

Abstract
Exposure to ambient air pollution contributes to the progression of cardiovascular disease, particularly in susceptible populations. The objective of the present study was to determine whether early life exposure to air pollution causes persistent cardiovascular consequences measured at adulthood. Pregnant FVB mice were exposed to filtered (FA) or concentrated ambient particulate matter (PM2.5) during gestation and nursing. Mice were exposed to PM2.5 at an average concentration of 51.69 μg/m3 from the Columbus, OH region for 6 h/day, 7 days/wk in utero until weaning at 3 wk of age. Birth weight was reduced in PM2.5 pups compared with FA (1.36 ± 0.12 g FA, n = 42 mice; 1.30 ± 0.15 g PM2.5, n = 67 P = 0.012). At adulthood, mice exposed to perinatal PM2.5 had reduced left ventricular fractional shortening compared with FA-exposed mice (43.6 ± 2.1% FA, 33.2 ± 1.6% PM2.5, P = 0.001) with greater left ventricular end systolic diameter. Pressure-volume loops showed reduced ejection fraction (79.1 ± 3.5% FA, 35.5 ± 9.5% PM2.5, P = 0.005), increased end-systolic volume (10.4 ± 2.5 μl FA, 39.5 ± 3.8 μl PM2.5, P = 0.001), and reduced dP/dt maximum (11,605 ± 200 μl/s FA, 9,569 ± 800 μl/s PM2.5, P = 0.05) and minimum (−9,203 ± 235 μl/s FA, −7,045 ± 189 μl/s PM2.5, P = 0.0005) in PM2.5-exposed mice. Isolated cardiomyocytes from the hearts of PM2.5-exposed mice had reduced peak shortening (%PS, 8.53 ± 2.82% FA, 6.82 ± 2.04% PM2.5, P = 0.003), slower calcium reuptake (τ, 0.22 ± 0.09 s FA, 0.26 ± 0.07 s PM2.5, P = 0.048), and reduced response to β-adrenergic stimulation compared with cardiomyocytes isolated from mice that were exposed to FA. Histological analyses revealed greater picro-sirius red-positive-stained areas in the PM2.5 vs. FA group, indicative of increased collagen deposition. We concluded that these data demonstrate the detrimental role of early life exposure to ambient particulate air pollution in programming of adult cardiovascular diseases and the potential for PM2.5 to induce persistent cardiac dysfunction at adulthood.
Keywords: maternal exposure, air pollution, cardiovascular development, inflammation
it has become increasingly evident that the environment plays a major role in the development of human systemic diseases. Current studies have indicated that inhalation of ambient particulate matter (PM) ranks as the ninth cause of overall disease (21). Well-established evidence that exposure to air pollution is linked with heart failure, myocardial infarction, lung dysfunction, lung cancer, and increased morbidity (5, 6, 14, 16, 20, 22, 24, 31) has caused a worldwide need for policy change. Further research regarding air pollution exposure in susceptible populations, including those with obesity, existing heart disease, cystic fibrosis, and more sensitive states is needed, particularly concerning exposure during development.
The sensitive nature of the developing fetus, termed “plasticity” (1), allows the fetus to adapt to an adverse intrauterine environment, e.g., nutritional deficits, maternal inflammation, or stress. The results of these adaptations can be evident into adulthood and have been linked to the development of various metabolic, neurological, pulmonary, and more recently, cardiovascular diseases. This “developmental origin of adult disease” could provide yet another explanation for the detrimental effects of maternal exposure to air pollution during fetal development on the hearts and lungs of offspring.
The consequences of exposure to air pollution during development are not well defined. Evidence from epidemiological studies indicates a strong association between ambient concentrations of air pollutants, including PM, and poor developmental outcomes, such as low birth weight (3, 19, 23) preterm birth (34, 37), and intrauterine growth restriction (10, 36). Animal studies have confirmed these outcomes (25, 38, 40, 44) and shown genetic and epigenetic changes in animals maternally exposed to air pollution (32). Consequences can be seen manifested as poor neurological outcomes (39, 49), increased airway inflammation, and respiratory dysfunction (18). The combination of these factors leads to the hypothesis that air pollution causes an adverse intrauterine environment, altering regular development, which increases the likelihood of subsequent adult diseases.
In the present study, we exposed mice to ambient PM with diameter less than 2.5 μm (PM2.5) during perinatal development, from gestation until weaning. Mice were subsequently assessed at adulthood for functional and structural deficits of the cardiovascular system compared with mice exposed to filtered air (FA) alone. This exposure paradigm allows us to examine the consequences at adulthood of early life exposure to relevant concentrations of ambient PM2.5.
MATERIALS AND METHODS
Animals and exposure.
All animals were handled according to NIH guidelines under Institutional Animal Care and Use Committee (IACUC) protocols approved at both The Research Institute at Nationwide Children's Hospital and The Ohio State University, Columbus, Ohio. FVB male and female mice were housed for at least 1 wk in our facility before breeding. Pregnancy was confirmed and time-dated with the presence of a vaginal plug. Dams were exposed to concentrated PM2.5 from the Columbus, OH region using the OASIS-1 aerosol concentration system, which has been previously described (47). Dams were placed into the exposure system for 6 h a day, 7 days a wk, separated into either PM2.5 exposure or air that has been filtered by a high-efficiency air filter (FA). The average PM2.5 concentration that the animals were exposed to in this study was 51.69 μg/m3. Because the exposure time represents one-quarter of the day, this is equivalent to a 24-h average of 12.92 μg/m3, below the national air quality standard of 15 μg/m3. Dams were paired overnight, and exposure was begun the day after a plug was observed. Exposures were continued until weaning at 3 wk of age. Following weaning, both groups of mice were returned to room air until they reached 3 mo of age, and male offspring were subjected to analyses as described below.
Echocardiography.
Cardiac function was analyzed by echocardiography (40 MHz transducer, Vevo 2100; Visualsonics, Toronto, Ontario, Canada). Internal temperature was maintained at 37°C, as mice were continuously sedated with 1% isoflurane (in 100% O2) during assessment. Prewarmed ultrasound gel (Aquasonic; Parker Laboratories, Fairfield, NJ) was used on the chest with a 15-MHz probe optimized for mice placed in the parasternal, short-axis orientation. Data were averaged from at least three analyses per mouse. Left ventricular (LV) dimensions (LV end-systolic and end-diastolic diameters, LVESd and LVEDd) and posterior wall thickness at systole and diastole (PWTs and PWTd) were assessed, using the leading-edge technique according to the American Society for Echocardiography. Fractional shortening (FS) was calculated using the equation: %FS = [(LVEDd − LVESd)/LVEDd * 100]. Additional images were acquired to obtain aortic and pulmonary dimension, and pulse-wave Doppler imaging was used to obtain aortic and pulmonary velocities. Stroke volume (SV) was estimated utilizing the velocity time integral trace multiplied by the cross-sectional area of the vessel, and this was used to calculate cardiac output (CO) as the product of SV and heart rate (HR).
Pressure-volume measurements.
LV hemodynamic parameters were assessed in vivo using a pressure-volume (PV) catheter according to the manufacturer's instructions (Transonic Scisense, London, Ontario, Canada). Mice were anesthetized and intubated, and a 1.2-French pressure-conductance catheter was retrogradely inserted through the right carotid artery into the LV. Following positioning of the catheter in the LV, mice were allowed to stabilize while anesthesia was maintained at 0.8 vol% isoflurane. The following systolic and diastolic hemodynamic parameters were acquired using 8–10 consecutive loops: HR, CO, LV end-diastolic volume (EDV), LV end-systolic volume (ESV), ejection fraction [EF, (EDV − ESV)/EDV * 100], maximum dP/dt (+dP/dt), minimum dP/dt (−dP/dt), LV end-diastolic pressure, LV end-systolic pressure, arterial elastance, and relaxation constant (τ Weiss).
Cardiomyocyte isolation and measurement of function and intracellular Ca2+ transients.
Cardiomyocytes were isolated as previously described (33, 42, 43, 46, 47). Briefly, hearts were removed and retrogradely perfused through the aorta with Liberase and trypsin until digested. Isolated cells were then plated on laminin-coated glass-bottom inserts (Cell MicroControls, Norfolk, VA) for functional analyses. Glass-bottom inserts were perfused with warm contractile buffer (27, 33, 42, 43, 46, 47) within a flow chamber attached to an Olympus IX-71 microscope. Cells were stimulated (1 Hz, 3-ms duration) with a Myopacer Field-Stimulator system (IonOptix, Milton, MA), and functional properties of the cells were evaluated with the Sarclen Sarcomere Length Acquisition Module using the Myocam-S Digital CCD camera video imaging system (IonOptix). Analyses of 40–45 cells from five mice per treatment provided the parameters of sarcomere percent peak shortening (normalized to baseline length, %PS; cellular equivalent of %FS), sarcomere maximal departure, return velocities (±dl/dt), and sarcomere time-to-90% PS and relengthening (TPS90 and TR90, respectively). For Ca2+ measurements, cardiomyocytes in glass-bottom dishes were loaded with fura-2 AM (0.5 μM) for 20 min at 25°C and then washed and treated with normal culture media for 20 min at 25°C. Fluorescence was recorded in stimulated cardiomyocytes using a dual-excitation, single-emission system (IonOptix). Transients were analyzed for values of calcium release (TPS90) and reuptake (τ, TR90). To measure the effect of β-adrenergic stimulation on contractility, contractility was measured in cells treated with in 10−7 M isoproterenol for 10 min.
Tissue analyses.
Mice were euthanized, and heart tissue was either fixed in 4% paraformaldehyde or snap frozen using liquid nitrogen. Fixed tissue was transferred to 70% ethyl alcohol after 24 h, paraffin embedded, and cut into 5-μm sections. Paraffin-embedded hearts were cut from the base to the apex. Five-micron-thick slides taken at the level below the papillary muscles were stained with picro-sirius red (12). Images were taken from the septum, anterior, posterior, and free wall, and quantitative planimetric analyses were performed. Percentage of picro-sirius red-positive-stained area was quantified as percentage of total myocardial area.
Statistical analyses.
All data are reported as means ± SE with n = 3–5 mice from at least five different litters. Cardiomyocyte data are reported with 40–45 cells from five mice for each value. Data were analyzed using a Student's t-test (two-tailed) with Prism 6 (GraphPad, San Diego, CA), and differences were considered statistically significant if P < 0.05.
RESULTS
PM2.5 exposure during perinatal development results in lower birth weight but greater body weight at adulthood.
To evaluate the consequences of PM2.5 exposure during fetal development, we assessed maternal weight gain and fetal birth weight. Maternal body weight was not different in PM2.5-exposed dams after 20 days of pregnancy, and there was no difference in weight gain compared with FA controls (data not shown). However, birth weights were significantly lower in pups that were born to PM2.5-exposed dams compared with pups born to FA-exposed control mice. At 3 mo of age, body weights were significantly greater in mice that were PM2.5-exposed during perinatal development compared with FA-exposed controls. Absolute heart weight and heart weight normalized to tibia length were increased in pups that were exposed to PM2.5 during perinatal development compared with FA-exposed control mice (Table 1).
Table 1.
Parameters from mice exposed to FA or PM2.5 during development
| FA | PM2.5 | P | |
|---|---|---|---|
| Birth weight, g | 1.36 ± 0.12 | 1.30 ± 0.15 | 0.012 |
| Body weight, g | 22.4 ± 0.5 | 24.0 ± 0.3 | 0.013 |
| Heart weight, mg | 107.6 ± 3.1 | 119.4 ± 2.7 | 0.014 |
| Heart weight/tibia length | 60.2 ± 1.5 | 66.9 ± 1.5 | 0.008 |
Values are means ± SE.
FA, filtered air; PM2.5, particulate matter with diameter less than 2.5 μm.
Perinatal PM2.5 exposure results in LV remodeling.
Three-month-old mice exposed to PM2.5 during perinatal development showed substantial cardiac remodeling as illustrated by increased LVESd and LVEDd dimensions and reduced PWTs (Fig. 1, A–E). Morphological alterations were associated with lower systolic function, as indicated by reduced %FS in PM2.5-exposed mice compared with FA controls (Fig. 1F).
Fig. 1.

Transthoracic echocardiographic morphological and functional parameters in 3-mo-old FVB mice that were exposed to filtered air (FA) or particulate matter with diameter less than 2.5 μm (PM2.5) during perinatal development. Representative M-mode images obtained from FA-exposed mice (A) and PM2.5-exposed mice (B). C: left ventricular end-systolic diameter (LVESd). D: LV end-diastolic diameter (LVEDd). E: posterior wall thickness at systole (PWTs), from n = 10 mice in each treatment group and no more than 2 mice per litter. F: fractional shortening (FS). 5 beat cycles were captured, and 3 loops were averaged per assessment. *P < 0.05 was considered statistically significant.
Perinatal PM2.5 exposure causes LV systolic dysfunction.
Transthoracic echocardiographic analyses revealed reduced systolic function in mice that were PM2.5 exposed during perinatal development (Fig. 1F). We performed PV analyses evaluating morphological changes and LV function (Fig. 2, A and B). LV ESV and EDV were both increased in 3-mo-old mice that were exposed to PM2.5 compared with FA-exposed controls, confirming the increased dimension observed by echocardiography (Fig. 2, C and D). Also consistent with echocardiographic findings, these structural alterations were associated with reduced LV contractility in mice exposed to PM2.5, as evident by reduced EF and dP/dtmax compared with FA-exposed controls (Fig. 2, E and F). HRs during PV analyses were not different between groups (501 ± 11 beats/min FA vs. 504 ± 6 beats/min PM2.5).
Fig. 2.

In vivo hemodynamic function in 3-mo-old FVB mice that were exposed to FA or PM2.5 during perinatal development. Representative pressure-volume loops from FA-exposed mice (A) and PM2.5-exposed mice (B). C: end-systolic volume (ESV). D: end-diastolic volume (EDV). E: ejection fraction (EF). F: dP/dtmax. G: dP/dtmin. H: arterial elastance (Ea). n = 5 mice per group from individual litters. *P < 0.05 was considered statistically significant.
LV diastolic dysfunction is evident in mice exposed to PM2.5 during perinatal development.
LV relaxation was decreased in mice that were PM2.5 exposed during perinatal development, as indicated by reduced dP/dtmin (Fig. 2G), which is common in other models of heart failure. Arterial load was increased in PM2.5-exposed mice compared with FA controls, as indicated by increased arterial elastance (Fig. 2H).
Perinatal exposure to PM2.5 results in in vitro cardiomyocyte dysfunction.
Perinatal PM2.5 exposure resulted in in vivo systolic and diastolic dysfunction in 3-mo-old mice. To further evaluate potential cell-specific mechanisms, we performed functional analyses in isolated cardiomyocytes. Cardiomyocytes isolated from mice that were exposed to PM2.5 during perinatal development showed alterations in sarcomere function evident at the cellular level, as indicated by a reduction in %PS (Fig. 3, A and B). TPS90, TR90, +dl/dR, and −dl/dt were not different between groups (Fig. 3, C and D).
Fig. 3.

In vitro cardiomyocyte sarcomere function. Contractility was obtained in isolated cardiomyocytes from 3-mo-old FVB mice that were exposed to FA or PM2.5 during perinatal development. A: representative sarcomere shortening. B: percentage of peak shortening (%PS) normalized to baseline sarcomere length. C: +dl/dt. D: −dl/dR. E: representative sarcomere shortening following isoproterenol (ISO) challenge. F: %PS. G: +dl/dt. H: −dl/dt. n = 40–45 cardiomyocytes isolated from 5 mice per group that were taken from individual litters. *P < 0.05 was considered statistically significant.
In vitro β-adrenergic response is suppressed in PM2.5-exposed mice.
Sarcomere shortening was altered in response to β-adrenergic stimulation in cardiomyocytes isolated from 3-mo-old mice that were exposed to PM2.5 during fetal development and nursing (Fig. 3E). Absolute sarcomeric contraction was not different following β-adrenergic stimulation with isoproterenol, as indicated by %PS of cardiomyocytes isolated from 3-mo-old mice that were exposed to PM2.5 or FA during fetal development and nursing (Fig. 3F). However, in contrast to nonstimulated cardiomyocytes, duration of contraction was increased in response to β-adrenergic stimulation in 3-mo-old mice that were exposed to PM2.5 during perinatal development, as indicated by reduced +dl/dt and −dl/dt compared with FA-exposed mice (Fig. 3, G and H).
Perinatal exposure to PM2.5 results in delayed calcium reuptake.
Calcium transient amplitude (%PS FURA) was not different in 3-mo-old mice that were exposed to PM2.5 or FA during perinatal development (Fig. 4A). However, calcium reuptake was delayed in mice from the PM2.5 group, as indicated by an increase in τ duration compared with the FA group (Fig. 4B).
Fig. 4.
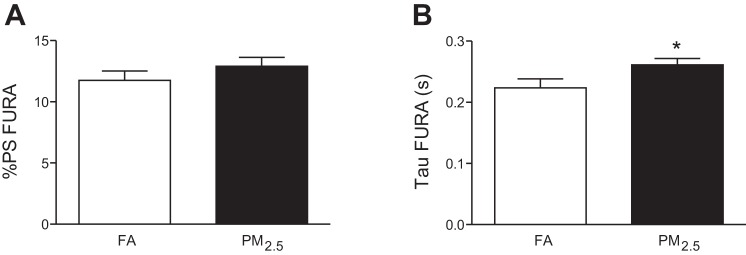
Calcium signaling in isolated cardiomyocytes from 3-mo-old mice that were exposed to FA or PM2.5 during perinatal development. A: fura-2 AM amplitude (%PS). B: calcium reuptake (τ) of n = 40–45 cardiomyocytes isolated out of 5 mice per group that were taken from individual litter. *P < 0.05 was considered statistically significant.
Perinatal PM2.5 exposure is associated with increased cardiac collagen deposition.
Morphometrical analyses of picro-sirius red-stained histological sections indicated an increase in collagen deposition in 3-mo-old mice hearts that were exposed to PM2.5 during perinatal development (Fig. 5). Quantitative real-time PCR analyses revealed no differences in inflammatory cytokines, chemokines, or fibrotic genes in 3-mo-old mice from either group (data not shown).
Fig. 5.

Collagen deposition from 3-mo-old mice (n = 3 mice from each group) that were exposed to FA (A) or PM2.5 during development (B). C: morphometric analyses of areas stained positive for picro-sirius red. *P < 0.05 was considered statistically significant.
DISCUSSION
In the current study, we found that exposure to environmentally relevant PM2.5 concentrations during early life can cause significant cardiovascular dysfunction at adulthood. Contractility defects in the perinatally exposed mice measured in isolated cardiomyocytes confirmed this observation and further established functional consequences on the calcium-handling system and contractile machinery of the heart, with impaired calcium reuptake and response to β-adrenergic stimulation. A profibrotic response was indicated by increased collagen deposition in the myocardium, but analyses at the molecular level did not indicate increased inflammatory or fibrotic markers.
This study is the first to show an incipient heart failure phenotype in mice exposed to PM2.5 during development both in vivo and in vitro. A recent study where mice were exposed to higher particulate concentrations of diesel exhaust (∼300 μg/m3) for 5 days a week until E17.5 also revealed increased collagen deposition, but a reduction in cardiovascular function was not observed in adult mice. However, this study revealed reduced blood pressure as measured using a tail cuff and an impaired response to transaortic constriction (45). Myocardial oxidative stress from animals maternally exposed to urban air pollution has also been shown (9), which could play a role the cardiovascular dysfunction observed in the adult mice in our study.
Cardiac inflammation resulting in matrix remodeling and fibrosis is a hallmark of various cardiac diseases in humans and experimental models (12, 13, 41). Inflammation during development may be sufficient to induce cardiovascular dysfunction later in life. In particular, rodent models investigating cardiac ischemia or pressure overload have reported a transient increase in inflammatory and fibrotic markers in response to the initial insult (12, 41). However, this insult results in a persistent collagen deposition, leading to cardiac fibrosis with sustained physiological consequences, including reduced contractility and relaxation.
The observed collagen deposition in our study is notably lower compared with other disease models. In a model of maternal inflammation induced by lipopolysaccharide injection, a cardiovascular phenotype was observed that was similar to what we found in mice exposed to both perinatal and in utero PM2.5 exposure (42, 43). Additionally, recent studies examining the placenta of pregnant mice exposed to PM2.5 have shown altered morphology, such as reduced maternal blood space area as well as recruitment of inflammatory cells to the placental vasculature (44). Neuroinflammation has also been observed in mice exposed to diesel exhaust during the prenatal period, evident by increased microglial activation and brain cytokine levels (4). Taken together, these studies suggest that exposure to PM2.5 causes maternal inflammation contributing to adulthood dysfunction in the resultant offspring; however, the mechanism(s) of how inflammation contributes to the development of cardiovascular dysfunction remain unclear.
Exposure to other contaminants during gestation has led to similar detrimental effects. In a study where exposure to polycyclic aromatic hydrocarbons (PAHs, a contaminant found in air pollution) was monitored in pregnant women, increased PAH exposure during the first trimester increased the risk for fetal growth ratio reduction (7). This was confirmed in controlled animal studies where exposure to PAH altered placental vasculature, increased apoptotic markers during gestation, and reduced survival of resultant adult mice (11). Prenatal exposure to environmental sidestream tobacco smoke has also been shown to increase atherogenesis, superoxide dismutase activity, and mitochondrial damage in the heart tissue of mice (48), as well as increase blood pressure reactivity in humans (8).
Another mechanism that may contribute to the adulthood consequences of developmental exposure to PM2.5 is the translocation of PM into the developing fetus. Although this has not been shown, the possibility that PM could translocate into the bloodstream of a pregnant mother through the lungs, and then enter the fetus through the placental vasculature is validated by several studies. In particular, radiolabeled particles can pass into the circulation from the lungs in both humans and rats, including iridium particles less than 100 nm (17, 28, 29). However, this research remains controversial, with conflicting research showing no translocation into the bloodstream when technicium-labeled 100-nm particles were inhaled following a single exposure (26). Regardless, PAHs and metals as components of PM can enter the bloodstream via inhalation and affect the developing fetus.
Lowered birth weights of mice exposed in utero to PM2.5 compared with FA is in concurrence with epidemiological studies linking higher PM exposure to decreases in birth weight (2, 19, 30, 35). This is confirmed in a similar study where mice were exposed to diesel exhaust during a portion of development (15). However, in another study where mice were exposed to high concentrations of diesel exhaust during gestation, body weight was not significantly reduced in 2-day-old pups (38). This disagreement could be due to the strain of mice used.
Further research is needed to identify a critical window in development in which exposure to ambient air pollution has the greatest effect. Research is also needed that will elucidate particular mechanisms that cause improper development or injury of the cardiovascular system during development. These studies will help to validate PM2.5 exposure as a unique and relevant model to evaluate the Barker hypothesis of the developmental programming of adult diseases.
GRANTS
This work was supported by funding provided in part by an American Heart Association Predoctoral Fellowship no. 16700011 to M. Gorr, the Bennett Memorial Scholarship from the College of Medicine at the Ohio State University to T. Nelin, and NIH grants no. R01ES019923 and R01NR012618 to L. E. Wold.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
Author contributions: M.W.G., M.V., T.N., and L.E.W. conception and design of research; M.W.G., M.V., T.N., D.J.Y., and L.E.W. performed experiments; M.W.G., M.V., T.N., D.J.Y., Q.S., and L.E.W. analyzed data; M.W.G., M.V., T.N., D.J.Y., Q.S., and L.E.W. interpreted results of experiments; M.W.G., M.V., and L.E.W. prepared figures; M.W.G., M.V., T.N., Q.S., and L.E.W. drafted manuscript; M.W.G., M.V., T.N., D.J.Y., and Q.S. edited and revised manuscript; M.W.G., M.V., T.N., D.J.Y., Q.S., and L.E.W. approved final version of manuscript.
REFERENCES
- 1.Barker DJ. Adult consequences of fetal growth restriction. Clin Obstet Gynecol 49: 270–283, 2006 [DOI] [PubMed] [Google Scholar]
- 2.Bell ML, Belanger K, Ebisu K, Gent JF, Lee HJ, Koutrakis P, Leaderer BP. Prenatal exposure to fine particulate matter and birth weight: variations by particulate constituents and sources. Epidemiology 21: 884–891, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bobak M. Outdoor air pollution, low birth weight, and prematurity. Environ Health Perspect 108: 173–176, 2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bolton JL, Smith SH, Huff NC, Gilmour MI, Foster WM, Auten RL, Bilbo SD. Prenatal air pollution exposure induces neuroinflammation and predisposes offspring to weight gain in adulthood in a sex-specific manner. FASEB J 26: 4743–4754, 2012 [DOI] [PubMed] [Google Scholar]
- 5.Brunekreef B, Janssen NA, de Hartog J, Harssema H, Knape M, van Vliet P. Air pollution from truck traffic and lung function in children living near motorways. Epidemiology 8: 298–303, 1997 [DOI] [PubMed] [Google Scholar]
- 6.Caiazzo F, Ashok A, Waitz IA, Yim SHL, Barrett SRH. Air pollution and early deaths in the United States. Part I: Quantifying the impact of major sectors in 2005. Atmos Environ 79: 198–208, 2013 [Google Scholar]
- 7.Choi H, Wang L, Lin X, Spengler JD, Perera FP. Fetal window of vulnerability to airborne polycyclic aromatic hydrocarbons on proportional intrauterine growth restriction. PloS One 7: e35464, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cohen G, Vella S, Jeffery H, Lagercrantz H, Katz-Salamon M. Cardiovascular stress hyperreactivity in babies of smokers and in babies born preterm. Circulation 118: 1848–1853, 2008 [DOI] [PubMed] [Google Scholar]
- 9.Damaceno-Rodrigues NR, Veras MM, Negri EM, Zanchi AC, Rhoden CR, Saldiva PH, Dolhnikoff M, Caldini EG. Effect of pre- and postnatal exposure to urban air pollution on myocardial lipid peroxidation levels in adult mice. Inhal Toxicol 21: 1129–1137, 2009 [DOI] [PubMed] [Google Scholar]
- 10.Dejmek J, Selevan SG, Benes I, Solansky I, Sram RJ. Fetal growth and maternal exposure to particulate matter during pregnancy. Environ Health Perspect 107: 475–480, 1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Detmar J, Rennie MY, Whiteley KJ, Qu D, Taniuchi Y, Shang X, Casper RF, Adamson SL, Sled JG, Jurisicova A. Fetal growth restriction triggered by polycyclic aromatic hydrocarbons is associated with altered placental vasculature and AhR-dependent changes in cell death. Am J Physiol Endocrinol Metab 295: E519–E530, 2008 [DOI] [PubMed] [Google Scholar]
- 12.Dewald O, Frangogiannis NG, Zoerlein M, Duerr GD, Klemm C, Knuefermann P, Taffet G, Michael LH, Crapo JD, Welz A, Entman ML. Development of murine ischemic cardiomyopathy is associated with a transient inflammatory reaction and depends on reactive oxygen species. Proc Natl Acad Sci USA 100: 2700–2705, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Duerr GD, Heinemann JC, Dunkel S, Zimmer A, Lutz B, Lerner R, Roell W, Mellert F, Probst C, Esmailzadeh B, Welz A, Dewald O. Myocardial hypertrophy is associated with inflammation and activation of endocannabinoid system in patients with aortic valve stenosis. Life Sci 92: 976–983, 2013 [DOI] [PubMed] [Google Scholar]
- 14.Gauderman WJ, McConnell R, Gilliland F, London S, Thomas D, Avol E, Vora H, Berhane K, Rappaport EB, Lurmann F, Margolis HG, Peters J. Association between air pollution and lung function growth in southern California children. Am J Respir Crit Care Med 162: 1383–1390, 2000 [DOI] [PubMed] [Google Scholar]
- 15.Hougaard KS, Jensen KA, Nordly P, Taxvig C, Vogel U, Saber AT, Wallin H. Effects of prenatal exposure to diesel exhaust particles on postnatal development, behavior, genotoxicity and inflammation in mice. Part Fibre Toxicol 5: 3, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Krewski D, Jerrett M, Burnett RT, Ma R, Hughes E, Shi Y, Turner MC, Pope CA, 3rd, Thurston G, Calle EE, Thun MJ, Beckerman B, DeLuca P, Finkelstein N, Ito K, Moore DK, Newbold KB, Ramsay T, Ross Z, Shin H, Tempalski B. Extended follow-up and spatial analysis of the American Cancer Society study linking particulate air pollution and mortality. Res Rep Health Eff Inst 140: 5–114; discussion 115–136, 2009 [PubMed] [Google Scholar]
- 17.Kreyling WG, Semmler M, Erbe F, Mayer P, Takenaka S, Schulz H, Oberdorster G, Ziesenis A. Translocation of ultrafine insoluble iridium particles from lung epithelium to extrapulmonary organs is size dependent but very low. J Toxicol Environ Health A 65: 1513–1530, 2002 [DOI] [PubMed] [Google Scholar]
- 18.Latzin P, Roosli M, Huss A, Kuehni CE, Frey U. Air pollution during pregnancy and lung function in newborns: a birth cohort study. Eur Respir J 33: 594–603, 2009 [DOI] [PubMed] [Google Scholar]
- 19.Lee BE, Ha EH, Park HS, Kim YJ, Hong YC, Kim H, Lee JT. Exposure to air pollution during different gestational phases contributes to risks of low birth weight. Hum Reprod 18: 638–643, 2003 [DOI] [PubMed] [Google Scholar]
- 20.Lepeule J, Laden F, Dockery D, Schwartz J. Chronic exposure to fine particles and mortality: an extended follow-up of the Harvard Six Cities study from 1974 to 2009. Environ Health Perspect 120: 965–970, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lim SS, Vos T, Flaxman AD, Danaei G, Shibuya K, Adair-Rohani H, Amann M, Anderson HR, Andrews KG, Aryee M, Atkinson C, Bacchus LJ, Bahalim AN, Balakrishnan K, Balmes J, Barker-Collo S, Baxter A, Bell ML, Blore JD, Blyth F, Bonner C, Borges G, Bourne R, Boussinesq M, Brauer M, Brooks P, Bruce NG, Brunekreef B, Bryan-Hancock C, Bucello C, Buchbinder R, Bull F, Burnett RT, Byers TE, Calabria B, Carapetis J, Carnahan E, Chafe Z, Charlson F, Chen H, Chen JS, Cheng AT, Child JC, Cohen A, Colson KE, Cowie BC, Darby S, Darling S, Davis A, Degenhardt L, Dentener F, Des Jarlais DC, Devries K, Dherani M, Ding EL, Dorsey ER, Driscoll T, Edmond K, Ali SE, Engell RE, Erwin PJ, Fahimi S, Falder G, Farzadfar F, Ferrari A, Finucane MM, Flaxman S, Fowkes FG, Freedman G, Freeman MK, Gakidou E, Ghosh S, Giovannucci E, Gmel G, Graham K, Grainger R, Grant B, Gunnell D, Gutierrez HR, Hall W, Hoek HW, Hogan A, Hosgood HD, 3rd, Hoy D, Hu H, Hubbell BJ, Hutchings SJ, Ibeanusi SE, Jacklyn GL, Jasrasaria R, Jonas JB, Kan H, Kanis JA, Kassebaum N, Kawakami N, Khang YH, Khatibzadeh S, Khoo JP, Kok C, Laden F, Lalloo R, Lan Q, Lathlean T, Leasher JL, Leigh J, Li Y, Lin JK, Lipshultz SE, London S, Lozano R, Lu Y, Mak J, Malekzadeh R, Mallinger L, Marcenes W, March L, Marks R, Martin R, McGale P, McGrath J, Mehta S, Mensah GA, Merriman TR, Micha R, Michaud C, Mishra V, Mohd Hanafiah K, Mokdad AA, Morawska L, Mozaffarian D, Murphy T, Naghavi M, Neal B, Nelson PK, Nolla JM, Norman R, Olives C, Omer SB, Orchard J, Osborne R, Ostro B, Page A, Pandey KD, Parry CD, Passmore E, Patra J, Pearce N, Pelizzari PM, Petzold M, Phillips MR, Pope D, Pope CA, 3rd, Powles J, Rao M, Razavi H, Rehfuess EA, Rehm JT, Ritz B, Rivara FP, Roberts T, Robinson C, Rodriguez-Portales JA, Romieu I, Room R, Rosenfeld LC, Roy A, Rushton L, Salomon JA, Sampson U, Sanchez-Riera L, Sanman E, Sapkota A, Seedat S, Shi P, Shield K, Shivakoti R, Singh GM, Sleet DA, Smith E, Smith KR, Stapelberg NJ, Steenland K, Stockl H, Stovner LJ, Straif K, Straney L, Thurston GD, Tran JH, Van Dingenen R, van Donkelaar A, Veerman JL, Vijayakumar L, Weintraub R, Weissman MM, White RA, Whiteford H, Wiersma ST, Wilkinson JD, Williams HC, Williams W, Wilson N, Woolf AD, Yip P, Zielinski JM, Lopez AD, Murray CJ, Ezzati M, AlMazroa MA, Memish ZA. A comparative risk assessment of burden of disease and injury attributable to 67 risk factors and risk factor clusters in 21 regions, 1990–2010: a systematic analysis for the Global Burden of Disease Study 2010. Lancet 380: 2224–2260, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lipsett MJ, Ostro BD, Reynolds P, Goldberg D, Hertz A, Jerrett M, Smith DF, Garcia C, Chang ET, Bernstein L. Long-term exposure to air pollution and cardiorespiratory disease in the California teachers study cohort. Am J Respir Crit Care Med 184: 828–835, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Maroziene L, Grazuleviciene R. Maternal exposure to low-level air pollution and pregnancy outcomes: a population-based study. Environ Health 1: 6, 2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Miller KA, Siscovick DS, Sheppard L, Shepherd K, Sullivan JH, Anderson GL, Kaufman JD. Long-term exposure to air pollution and incidence of cardiovascular events in women. N Engl J Med 356: 447–458, 2007 [DOI] [PubMed] [Google Scholar]
- 25.Mohallem SV, de Araujo Lobo DJ, Pesquero CR, Assuncao JV, de Andre PA, Saldiva PH, Dolhnikoff M. Decreased fertility in mice exposed to environmental air pollution in the city of Sao Paulo. Environ Res 98: 196–202, 2005 [DOI] [PubMed] [Google Scholar]
- 26.Moller W, Felten K, Sommerer K, Scheuch G, Meyer G, Meyer P, Haussinger K, Kreyling WG. Deposition, retention, and translocation of ultrafine particles from the central airways and lung periphery. Am J Respir Crit Care Med 177: 426–432, 2008 [DOI] [PubMed] [Google Scholar]
- 27.Monreal G, Nicholson LM, Han B, Joshi MS, Phillips AB, Wold LE, Bauer JA, Gerhardt MA. Cytoskeletal remodeling of desmin is a more accurate measure of cardiac dysfunction than fibrosis or myocyte hypertrophy. Life Sci 83: 786–794, 2008 [DOI] [PubMed] [Google Scholar]
- 28.Nemmar A, Hoet PH, Vanquickenborne B, Dinsdale D, Thomeer M, Hoylaerts MF, Vanbilloen H, Mortelmans L, Nemery B. Passage of inhaled particles into the blood circulation in humans. Circulation 105: 411–414, 2002 [DOI] [PubMed] [Google Scholar]
- 29.Oberdorster G, Sharp Z, Atudorei V, Elder A, Gelein R, Lunts A, Kreyling W, Cox C. Extrapulmonary translocation of ultrafine carbon particles following whole-body inhalation exposure of rats. J Toxicol Environ Health A 65: 1531–1543, 2002 [DOI] [PubMed] [Google Scholar]
- 30.Parker JD, Woodruff TJ, Basu R, Schoendorf KC. Air pollution and birth weight among term infants in California. Pediatrics 115: 121–128, 2005 [DOI] [PubMed] [Google Scholar]
- 31.Pope CA, 3rd, Burnett RT, Thun MJ, Calle EE, Krewski D, Ito K, Thurston GD. Lung cancer, cardiopulmonary mortality, and long-term exposure to fine particulate air pollution. JAMA 287: 1132–1141, 2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Reliene R, Hlavacova A, Mahadevan B, Baird WM, Schiestl RH. Diesel exhaust particles cause increased levels of DNA deletions after transplacental exposure in mice. Mutat Res 570: 245–252, 2005 [DOI] [PubMed] [Google Scholar]
- 33.Ren J, Roughead ZK, Wold LE, Norby FL, Rakoczy S, Mabey RL, Brown-Borg HM. Increases in insulin-like growth factor-1 level and peroxidative damage after gestational ethanol exposure in rats. Pharmacol Res 47: 341–347, 2003 [DOI] [PubMed] [Google Scholar]
- 34.Ritz B, Yu F, Chapa G, Fruin S. Effect of air pollution on preterm birth among children born in Southern California between 1989 and 1993. Epidemiology 11: 502–511, 2000 [DOI] [PubMed] [Google Scholar]
- 35.Rudra CB, Williams MA, Sheppard L, Koenig JQ, Schiff MA. Ambient carbon monoxide and fine particulate matter in relation to preeclampsia and preterm delivery in western Washington State. Environ Health Perspect 119: 886–892, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Salam MT, Millstein J, Li YF, Lurmann FW, Margolis HG, Gilliland FD. Birth outcomes and prenatal exposure to ozone, carbon monoxide, and particulate matter: results from the Children's Health Study. Environ Health Perspect 113: 1638–1644, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Suh YJ, Ha EH, Park H, Kim YJ, Kim H, Hong YC. GSTM1 polymorphism along with PM10 exposure contributes to the risk of preterm delivery. Mutat Res 656: 62–67, 2008 [DOI] [PubMed] [Google Scholar]
- 38.Tsukue N, Tsubone H, Suzuki AK. Diesel exhaust affects the abnormal delivery in pregnant mice and the growth of their young. Inhal Toxicol 14: 635–651, 2002 [DOI] [PubMed] [Google Scholar]
- 39.Tsukue N, Watanabe M, Kumamoto T, Takano H, Takeda K. Perinatal exposure to diesel exhaust affects gene expression in mouse cerebrum. Arch Toxicol 83: 985–1000, 2009 [DOI] [PubMed] [Google Scholar]
- 40.Umezawa M, Sakata C, Tabata M, Tanaka N, Kudo S, Takeda K, Ihara T, Sugamata M. Diesel exhaust exposure enhances the persistence of endometriosis model in rats. J Health Sci 54: 503–507, 2008 [Google Scholar]
- 41.Velten M, Duerr GD, Pessies T, Schild J, Lohner R, Mersmann J, Dewald O, Zacharowski K, Klaschik S, Hilbert T, Hoeft A, Baumgarten G, Meyer R, Boehm O, Knuefermann P. Priming with synthetic oligonucleotides attenuates pressure overload-induced inflammation and cardiac hypertrophy in mice. Cardiovasc Res 96: 422–432, 2012 [DOI] [PubMed] [Google Scholar]
- 42.Velten M, Gorr MW, Youtz DJ, Velten C, Rogers LK, Wold LE. Adverse perinatal environment contributes to altered cardiac development and function. Am J Physiol Heart Circ Physiol 306: H1334–H1340, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Velten M, Hutchinson KR, Gorr MW, Wold LE, Lucchesi PA, Rogers LK. Systemic maternal inflammation and neonatal hyperoxia induces remodeling and left ventricular dysfunction in mice. PloS One 6: e24544, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Veras MM, Damaceno-Rodrigues NR, Caldini EG, Maciel Ribeiro AA, Mayhew TM, Saldiva PH, Dolhnikoff M. Particulate urban air pollution affects the functional morphology of mouse placenta. Biol Reprod 79: 578–584, 2008 [DOI] [PubMed] [Google Scholar]
- 45.Weldy CS, Liu Y, Liggitt HD, Chin MT. In utero exposure to diesel exhaust air pollution promotes adverse intrauterine conditions, resulting in weight gain, altered blood pressure, and increased susceptibility to heart failure in adult mice. PloS One 9: e88582, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wold LE, Muralikrishnan D, Albano CB, Norby FL, Ebadi M, Ren J. Insulin-like growth factor I (IGF-1) supplementation prevents diabetes-induced alterations in coenzymes Q9 and Q10. Acta Diabetol 40: 85–90, 2003 [DOI] [PubMed] [Google Scholar]
- 47.Wold LE, Ying Z, Hutchinson KR, Velten M, Gorr MW, Velten C, Youtz DJ, Wang A, Lucchesi PA, Sun Q, Rajagopalan S. Cardiovascular remodeling in response to long-term exposure to fine particulate matter air pollution. Circ Heart Fail 5: 452–461, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yang Z, Knight CA, Mamerow MM, Vickers K, Penn A, Postlethwait EM, Ballinger SW. Prenatal environmental tobacco smoke exposure promotes adult atherogenesis and mitochondrial damage in apolipoprotein E-/- mice fed a chow diet. Circulation 110: 3715–3720, 2004 [DOI] [PubMed] [Google Scholar]
- 49.Yokota S, Mizuo K, Moriya N, Oshio S, Sugawara I, Takeda K. Effect of prenatal exposure to diesel exhaust on dopaminergic system in mice. Neurosci Lett 449: 38–41, 2009 [DOI] [PubMed] [Google Scholar]