

Abstract
Recently, we described a peptide-modified AAV2 vector (AAV-GMN) containing a capsid-displayed peptide that directs in vivo brain vascular targeting and transduction when delivered intravenously. In this study, we sought to identify the receptor that mediates transduction by AAV-GMN. We found that AAV-GMN, but not AAV2, readily transduces the murine brain endothelial cell line bEnd.3, a result that mirrors previously observed in vivo transduction profiles of brain vasculature. Studies in vitro revealed that the glycosaminoglycan, chondroitin sulfate C, acts as the primary receptor for AAV-GMN. Unlike AAV2, chondroitin sulfate expression is required for cell transduction by AAV-GMN, and soluble chondroitin sulfate C can robustly inhibit AAV-GMN transduction of brain endothelial cells. Interestingly, AAV-GMN retains heparin-binding properties, though in contrast to AAV2, it poorly transduces cells that express heparan sulfate but not chondroitin sulfate, indicating that the peptide insertion negatively impacts heparan-mediated transduction. Lastly, when delivered directly, this modified virus can transduce multiple brain regions, indicating that the potential of AAV-GMN as a therapeutic gene delivery vector for central nervous system disorders is not restricted to brain vascular endothelium.
Keywords: AAV, brain vasculature, capsid engineering, chondroitin sulfate, endothelium, phage display, receptor
Introduction
Adeno-associated virus (AAV) has been used extensively as a vector for gene transfer to a wide array of tissues in vivo.1 Of the many serotypes that exist, only a handful are used experimentally and each displays a unique profile of tissue tropism, dictated by the receptor(s) utilized for cell binding and transduction.1 Several AAV serotypes are efficient at gene transfer to the central nervous system (CNS), with several (e.g., AAV9, rAAVrh.8, rAAVrh.10) showing robust brain delivery after intravenous injection.2,3 One approach to expand the tropism or re-target AAV to specific tissues of therapeutic interest has been to modify the capsid surface by introducing peptides that bind to tissue-specific or tissue-enriched cell surface receptors.4 These so-called peptide-modified AAVs have the capacity to deliver genes to cells that are not normally transduced by the unmodified parent AAV serotype.5,6
Recently, our lab generated several peptide-modified AAV serotype 2 (AAV2) vectors that can home to and transduce brain vascular endothelium when delivered systemically.7 This was achieved by modifying the AAV2 capsid with peptides, discovered through in vivo phage display biopanning, which have binding affinity for receptors on the luminal surface of brain vascular endothelial cells. One of the engineered vectors, AAV-GMN, displays the heptamer peptide GMNAFRA.7 This peptide was identified from biopanning in a mouse model of the lysosomal storage disease, late infantile neuronal ceroid lipofuscinosis (LINCL), which is a fatal, childhood-onset, neurodegenerative disorder caused by the loss of expression of the lysosomal enzyme tri-peptidyl peptidase I (TPP1).8,9 When delivered intravenously, AAV-GMN encoding TPP1 robustly transduces brain vascular endothelium, resulting in expression and secretion of TPP1 into the brain and reversal of the disease phenotypes in the mouse model of LINCL.7
In order to evaluate the utility of the AAV-GMN vector in large animal models and to ultimately translate its use to the clinic for treatment of LINCL patients, an understanding of the molecular mechanism of brain vascular tropism is required. Specifically, the receptor or receptors that mediate brain endothelial cell binding and transduction, via the GMN peptide, must be elucidated. Only a small number of receptors for biopanning-derived peptides have been identified, and typically they are a single membrane or extracellular matrix-associated protein.10,11,12 In this study, we describe experiments revealing that the AAV-GMN vector, unlike AAV2, utilizes chondroitin sulfate as its primary cellular receptor. Interestingly, we discovered that while both AAV-GMN and AAV2 can bind to heparin, only AAV2 efficiently utilizes heparan sulfate proteoglycans as a functional receptor for transduction. This finding provides important insight into the transduction biology of AAV2 and indicates that targeting chondroitin sulfate, but not heparan sulfate, is an effective strategy for in vivo gene delivery to brain vascular endothelium. Lastly, we also show that AAV-GMN can transduce regions throughout the CNS, warranting further investigation of this engineered vector for therapeutic gene delivery in neurological disorders.
Results
AAV-GMN but not AAV2 transduces brain endothelial cells in vitro using a proteinaceous receptor
In our previous studies AAV-GMN, but not AAV2, transduced brain vasculature of CLN2−/− mice following intravenous delivery.7 To test whether this transduction profile could be recapitulated in vitro, we evaluated transduction of immortalized brain endothelial cell lines. We discovered that the mouse brain endothelial cell line, bEnd.3, was readily transduced by AAV-GMN but not AAV2 or a control peptide-modified virus, AAV-PPS, which homes to brain vascular endothelium in wild type mice7,13,14 (Figure 1a,b) (see Supplementary Figure S1 for confirmation of AAV2 bioactivity). These results, which mirrored the in vivo findings,7 suggest that bEnd.3 cells express the receptor that mediates AAV-GMN transduction of brain endothelial cells and that the transduction is conferred by the presence of the GMN peptide. To determine whether the AAV-GMN receptor is proteinaceous or contains a carbohydrate structure, we modified the surface of bEnd.3 cells with different enzymes before assaying transduction. Pre-treatment with trypsin to remove cell-surface proteins reduced transduction >80% (Figure 1b). Treatment with Endo H, an enzyme that removes core mannose structures from N-linked glycans, did not affect transduction (Figure 1b). In contrast, removal of N-linked glycans by treatment with PNGase F, increased transduction by ~70% (Figure 1b). These results support the hypothesis that the receptor is proteinaceous and is not an N-linked glycan; in fact, N-linked carbohydrate groups may inhibit transduction.
Figure 1.
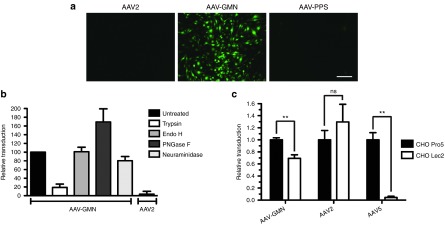
The GMN peptide modification confers unique in vitro transduction profiles compared to the parent vector, AAV2. (a) bEnd.3 mouse brain endothelial cells were transduced with AAV2, AAV-GMN, or control AAV2-PPS encoding eGFP reporter genes at an MOI of 105 vg/cell. 48 hours post-transduction, eGFP expression was measured by fluorescence microscopy. Scale bar is 250 μm (b) Prior to transduction, bEnd.3 cells were treated with the indicated enzymes to modify cell surface receptors. 48 hours after transduction, relative transduction was measured by fluorometric quantitation of eGFP expression. (c) CHO Pro5 and sialic acid deficient Lec2 cells were transduced with AAV-GMN, AAV2, or AAV5. 24 hours later, transduction was measured by eGFP fluorometry. In both graphs, transduction is presented as relative fluorescence units (RFU) of eGFP signal per μg of protein extract. Data shown are derived from ≥3 independent experiments. Error bars are mean ± SD. **P value <0.01; ns, not significant.
Sialic acid moieties associated with cell-surface carbohydrate chain termini act as receptors for several AAV serotypes and could potentially contribute to transduction by AAV-GMN.15,16 Enzymatic removal of sialic acid from bEnd.3 cells using neuraminidase yielded an ~25% decrease in transduction (Figure 1b). To further investigate the dependence of sialic acid for AAV-GMN transduction, we compared transduction of the mutant CHO cell line Lec2, which is deficient in sialic acid addition to N and O-linked carbohydrates, to the wild-type parent cell line, CHO Pro5.17 Similar to the effect of neuraminidase treatment on bEnd.3 cells, AAV-GMN transduced Lec2 cells significantly lower (~30% less) than Pro5 cells (Figure 1c). In contrast, AAV2 transduced Lec2 cells greater than Pro5 cells, though the difference was not statistically significant (Figure 1c). As expected, AAV5, which utilizes sialic acid as its primary receptor, transduced Lec2 cells significantly lower (~95% less) than Pro5 cells (Figure 1c). These findings indicate that sialic acid moieties can mediate, to a minor extent, AAV-GMN transduction.
AAV-GMN transduces a broad range of cultured cell lines
Our observation that AAV-GMN can transduce CHO cells indicated that expression of the putative receptor required for transduction is not restricted to brain endothelial cells, as evidenced by our earlier findings.7 To further evaluate the tropism of AAV-GMN, we measured transduction of several genetically and phenotypically diverse cell lines. All cells lines tested, including an additional immortalized mouse brain endothelial cell (MBEC) line generated in our lab,18 mouse neuronal-like cells (N2a), as well as human fibroblasts and tumor cell lines (HeLa, PC-3), were readily transduced by AAV-GMN (Figure 2a). This broad tropism indicates that the receptor is expressed across multiple tissue types, at least when cells are grown in vitro.
Figure 2.

AAV-GMN has broad tissue tropism and retains heparin-binding activity. (a) The indicated cell lines were transduced with AAV-GMN encoding an eGFP reporter at an MOI of 105 and 24–48 hours later, transduction was assessed by eGFP fluorescence. Scale bars are 250 μm (PC-3 scale bar corresponds to all images except fibroblasts).(b) AAV2, AAV-GMN, or AAV5 were incubated with heparin-agarose and unbound flow-through (FT) and wash fractions were retained. Bound virus was eluted with high salt buffer and all fractions were analyzed by slot blot and immunodetection with anti-capsid antibody.
The GMN peptide modification does not ablate heparin-binding activity
The primary cellular receptor for AAV2 is heparan sulfate.19 Insertion of targeting peptides into the heparan-binding loop of AAV2 often induces a disruption of the local capsid structure and ablation of heparan-binding activity.4,5 However, in some instances where the inserted peptide is composed of non-bulky amino acids and/or contains arginine residues, heparan binding is maintained.20 As a result, these peptide-modified vectors can bind to both heparan sulfate as well as the inserted peptide's cognate receptor. Systemic delivery of peptide-modified AAV2 that lacks heparan-binding activity usually results in de-targeting of AAV2 to the liver and spleen.21,22 In contrast, modified AAV2 vectors that retain heparan binding typically do not display this de-targeting phenotype.20 In our in vivo studies, systemic delivery of AAV-GMN resulted in biodistribution profiles that were similar to AAV2 except for the brain vascular targeting; thus, de-targeting to liver, spleen, and other organs was not observed.7 This was in stark contrast to other, heparan binding-deficient, peptide-modified AAV2 vectors that we evaluated, which showed significant reductions in targeting to liver, spleen, and lung.7 Collectively, these in vivo results and the broad in vitro tropism of AAV-GMN suggested that this vector retains heparan-binding activity. To test this, we performed a solid-phase binding assay using a column composed of agarose resin conjugated to heparin, a chemical analogue of heparan. AAV5, which does not bind heparan/heparin, was only detected in the flow-through and wash fractions (Figure 2b). As expected, AAV2 bound to the heparin column and was present in the elution (Figure 2b). Similar to AAV2, AAV-GMN also bound and was eluted from the column (Figure 2b). This result demonstrates that AAV-GMN maintains heparin-binding activity despite the presence of the GMN peptide within the heparan-binding loop.
Unexpectedly, we observed in multiple experiments that the input AAV-GMN signal was lower compared to the elution fraction (Figure 2b, middle row). While it is unclear why this discrepancy occurred, it might be that exposure of the virus to high salt concentrations during the elution either promotes subsequent adsorption to the membrane and/or epitope exposure prior to antibody detection.
AAV-GMN transduction is blocked by heparin and chondroitin sulfate C
Given our finding that AAV-GMN can interact with heparin, we tested the effect of heparin binding on transduction of bEnd.3 cells. Pre-incubating AAV-GMN with varying concentrations of heparin prior to transduction resulted in a dose-dependent increase in inhibition of transduction (Figure 3a). Greater than 95% of transduction was blocked by heparin at concentrations of 10 μg/ml or higher (Figure 3a). In previous studies it was shown that, unlike heparin, other GAGs such as chondroitin sulfate do not block AAV2 transduction or cell-surface binding.19 Interestingly, we found that chondroitin sulfate C was also a potent inhibitor of AAV-GMN transduction (Figure 3a). Chondroitin sulfate A displayed some blocking activity, though it was significantly lower than with chondroitin sulfate C. Furthermore, this commercial chondroitin sulfate A preparation is not pure, and contains 30–40% chondroitin sulfate C (as opposed to the chondroitin sulfate C preparation, which is >90% pure). Therefore, the inhibition resulting from the chondroitin sulfate A may actually be due to the contaminating chondroitin sulfate C. We also tested dermatan sulfate, which only inhibited transduction 20–30% (Figure 3a).
Figure 3.
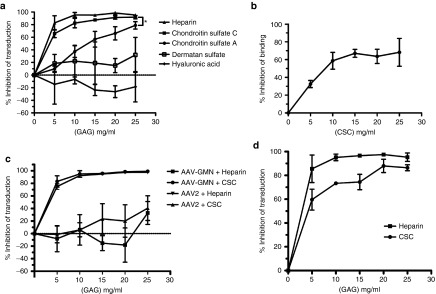
Heparin and chondroitin sulfate C (CSC) potently block AAV-GMN transduction and binding. (a,c,d) AAV-GMN or (c) AAV2 encoding an eGFP reporter was pre-incubated alone or with varying concentrations of the indicated GAGs for one hour prior to transduction of (a) bEnd.3, (c) N2a, or (d) MBEC cells. 48 hours later, transduction was measured by fluorometric quantitation of eGFP expression. Data are represented as inhibition of transduction (relative to the maximum RFU/μg of protein extract) relative to cells transduced with AAV-GMN alone. (b) AAV-GMN was pre-incubated alone or with varying concentrations of CSC on ice for one hour before incubation with ice-cold bEnd.3 cells for an additional hour to allow surface binding. After washing away unbound virus, bound AAV-GMN was assayed by quantitative real-time PCR measurements of viral genomes. Data are represented as inhibition of binding (virion genomes/ml of extracted DNA) relative to the binding by AAV-GMN alone. Data shown in all figures are derived from ≥3 independent experiments. Error bars are mean ± SD. *P value <0.05.
Lastly, we tested hyaluronic acid, which is the only non-sulfated GAG. It did not inhibit transduction; in contrast, it enhanced transduction at the higher doses (Figure 3a). In order to evaluate whether chondroitin sulfate C blocks transduction by preventing binding of AAV-GMN to the cell surface, we measured virus binding after chondroitin pre-incubation. Similar to the effect on transduction, chondroitin sulfate C pre-incubation caused a dose-dependent increase in blocking of surface binding (Figure 3b). However, at the highest dose, blocking of binding was ~68%, which is ~20% less compared to blocking of transduction at the same concentration (Figure 3b versus 3a). This indicates that some of the AAV-GMN virions bind to bEnd.3 cells in manner not mediated by chondroitin sulfate C, but that this binding does not necessarily lead to productive transduction. A similar discrepancy between blocking of transduction and blocking of binding was observed when AAV2 transduction of HeLa cells was inhibited with heparin.19
It is possible that the observed chondroitin sulfate C blocking activity is specific to bEnd.3 cells. To test whether this finding could be extended to other cell lines, we evaluated blocking of AAV-GMN transduction in MBEC and N2a cells. In MBEC, both heparin and chondroitin sulfate C blocked transduction in a manner comparable to in bEnd.3 cells (Figure 3d). Chondroitin sulfate C also blocked AAV-GMN transduction of N2a cells (Figure 3c). However, in contrast to the results with bEnd.3 and MBEC cells, heparin did not block transduction by AAV-GMN. The inverse was true for AAV2; as expected, heparin blocked transduction whereas chondroitin sulfate C showed minimal blocking (Figure 3c). The mechanism underlying differential blocking by heparin might be related to cell-type specific differences in the structure or heterogeneity of chondroitin sulfate proteoglycans that yield a distinct binding interaction with N2a-expressed chondroitin sulfate that cannot be readily disrupted by heparin.
Chondroitin sulfate expression is required for AAV-GMN transduction
Our finding that both soluble heparin and chondroitin sulfate can block transduction of bEnd.3 cells suggested that either of these GAGs could potentially act as a cell surface receptor to mediate AAV-GMN transduction. To determine whether the expression of these GAGs is required for transduction, we utilized CHO cell lines that are deficient in the biosynthesis of specific GAGs (Figure 4a). The mutant cell line CHO pgsA-745 lacks cell surface expression of both heparan and chondroitin sulfate.23,24 As expected, AAV2 transduced these cells at a very low level (~18%) relative to the parent cell line, K1, which displays normal GAG expression (Figure 4b). Similarly, transduction of pgsA-745 by AAV-GMN was also very low (~24%) relative to K1 cells (Figure 4b). These results suggest that heparan and/or chondroitin sulfate are required for AAV-GMN transduction. Compared to K1 cells, AAV2 also showed little transduction of CHO pgsD-677 mutant cells, which only lack expression of cell surface heparan sulfate23 (Figure 4b). In contrast, relative to K1 cells AAV-GMN transduced pgsD-677 cells significantly higher than AAV2 (~82% versus ~21% of K1) (Figure 4b). Although transduction of pgsD-677 cells is not 100% of K1 as measured in our assay (where transduction = RFU/μg), the fluorescence per cell is actually higher in pgsD-677 cells (Figure 4c). This discrepancy in transduction measurements is likely due to the fact that, unlike the other mutant lines, pgsD-677 cells are larger than K1 cells and thus contain more protein per cell (data not shown), which negatively biases the transduction measurement (Figure 4c). Regardless, these findings indicate that heparan sulfate expression is required for transduction by AAV2 but not AAV-GMN, and suggest that chondroitin sulfate expression is sufficient for AAV-GMN transduction. Lastly, we compared transduction of CHO K1 cells and a mutant derivative ldlD, which, under the culture conditions used, is deficient in the expression of chondroitin sulfate but not heparan sulfate24,25. AAV2 transduced these cells to near normal levels (~89% of K1), which is consistent with previous results indicating that heparan and not chondroitin sulfate is its primary receptor19 (Figure 4b). Interestingly, AAV-GMN transduced ldlD cells at only ~24% relative to K1, which was significantly lower compared to AAV2 and a reduction equivalent to the transduction of pgsA-745 cells, which lack both heparan and chondroitin sulfate (Figure 4b). This result suggests that heparan sulfate expression is not sufficient for AAV-GMN transduction. To further validate the dependence of chondroitin sulfate for AAV-GMN transduction, we treated pgsD-677 cells with chondroitinase ABC, an enzyme that can cleave cell surface chondroitin sulfate, prior to transduction. Relative to untreated cells pgsD-677 cells, AAV-GMN transduced chondroitinase treated cells ~62% less (Figure 4d). In order to test if the inability of AAV-GMN transduction of ldlD cells is due to impaired binding and/or a failure to undergo endocytosis, we measured virus binding versus internalization. Unexpectedly, in contrast to the difference in transduction, comparable amounts of AAV2 and AAV-GMN both bound and internalized (30.6% versus 38.%) into ldlD cells (Figure 4e).
Figure 4.
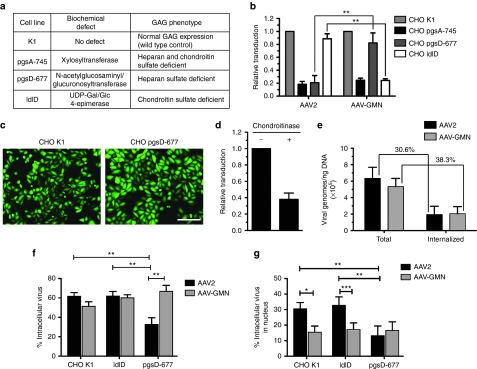
AAV-GMN transduction requires chondroitin sulfate expression. (a) Wild type and mutant CHO cell lines used for transduction experiments. The specific GAG biosynthesis phenotypes and underlying biochemical defects are listed. (b) Each CHO cell line was transduced with either AAV2 or AAV-GMN encoding an eGFP reporter, and transduction was measured by eGFP fluorometry 24 hours later. Data are presented as relative to transduction (RFU/μg of protein extract) of the wild type parent CHO K1 cell line. (c) Representative fluorescence microscopy image of CHO K1 and pgsD-677 cells transduced with AAV-GMN showing that pgsD-677 cells are larger and display greater eGFP fluorescence compared to the transduced K1 cells. (d) CHO pgsD-677 cells were treated with chondroitinase ABC prior to transduction with AAV-GMN. Transduction was measured by eGFP fluorometry 24 hours after transduction and results are shown as relative to mock-treated cells. (e) Comparison of the amount of AAV2 and AAV-GMN that binds to the cell surface and internalizes when incubated with ldlD cells. Virus was incubated for 1 hour on ice-cold cells, washed, and then incubated at 37 °C for 1 hour. Internalized virus was measured in cells treated with trypsin to remove extracellular, bound virus whereas bound and internalized (Total) virus was measured using cells that were not trypsin treated. The amount of recovered virus was measured by quantitative PCR for viral genomes. (f,g) AAV sub-cellular localization. The indicated CHO cells were transduced for 4 hours with fluorescently labeled AAV2 and AAV-GMN, and the percent of internalized and nuclear-localized AAV relative to total AAV (surface localized + internalized) was quantified. AAV2 entry is dependent on heparin expression, whereas AAV-GMN enters all cell types equivalently. In all figures, data shown are derived from ≥3 independent experiments. For b error bars are mean ± SD. **P value <0.01. For f and g error bars are mean ± SEM; *P < 0.05, **P < 0.01, and ***P < 0.001 by Mann–Whitney U–test.
Although total internalized virus was comparable in ldlD cells, it is possible that differences in nuclear entry and/or subcellular localization between AAV2 and AAV-GMN can explain their distinct transduction profiles. To investigate potential differences in intracellular trafficking, we tracked fluorescently labeled AAV by confocal microscopy after four hours of transduction of CHO cells (Figure 4f,g). As expected, the percentage of both intracellular and nuclear-localized AAV2 was similar in K1 and ldlD cells, but significantly lower in pgsD-677 cells, which lack heparan sulfate expression (Figure 4f,g). In contrast, the amount of intracellular and nuclear-localized AAV-GMN was equivalent across all three cells lines (Figure 4f,g, and Supplementary Figure S3). This finding indicates that the lower relative transduction of ldlD cells by AAV-GMN is not due to inefficient entry into either the cell or nucleus.
Collectively, these results indicate that chondroitin sulfate expression is required for AAV-GMN transduction and despite the fact that AAV-GMN can bind heparin, heparan sulfate expression alone cannot compensate for chondroitin sulfate, at least in CHO ldlD cells.
CLN2 null mice do not aberrantly express GAGs or sialic acid in brain tissue
A hallmark of lysosomal storage disorders is the accumulation of protein and lipids in response to loss of expression of specific lysosomal enzymes.26 An abnormal accumulation of GAGs, including heparan and chondroitin sulfate, is a well-known phenotype of the LSD subtype, mucopolysaccharidoses, which are caused by deficiencies in enzymes that metabolize GAGs.27 The in vivo substrates of TPP-1 are not well defined so it is unknown whether, either directly or indirectly, loss of this enzyme could result in the accumulation of GAGs. To test the hypothesis that an accumulation of GAGs in CLN2 null mouse brain pre-disposed the selection of the chondroitin sulfate-binding GMN-expressing phage, we measured total sulfated GAG content in whole brain and brain vasculature preps. The total level of GAGs was significantly higher in brain vasculature compared to whole brain (Figure 5a). However, we found that the level of GAGs in CLN2 heterozygote mouse brain was not significantly different than CLN2 knockout mice, though the knockout mice did trend towards having a higher GAG concentration (Figure 5a). Similarly, there was no discernable difference in GAG content in brain vasculature between CLN2 heterozygote and knockout mice (Figure 5a).
Figure 5.

Total sulfated GAG content and chondroitin sulfate expression are not significantly different in CLN2 knockout versus normal brain tissue. (a) GAG extracts from either whole brain or brain vasculature were prepared from CLN2 knockout and control heterozygote mice and GAG concentrations were measured using Farndale reagent. As an assay positive control, KO tissue was spiked with 50 μg/ml chondroitin sulfate C before processing (Spiked KO Brain). N = 3 for all conditions except the Spiked KO (N = 1); error bars are mean ± SEM. *P value ≤0.05. (b) Tissue sections from forebrain of age-matched CLN2 knockout and control heterozygote mouse brain immunostained for chondroitin sulfate. Scale bar is 250 μm.
It is possible that the total content of all sulfated GAGs could be the same in both mouse genotypes, but that the relative proportion of distinct GAGs could change in the knockout mouse. To evaluate if there is elevated chondroitin sulfate in the knockout mouse relative to the heterozygote, we performed immunohistochemical staining of brain tissue sections. However, in contrast to other LSD mouse models such as for mucopolysaccharidoses type VII (MPSVII),28 there was no apparent difference in chondroitin sulfate staining between knockout and heterozygous mice (Figure 5b).
In previous work, we unexpectedly discovered that sialic acid levels were elevated in brain tissue in a mouse model of MPSVII.29 Given this observation, and that sialic acid partially mediates transduction by AAV-GMN, we performed lectin staining to assess the level of sialic acid in CLN2 heterozygote and knockout brain tissue. Unlike the MPSVII model, however, there was no qualitative difference in the level of sialic acid staining between normal and CLN2 knockout mouse brain (Supplementary Figure S2). However, there could still be a subtle increase in sialic acid in CLN2 knockout tissue, but a more sensitive assay would be required to test this.
AAV-GMN transduces multiple brain regions when delivered directly
Chondroitin sulfate is the most abundant GAG expressed in the body,30 and a subset of chondroitin sulfate proteoglycans including neurocan, brevican, and versican V2 are exclusively expressed or highly enriched in the brain (for review of brain GAG expression see ref. 31). Although AAV-GMN was originally engineered for intravenous delivery for brain vascular transduction, we evaluated its ability to transduce cells when delivered directly into different brain regions. AAV-GMN encoding an eGFP reporter was delivered by stereotaxic injection into mice deep cerebellar nuclei (DCN), cortex, striatum, and thalamus, or into the lateral ventricle. Three weeks post-injection into the DCN, AAV-GMN displayed robust and widespread transduction throughout the cerebellar cortex, including the Purkinje cell layer (Figure 6a,b). Additionally, transduction was evident throughout the brain stem (Figure 6a). AAV-GMN also displayed prominent transduction when delivered to the cortex, thalamus, and striatum, including portions of the descending nigrostriatal track (Figure 6c–f). AAV serotypes that can transduce ependymal cells, which line brain ventricles, are of therapeutic value because delivered genes can enable secretion of therapeutic proteins into cerebral spinal fluid, which promotes bioavailability throughout the CNS.28 Ependymal cells of the lateral ventricle were clearly transduced, as compared to non-injected ventricles (Figure 6g,h). These findings reveal that AAV-GMN's utility for central nervous system gene transfer is not limited to brain vascular endothelium.
Figure 6.

AAV-GMN displays broad transduction in regions throughout the brain following direct delivery. (a–h) Wild-type mice were injected with AAV-GMN encoding an eGFP reporter into either the deep cerebellar nuclei of the cerebellum, the frontal cortex, the striatum, the thalamus, or the lateral ventricle. Three weeks after injection, brains were sagitally (a–d,f–h) or coronally (e) sectioned and examined for eGFP expression by fluorescence microscopy. Brainstem (*), Purkinje cells (arrowhead), ependymal cells (arrow). Scale bar: (a,c) 1 mm, (b,d) 200 μm, (g,h) 250 μm, and (e,f) 500 μm.
Discussion
In this study, we determined that an AAV2-based vector designed to transduce brain endothelium after intravenous delivery utilizes chondroitin sulfate as its primary cell surface receptor. Furthermore, the interaction with chondroitin sulfate is mediated by capsid display of the peptide sequence GMNAFRA, originally discovered in our lab by in vivo phage biopanning.7 Previous in vivo biopanning studies have identified peptides with affinity for different vascular beds and/or organs, including brain; however, in most cases the peptide receptors have not been identified.32,33,34 Furthermore, in those cases where cell surface receptors have been found to be membrane-associated proteins, it could be that phage peptide binding is at least partially mediated by post-translational modifications of the identified protein.
Chondroitin sulfate is a broadly expressed molecule occurring as a covalent modification to multiple core membrane proteins.30 Given its relative abundance, it is not surprising that our previous in vivo biopanning procedure isolated peptides that have binding affinity for cell surface chondroitin sulfate. In fact, we previously identified a distinct phage clone from biopanning in a MPSVII mouse model whose cell binding was at least partially mediated by chondroitin sulfate.7 In addition, there is evidence that several naturally occurring viruses utilize chondroitin sulfate as receptors or attachment factors to mediate infection. These include porcine circovirus 2,35 herpes simplex virus,36 as well as human immunodeficiency virus type I, which has been shown in vitro to utilize chondroitin sulfate proteoglycans to facilitate infection of brain microvascular endothelial cells.37 Lastly, our finding is consistent with the notion that phage display panning against tissue is biased towards isolating peptides that bind to the most abundant cell surface molecules, unless pre-clearing or subtractive procedures are performed to restrict binding to unique targets.
The discovery that chondroitin sulfate C blocks AAV-GMN transduction more potently than chondroitin sulfate A and dermatan sulfate indicates that there is structural specificity in the interaction with the virus, rather than just general binding to any negatively charged glycosaminoglycan or carbohydrate chain. On the other hand, the more limited effect of sialic acid, which is also negatively charged, might occur via non-specific electrostatic interaction, which in turn promotes binding to proximal chondroitin sulfate chains. The fact that hyaluronic acid did not inhibit AAV-GMN transduction of brain endothelial cells indicates that sulfation of monosaccharides is likely required for binding of the virus to the carbohydrate chain, and presumably the negative charge associated with the sulfate group is an important determinant of this interaction. With respect to virus binding, it would be interesting to evaluate the relative contribution and requirement of specific monosaccharides, as well as the degree and position of sulfation, in the GAG chains.
Coincident with our studies, an independent group reported an in vitro phage display panning experiment in which the same GMN peptide was isolated. In that study, peptides displayed by phage in a cyclic format (i.e., constrained in a loop by flanking disulfide-bonded cysteines) were selected for binding to an artificial dermal graft composed of collagen and chondroitin sulfate C.38 In a secondary assay, it was demonstrated that GMN phage bind directly to immobilized chondroitin sulfate C.38 This result is consistent with our findings and, given that our phage library displays peptides linearly, supports the conclusion that the GMN peptide can bind chondroitin sulfate C in multiple structural formats (i.e., linear, cyclic, or as a fusion with the AAV capsid).
It has long been appreciated that AAV2 poorly transduces endothelial cells, despite cell surface expression of heparan sulfate proteoglycans.39 Given that intravenously delivered AAV2 does not target and transduce brain endothelium, it is intriguing that introducing chondroitin sulfate binding activity to AAV2 confers brain vascular tropism. There are many possible explanations for why AAV-GMN but not AAV2 can transduce brain endothelium. It could be that in vivo, brain endothelium expresses cell-surface heparan sulfate, which allows AAV2 surface binding, but that the necessary co-receptors for AAV2 internalization (e.g., αVβ5 integrin)40 and subsequent transduction are not sufficiently expressed. It is unclear whether chondroitin sulfate alone is sufficient for transduction or if AAV-GMN still interacts with AAV2's native co-receptors. It could also be that chondroitin sulfate is much more highly expressed on brain endothelium than heparan sulfate, and is therefore more available to act as receptor for transduction. Unfortunately, to our knowledge, there has not been a rigorous in vivo evaluation of the repertoire of glycosaminoglycans and associated core proteins in brain endothelium so it is difficult to know the degree to which differential expression of receptors and co-receptors underlies the disparity between AAV2 and AAV-GMN transduction.
Given the accumulation of GAGs in several other lysosomal storage diseases, we had hypothesized that general lysosomal dysfunction in CLN2 null mice could possibly lead to accumulated GAGs, thereby making chondroitin sulfate an even more dominant cell surface epitope. However, while brain vasculature was enriched in GAGs, we did not observe appreciable elevation of GAGs in brain tissue from CLN2 null mice, supporting the conclusion that lack of TPP1 enzyme expression does not affect GAG metabolism. It could be, however, that our assays did not detect more subtle alterations in the relative abundance of different GAGs or the degree of GAG sulfation, which was shown to be elevated on heparan chains in a MPSI lysosomal storage disease mouse model.41
Our studies evaluating AAV-GMN transduction both in vitro using cell lines of various origin, and in vivo by direct delivery into the brain, reveal that this virus has broad tropism beyond brain endothelium. While our results from experiments with GAG mutant CHO cells strongly suggest that AAV-GMN utilizes chondroitin sulfate as its functional receptor, it is still possible that in other cell types, the virus can utilize both chondroitin sulfate and heparan sulfate. It would be interesting to evaluate in vivo virus biodistribution after ablating the heparan binding activity by mutation of critical arginine residues in the heparan-binding loop (R585 and R588).20 It may be that a heparan-binding deficient AAV-GMN variant shows enhanced distribution to the brain and diminished distribution to non-CNS organs such as the liver, spleen, and lung, thereby making it a more effective brain delivery vector. Furthermore, given recent advances in achieving widespread AAV delivery and transduction of the non-human primate brain, it would be beneficial to evaluate additional routes of CNS delivery such as into the CSF via the cisterna magna.42 Overall, the broad transduction profile warrants further study of AAV-GMN as a vector for gene delivery in the CNS as well as organs beyond the brain.
An important aspect of AAV transduction biology raised by our study is how the peptide modification might modulate both pre- and post-internalization events, such as intracellular trafficking and targeting for proteosomal degradation. Our observation that AAV-GMN can internalize into chondroitin-deficient CHO ldlD cells to a level similar to AAV2, but displays much lower relative transduction, suggests that in the absence of chondroitin sulfate the virus utilizes a receptor and subsequent nuclear trafficking pathway that is inhibitory to transduction. It is not unprecedented for AAV to enter the nucleus but fail to transduce; for example, AAV1 fails to transduce HeLa cells despite nuclear localization.43 It may be that the pathway AAV-GMN follows in ldlD cells leads to non-productive uncoating within the nucleus. Furthermore, the idea that cell entry mechanisms affect transduction is supported by findings showing that AAV2 internalization via an integrin-mediated route leads to more efficient transduction as compared to endocytosis using competing co-receptors.44 How the GMN peptide might affect AAV co-receptor use is unclear and would require further experimentation; investigating this question could provide valuable insight into endocytosis events that control AAV transduction. It is possible that the GMN peptide could also affect transduction events post-internalization. Studies have shown that selecting for capsid modified AAV with improved transduction capacity yields peptides that improve subcellular localization and intracellular processing relative to AAV2.45 Although the GMN peptide was isolated for endothelial binding and not transduction, it is still possible that it could beneficially modulate restrictive events such as evasion of proteosomal degradation, nuclear trafficking, and capsid uncoating. Furthermore, if the GMN modification does affect these processes, it may provide a partial mechanistic explanation for why AAV-GMN but not AAV2 robustly transduces brain endothelium. Experimentally addressing these possibilities will be an important avenue of future research that will contribute to development of next-generation AAV vectors with novel organ-targeting properties.
The primary motivation for identifying the AAV-GMN endothelial receptor was to advance this vector towards clinical trials for treating human patients with LINCL. Previous research has shown that glycosaminoglycans, including chondroitin sulfate, are expressed throughout the brain.31 Although relatively few studies have examined proteoglycan expression in blood-brain barrier tissue, there are several reports confirming chondroitin sulfate expression in human brain vascular endothelial cells.37,46,47 This supports that AAV-GMN transduction of human brain endothelium is a possible approach for therapeutic gene delivery. Given our findings, it will be exciting to evaluate the efficacy of this vector in a large animal model of LINCL,48 and ultimately in the clinic.
Materials and Methods
Cell lines and recombinant AAV. bEnd.3, N2a, and CHO pgsD-677 cells were obtained from American Type Culture Collection (Manassas, VA). CHO K1 and ldlD cells were kindly provided by Monty Krieger (MIT, Cambridge, MA). CHO Pro5, Lec2, and pgsA-475 cells were provided by the University of Iowa Cells and Tissue Core facility (Iowa City, IA). PC-3 cells were provided by Michael Wright (University of Iowa, Iowa City, IA). HEK293T and HeLa cells were provided by the University of Iowa Gene Transfer Vector Core (Iowa City, IA). MBEC were developed in the Davidson lab.18 Human fibroblasts were obtained from the Coriell Institute (Camden, NJ). All cells except CHO and fibroblasts were grown in Dulbecco modified Eagle medium (Life Technologies, Carlsbad CA) containing 10% fetal bovine serum. CHO cells were maintained in Ham's F12 media (Life Technologies) containing 10% fetal bovine serum and penicillin-streptomycin (Life Technologies). Fibroblasts were maintained in modified Eagle medium (Life Technologies) supplemented with 10% fetal bovine serum, L-glutamine, non-essential amino acids (Life Technologies) and penicillin-streptomycin. All cells were grown at 37 °C in a humidified 5% CO2 environment. The recombinant AAV-GMN and AAV2-PPS vectors were previously described.7 All vectors including AAV2 and AAV5 encoded an eGFP reporter gene driven by the CMV promoter. AAV were produced at the University of Iowa Gene Transfer Vector Core facility by either triple plasmid co-transfection of HEK293 cells or in Sf9 insect cells using the Bac-to-Bac baculovirus expression system (Life Technologies), following previously described methods.49,50 AAV vectors were resuspended in Formulation Buffer 18 (University of Iowa Gene Transfer Vector Core) before use for in vivo experiments.
AAV transduction assays. One day prior to transduction, bEnd.3 or CHO cells were plated at 3 × 104 or 2 × 104 cells/well in 48-well dishes, respectively. The following day, bEnd.3 cells were transduced at an MOI of 105 virion genomes (vg)/cell for 3 hours in Transduction Media (basal growth media containing 2% serum and 2 μmol/l Hoechst 3342 (Life Technologies)). CHO cells were transduced for 2 hours. After transduction, cells were washed twice with media and then cultured in regular growth media (10% serum) containing 2 μmol/l Hoechst 3342 for either 24 hours for CHO cells, or 48 hours for bEnd.3 cells. To assay transduction (i.e., eGFP expression), cells were harvested with PBS+0.5% Triton X-100 containing complete mini protease inhibitors (Roche, Basal, Switzerland). Lysates were clarified by centrifugation at 16K g for 5 minutes at 4 °C and fluorometric analysis of eGFP amount in the supernatants was measured at 535 nm using a Wallac Victorplate reader (Perkin Elmer, Waltham, MA). Protein concentrations of each lysate were measured using a DC protein assay (Bio-Rad, Hercules, CA). Transduction was calculated by normalizing the eGFP fluorescence to the total protein concentration of each sample (i.e., transduction = relative fluorescence units/μg of protein). Transduction of cell lines other than bEnd.3 or CHO were performed the same way but at varying plating densities, dependent on the particular cell type. Statistical analysis was performed comparing the relative transduction values calculated from at least three independent experiments. Fluorescence microscopy images were taken using an Olympus BX60 fluorescent microscope.
Cell surface enzyme treatments. For the trypsin treatment experiment, bEnd.3 cells were treated with either 5 mmol/l EDTA in Dulbecco's PBS, without calcium or magnesium (DPBS) to lift the cells from the culture plate, or treated with EDTA followed by 0.25% trypsin-EDTA (Life Technologies) to lift the cells and digest cell-surface proteins. Cells were suspended in Transduction Media containing AAV-GMN at an MOI of 105 vg/cell and allowed to incubate for 45 minutes at room temperature. Cells were then washed and re-plated in regular growth media containing 2 μmol/l Hoechst 3342. 48 hours after transduction eGFP was measured by fluorometry as described above. For Endo H treatment, bEnd.3 cells plated in a 48-well dish were treated with 10,000 U/well of Endo H (New England Biolabs, Ipswich, MA) for 1 hour at 37 °C, washed with DPBS and then transduced with AAV-GMN as described above. For PNGase F treatment, cells were treated the same as with Endo H except with 10 U/ml of PNGase F (New England Biolabs). For sialic acid digestion, bEnd.3 cells were treated the same way except with 0.2 U (1 U/ml final) of neuraminidase (Sigma-Aldrich, St Louis, MO). For chondroitin sulfate digestion, CHO pgsD-677 cells plated in 48-well dishes were treated with 0.1 U/well of chondroitinase ABC (Sigma-Aldrich) for 1 hour at 37 °C, washed with regular growth media, and then transduced as described above. For all experiments, control cells were transduced that were treated the same way except with buffer instead of the enzyme.
Heparin agarose binding assay. 200 μl of heparin-bound agarose resin (Sigma-Aldrich) pre-equilibrated in Binding Buffer (DPBS, 5 mmol/l MgCl2) was combined with 2 × 1010 vg of each virus in 300 μl of Binding Buffer. Samples were allowed to incubate with end-over-end rotation for 30 minutes at room temperature. After incubation, the unbound (flow-through) fraction was collected by centrifugation and the agarose resin was washed four times with 300 μl of Binding Buffer (1.2 ml total wash volume). Bound virus was eluted from the heparin agarose resin by incubation in 600 μl of Elution Buffer (DPBS, 5 mmol/l MgCl2, 2 mol/l NaCl) for 10 minutes at room temperature. Sodium dodecyl sulfate was added to all collected fractions to a final concentration of 1% and all samples were denatured by heating at 95 °C for 10 minutes (Note: the elution samples were diluted with 600 μl of water prior to heating in order to avoid sodium dodecyl sulfate precipitation). The entire volume of all fractions was applied to a nitrocellulose membrane (Bio-Rad) pre-wetted with Tris-buffered saline (TBS) using a slot-blot apparatus (Schleicher & Schuell, Keene, NH). Input samples contained the same amount of AAV incubated with the heparin agarose. After adsorption to nitrocellulose, the membrane was blocked with Blocking Buffer (TBS+0.05% Tween-20, 5% non-fat dried milk) for 30 minutes at room temperature and then incubated overnight at 4 °C in Antibody Buffer (TBS+0.05% Tween-20, 2% non-fat dried milk) containing 1:200 diluted mouse monoclonal anti-AAV VP1, VP2, VP3 antibody (Clone B1, American Research Products, Waltham, MA). The membrane was washed and then incubated for 1 hour at room temperature with HRP conjugated anti-mouse IgG secondary antibody (Cell Signaling Technology, Danvers, MA) diluted 1:2,000 into Antibody Buffer. After washing, the blot was developed using enhanced chemiluminescence reagent (Thermo Scientific, Waltham, MA) and imaged on x-ray film (RPI, Mount Prospect, IL) and digitally scanned to prepare the figure.
Glycosaminoglycan transduction and binding blocking assays. Mouse bEnd.3 cells were plated at 3 × 104 cells/well 24 hours before transduction. Heparin sulfate (Sigma-Aldrich#H3393), chondroitin sulfate A (Sigma-Aldrich#C9819), chondroitin sulfate C (Sigma-Aldrich#C4384), dermatan sulfate (Sigma-Aldrich #C3788), and hyaluronic acid (Sigma-Aldrich #53747) were prepared in DPBS. AAV-GMN was combined and pre-incubated with each glycosaminoglycan at various concentrations in DMEM containing 2% FBS and 2 μmol/l Hoechst 3342, for 1 hour at 37 °C and then applied to cells and transduction was performed as described above. For the binding experiments, 0.5 × 104 vg/cell were pre-incubated with chondroitin sulfate C in DPBS for 1 hour on ice and then applied to cells on ice that were pre-washed with ice-cold DPBS. After incubation for 1 hour, cells were washed three times with DPBS and DNA was extracted using a DNeasy Kit (Qiagen, Germantown, MD). Recovered viral genomes were measured by quantitative PCR using a TaqMan assay for the eGFP reporter gene (ABI/Life Technologies). Viral genomes were calculated using a standard curve generated from known quantities of a plasmid template encoding the eGFP reporter gene.
AAV internalization assay. CHO ldlD cells were plated at 5 × 104 cells/well and 24 hours later, 5 × 105 vg/cell were incubated for 1 hour with the cells (pre-chilled on ice) in ice-cold Ham's F12 media containing 2% FBS. Cells were washed twice with media and then incubated at 37 °C for 1 hour to allow for endocytosis of bound virus. For measurement of internalized virus, cells were treated with 0.25% trypsin-EDTA for 20 minutes at 37 °C, and washed by resuspending in Ham's F12 containing 10% FBS and then pelleting by centrifugation at 1,000g for 5 minutes at 4 °C. For measurement of total virus, both bound and internalized, cells were not treated with trypsin. Both trypsin-treated cell pellets and non-trypsinized cells (still in the tissue culture dish), were harvested for DNA extraction using the DNeasy kit. Recovered viral genomes were quantified as described above by quantitative PCR. Recovered genomes were normalized to the total amount of DNA recovered from the treated cells to account for any variation in extraction due to trypsin treatment, handling, etc.
Fluorescent labeling of AAV and image analysis. AAV2 and AAV2-GMN particles were labeled with Alexa Fluor 488 and 568 Protein Labeling Kits purchased from Invitrogen Life Technologies (Grand Island, NY) as previously described.43 Fluorescently labeled viral particles (105 vector genome copies/cell) were bound to CHO cells on glass coverslip dishes at 4 °C in media containing 2% FBS for 1 hour, followed by washing in 2% media to removed unbound particles and shifting to 37 °C in warm media for 4 hours. To stop the infection, media was replaced with ice-cold PBS for 15 minutes. Plasma membranes were labeled with Alexa 647-WGA (wheat germ agglutinin) (1:200, Invitrogen Life Technologies, Grand Island, NY) by binding in PBS for 1 hour at 4 °C. Cells were then fixed for 15 minutes in 4% paraformaldehyde, washed in PBS, and covered with Vectashield mounting medium with DAPI (Vector Biolabs, Burlingame, CA). Cells were imaged using a Zeiss LSM 710 laser scanning confocal microscope in three dimensions using 0.39 µm z-step sizes. Image analysis was performed on single planes through the center of the cells using Metamorph Software (Molecular Devices, Downingtown, PA) by drawing regions around the entire cell, the intracellular space, and the nucleus using the WGA and DAPI signals as a guide. Total integrated intensities for the AAV2 and AAV2-GMN were then calculated for each region using image thresholds (20,000 for AAV2-Alexa 488, 3,500 for AAV2-GMN-Alexa488) and used to calculate the percentage of virus in each compartment per cell. A total of 20–50 cells were analyzed for each condition, and statistical differences between percent virus per compartment was tested for significant differences using the Mann–Whitney U–test.
Analysis of glycosaminoglycans and sialic acid in brain tissue. Measurement of total sulfated GAG content was performed by Farndale assay as previously described, but with some minor modifications.51 CLN2 heterozygote and knockout mice8 at ~9 weeks of age were sacrificed and brains were perfused with saline before dissection. Animal maintenance conditions and experimental protocols were approved by the University of Iowa Animal Care and Use Committee. For vasculature analysis, brain vessels were first isolated by dextran gradient centrifugation as previously described.52 Whole brain and brain vessel tissues were homogenized in 100 mmol/l K2HPO4 buffer containing 50 μg/ml proteinase K (Research Products International, Mt. Pleasant, IL) and digested overnight at 56 °C. For the spiked positive control, chondroitin sulfate C (Sigma) was added to an aliquot of KO brain homogenate at a concentration of 50 μg/ml prior to the overnight incubation step. The proteinase was inactivated by incubation at 90 °C for 10 minutes and the samples were clarified by centrifugation at 16K g for 10 minutes at 4 °C and then filtered by centrifugation through 0.45 μm cellulose acetate spin columns (Corning, Tewksbury, MA). The filtrates containing extracted GAGs were diluted tenfold into 100 mmol/l K2HPO4 buffer before analysis. A twofold standard curve dilution series ranging from 0 to 10 μg/ml was prepared in 100 mmol/l K2HPO4 buffer using purified chondroitin sulfate C (Sigma-Aldrich #C4384). 100 μl of all samples were combined with 100 μl of Farndale reagent (38.45 μmol/l 1,9-dimethyl-methylene blue (Sigma-Aldrich #341088), 40.5 mmol/l NaCl, 40.5 mmol/l glycine; pH 3.0) and absorbance at 525 nm was immediately measured using a Tecan Safire II plate reader (Tecan Systems, San Jose, CA). GAG concentration in tissue samples was calculated against the standard curve after subtracting background absorbance. For immunostaining of chondroitin sulfate, snap-frozen brains were imbedded in OCT compound (Sakura Fineteck, Torrance, CA) and 20 μm sections were prepared on a cryostat. Sections were fixed with 4% paraformaldehyde (Electron Microscopy Sciences, Hatfield, PA) and stained overnight with 1:200 diluted mouse anti-chondroitin sulfate antibody (Thermo Scientific, #MA1-3055). Sections were then incubated with 1:200 diluted biotinylated goat anti-mouse IgM secondary F (ab')2 antibody fragment (Jackson ImmunoResearch, West Grove, PA) for 1 hour and staining was developed using a DAB kit (Vector Laboratories, Burlingame, CA) following the manufacturer's protocol. Background staining was assessed by only probing tissue with secondary antibody. Lectin staining for sialic acid with fluorescein-labeled SNA (Vector Laboratories) was performed as previously described.29
In vivo brain transduction with AAV-GMN. Adult C57BL/6 mice (male and female, n = 2–3 per injection group) were injected with 5 μl unilaterally (lateral ventricle and striatum), 3 μl unilaterally (deep cerebellar nuclei), or 1.5 μl unilaterally (cortex) of AAV-GMN (stock titer = 4 × 1012 vg/ml). For thalamus injections, 3 μl of virus was injected bilaterally. Stereotaxic coordinates used were as follows: cortex (+0.86 mm anterior/posterior, ±1.3 mm medial/lateral, −0.8 mm dorsal/ventral from brain surface), striatum (+0.86 mm anterior/posterior, ±1.8 mm medial/lateral, −2.5 mm dorsal/ventral from brain surface), lateral ventricle (−0.4 mm anterior/posterior, ±1.0 mm medial/lateral, −2.0 mm dorsal/ventral from brain surface), deep cerebellar nuclei (−6.0 mm anterior/posterior, ±2.0 mm medial/lateral, −2.2 mm dorsal/ventral from brain surface), and thalamus (−2.0 mm anterior/posterior, ±1.5 mm medial/lateral, −3.0 mm dorsal/ventral from brain surface). Three weeks after injection, mice were sacrificed and perfused and fixed with paraformaldehyde. Sagittal and coronal sections (20–30 μm) were analyzed for eGFP expression by fluorescence microscopy. Some images are a composite of several over laid pictures prepared using Adobe Photoshop CS6 (Adobe, San Jose, CA).
Statistical analysis. Unless otherwise indicated, we analyzed data by unpaired Student's t test using GraphPad Prism software (La Jolla, CA).
Figure preparation. All graphs were prepared using GraphPad Prism software. Figures were prepared using Adobe Illustrator CS6.
SUPPLEMENTARY MATERIAL Figure S1. The AAV2 preparation used for transfection of bEnd.3 cells in Figure 1 is infectious. Figure S2. CLN2 knockout mouse brain does not have elevated sialic acid levels. Figure S3. AAV-GMN cell and nuclear entry is equivalent in wild type and mutant CHO cells.
Acknowledgments
The authors acknowledge the members of the Davidson Lab and Michael Wright (University of Iowa) for discussion and technical advice. They also thank the University of Iowa Gene Transfer Vector Core for producing the AAV vectors. This work was supported by NIH grant NINDS R21NS077516, the Batten Disease Support and Research Foundation, and the Our Promise to Nicholas Foundation. B.L.D. is a founder of Spark Therapeutics, Inc., a gene therapy company. The other authors declare no conflict of interest.
Supplementary Material
References
- Asokan A, Schaffer DV, Samulski RJ. The AAV vector toolkit: poised at the clinical crossroads. Mol Ther. 2012;20:699–708. doi: 10.1038/mt.2011.287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foust KD, Nurre E, Montgomery CL, Hernandez A, Chan CM, Kaspar BK. Intravascular AAV9 preferentially targets neonatal neurons and adult astrocytes. Nat Biotechnol. 2009;27:59–65. doi: 10.1038/nbt.1515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang B, Li S, Wang H, Guo Y, Gessler DJ, Cao C, et al. Global CNS transduction of adult mice by intravenously delivered rAAVrh.8 and rAAVrh.10 and nonhuman primates by rAAVrh.10. Mol Ther. 2014;22:1299–1309. doi: 10.1038/mt.2014.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Girod A, Ried M, Wobus C, Lahm H, Leike K, Kleinschmidt J, et al. Genetic capsid modifications allow efficient re-targeting of adeno-associated virus type 2. Nat Med. 1999;5:1438. doi: 10.1038/71021. [DOI] [PubMed] [Google Scholar]
- Grifman M, Trepel M, Speece P, Gilbert LB, Arap W, Pasqualini R, et al. Incorporation of tumor-targeting peptides into recombinant adeno-associated virus capsids. Mol Ther. 2001;3:964–975. doi: 10.1006/mthe.2001.0345. [DOI] [PubMed] [Google Scholar]
- Nicklin SA, Buening H, Dishart KL, de Alwis M, Girod A, Hacker U, et al. Efficient and selective AAV2-mediated gene transfer directed to human vascular endothelial cells. Mol Ther. 2001;4:174–181. doi: 10.1006/mthe.2001.0424. [DOI] [PubMed] [Google Scholar]
- Chen YH, Chang M, Davidson BL. Molecular signatures of disease brain endothelia provide new sites for CNS-directed enzyme therapy. Nat Med. 2009;15:1215–1218. doi: 10.1038/nm.2025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sleat DE, Wiseman JA, El-Banna M, Kim KH, Mao Q, Price S, et al. A mouse model of classical late-infantile neuronal ceroid lipofuscinosis based on targeted disruption of the CLN2 gene results in a loss of tripeptidyl-peptidase I activity and progressive neurodegeneration. J Neurosci. 2004;24:9117–9126. doi: 10.1523/JNEUROSCI.2729-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sleat DE, Donnelly RJ, Lackland H, Liu CG, Sohar I, Pullarkat RK, et al. Association of mutations in a lysosomal protein with classical late-infantile neuronal ceroid lipofuscinosis. Science. 1997;277:1802–1805. doi: 10.1126/science.277.5333.1802. [DOI] [PubMed] [Google Scholar]
- Balestrieri ML, Napoli C. Novel challenges in exploring peptide ligands and corresponding tissue-specific endothelial receptors. Eur J Cancer. 2007;43:1242–1250. doi: 10.1016/j.ejca.2007.02.006. [DOI] [PubMed] [Google Scholar]
- Rajotte D, Ruoslahti E. Membrane dipeptidase is the receptor for a lung-targeting peptide identified by in vivo phage display. J Biol Chem. 1999;274:11593–11598. doi: 10.1074/jbc.274.17.11593. [DOI] [PubMed] [Google Scholar]
- Daquinag AC, Zhang Y, Amaya-Manzanares F, Simmons PJ, Kolonin MG. An isoform of decorin is a resistin receptor on the surface of adipose progenitor cells. Cell Stem Cell. 2011;9:74–86. doi: 10.1016/j.stem.2011.05.017. [DOI] [PubMed] [Google Scholar]
- Brown RC, Morris AP, O'Neil RG. Tight junction protein expression and barrier properties of immortalized mouse brain microvessel endothelial cells. Brain Res. 2007;1130:17–30. doi: 10.1016/j.brainres.2006.10.083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montesano R, Pepper MS, Möhle-Steinlein U, Risau W, Wagner EF, Orci L. Increased proteolytic activity is responsible for the aberrant morphogenetic behavior of endothelial cells expressing the middle T oncogene. Cell. 1990;62:435–445. doi: 10.1016/0092-8674(90)90009-4. [DOI] [PubMed] [Google Scholar]
- Kaludov N, Brown KE, Walters RW, Zabner J, Chiorini JA. Adeno-associated virus serotype 4 (AAV4) and AAV5 both require sialic acid binding for hemagglutination and efficient transduction but differ in sialic acid linkage specificity. J Virol. 2001;75:6884–6893. doi: 10.1128/JVI.75.15.6884-6893.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Z, Miller E, Agbandje-McKenna M, Samulski RJ. Alpha2,3 and alpha2,6 N-linked sialic acids facilitate efficient binding and transduction by adeno-associated virus types 1 and 6. J Virol. 2006;80:9093–9103. doi: 10.1128/JVI.00895-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robertson MA, Etchison JR, Robertson JS, Summers DF, Stanley P. Specific changes in the oligosaccharide moieties of VSV grown in different lectin-resistnat CHO cells. Cell. 1978;13:515–526. doi: 10.1016/0092-8674(78)90325-2. [DOI] [PubMed] [Google Scholar]
- Tecedor L, Stein CS, Schultz ML, Farwanah H, Sandhoff K, Davidson BL. CLN3 loss disturbs membrane microdomain properties and protein transport in brain endothelial cells. J Neurosci. 2013;33:18065–18079. doi: 10.1523/JNEUROSCI.0498-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Summerford C, Samulski RJ. Membrane-associated heparan sulfate proteoglycan is a receptor for adeno-associated virus type 2 virions. J Virol. 1998;72:1438–1445. doi: 10.1128/jvi.72.2.1438-1445.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perabo L, Goldnau D, White K, Endell J, Boucas J, Humme S, et al. Heparan sulfate proteoglycan binding properties of adeno-associated virus retargeting mutants and consequences for their in vivo tropism. J Virol. 2006;80:7265–7269. doi: 10.1128/JVI.00076-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kern A, Schmidt K, Leder C, Müller OJ, Wobus CE, Bettinger K, et al. Identification of a heparin-binding motif on adeno-associated virus type 2 capsids. J Virol. 2003;77:11072–11081. doi: 10.1128/JVI.77.20.11072-11081.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White SJ, Nicklin SA, Büning H, Brosnan MJ, Leike K, Papadakis ED, et al. Targeted gene delivery to vascular tissue in vivo by tropism-modified adeno-associated virus vectors. Circulation. 2004;109:513–519. doi: 10.1161/01.CIR.0000109697.68832.5D. [DOI] [PubMed] [Google Scholar]
- Esko JD, Stewart TE, Taylor WH. Animal cell mutants defective in glycosaminoglycan biosynthesis. Proc Natl Acad Sci USA. 1985;82:3197–3201. doi: 10.1073/pnas.82.10.3197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esko JD, Rostand KS, Weinke JL. Tumor formation dependent on proteoglycan biosynthesis. Science. 1988;241:1092–1096. doi: 10.1126/science.3137658. [DOI] [PubMed] [Google Scholar]
- Kingsley DM, Kozarsky KF, Hobbie L, Krieger M. Reversible defects in O-linked glycosylation and LDL receptor expression in a UDP-Gal/UDP-GalNAc 4-epimerase deficient mutant. Cell. 1986;44:749–759. doi: 10.1016/0092-8674(86)90841-x. [DOI] [PubMed] [Google Scholar]
- Schultz ML, Tecedor L, Chang M, Davidson BL. Clarifying lysosomal storage diseases. Trends Neurosci. 2011;34:401–410. doi: 10.1016/j.tins.2011.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muenzer J. Overview of the mucopolysaccharidoses. Rheumatology (Oxford) 2011;50 (suppl. 5):v4–12. doi: 10.1093/rheumatology/ker394. [DOI] [PubMed] [Google Scholar]
- Liu G, Martins I, Wemmie JA, Chiorini JA, Davidson BL. Functional correction of CNS phenotypes in a lysosomal storage disease model using adeno-associated virus type 4 vectors. J Neurosci. 2005;25:9321–9327. doi: 10.1523/JNEUROSCI.2936-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen YH, Claflin K, Geoghegan JC, Davidson BL. Sialic acid deposition impairs the utility of AAV9, but not peptide-modified AAVs for brain gene therapy in a mouse model of lysosomal storage disease. Mol Ther. 2012;20:1393–1399. doi: 10.1038/mt.2012.100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gandhi NS, Mancera RL. The structure of glycosaminoglycans and their interactions with proteins. Chem Biol Drug Des. 2008;72:455–482. doi: 10.1111/j.1747-0285.2008.00741.x. [DOI] [PubMed] [Google Scholar]
- Zimmermann DR, Dours-Zimmermann MT. Extracellular matrix of the central nervous system: from neglect to challenge. Histochem Cell Biol. 2008;130:635–653. doi: 10.1007/s00418-008-0485-9. [DOI] [PubMed] [Google Scholar]
- Pasqualini R, Ruoslahti E. Organ targeting in vivo using phage display peptide libraries. Nature. 1996;380:364–366. doi: 10.1038/380364a0. [DOI] [PubMed] [Google Scholar]
- Rajotte D, Arap W, Hagedorn M, Koivunen E, Pasqualini R, Ruoslahti E. Molecular heterogeneity of the vascular endothelium revealed by in vivo phage display. J Clin Invest. 1998;102:430–437. doi: 10.1172/JCI3008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arap W, Kolonin MG, Trepel M, Lahdenranta J, Cardó-Vila M, Giordano RJ, et al. Steps toward mapping the human vasculature by phage display. Nat Med. 2002;8:121–127. doi: 10.1038/nm0202-121. [DOI] [PubMed] [Google Scholar]
- Misinzo G, Delputte PL, Meerts P, Lefebvre DJ, Nauwynck HJ. Porcine circovirus 2 uses heparan sulfate and chondroitin sulfate B glycosaminoglycans as receptors for its attachment to host cells. J Virol. 2006;80:3487–3494. doi: 10.1128/JVI.80.7.3487-3494.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergefall K, Trybala E, Johansson M, Uyama T, Naito S, Yamada S, et al. Chondroitin sulfate characterized by the E-disaccharide unit is a potent inhibitor of herpes simplex virus infectivity and provides the virus binding sites on gro2C cells. J Biol Chem. 2005;280:32193–32199. doi: 10.1074/jbc.M503645200. [DOI] [PubMed] [Google Scholar]
- Argyris EG, Acheampong E, Nunnari G, Mukhtar M, Williams KJ, Pomerantz RJ. Human immunodeficiency virus type 1 enters primary human brain microvascular endothelial cells by a mechanism involving cell surface proteoglycans independent of lipid rafts. J Virol. 2003;77:12140–12151. doi: 10.1128/JVI.77.22.12140-12151.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawada R, Peterson CY, Gonzalez AM, Potenza BM, Mueller B, Coimbra R, et al. A phage-targeting strategy for the design of spatiotemporal drug delivery from grafted matrices. Fibrogenesis Tissue Repair. 2011;4:7. doi: 10.1186/1755-1536-4-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pajusola K, Gruchala M, Joch H, Lüscher TF, Ylä-Herttuala S, Büeler H. Cell-type-specific characteristics modulate the transduction efficiency of adeno-associated virus type 2 and restrain infection of endothelial cells. J Virol. 2002;76:11530–11540. doi: 10.1128/JVI.76.22.11530-11540.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Summerford C, Bartlett JS, Samulski RJ. AlphaVbeta5 integrin: a co-receptor for adeno-associated virus type 2 infection. Nat Med. 1999;5:78–82. doi: 10.1038/4768. [DOI] [PubMed] [Google Scholar]
- Holley RJ, Deligny A, Wei W, Watson HA, Niñonuevo MR, Dagälv A, et al. Mucopolysaccharidosis type I, unique structure of accumulated heparan sulfate and increased N-sulfotransferase activity in mice lacking a-l-iduronidase. J Biol Chem. 2011;286:37515–37524. doi: 10.1074/jbc.M111.287474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray SJ, Nagabhushan Kalburgi S, McCown TJ, Jude Samulski R. Global CNS gene delivery and evasion of anti-AAV-neutralizing antibodies by intrathecal AAV administration in non-human primates. Gene Ther. 2013;20:450–459. doi: 10.1038/gt.2012.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keiser NW, Yan Z, Zhang Y, Lei-Butters DC, Engelhardt JF. Unique characteristics of AAV1, 2, and 5 viral entry, intracellular trafficking, and nuclear import define transduction efficiency in HeLa cells. Hum Gene Ther. 2011;22:1433–1444. doi: 10.1089/hum.2011.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaminsky PM, Keiser NW, Yan Z, Lei-Butters DC, Engelhardt JF. Directing integrin-linked endocytosis of recombinant AAV enhances productive FAK-dependent transduction. Mol Ther. 2012;20:972–983. doi: 10.1038/mt.2011.295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naumer M, Popa-Wagner R, Kleinschmidt JA. Impact of capsid modifications by selected peptide ligands on recombinant adeno-associated virus serotype 2-mediated gene transduction. J Gen Virol. 2012;93 Pt 10:2131–2141. doi: 10.1099/vir.0.044735-0. [DOI] [PubMed] [Google Scholar]
- Bobardt MD, Salmon P, Wang L, Esko JD, Gabuzda D, Fiala M, et al. Contribution of proteoglycans to human immunodeficiency virus type 1 brain invasion. J Virol. 2004;78:6567–6584. doi: 10.1128/JVI.78.12.6567-6584.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto C, Deng X, Fujiwara Y, Kaji T. Proteoglycans predominantly synthesized by human brain microvascular endothelial cells in culture are perlecan and biglycan. J Health Sci. 2005;51:576–583. [Google Scholar]
- Awano T, Katz ML, O'Brien DP, Sohar I, Lobel P, Coates JR, et al. A frame shift mutation in canine TPP1 (the ortholog of human CLN2) in a juvenile Dachshund with neuronal ceroid lipofuscinosis. Mol Genet Metab. 2006;89:254–260. doi: 10.1016/j.ymgme.2006.02.016. [DOI] [PubMed] [Google Scholar]
- Urabe M, Ding C, Kotin RM. Insect cells as a factory to produce adeno-associated virus type 2 vectors. Hum Gene Ther. 2002;13:1935–1943. doi: 10.1089/10430340260355347. [DOI] [PubMed] [Google Scholar]
- Smith RH, Levy JR, Kotin RM. A simplified baculovirus-AAV expression vector system coupled with one-step affinity purification yields high-titer rAAV stocks from insect cells. Mol Ther. 2009;17:1888–1896. doi: 10.1038/mt.2009.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farndale RW, Buttle DJ, Barrett AJ. Improved quantitation and discrimination of sulphated glycosaminoglycans by use of dimethylmethylene blue. Biochim Biophys Acta. 1986;883:173–177. doi: 10.1016/0304-4165(86)90306-5. [DOI] [PubMed] [Google Scholar]
- Song L, Pachter JS. Culture of murine brain microvascular endothelial cells that maintain expression and cytoskeletal association of tight junction-associated proteins. In Vitro Cell Dev Biol Anim. 2003;39:313–320. doi: 10.1290/1543-706X(2003)039<0313:COMBME>2.0.CO;2. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.