, two independent pathways can induce hepatic SHP. First, bile acids via their nuclear receptor (FXR) in the hepatocytes may directly induce the expression of SHP in the liver. Second, FGF15/19 secreted from the ileum and via portal vein circulation to the liver may also induce SHP expression in the hepatocytes (8, 12).
The normal control mice fasting for 10 h were given a single dose of calcitriol and then killed 12 h later. As expected, acute injection of calcitriol induced ileal FGF15 expression (Fig. 4A). Whereas the mRNA of ileal FXR expression was suppressed by calcitriol, the hepatic FXR in its mRNA levels was increased, suggesting that calcitriol might increase the responsiveness of FXR to bile acids in the liver (Fig. 4B). Importantly, calcitriol significantly stimulated hepatic SHP expression, along with the suppression of hepatic SREBP-1c and consequent suppression of FAS expression. Cyp7A1, which is another targeting gene for SHP, was also suppressed by the calcitriol administration, which explained well the downregulated bile acids in the liver (Fig. 4C). Conversely, Cy3A11 (Cyp3A4 for humans), a prominent P450 monooxygenase for breakdown xenobiotics including vitamin D and bile acids, was increasingly expressed by calcitriol treatment, in an expected manner. Moreover, calcitriol treatment elevated ileal FGF15 expression, which was in line with suppression of the ileal ASBT expression at the physiological condition (Fig. 4A). Finally, we measured the retention of bile acids in the liver and gallbladder by calcitriol treatment at the physiological condition. As shown, the hepatic levels of bile acids were suppressed by calcitriol treatment, whereas bile acid filling in the gallbladder was upregulated (Fig. 4D), which is in line with a previous report showing that FGF15 promoted bile filling in the gallbladder (7). In summary, at the physiological condition, vitamin D signaling, through induction of ileal FGF15 to induce hepatic SHP that in turn suppresses hepatic SREBP-1c and FAS, may ultimately restrain fatty acid synthesis.
Fig. 4.

Calcitriol induces ileal FGF15 and hepatic SHP pathway to suppress lipogenic machinery and module enterohepatic circulation in the physiological condition. Balb/C mice (n = 8) were given a single dose of calcitriol or control. A: ileal expression of FGF15, FXR, and ASBT was determined by qRT-PCR analysis. B: machinery for hepatic lipogenesis, including FXR, SHP, SREBP-1c, and FAS expressed as the mRNA in the liver, was measured by qRT-PCR analysis. C: Cyp7A1 and Cyp3A11, for bile acid synthesis and catabolism, respectively, in the form of mRNA levels expressed in the liver, were determined by qRT-PCR analysis. D: total bile acids in the liver and gallbladder were determined. The data are presented as means ± SE. *P < 0.05, **P < 0.01.
Manipulating enterohepatic circulation by bile acid sequestration attenuates steatosis.
To further investigate our hypothesis that vitamin D regulates the levels of bile acids in enterohepatic circulation, which prevents hepatic steatosis, we administered cholestyramine in the HFD-fed animals to sequester the bile acids and determined its roles in steatosis treatment. As shown, cholestyramine significantly attenuated the HFD-induced steatosis, characterized by clearance of the ballooning cells around the portal veins as measured by H and E and Oil-Red-O staining (Fig. 5A). Hepatic TG levels, increased by HFD feeding, were efficiently suppressed by cholestyramine treatment (Fig. 5B). Body weight was also attenuated by cholestyramine treatment (Fig. 5C).
Fig. 5.

Manipulating enterohepatic circulation by bile acid sequestration (BAS) attenuates steatosis. Balb/C mice (n = 8) were fed with HFD or not for 2 wk, followed by addition of cholestyramine to the diet or not for a consecutive 6 wk. A: representative slides of H&E staining and Oil-Red-O staining. B: TG levels in the liver tissues, in milligrams per gram of liver weight. C: body weight of the subjects. D: total bile acid levels in the liver and gallbladder, in micromoles per gram of liver weight and micromoles per gram of body weight, respectively. E: SHP, SREBP-1c, FAS, Cyp7A1, TNF-α, and IL-6 expressed in the liver as mRNA were determined by qRT-PCR analysis. F: FGF15 and ASBT expressed in the ileum as the mRNA levels were determined by qRT-PCR analysis. *P < 0.05, **P < 0.01.
Bile acid sequestrants were originally used to lower plasma cholesterol, through a mechanism of the compensative de novo synthesis of bile acids, leading to consumption of cholesterol, the precursor for bile acids. Indeed, the levels of bile acids in the liver and gallbladder were significantly elevated by cholestyramine treatment as expected (Fig. 5D). Furthermore, the expression of Cyp7A1, being suppressed by HFD feeding, was elevated by the bile sequestration, which is in agreement with the increased pool of bile acids in the liver and gallbladder (Fig. 5, D and E). In addition to vitamin D, bile acids through ileal FXR can also induce ileal FGF15, which may consequently suppress the hepatic Cyp7A1 and lipogenesis. The expression of ileal FGF15, which was elevated in the HFD feeding, was significantly suppressed by bile acid sequestration (Fig. 5F). Downregulation of ileal FGF15 was associated with downregulation of hepatic SHP and consequent elevation of hepatic Cyp7A1, SREBP-1c, and FAS (Fig. 5E). Moreover, the iASBT levels were upregulated through bile acid sequestration, which is also related to the downregulation of FGF15. These results suggest that the long-term bile acid sequestration may resume endogenous lipogenesis in a way to compensate the lost lipid adsorption. Inflammatory cytokines, such as TNF-α and IL-6 being elevated by HFD, were moderately suppressed by bile acid sequestration (Fig. 5E).
DISCUSSION
The current work highlights the roles of vitamin D signaling in maintaining hepatic lipid homeostasis and its deficiency in promotion of NASH. In particular, we found that 1) VDD exacerbates simple steatosis into NASH in animals under HFD feeding (VDD + HFD), demonstrating VDD as a critical second hit, promoting inflammation and fatty degeneration in the liver; 2) the VDD + HFD-induced fatty liver is mediated at least in part by the impaired enterohepatic circulation; 3) the impaired enterohepatic circulation in the NASH model is mediated by downregulation iASBT; and 4) bile acid sequestration can attenuate steatosis in part through upregulation of iASBT. A summary of these findings is depicted in Fig. 6.
Fig. 6.
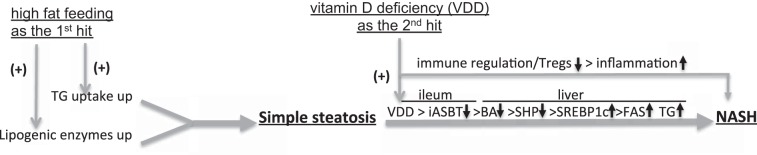
A potential mechanism for experimental NASH development. We hypothesized that, based on the current results and others, VDD may serve as a second hit to accelerate simple steatosis into NASH through two distinct pathways. First, VDD may promote additional hepatic lipogenesis in part through impaired ileal ASBT expression and poor bile filling, leading to upregulation of hepatic lipogenesis. Second, VDD may impede immune homeostasis, in part by impaired Tregs induction and loss of immune regulation, leading to hepatic inflammation in NASH.
Whereas HFD-induced steatosis is understandable, how VDD exacerbates TG in the fatty liver is intriguing. Our data showing that VDD does not boost additional calorie uptake, while TG deposition is conspicuously increased, indicate that increased lipogenesis and/or decreased energy expenditure may contribute to the NASH development. Previous works showed a paradox that, despite HFD feeding, the endogenous lipogenesis machinery is still highly active rather than downregulated (21, 27). Our study confirmed such finding, showing increased expression of hepatic SREBP-1c and FAS along with the increased TG deposition even in the presence of HFD feeding. Moreover, in the present study, we provide novel evidence showing a mechanism by which VDD drives upregulation of lipogenic enzymes in HFD feeding. In particular, we show that SREBP-1c and FAS are upregulated in VDD under HFD feeding, which are further linked to the failed bile filling in the liver and biliary tree.
In addition, our findings show a potential role for vitamin D signaling on mitochondrial fatty acid oxidation. CPT-1α that is necessary for fatty acid β-oxidation and PPAR-α that is an upstream transcription for CPT-1 were both inhibited in the mice under VDD + HFD feeding. Importantly, administration of calcitriol restored the expression of PPAR-α and CPT-1α along with ameliorating steatosis. Thus vitamin D, through two independent pathways by suppressing SREBP-1c to restrain TG synthesis and inducing CPT-1 to promote energy expenditure, may maintain balanced TG levels in the healthy liver.
To explore the molecular mechanisms, we tested the effects of calcitriol on FGF15 expression in the ileum. FGF15 is a known regulator of iASBT. We confirmed a previous report showing that transient administration of calcitriol can promptly upregulate FGF15 expression in the ileum (25), which is consistent with suppression of iASBT, indicating a causative relationship. Moreover, the upregulation of ileal FGF-15 by administration of calcitriol in the healthy mice may further induce hepatic SHP that in turn suppresses the expression of FAS and Cyp7A1 in the liver.
At the same time, we discovered that calcitriol could promote gallbladder filling, presumably through induction of ileal FGF15 that acts on the gallbladder. Such speculation is supported by a previous report showing that FGF15 can promote gallbladder filling (5). These lines of results indicate that vitamin D at the physiological condition may simultaneously balance enterohepatic circulation and hepatic lipogenesis, suggesting a causal relationship from enterohepatic circulation to lipogenesis.
Although administration of calcitriol to the healthy and NASH mice could suppress SREBP-1c and FAS, alternative upstream machineries might be applied for each case. For the healthy mice, calcitriol upregulated ileal FGF15 followed by induction of hepatic SHP, leading to suppression of SREBP-1c and FAS (Fig. 4). In contrast, under the long-term VDD + HFD, ileal FGF15 expression was upregulated, whereas SREBP-1c and FAS were upregulated as well, in line with TG accumulation. Thus the ileal FGF15 and subsequent hepatic SHP were apparently disconnected by the mice under VDD + HFD feeding. In such context, repeating administration of calcitriol suppressed ileal FGF15, along with suppression of hepatic SREBP-1c and FAS. For such regard, the calcitriol-exerted amelioration of steatosis is likely mediated by an alternative pathway, namely increased bile filling in the liver, which through FXR activation may suppress SREBP-1c and FAS and consequently resolve the NASH phenotypes. On the other hand, given the fact of SHP-independent suppression of Cyp7A1 by bile acids (13), it is likely that a similar mechanism may also apply to the calcitriol-mediated suppression of NASH in our model. Above all, SREBP-1c is suppressed by calcitriol in both the physiological acute treatment and repeating administration for NASH.
Not addressed is how vitamin D treatment alleviates inflammation in the NASH model. Low-grade and persistent inflammation is a driving force for NASH from simple steatosis. It has been known that calcitriol has profound immune regulatory functions to limit excessive immune response (24). Additional works showed that vitamin D through induction of CD4+CD25+FoxP3 Tregs can restrain Th1 and Th2 development in various disease models (9, 10) (15). Whether Tregs are downregulated by VDD + HFD in the NASH model and whether calcitriol treatment can restore Tregs are two subjects of our future work. Finally, how much our finding can be applied to human NASH should be further explored. In particular, we used chaw containing 60% calories from fat, which is beyond the scope of human diet. On the other hand, insulin resistance is critical for NASH in humans. Whether and how VDD can induce insulin resistance is the subject of our current investigation. In closing, our findings may provide potential therapeutic strategies based on vitamin D supplementation as well as bile acid sequestration to treat or prevent NASH.
GRANTS
The study was supported in part by National Science and Technology Major Project (2012ZX10002004-006, 2012ZX10004904-003-001, 2013ZX10002002-006-001) to Z. Duan, NIH grants (R01 DK069418) and startup fund from Sichuan University to Y.-P. Han, and Department of Veterans Affairs and NIH grants P01 CA163200 and P50 AA11999 to S. Pandol.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
Author contributions: M.K. and L.Z. performed experiments; L.B., X.Z., and S.L. analyzed data; Y.C. and S.Z. interpreted results of experiments; S.J.P. edited and revised manuscript; Y.-P.H. and Z.D. conception and design of research; Z.D. approved final version of manuscript.
REFERENCES
- 1.Adorini L, Daniel KC, Penna G. Vitamin D receptor agonists, cancer and the immune system: an intricate relationship. Curr Top Med Chem 6: 1297–1301, 2006. [DOI] [PubMed] [Google Scholar]
- 2.Angulo P. GI epidemiology: nonalcoholic fatty liver disease. Aliment Pharmacol Ther 25: 883–889, 2007. [DOI] [PubMed] [Google Scholar]
- 3.Barchetta I, Angelico F, Del Ben M, Baroni MG, Pozzilli P, Morini S, Cavallo MG. Strong association between nonalcoholic fatty liver disease (NAFLD) and low 25(OH) vitamin D levels in an adult population with normal serum liver enzymes. BMC Med 9: 85, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bateson MC, Maclean D, Evans JR, Bouchier IA. Chenodeoxycholic acid therapy for hypertriglyceridaemia in men. Br J Clin Pharmacol 5: 249–254, 1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Choi M, Moschetta A, Bookout AL, Peng L, Umetani M, Holmstrom SR, Suino-Powell K, Xu HE, Richardson JA, Gerard RD, Mangelsdorf DJ, Kliewer SA. Identification of a hormonal basis for gallbladder filling. Nat Med 12: 1253–1255, 2006. [DOI] [PubMed] [Google Scholar]
- 6.Day CP, James OF. Steatohepatitis: a tale of two “hits”? Gastroenterology 114: 842–845, 1998. [DOI] [PubMed] [Google Scholar]
- 7.Eliades M, Spyrou E, Agrawal N, Lazo M, Brancati FL, Potter JJ, Koteish AA, Clark JM, Guallar E, Hernaez R. Meta-analysis: vitamin D and non-alcoholic fatty liver disease. Aliment Pharmacol Ther 38: 246–254, 2013. [DOI] [PubMed] [Google Scholar]
- 8.Goodwin B, Jones SA, Price RR, Watson MA, McKee DD, Moore LB, Galardi C, Wilson JG, Lewis MC, Roth ME, Maloney PR, Willson TM, Kliewer SA. A regulatory cascade of the nuclear receptors FXR, SHP-1, and LRH-1 represses bile acid biosynthesis. Mol Cell 6: 517–526, 2000. [DOI] [PubMed] [Google Scholar]
- 9.Gorman S, Judge MA, Burchell JT, Turner DJ, Hart PH. 1,25-dihydroxyvitamin D3 enhances the ability of transferred CD4+ CD25+ cells to modulate T helper type 2-driven asthmatic responses. Immunology 130: 181–192, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gregori S, Giarratana N, Smiroldo S, Uskokovic M, Adorini L. A 1alpha,25-dihydroxyvitamin D(3) analog enhances regulatory T-cells and arrests autoimmune diabetes in NOD mice. Diabetes 51: 1367–1374, 2002. [DOI] [PubMed] [Google Scholar]
- 11.Ikemoto S, Takahashi M, Tsunoda N, Maruyama K, Itakura H, Kawanaka K, Tabata I, Higuchi M, Tange T, Yamamoto TT, Ezaki O. Cholate inhibits high-fat diet-induced hyperglycemia and obesity with acyl-CoA synthetase mRNA decrease. Am J Physiol Endocrinol Metab 273: E37–E45, 1997. [DOI] [PubMed] [Google Scholar]
- 12.Inagaki T, Choi M, Moschetta A, Peng L, Cummins CL, McDonald JG, Luo G, Jones SA, Goodwin B, Richardson JA, Gerard RD, Repa JJ, Mangelsdorf DJ, Kliewer SA. Fibroblast growth factor 15 functions as an enterohepatic signal to regulate bile acid homeostasis. Cell Metab 2: 217–225, 2005. [DOI] [PubMed] [Google Scholar]
- 13.Kerr TA, Saeki S, Schneider M, Schaefer K, Berdy S, Redder T, Shan B, Russell DW, Schwarz M. Loss of nuclear receptor SHP impairs but does not eliminate negative feedback regulation of bile acid synthesis. Dev Cell 2: 713–720, 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kitson MT, Roberts SK. D-livering the message: the importance of vitamin D status in chronic liver disease. J Hepatol 57: 897–909, 2012. [DOI] [PubMed] [Google Scholar]
- 15.Kreindler JL, Steele C, Nguyen N, Chan YR, Pilewski JM, Alcorn JF, Vyas YM, Aujla SJ, Finelli P, Blanchard M, Zeigler SF, Logar A, Hartigan E, Kurs-Lasky M, Rockette H, Ray A, Kolls JK. Vitamin D3 attenuates Th2 responses to Aspergillus fumigatus mounted by CD4+ T cells from cystic fibrosis patients with allergic bronchopulmonary aspergillosis. J Clin Invest 120: 3242–3254, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lefebvre P, Cariou B, Lien F, Kuipers F, Staels B. Role of bile acids and bile acid receptors in metabolic regulation. Physiol Rev 89: 147–191, 2009. [DOI] [PubMed] [Google Scholar]
- 17.Lu L, Feng M, Gu J, Xia Z, Zhang H, Zheng S, Duan Z, Hu R, Wang J, Shi W, Ji C, Shen Y, Chen G, Zheng SG, Han YP. Restoration of intrahepatic regulatory T cells through MMP-9/13-dependent activation of TGF-beta is critical for immune homeostasis following acute liver injury. J Mol Cell Biol 5: 369–379, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ludwig J, Viggiano TR, McGill DB, Oh BJ. Nonalcoholic steatohepatitis: Mayo Clinic experiences with a hitherto unnamed disease. Mayo Clin Proc 55: 434–438, 1980. [PubMed] [Google Scholar]
- 19.Morgan K, Uyuni A, Nandgiri G, Mao L, Castaneda L, Kathirvel E, French SW, Morgan TR. Altered expression of transcription factors and genes regulating lipogenesis in liver and adipose tissue of mice with high fat diet-induced obesity and nonalcoholic fatty liver disease. Eur J Gastroenterol Hepatol 20: 843–854, 2008. [DOI] [PubMed] [Google Scholar]
- 20.Neuschwander-Tetri BA, Caldwell SH. Nonalcoholic steatohepatitis: summary of an AASLD Single Topic Conference. Hepatology 37: 1202–1219, 2003. [DOI] [PubMed] [Google Scholar]
- 21.Oosterveer MH, van Dijk TH, Tietge UJ, Boer T, Havinga R, Stellaard F, Groen AK, Kuipers F, Reijngoud DJ. High fat feeding induces hepatic fatty acid elongation in mice. PLoS One 4: e6066, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Petta S, Camma C, Scazzone C, Tripodo C, Di Marco V, Bono A, Cabibi D, Licata G, Porcasi R, Marchesini G, Craxi A. Low vitamin D serum level is related to severe fibrosis and low responsiveness to interferon-based therapy in genotype 1 chronic hepatitis C. Hepatology 51: 1158–1167, 2010. [DOI] [PubMed] [Google Scholar]
- 23.Qin L, Han YP. Epigenetic repression of matrix metalloproteinases in myofibroblastic hepatic stellate cells through histone deacetylases 4: implication in tissue fibrosis. Am J Pathol 177: 1915–1928, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rigby WF, Stacy T, Fanger MW. Inhibition of T lymphocyte mitogenesis by 1,25-dihydroxyvitamin D3 (calcitriol). J Clin Invest 74: 1451–1455, 1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schmidt DR, Holmstrom SR, Fon Tacer K, Bookout AL, Kliewer SA, Mangelsdorf DJ. Regulation of bile acid synthesis by fat-soluble vitamins A and D. J Biol Chem 285: 14486–14494, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sinha J, Chen F, Miloh T, Burns RC, Yu Z, Shneider BL. β-Klotho and FGF-15/19 inhibit the apical sodium-dependent bile acid transporter in enterocytes and cholangiocytes. Am J Physiol Gastrointest Liver Physiol 295: G996–G1003, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Strable MS, Ntambi JM. Genetic control of de novo lipogenesis: role in diet-induced obesity. Crit Rev Biochem Mol Biol 45: 199–214, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Watanabe M, Houten SM, Wang L, Moschetta A, Mangelsdorf DJ, Heyman RA, Moore DD, Auwerx J. Bile acids lower triglyceride levels via a pathway involving FXR, SHP, and SREBP-1c. J Clin Invest 113: 1408–1418, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yang CY, Leung PS, Adamopoulos IE, Gershwin ME. The implication of vitamin D and autoimmunity: a comprehensive review. Clin Rev Allergy Immunol 45: 217–226, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]