

Abstract
Gene knockdown using micro RNA (miRNA)-based vector constructs is likely to become a prominent gene therapy approach. It was the aim of this study to improve the efficiency of gene knockdown through optimizing the structure of miRNA mimics. Knockdown of two target genes was analyzed: CCR5 and green fluorescent protein. We describe here a novel and optimized miRNA mimic design called mirGE comprising a lower stem length of 13 base pairs (bp), positioning of the targeting strand on the 5′ side of the miRNA, together with nucleotide mismatches in upper stem positions 1 and 12 placed on the passenger strand. Our mirGE proved superior to miR-30 in four aspects: yield of targeting strand incorporation into RNA-induced silencing complex (RISC); incorporation into RISC of correct targeting strand; precision of cleavage by Drosha; and ratio of targeting strand over passenger strand. A triple mirGE hairpin cassette targeting CCR5 was constructed. It allowed CCR5 knockdown with an efficiency of over 90% upon single-copy transduction. Importantly, single-copy expression of this construct rendered transduced target cells, including primary human macrophages, resistant to infection with a CCR5-tropic strain of HIV. Our results provide new insights for a better knockdown efficiency of constructs containing miRNA. Our results also provide the proof-of-principle that cells can be rendered HIV resistant through single-copy vector transduction, rendering this approach more compatible with clinical applications.
Introduction
Micro RNAs (miRNAs) are naturally occurring, noncoding small RNAs, which regulate the expression of target genes1 by degradation of their mRNA and/or block of their translation. The generation and processing of miRNA from miRNA genes follows a defined pattern.2 Briefly, miRNAs are typically transcribed by RNA polymerase II as a primary miRNA (pri-miRNA) of several hundred nucleotides comprising a ~70 bp stem-loop structure. The stem-loop structure is then cleaved from the pri-miRNA by a “microprocessor complex” formed by the RNAse III enzyme Drosha and its subunit protein DGCR8, generating the precursor miRNA (pre-miRNA), which is similar in structure to short hairpin RNAs (shRNAs).3,4 The pre-miRNAs are further processed in the cytoplasm by the endoribonuclease Dicer, which removes the loop of the hairpin, yielding a miRNA duplex of ~22 bp. The antisense strand (targeting strand in this paper) of the miRNA duplex will be integrated into the RNA-induced silencing complex (RISC), where it blocks translation through interaction with its target mRNA.
The elegance and efficiency of RNA interference has rapidly led to knockdown vectors designed from naturally occurring miRNAs. A first generation of lentivectors directly expressed shRNAs as a simple stem loop structure with no flanking sequences. When transcribed, they immediately form a thermodynamically stable stem loop and are directly exported to the cytoplasm where they are processed by DICER bypassing Drosha. In order to express shRNAs without flanking sequences, H1 (ref. 5) or U6 (ref. 6) RNA Pol III promoters or snRNA U1 Pol II promoters7 were initially used. However, these promoters suffer from (i) their constitutive expression and (ii) potential toxicity due to competition of the artificial shRNA with endogenous miRNAs and an eventual subsequent saturation of the RNAi machinery, specifically the karyopherin exportin-5.8,9 Also, overexpression of shRNAs may stimulate the innate immune system through activation of the RNA-dependent protein kinase/interferon response.10 On the other hand, miRNA mimics are compatible with expression from RNA pol II tissue-specific or inducible promoters and produce less processed antisense/targeting strand RNA, which if in excess can lead to cellular toxicity.11 Thus, constructs reproducing natural miRNA synthesis and processing for the purpose of gene knockdown could be preferred to shRNA constructs. Indeed, artificial pri-miRNA mimics have been used for silencing a variety of target genes.12,13,14,15 Most of these constructs were based on a naturally occurring miRNA miR-30 backbone with various flanking region, stem and loop modifications.1,12,13,14,15,16,17,18,19,20,21 A similar version of this miR-30-based miRNA mimic is also commercially available (GIPZ & TRIPZ shRNAmir lentivector expression systems; Open Biosystems; GE Dharmacon, Lafayette, CO).
Lentiviral vectors containing either shRNA or miRNA are very promising tools for gene therapy involving gene repression. RNA interference as a tool for gene therapy has been explored in vitro and in vivo using both shRNA and miRNA mimics. Focusing on miRNA in lentivectors, the main targets involved were HIV,22,23 hepatitis B virus,24 cancer,25 and Alzheimer disease,26 only to mention a few. But the standards for such vector systems are high since they should achieve the required knockdown effect (ideally at a single transgene integration level) without affecting normal cell functions. Indeed, multiple transgene insertions into the host cell genome resulting from high vector-mediated transduction rates can increase the risk of alteration of functionally relevant parts of the genome and in particular increase the risk of oncogenesis.8,27
In this study, we optimized miRNA-mimic design by adjusting complementary sequences and stem lengths (mirGE). We then chose the most potent variant, called mirGE herein after, and compared it with the original miR-30 in its efficiency to knockdown the CCR5 HIV coreceptor. Our mirGE proved largely more potent than miR-30. In particular, high-throughput sequencing revealed that mirGE is superior to miR-30 in four aspects: yield of targeting strand incorporation into RISC; incorporation into RISC of correct targeting strand; precision of cleavage by Drosha; and ratio of targeting strand over passenger strand.
We increased mirGE efficiency by expressing it as a multiple hairpin structure and found that a triple hairpin anti-CCR5 mirGE downregulates CCR5 expression by more than 90% with a single copy of lentivector. This level of CCR5 knockdown efficiency by a single copy of anti-CCR5 lentivector protected HeLa cells as well as primary human macrophages against in vitro infection with a CCR5-tropic HIV strain. To the best of our knowledge, this is the most efficient miRNA-based lentivector described so far and the first demonstration that miRNA-based CCR5 knockdown by a single copy of a lentivector can confer intracellular immunization against HIV.
Our results thus pave the way toward an efficient yet clinically compatible gene therapy approach aimed at generating HIV-resistant immune cells in human patients.
Results
Effect of miRNA hairpin features on green fluorescent protein knockdown
To test miRNA hairpin features critical for efficient gene knockdown, we constructed a lentivector containing two independent expression units (Supplementary Figure S1). One unit is expressing, under the control of the human Ubiquitin promoter, an mRNA containing the human microsomal glutathione S-transferase 2 (MGST2) coding sequence followed by a variable miRNA structure targeting the green fluorescent protein (GFP) living color. The MGST2 gene was used as spacer as we initially observed that repression activity was dramatically reduced when miRNAs were placed immediately after the transcription start site (data not shown). A similar finding has been reported that absence of intervening sequence between the 5′ end of the mRNA and miRNA hairpin was detrimental to knockdown activity.12 The other unit is expressing the mCherry living color under the control of the human phosphoglycerate kinase 1 promoter. This dual cassette design allowed us to reliably follow transduced cells expressing constant amounts of red mCherry protein, while GFP expression was downregulated by the miRNA.
We then transduced a clone of HeLa cells stably expressing GFP (4.5 cells),28 with lentivectors containing seven versions of a composite miRNA backbone containing the same 22 nucleotide segment targeting the GFP living color (Figure 1). This miRNA backbone contains the miR-16 lower stem as well as flanking regions, the target-specific stem containing a wooble at position +12 (as in miR-16) and the miR-30 loop. We kept the miR-30 loop since the terminal loop seems to be dispensable for pri-miRNA processing.4 Variations on this backbone were made in order to test for two features, i.e., the length of the lower stem and the side of the mismatch on the targeting strand. D13M5 refers to a 13 bp long lower stem and a single nucleotide mismatch at the 5′ end of the targeting strand and is thus analogous to miR-16 (ref. 4) in terms of lower stem length and targeting strand mismatch orientation. Other hairpin designs follow the same numbering code, where the number after D represents the length of the lower stem, and the number after M represents the mismatched end of the targeting strand. GFP-positive 4.5 cells were transduced with similar multiplicity of infections (MOIs) of each vector, and the percentage of GFP-negative cells generated within the total of mCherry-positive transduced cells was analyzed by flow cytometry (Figure 1b). Percentages were then plotted for statistical analysis (Figure 1c). The most obvious observation is that the length of the lower stem is critical for overall efficiency, as hairpins with 13 or 15 nucleotides in their lower stem were the most potent at generating GFP-negative cells. Longer (D17) or shorter (D11, D9) stem lengths show negligible effect on GFP downregulation. Such an impact of lower stem length on knockdown efficiency was reported previously15 and is likely due to the generation by Drosha processing of hairpins with lengths that are inappropriate for further steps in RISC processing.29 Indeed, it was shown that the Drosha cleavage site is determined mostly by the junction between the flanking sequences and the lower stem and that cleavage occurs ~11 bp (when counting on the bottom strand, i.e., 13 bp on the top strand as we number in this paper) up from this junction.4 Comparison of D13M5 and D13M3 shows a difference; however, it is not statistically significant. Increasing the number of experiments may reveal a significant difference which will be in accordance with the current understanding of preferential strand incorporation into RISC.30,31 We thus chose the D13M5 hairpin (hereinafter called mirGE) as the best miRNA configuration to compare to the miR-30 hairpin in knockdown experiments targeting CCR5.
Figure 1.

Downregulation of GFP in 4.5 cells using lentivectors expressing various anti-GFP miRNAs constructs. (a) Schematic diagram of variations introduced in the miRNA design, based on features described in Supplementary Figure S2. (b) FACS plots representative of independent experiments: control, D9M3, D9M5, D11M5, and D13M3 (n = 4), D13M5 (n = 7). D15M5, D17M5 (n = 3). 4.5 GFP expressing cells were transduced with equivalent quantities of lentivectors expressing mCherry alone (GFP control, upper plot), or mCherry together with the corresponding miRNA hairpin displayed alongside in panel a. For analysis, FACS plots were analyzed using quadrants to delineate Cherry-positive transduced cells. Then, the ratio was calculated as the fraction of GFP-negative/Cherry-positive cells over the total of Cherry-positive cells. (c) Bar graph diagram corresponding to the different experiments as illustrated in the FACS plots alongside in panel b, showing the percentage of GFP-negative cells within total Cherry-positive cells for each corresponding miRNA design. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001, using one-way ANOVA followed by Bonferroni's mulitple comparison test. ANOVA, analysis of variance; GFP, green fluorescent protein; miRNA, micro RNA.
Effect of targeting sequence and hairpin context on CCR5 knockdown
We then compared the efficiency of mirGE and miR-30 hairpin (for details, see Figure 2), context for the knockdown of a second target gene, namely CCR5. Two CCR5-targeting sequences, T1 and T7, were identified in silico (see Supplementary Material). The T7 target sequence has also previously been described as a very potent target.32 T1 and T7 were cloned as a single hairpin in either the miR-30 or miGE backbones. As shown in Supplementary Figure S3, the lentivector expression cassette transcribes a single mRNA that encodes for the GFP living color followed by the CCR5-targeting miRNAs. Lentivectors are then used to transduce a clone of HeLa cells constitutively expressing CCR5 (HR5 cells), and CCR5 knockdown is measured in GFP-positive transduced cells (Figure 3a). We found that both T1 and T7 targeting sequences were much more efficient in downregulating CCR5 in the mirGE than that in the miR-30 backbone (Figure 3b) and that T7 was superior to T1 in the same hairpin context.
Figure 2.

Schematic representations of miR-30 and mirGE designs. (a) miR-30 design as it has been previously described.1,14,17 Main features include 11 bp lower stem, upper double-strand stem composed of the fully complementary targeting strand (bottom strand, dotted line), 3′ mismatch of targeting strand as well as a weak AU bond on the 5′ of the targeting strand. (b) mirGE backbone developed in this study. Main features include miR-16 flanking regions, upper double-strand stem composed of the fully complementary targeting strand (top strand, dotted line), a 13 bp lower stem, a wobble at position 12 of the upper stem (position 1 is the first 5′ base of the targeting strand), a 5′ mismatch, and no 3′ mismatch on the targeting strand.
Figure 3.
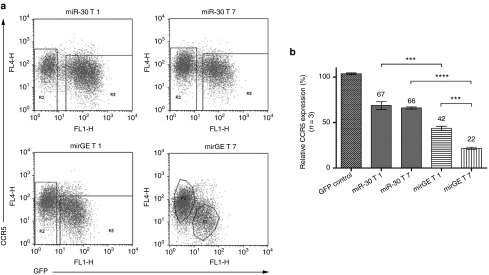
Downregulation of CCR5 in HR5 cells using lentivectors expressing different anti-CCR5 miRNA target sequences in different hairpin contexts. (a) FACS plots showing representative data from three independent experiments performed with three independent lentivector preparations. HR5 cells were transduced with lentivectors expressing either GFP alone (GFP control vector, not shown) or GFP followed by miR-30 or mirGE hairpins containing the T1 or the T7 targeting sequence (see Results and Materials and Methods for details). For determination of MFI (mean of fluorescence intensity) of transduced versus untransduced cells, square gates were used by default, and polygonal gates were used when populations were overlapping in one or two axes. MFI of GFP for the transduced population (R3) for the miR-30 T1 condition was: 150 (percentage transduction 66%), miR-30 T7 condition: 103.5 (percentage trandsuction 46%), mirGE T1 condition: 47.8 (percentage transduction 48%), and mirGE T7 condition: 30.4 (percentage transduction 23%). (b) Bar graph analysis of data in a. CCR5 downregulation was calculated by dividing the CCR5 MFI value of GFP-positive cells (transduced cells, gate R3) by the CCR5 MFI value of GFP-negative cells (untransduced cells, gate R2), within each sample. This allows for compensation of sample-to-sample variations due to slight changes in CCR5 antibody-to-cell ratios. The relative CCR5 expression in transduced cells was then displayed as percentage of CCR5 expression normalized to the internal control provided by untransduced cells. Values represent average from n = 3 independent experiments. ***P < 0.001, ****P < 0.0001 using one-way ANOVA followed by Bonferroni's mulitple comparison test. ANOVA, analysis of variance; GFP, green fluorescent protein.
Note that the mean fluorescence intensity (MFI) of GFP decreases more in parallel with the knockdown efficiency (Figure 3, in particular in mirGE-transduced cells). This is due to the fact that—in this construct—the hairpins are within the 3′UTR of the mRNAs coding for GFP (see Supplementary Figure S3). Therefore, efficient miRNA processing removes poly-A tail leading to destabilization of mRNAs coding for GFP.33 A similar finding was described by Sun et al.20
This set of experiments indicates that T7 is a better CCR5 target than T1 and that the mirGE backbone is more efficiently processed by Drosha than the miR-30 backbone.
Effect of miRNA backbone and hairpin number on CCR5 knockdown
We then analyzed the knockdown potential of T7 targeting sequence in HR5 cells when miRNA hairpins are expressed as multiple copies on a single mRNA, either in miR-30 or mirGE context. For this, we used a system that allowed us to clone several copies of miRNA hairpins in tandem in a single lentivector backbone. This strategy, depicted in Supplementary Figure S3, is largely inspired from Sun et al.20
Since our ultimate goal is to apply miRNA-based gene silencing for clinical applications, we analyzed experiments performed in conditions which represent the safest transduction rate for gene therapy, i.e., one copy of transgene per cell. According to Poisson's law, when less than 20% of cells are transduced, the majority of transduced cells contain only one copy of the lentivector transgene. So, not only it reproduces the most desirable situation for clinical application but also it facilitates the comparison of different constructs between each other.
We first assessed the efficiency of the miR-30 backbone containing one or two T7 constructs (Figure 4). They gave unsatisfactory results with a knockdown of CCR5 of 16 and 33%, respectively. In contrast, a single hairpin of T7 in the mirGE context was already more efficient than two copies of miR-30 with a knockdown of 45 compared with 33%. Increasing the numbers of hairpins in mirGE to two or three resulted in 80 or 91% knockdown, respectively.
Figure 4.
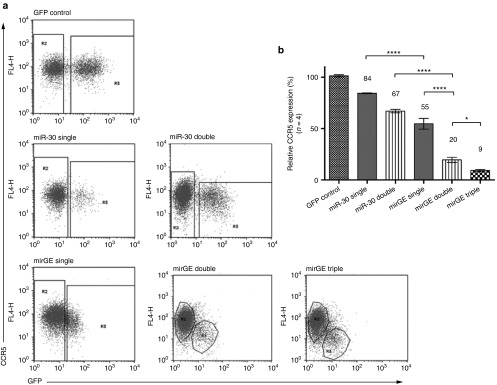
Downregulation of CCR5 in HR5 cells using lentivectors containing various copies of different anti-CCR5 miRNA constructs. (a) FACS plots showing representative data from four independent experiments (n = 4). HR5 cells were transduced with various concentrations of lentivectors expressing either GFP alone (GFP control vector) or GFP followed by one or several copies of a given miRNA backbone containing the T7 targeting sequence. The T7 targeting sequence was inserted in one or two copies in the miR-30 context, and in one, two, or three copies in the mirGE context. For further analysis, only FACS plots displaying less than 20% transduced cells (hence mostly single-copy GFP-positive cells) were retained. For determination of MFI (mean of fluorescence intensity) of transduced versus untransduced cells, square gates were used by default and polygonal gates were used when populations were overlapping in one or two axes. MFI of GFP for the transduced population (R3) for the GFP control condition was: 195.7 (percentage transduction 38.3%), miR-30 single: 98.8 (percentage transduction 7.6%), miR-30 double: 62.37 (percentage transduction 19.7%), mirGE single: 36 (percentage transduction 13.7%), mirGE double: 18.5 (percentage transduction 7.1%), and mirGE triple: 12.9 (percentage transduction 9.1%). (b) Bar graph analysis of data in a. CCR5 downregulation was calculated as described in Figure 3. The relative CCR5 expression in transduced cells is displayed as percentage of CCR5 expression normalized to internal control untransduced cells. Values represent average from n = 4 independent experiments. ****P < 0.0001 using one-way ANOVA followed by Bonferroni's mulitple comparison test. ANOVA, analysis of variance; GFP, green fluorescent protein.
As observed in Figure 3, higher knockdown efficiency of mirGE backbones was correlated with a more important decrease in GFP fluorescence as measured by MFI of GFP, again suggesting that a better Drosha processing results in fewer copies of translatable mRNA. When the mRNA only contains the GFP coding sequence, GFP MFI is 195. When containing GFP and one copy of miR-30 hairpin, the GFP MFI drops to 99 and when containing GFP and one copy of mirGE, GFP MFI drops to 36. From this, one can estimate that only 50% of all mRNA containing one miR-30 hairpin is processed and available for RNA interference, whereas when one copy of mirGE is present, more than 80% of mRNAs are processed and available for RNA interference.
High-throughput sequencing of mirGE and miR-30 small RNA products
The lower Drosha processing of miR-30 could suffice to account for its lower knockdown efficiency as compared with mirGE. However, another scenario can also contribute to the lower efficiency of miR-30. As discussed for Figure 1, if Drosha processes miR-30 after ~13 bp of lower stem structure, it will generate a targeting strand that will miss two nucleotides complementary to the target sequence at its 3′ end and will also have an extra AU artificial sequence at its 5′ end (Figure 2a).
To investigate this cleavage issue, we performed a high-throughput sequencing analysis of the small RNAs generated by miR-30 and mirGE constructs. As described in Materials and Methods, we purified 15 to 40 nucleotide long fragments before sequencing, in order to analyze mostly RNAs that are incorporated into RISC. The raw counts of reads from this sequencing are shown in Supplementary Table S1 and can also be downloaded as described in Materials and Methods. As shown in Supplementary Table S2, the percentage of reads assigned to either miR-30 or mirGE are comparable, i.e., 2.33 versus 2.21%, respectively. Since mirGE cells are transduced at 23% and miR-30 cells are transduced at 43%, and according to Poisson's law, the average copy number in mirGE cells can be estimated at 0.23, whereas the average copy number in miR-30 cells can be estimated at more than 0.4. This indicates that mirGE can produce approximately two times more targeting strands than miR-30. This is in accordance with GFP intensities observed in Figure 4, suggesting that Drosha cleavage is more efficient on mRNAs containing mirGE hairpins than on mRNAs containing miR-30 hairpins.
Moreover, the percentage of reads where the targeting strand has an incorrect start position is 7.53 for miR-30 and 0.16 for mirGE, indicating that overall processing of RISC-incorporated targeting strands is ~50 times better for mirGE than that for miR-30. This is confirmed in the mirGE_triple sample, where the transduction rate is high, 90% positive cells corresponding to an estimated average copy number of more than five. In this case, even though the reads assigned to mirGE represent more than two-thirds of all cellular reads (69.8%), the percentage of reads with an incorrect start position is even lower than in mirGE_single (0.06 versus 0.16%, respectively). This suggests that the mirGE backbone can be processed with higher efficiency and higher precision than miR-30, even when representing the majority of all Drosha-processed miRNAs. We further investigated the issue of cleavage accuracy for the mirGE or miR-30 backbone. As shown in Figure 5, the Drosha cleavage occurs at the correct position both in mirGE and miR-30 although the accuracy with which this occurs is very different when comparing the two backbones (discussed below). This is surprising for miR-30 since its lower stem is two nucleotides shorter than that usually seen in miRNAs.4 Also, Dicer cutting occurs mostly at the expected site so the vast majority of the reads covers the targeting strand, whether it is generated by mirGE or by miR-30.
Figure 5.

High-throughput sequencing of short RNAs generated by miR-30 and mirGE and miRNA constructs. (a) Schematic diagram of the miR-30 miRNA containing the CCR5 T7 target. Targeting strand is in red (bottom strand). Position and numbering of reads major starts and reads major ends of coverage (see Supplementary Table S1 for details) are indicated by arrows. (b) Schematic diagram of the mirGE miRNA containing the CCR5 T7 target. Targeting strand is in red (top strand). Position and numbering of reads major starts and reads major ends of coverage (see Supplementary Table S1 for details) are indicated by arrows. (c) Coverage plot of reads aligning to the targeting strands of miR-30 or mirGE hairpins sequences. Base 1 corresponds to nucleotide 72 for miR-30 and to nucleotide 26 for mirGE. (d) Summary of analysis of reads covering the targeting or passenger strand sequences (see text and Supplementary Table S1 for details).
However, this analysis identified two further points where mirGE was superior to miR-30. First, there is a 10-fold difference in the ratio of RISC incorporation of targeting versus passenger in favor of mirGE over miR-30. As shown in Supplementary Table S1 and Figure 5, for mirGE_single sample, there is a count of 18,253 reads at position 26 versus no read at position 65 or around. Thus, a ratio of 14,823 could only be calculated with the mirGE_triple sample (2,638,464 reads starting at position 26 over 178 reads starting at position 65). For miR-30, there are 44,853 reads of starting at position 72 over 33 reads starting at position 33, giving a ratio of targeting strand/passenger strand of 1,359. Second, Drosha cutting is 500–1,000-fold more precise in mirGE than that in miR-30. As shown in Supplementary Table S1 and Figure 5, for mirGE_single sample, there is a count of 18,253 reads at position 26 and a total of five reads proximal but outside position 26, giving a ratio of 0.00027 of incorrect cuts. A similar ratio was obtained from the mirGE_triple sample, with 2,638,464 reads at position 26 and a total of 1,194 reads proximal but outside position 26, giving a ratio of 0.00045. Of note, even when reads are coming from mirGE processing of a triple hairpin and amount to more than two-thirds of all trimmed reads, Drosha still manages to be extremely accurate in mirGE processing. On the contrary, miR-30 processing by Drosha is 500–1,000 times less precise with a ratio of 0.21 of incorrect cuts (33 reads at position 33 and 7 reads at position 34). When applying this calculation to Dicer processing, miR-30 appears to be processed with an accuracy that is six times higher than that for mirGE, with ratios of incorrect cuts of 0.08 and 0.49, respectively. Such a difference is hard to explain since both hairpins have the same loop to present to Dicer.
Taken together, this high-throughput sequencing analysis shows that mirGE is superior to miR-30 in four aspects: yield of targeting strand incorporation into RISC; percentage of RISC-incorporated targeting strands with the correct sequence; precision of cleavage by Drosha; and ratio of targeting strand over passenger strand. It thus provides molecular basis to account for the superiority of mirGE over miR-30 in knockdown efficiency.
Effect of CCR5 knockdown on HIV infection in vitro
Finally, we wanted to determine whether the CCR5 knockdown induced by transduction of cells with a single copy of lentivector containing an optimized anti-CCR5 miRNA design was efficient enough to render cells resistant to HIV infection. For this, we used a HIV-permissive clone of HeLa cells expressing both CD4 and CCR5 (HR5 cells, see Materials and Methods section). We transduced the HR5 cells with varying amounts of lentivectors, containing GFP and triple hairpin anti-CCR5 mirGE, or GFP only as negative control (Figure 6a). Again, we included a condition with ≤20% of the cells transduced (mirGE 2 µl = 13% and 1 µl = 6%), hence observing the effect of single-copy transgene expression. Cells were seeded at 20% confluence 1 day prior to infection with the CCR5 tropic HIV strain, YU-2. All cells which were either mock-transduced or transduced with the GFP control lentiviral vector were either dead or showed massive syncytia induction 5 days postinfection (Figure 6a). In contrast, a subpopulation of the cells previously transduced with the anti-CCR5 mirGE lentivector survived the HIV infection and further proliferated. Monitoring of the HIV p24 antigen in the supernatants reflected the microscopic findings with lower levels of p24 antigen in the cells transduced with the anti-CCR5 mirGE (Figure 6b). At day 5, we observed syncytia induction in close proximity to apparently healthy cells. At day 7, the positive selection for the HIV resistant cells is evident. Phenotypic analysis of these surviving cells shows that they have lost CCR5 expression (Figure 6g). The cells which survived this first infection with R5-tropic HIV were rechallenged with the same HIV strain. Corroborating our observation that these cells have lost CCR5 expression, they were entirely HIV resistant (Figure 6c). Finally, we wanted to verify that the two rounds of R5-tropic HIV infection did not select for cells that were resistant to HIV infection in general. For this, we infected the surviving cells with an X4-tropic HIV strain. As shown in Figure 6d, these cells can support X4-tropic HIV replication at the same level as naive HR5 cells. Thus, no nonspecific HIV resistance appears to have developed in the process. We also performed a phenotypic analysis of cells that survived R5-tropic infection and compared them with the subpopulation of cells transduced with the anti-CCR5 lentivector before infection. As shown in Figure 6f, the transduced cells represent 6% of the total population, which implies that the vast majority contain only one copy of the transgene. When compared with the internal negative control provided by the untransduced cells whose CCR5 MFI is 114, the knockdown ratio is more than 10-fold, as also shown in Figure 4b. The same phenotypic analysis performed 7 days after HIV R5 infection shows a dramatic enrichment, yielding to a population where CCR5 expression is virtually absent (Figure 6g).
Figure 6.

Resistance to R5-tropic HIV infection of HR5 cells transduced with anti-CCR5 mirGE lentivector. (a) Representative images of control parental HeLa cells (HeLa), untransduced HR5 (HR5), HR5 cells transduced with a lentivector expressing GFP alone (GFP), and HR5 cells transduced with a lentivector expressing GFP and three hairpins of anti-CCR5 mirGE (mirGE). Pictures of these four cell lines were taken at day 0, day 5, and day 7 of HIV-R5 infection. Only parental HeLa cells and mirGE-transduced cells survive the infection. Note that mirGE 1 µl indicates cells transduced with the lowest volume of vector corresponding to a 7% transduction rate. (b) Values of HIV p24 antigen in supernatants of cells showed in Figure 6a, collected over 6 days post-R5 HIV infection. (c) Values of HIV p24 antigen collected over 6 days post-R5 HIV infection, in supernatants of mirGE cells that survived the first HIV challenge in Figure 6b. Negative and positive controls for HIV replication were provided by naive HeLa and HR5 cells, respectively. (d) Values of HIV p24 antigen collected over 6 days post X4 HIV infection, in supernatants of mirGE cells that survived the first and second HIV challenges in Figure 6b,c. Negative control for X4 HIV infection was provided by naive CD4-negative parental HeLa cells and a fresh batch of naive HR5 cells served as the positive control for infection. (e) Phenotypic analysis of HR5 cells transduced with GFP control lentivector (no miRNA). (f) Phenotypic analysis of HR5 cells transduced with GFP-anti-CCR5 mirGE lentivector prior to R5-tropic HIV infection. Within the untransduced cells in gate R2 (89%), the CCR5 and GFP MFIs are 144 and 2.4, respectively. Within the transduced cells in gate R3 (6%), the CCR5 and GFP MFIs are 14 and 18, respectively. (g) Phenotypic analysis of HR5 cells from (F) 7 days post infection with R5-tropic HIV. The majority of cells (81%) are CCR5-negative. The CCR5 and GFP MFIs in this population (gate R3) are 3 and 48, respectively.
To further check if our anti-CCR5 lentivector could inhibit HIV infection in natural target of the virus, we performed HIV-R5 infection in human monocyte-derived macrophages (MDMs) with or without prior transduction with the anti-CCR5 T7 triple hairpin lentivector described above. MDMs were matured in culture for 8 days as described in the Materials and Methods section. In order to maximize R5-tropic HIV infection, MDMs were treated with VPX-virus-like particles for 2 hours prior to transduction, as described previously.34 The MDMs were then transduced with various quantities of either a control GFP vector or the anti-CCR5 lentivector, followed by infection with R5-tropic HIV-luciferase virions after 4 days (see Materials and Methods for details). As shown in Figure 7, we can see a strong inhibition of HIV infection using anti-CCR5 lentivector at a MOI of 1.7. Other conditions using lentivectors, i.e., lower MOI of anti-CCR5 lentivector or control GFP lentivector, display a moderate inhibition of HIV infection, as compared with the no vector condition. This is most likely due to the triggering by defective particle of cell-autonomous innate immune systems independent of SAMHD1 (see ref. 35 for review).
Figure 7.

Resistance to R5-tropic HIV infection of monocyte-derived macrophages (MDMs) transduced with anti-CCR5 mirGE lentivector. (a) Bar graphs showing the level of infection of MDMs by recombinant R5-tropic HIV particles encoding luciferase (see Materials and Methods for details). Prior to infection, MDMs were either not transduced (no vector) or transduced with various amounts of anti-CCR5 mirGE lentivector (CCR5 KD) or with a control vector expressing only GFP (GFP). Vector stocks (CCR5 KD and GFP) were normalized and 1 µl of vector corresponds to a MOI of 1.7. Experiments consisted of a minimum of three technical replicates. Luciferase activity: no vector 1,250 (SEM ± 248), 0.1 µl CCR5 knockdown (KD) vector 811 (SEM ± 195), 0.1 µl GFP vector 711 (SEM ± 137), 1 µl CCR5 KD vector 69 (SEM ± 44), and 1 µl GFP vector 791 (SEM ± 18). P values (**P < 0.0099, **P < 0.0038, and ***P < 0.0004) were obtained using a two-tailed unpaired t-test. (b,c) Light microscope images of macrophages 4 days posttransduction with 1 µl of anti-CCR mirGE vector or 1 µl of GFP vector, respectively (×20 magnification).
Altogether, these data show that our anti-CCR5 lentivector can efficiently block HIV infection at low copy number in both artificial and natural target cells.
Discussion
The work presented here is the result of an ambitious goal: efficient gene knockdown with a single copy of miRNA-containing lentivector. We could achieve this goal by optimization of critical miRNA features, combined with multimerization of miRNA hairpins in a single cassette. When applied to human CCR5, we reached a knockdown efficiency which provides the ground for a clinically applicable approach toward cellular immunization against HIV.
When we designed our knockdown lentivector constructs by implementing the miR-30 protocols from institutional online tools (http://hannonlab.cshl.edu), these constructs hardly reduced the level of CCR5 expression by one-third (Figure 3, miR-30 T1 and miR-30 T7). This was surprising and disappointing since T7 has been described as one of the most potent CCR5 RNAi targets.32 Notably, cells with multiple copies of anti-CCR5 T7 miR-30 did show efficient CCR5 knockdown. Clearly, one cannot expect any phenotype with this level of knockdown and therefore no clinical applicability.
A careful review of articles about Drosha processing and RISC incorporation identified three potential features critical for optimal miRNA-based silencing: (i) length of the lower stem for optimal Drosha processing, (ii) destabilization of the 5′ end of the targeting strand for optimal RISC incorporation, and (iii) a mismatch in the middle of the upper stem to avoid abortive processing by Drosha.4,12,15,19,20,36,37,38
When these three features were incorporated in our mirGE hairpin design, the knockdown efficiency improved dramatically compared with miR-30-based hairpin designs, whether we targeted GFP or CCR5 (Figures 1 and 3). The level of knockdown with a single mirGE hairpin construct, however, still did not reach a level of downregulation compatible with clinical applications, i.e., >90%. We reached this threshold by tandem addition of hairpins, achieving 90% knockdown with a single copy of a lentivector containing a triple anti-CCR5 mirGE construct (Figure 4). These tandem repeats do not seem to be subject to recombination (data not shown) and are thus a safe alternative to the accumulation of integrated proviral copies in the target cell genome.
To investigate the mechanisms underlying the superiority of mirGE over miR-30, we performed a high-throughput sequencing of short RNAs generated by the two miRNA backbones (Figure 5). We found that mirGE performed better than miR-30 in terms of yield of targeting strand incorporation into RISC as well as percentage of correct targeting strands incorporated into RISC. Also, the ratio of targeting strand over passenger strand is higher for mirGE than that for miR-30. We also found that Drosha cutting on miR-30 was quite imprecise, with 21% of cuts outside of the correct position. On the contrary, Drosha cutting on mirGE was very precise (500–1,000-fold more precise than that for miR-30) and keeps the same level of accuracy even when mirGE is expressed at high levels and in a triple hairpin context.
Surprisingly, Dicer cutting was not very precise, ranging from 8 to 49% of incorrect cuts for miR-30 and mirGE, respectively. This level of inaccuracy is comparable to Drosha cutting on miR-30 (21%). One possibility is that the miR-30 loop that we kept in our mirGE design is also not optimal. We plan on testing other loop sequences based on features affecting enzymatic processing of hairpin transcripts as described elsewhere.39
Taken together, our results show a clear superiority of mirGE over miR-30 in terms of processing, cleavage accuracy, targeting strand incorporation, and, most of all, knockdown efficiency. Also, in mirGE, the upper strand is incorporated in RISC, whereas in miR-30, it is the lower strand. In mirGE, Drosha cutting generates the 5′ end of the guide strand and is extremely precise, whereas in miR-30, the terminal loop processing, whether done by Dicer or single-strand RNAses, generates the 5′ end of the guide strand and is imprecise and requires further optimization. Finally, in the meantime, we have applied the mirGE design to other target genes (Caveolin-1, VEGF, and Wnt5a, data not shown) and found equally efficient knockdown efficiency, allowing gene repression at low copy number. We thus propose to use the mirGE design as default design for miRNA-based knockdown applications.
Using a triple anti-CCR5 mirGE construct to protect Hela R5 cells against HIV infection in vitro, we showed that a subpopulation of cells transduced at levels lower than 10%, hence mostly containing only one copy of anti-CCR5 mirGE,27 could resist HIV R5 infection (Figure 6). Rechallenging these surviving cells with HIV R5 did not show any virus replication, whereas the same cells remained permissive to HIV X4 infection, proving the specificity of the anti-CCR5 cellular immunization. To further validate the protective effect of our anti-CCR5 constructs, we tested it in natural targets of HIV R5, i.e., human macrophages (Figure 7). We found that our construct, even at low MOI, efficiently and specifically protected macrophages against HIV-R5 infection. This protective effect in primary human cells was later confirmed in humanized mice (Myburgh et al., manuscript in preparation).
Altogether, these results can be compared in terms of efficiency to the work of Ringpis et al.40 However, they used an shRNA-based vector which may be less desirable for clinical application due to potential adverse effect of shRNA-related toxicity.8,9,11
In conclusion, this work provides the ground to ultimately build a genetic tool that can be used to confer HIV resistance in patients. Also, the efficiency of our system provides a universal tool to knockdown any clinically relevant target gene without sacrificing safety by requiring multiple transgene integrations. Finally, we provide here an algorithm (see Supplementary Material) as well as a set of genetic tools for everyone to design “optimized” miRNA against any target gene.
Materials and methods
Construction of miRNA-containing plasmids and lentiviral vectors. To construct the plasmid intermediates containing the various miRNA hairpins, we followed a protocol largely inspired by Sun et al.20 with the following modifications. An amplicon containing the miRNA hairpin flanked by sequences for digestion with restriction enzymes was generated by high-fidelity PCR using Herculase II polymerase (Agilent, Santa Clara, CA). The oligos for PCR template and primers were obtained from Microsynth (Balgach, Switzerland) and then Sigma (St Louis, MO). The templates for miR-30 hairpins had inside sequences specific for gene targeting and common outside sequences for annealing with miR-30 primers (see Supplementary Material). The mirGE hairpins were amplified from similar templates but with common outside sequences for annealing with mirGE primers (see Supplementary Material). PCR products were digested with BamHI and XbaI restriction enzymes (New England Biolabs, Ipswich, MA) and ligated into a pENTR-derived plasmid (Invitrogen) using BamHI and XbaI sites located directly, either downstream of the MGST2 gene (Genbank Access: U77604.1) for miRNAs targeting GFP (see Supplementary Figure S1a) or downstream of the GFP gene for miRNAs targeting hCCR5 (Supplementary Figure S3a). Ligation was performed using T4 DNA ligase (New England Biolabs). The pENTR-GFPmir plasmid maps and sequences are available at our institutional website (http://lentilab.unige.ch). The amplicon parts of each clone were verified by sequencing. For constructs containing multiple hairpins, PCR products were inserted using BamHI and SpeI sites as depicted in Supplementary Figure S3a and described before by Sun et al.20 (CCR5 knockdown). This allows for the insertion of multiple miRNAs where each new addition was verified by sequencing. The final lentivector plasmid was generated by an LR Clonase II (Invitrogen, Carlsbad, CA)-mediated recombination of a pENTR plasmid containing the human ubiquitin promoter (pENTR-L4-UBI-L1R) and a pCLX-R4-DEST-R2 lentivector destination cassette (Supplementary Figures S1b and S3b). For GFP knockdown experiments, a specific lentivector destination cassette (pCWX-R4-DEST-R2-PC) was used, containing an additional transcription unit with the human phosphoglycerate kinase 1 promoter driving the mCherry living color, as described in the text. All maps and sequences of the plasmids used in this study are available at our institutional website (http://lentilab.unige.ch). The GFP target sequence—AAGAACGGCATCAAGGTGAACT—was taken from previous publication.41 The human CCR5 (Genbank NM_000579.3) target sequences were chosen using the online web server at http://rna.tbi.univie.ac.at/cgi-bin/RNAxs and from previously published article.32 The two CCR5 target sequences retained were named target 1 (T1) 5′-TTTCCATACAGTCAGTATCAAT and target 7 (T7) 5′-AAGTGTCAAGTCCAATCTATGA. Sequences of templates for various anti-GFP miRNA, anti-CCR5-T1, and anti-CCR5-T7, used for PCR amplification, as well as the full algorithm used to design optimized upper and lower strands, are given in Supplementary Material section.
Lentiviral vector production and titration. Lentiviral vector stocks were generated using transient transfection of HEK 293T/17 cells with the specific lentivector transfer plasmid, the psPAX2 plasmid encoding gag/pol and the pCAG-VSVG envelope plasmid, as previously described.42,43 Lentivector titer was performed using transduction of HT-1080 cells followed by flow cytometry quantification of GFP-positive (or mCherry positive) cells 5 days after infection, as previously described.42,43
Cell culture and knockdown analysis. All cell lines were cultured in high-glucose Dulbecco's modified eagle medium (Sigma) supplemented with 10% fetal calf serum, 1% Penicillin, 1% Streptomycin, and 1% l-glutamine. For GFP knockdown studies, the 4.5 cell line28 containing one copy of a GFP-expressing lentivector was used. For each knockdown assay, cells were analyzed 5 days after transduction. For CCR5 knockdown studies, a subclone of Hela-derived TZM-bl cells (AIDS Repository, Germantown, MD), expressing CD4 and high levels of human CCR5, named here HR5, was used. CCR5 and CD4 quantification was performed using an APC and PE Cy7-labeled antibody, respectively (BD Pharmingen Cat. 550856; Franklin Lakes, NJ, Biolegend Cat. 300511; San Diego, CA), followed by flow cytometry analysis using FACS Vantage (Becton Dickinson, Franklin Lakes, NJ) or MoFlow Astrios (Beckman Coulter, Brea, CA).
High-throughput sequencing of miRNA cleavage products. The samples were processed following the illumina TruSeq Small RNA protocol (www.illumina.com). In short, total RNA (2 µg per sample) from R5 cells transduced with lentivectors expressing either mirGE-T7 or miR-30 T7 miRNAs was extracted using TRIzol (Life Technologies, Carlsbad, CA). After validation of RNAs, 1 µg of total RNA from each sample was ligated to specific 3′ and 5′ adapters. In a subsequent reverse transcription step, single-stranded cDNA was generated, followed by PCR amplification and gel purification that selecting constructs holding specific RNA fragments with an approximate length of 22 nucleotides (range: 15–40). Reads were sequenced on the illumina HiSeq platform with a length of 50 bp. Partial adapter sequences were trimmed using Flexbar v2.4 (http://sourceforge.net/projects/flexbar/?source=navbar). The trimmed reads were aligned to the hairpin sequences of miRBase v20 (http://www.mirbase.org) and the sequences of the mirGE and miR-30 constructs. Alignment was done with Bowtie with the options “--chunkmbs 1024 -e 50 -a -m 50 --best --strata --nomaqround –y”. We counted only the number of reads that aligned to the positive strand of the sequences. Raw data of this study have been uploaded to the European Nucleotide Archive and can be accessed at this link: http://www.ebi.ac.uk/ena/data/view/PRJEB7175.
R5-tropic and X4-tropic HIV infection of HR5 cells. Viral stocks were obtained by polyethylenimine-mediated transfection (Polysciences, Warrington, PA) of 293T cells with pYU-2 or pNL4-3 (provided through the NIH AIDS Research and Reference Reagent Program). Forty-eight hours after transfection, virus was harvested, filtered (0.45 µm), and frozen at −80 °C until use. Virus titers were determined as previously described.44 Briefly, TCID50 (tissue culture infectious dose 50%) was determined by infecting human CD8+T cell depleted peripheral blood mononucleated cell from three donors which were stimulated by PHA and anti-CD3 beads (Dynal 11131D; Life Technologies). Then, viral stocks were adjusted to 1 × 106 TCID50/ml, aliquoted, and frozen at −80 °C before use. HR5 cells were infected for 6 hours at an MOI of 2.5, washed three times with phosphate-buffered saline, and then cultured in Dulbecco's modified eagle medium (Invitrogen) with 10% fetal calf serum (PAA Laboratories, Piscataway, NJ), 1% Penicillin/Streptomycin (Invitrogen Life Technologies), and 1% l-glutamine (Invitrogen Life Technologies). Assays for HIV p24 antigen levels in cell culture supernatants were performed using an in-house enzyme-linked immunosorbent assay as previously described.45
R5-tropic HIV-luciferase infection of MDM. VPX-encoding virus-like particles were produced essentially as described previously.46 Briefly, 293T cells were transfected using polyethylenimine (Sigma-Aldrich) with the VPX-containing pSIV3+ plasmid (provided by A. Cimarelli, Lyon) and the pMD2.G (VSV-G envelope) plasmid. The medium was changed after 16 hours, and supernatants were collected after 48 hours and filtered through 0.45 µm low protein binding syringe filters. MDMs were prepared as follows: monocytes from HIV seronegative donors were isolated from total peripheral blood after ACK lysis (Lonza, Basel, Switzerland) by CD14 microbeads (Miltenyi Biotec, Bergisch Gladbach, Germany). The isolated monocytes were matured to produce macrophages for 6 days with X-vivo-10 medium (Lonza) supplemented with 1,000 U/ml granulocyte-macrophage colony-stimulating factor and 100 ng/ml macrophage colony-stimulating factor. After 6 days, the medium was changed to X-vivo-10 w/o Cytokines for 2 days. VPX-encoding virus-like particles were added onto the cells for 2 hours, cells were washed, and then transduced overnight with the anti-CCR5 mirGE or control GFP lentivectors. The next day, the cells were washed and kept in culture for 4 days prior to infection with R5 tropic NL4-3/luciferase virus pseudotyped with ADA envelope virus for 6 hours. After 6 hours, cells were washed, and new medium added. Cells were lysed 24 hours after infection with the Cell Culture lysis reagent (Luciferase assay System; Promega, Madison, WI), and luciferase was measured with the MLX microplate luminometer (Dynex Technologies, Chantilly, VA).
Statistical analysis. The statistical analyses were performed using GraphPad Prism 5.04 (GraphPad Software, La Jolla, CA). We used one-way analysis of variance followed by Bonferroni's multiple comparison test as well as t-test (non parametric, Mann–Whitney U-test). In Supplementary Table S2, significant difference of incorrect targeting strand processing was determined using a proportion test (R prop.test; two-sample test for equality of proportions with continuity correction). For all tests, P < 0.05 was taken as statistically significant.
Algorithm for the design of mirGE-based amplicons for cloning. This algorithm is a compilation of several published and on-line references including refs. (47) and (48).
Copy your target sequence (ORF, 3′UTR, etc).
Use the three following web tools to select at least ten target sequences (http://www5.appliedbiosystems.com/tools/siDesign/; http://www.med.nagoya-u.ac.jp/neurogenetics/i_Score/i_score.html; http://rna.tbi.univie.ac.at/cgi-bin/RNAxs
Select candidates that are in overlapping pools.
Blast each 19mer to check that they are targeting no other gene other than your target gene.
Exclude “bad” candidates by following instructions of ref. (48) page 12.
Extend 19mer to 22mer by adding nucleotides from the original target sequence, either upstream or downstream, favoring a resulting complementary sequence that will have a T on the 5′ end.47 This 22mer is the basis for the following shRNA design. For human CCR5 mRNA (GenBank: NM_000579.3), the result is TTTCCATACAGTCAGTATCAAT (22mer).
Make a complementary strand to this 22mer, which will be the top strand of mirGE context, i.e the targeting strand. To be efficiently incorporated into RISC, the 5′ terminus A needs to flap, thus make a wobble on the complementary strand (Z) 5′-ATTGATACTGACTGTATGGAAA 3′-ZAACTATGACTGACATACCTTT
Make another wobble on the complementary strand corresponding to position +12 of the targeting strand, to suppress abortive processing.4 As a general rule: Antiparallel (targeting) sequence being in blue, 100% complementary to target mRNA (in red) 5′- NNNNNNNNNNNNNNNNNNNNNN 3′- ZNNNNNNNNNNZNNNNNNNNNN
Then choose Z to add to bottom oligo according to the N in top oligo facing Z. If N is T, make Z a C. If N is C, make Z an A. If N is A, make Z a C. If N is G, make Z an A.
Replace NNN stretches by sense and antisense oligos to make the PCR template oligo
Amplify this template with the 5′ and 3′ PCR oligos, as described above.
SUPPLEMENTARY MATERIAL Figure S1. Schematic diagram of the lentiviral vector plasmid used for the GFP knock-down experiments. Figure S2. Schematic representations GFP knock-down miRNA designs. Figure S3. Cloning strategy used to easily clone multiple miRNA hairpins in a lentiviral expression cassette. Table S1. Positions of starts and ends of coverage of short RNAs generated by mirGE and miR-30 miRNAs. Table S2. Numbers of total reads, reads assigned to miRNAs, and reads aligned to miR-30 or mirGE. Supplementary Material.
Acknowledgments
We thank Marc Giry-Laterrière for preliminary clonings and experiments involving the miR-30 constructs, Erika Schlaepfer for providing technical advice regarding in vitro HIV infection, Dominique Wohlwend for invaluable help in flow cytometry, Silvia Sorce for helpful comments and advice as well as preliminary experiments in hCCR5 cloning, Vincent Jaquet for helpful comments and advice, Jozsef Kiss for scientific and logistical support and members of the Kiss lab for helpful discussions. We also wish to thank Mylene Docquier (Genomics Platform of Geneva) for helpful discussion regarding the high-throughput sequencing. Marc Giry-Laterrière and P.S. were supported by the Chicago Diabetes Project (www.chicagodiabetesproject.org) in the seeding phase of the mirGE design. R.M. was supported by the University of Pretoria (South African National Research Foundation and the Medical Research Council of South Africa), Geneva and Zurich (Forschungskredit der Universität Zürich). K.H.K. was supported by the Swiss National Foundation Grant No. 3200A0-103725. R.F.S. is supported by the Clinical Research Focus Program of the University Hospital of Zurich.
The authors declare no competing financial interests.
Supplementary Material
References
- Zeng Y, Wagner EJ, Cullen BR. Both natural and designed micro RNAs can inhibit the expression of cognate mRNAs when expressed in human cells. Mol Cell. 2002;9:1327–1333. doi: 10.1016/s1097-2765(02)00541-5. [DOI] [PubMed] [Google Scholar]
- Han J, Lee Y, Yeom KH, Kim YK, Jin H, Kim VN. The Drosha-DGCR8 complex in primary microRNA processing. Genes Dev. 2004;18:3016–3027. doi: 10.1101/gad.1262504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cullen BR. RNAi the natural way. Nat Genet. 2005;37:1163–1165. doi: 10.1038/ng1105-1163. [DOI] [PubMed] [Google Scholar]
- Han J, Lee Y, Yeom KH, Nam JW, Heo I, Rhee JK, et al. Molecular basis for the recognition of primary microRNAs by the Drosha-DGCR8 complex. Cell. 2006;125:887–901. doi: 10.1016/j.cell.2006.03.043. [DOI] [PubMed] [Google Scholar]
- Abbas-Terki T, Blanco-Bose W, Déglon N, Pralong W, Aebischer P. Lentiviral-mediated RNA interference. Hum Gene Ther. 2002;13:2197–2201. doi: 10.1089/104303402320987888. [DOI] [PubMed] [Google Scholar]
- An DS, Xie Y, Mao SH, Morizono K, Kung SK, Chen IS. Efficient lentiviral vectors for short hairpin RNA delivery into human cells. Hum Gene Ther. 2003;14:1207–1212. doi: 10.1089/104303403322168037. [DOI] [PubMed] [Google Scholar]
- ter Brake O, ‘t Hooft K, Liu YP, Centlivre M, von Eije KJ, Berkhout B. Lentiviral vector design for multiple shRNA expression and durable HIV-1 inhibition. Mol Ther. 2008;16:557–564. doi: 10.1038/sj.mt.6300382. [DOI] [PubMed] [Google Scholar]
- Grimm D, Streetz KL, Jopling CL, Storm TA, Pandey K, Davis CR, et al. Fatality in mice due to oversaturation of cellular microRNA/short hairpin RNA pathways. Nature. 2006;441:537–541. doi: 10.1038/nature04791. [DOI] [PubMed] [Google Scholar]
- An DS, Qin FX, Auyeung VC, Mao SH, Kung SK, Baltimore D, et al. Optimization and functional effects of stable short hairpin RNA expression in primary human lymphocytes via lentiviral vectors. Mol Ther. 2006;14:494–504. doi: 10.1016/j.ymthe.2006.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson AL, Linsley PS. Noise amidst the silence: off-target effects of siRNAs. Trends Genet. 2004;20:521–524. doi: 10.1016/j.tig.2004.08.006. [DOI] [PubMed] [Google Scholar]
- Boudreau RL, Martins I, Davidson BL. Artificial microRNAs as siRNA shuttles: improved safety as compared to shRNAs in vitro and in vivo. Mol Ther. 2009;17:169–175. doi: 10.1038/mt.2008.231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stegmeier F, Hu G, Rickles RJ, Hannon GJ, Elledge SJ. A lentiviral microRNA-based system for single-copy polymerase II-regulated RNA interference in mammalian cells. Proc Natl Acad Sci USA. 2005;102:13212–13217. doi: 10.1073/pnas.0506306102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickins RA, Hemann MT, Zilfou JT, Simpson DR, Ibarra I, Hannon GJ, et al. Probing tumor phenotypes using stable and regulated synthetic microRNA precursors. Nat Genet. 2005;37:1289–1295. doi: 10.1038/ng1651. [DOI] [PubMed] [Google Scholar]
- Silva JM, Li MZ, Chang K, Ge W, Golding MC, Rickles RJ, et al. Second-generation shRNA libraries covering the mouse and human genomes. Nat Genet. 2005;37:1281–1288. doi: 10.1038/ng1650. [DOI] [PubMed] [Google Scholar]
- Zhou H, Xia XG, Xu Z. An RNA polymerase II construct synthesizes short-hairpin RNA with a quantitative indicator and mediates highly efficient RNAi. Nucleic Acids Res. 2005;33:e62. doi: 10.1093/nar/gni061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McManus MT, Petersen CP, Haines BB, Chen J, Sharp PA. Gene silencing using micro-RNA designed hairpins. RNA. 2002;8:842–850. doi: 10.1017/s1355838202024032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boden D, Pusch O, Silbermann R, Lee F, Tucker L, Ramratnam B. Enhanced gene silencing of HIV-1 specific siRNA using microRNA designed hairpins. Nucleic Acids Res. 2004;32:1154–1158. doi: 10.1093/nar/gkh278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng Y, Cullen BR. Structural requirements for pre-microRNA binding and nuclear export by Exportin 5. Nucleic Acids Res. 2004;32:4776–4785. doi: 10.1093/nar/gkh824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng Y, Cai X, Cullen BR. Use of RNA polymerase II to transcribe artificial microRNAs. Methods Enzymol. 2005;392:371–380. doi: 10.1016/S0076-6879(04)92022-8. [DOI] [PubMed] [Google Scholar]
- Sun D, Melegari M, Sridhar S, Rogler CE, Zhu L. Multi-miRNA hairpin method that improves gene knockdown efficiency and provides linked multi-gene knockdown. Biotechniques. 2006;41:59–63. doi: 10.2144/000112203. [DOI] [PubMed] [Google Scholar]
- Bauer M, Kinkl N, Meixner A, Kremmer E, Riemenschneider M, Förstl H, et al. Prevention of interferon-stimulated gene expression using microRNA-designed hairpins. Gene Ther. 2009;16:142–147. doi: 10.1038/gt.2008.123. [DOI] [PubMed] [Google Scholar]
- Glazkova DV, Vetchinova AS, Bogoslovskaia EV, Zhogina IuA, Markelov ML, Shipulin GA. [Downregulation of human CCR5 receptor gene expression using artificial microRNAs] Mol Biol (Mosk) 2013;47:475–485. doi: 10.7868/s002689841303004x. [DOI] [PubMed] [Google Scholar]
- Liu YP, Westerink JT, ter Brake O, Berkhout B. RNAi-inducing lentiviral vectors for anti-HIV-1 gene therapy. Methods Mol Biol. 2011;721:293–311. doi: 10.1007/978-1-61779-037-9_18. [DOI] [PubMed] [Google Scholar]
- Kumar M, Follenzi A, Garforth S, Gupta S. Control of HBV replication by antiviral microRNAs transferred by lentiviral vectors for potential cell and gene therapy approaches. Antivir Ther. 2012;17:519–528. doi: 10.3851/IMP2014. [DOI] [PubMed] [Google Scholar]
- Huang X, Jia Z. Construction of HCC-targeting artificial miRNAs using natural miRNA precursors. Exp Ther Med. 2013;6:209–215. doi: 10.3892/etm.2013.1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy SR, Chang CC, Dogbevia G, Bryleva EY, Bowen Z, Hasan MT, et al. Acat1 knockdown gene therapy decreases amyloid-β in a mouse model of Alzheimer's disease. Mol Ther. 2013;21:1497–1506. doi: 10.1038/mt.2013.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fehse B, Kustikova OS, Bubenheim M, Baum C. Pois(s)on–it's a question of dose. Gene Ther. 2004;11:879–881. doi: 10.1038/sj.gt.3302270. [DOI] [PubMed] [Google Scholar]
- Salmon P, Oberholzer J, Occhiodoro T, Morel P, Lou J, Trono D. Reversible immortalization of human primary cells by lentivector-mediated transfer of specific genes. Mol Ther. 2000;2:404–414. doi: 10.1006/mthe.2000.0141. [DOI] [PubMed] [Google Scholar]
- Lewis BP, Shih IH, Jones-Rhoades MW, Bartel DP, Burge CB. Prediction of mammalian microRNA targets. Cell. 2003;115:787–798. doi: 10.1016/s0092-8674(03)01018-3. [DOI] [PubMed] [Google Scholar]
- Schwarz DS, Hutvágner G, Du T, Xu Z, Aronin N, Zamore PD. Asymmetry in the assembly of the RNAi enzyme complex. Cell. 2003;115:199–208. doi: 10.1016/s0092-8674(03)00759-1. [DOI] [PubMed] [Google Scholar]
- Hibio N, Hino K, Shimizu E, Nagata Y, Ui-Tei K. Stability of miRNA 5'terminal and seed regions is correlated with experimentally observed miRNA-mediated silencing efficacy. Sci Rep. 2012;2:996. doi: 10.1038/srep00996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson J, Akkina R. Complete knockdown of CCR5 by lentiviral vector-expressed siRNAs and protection of transgenic macrophages against HIV-1 infection. Gene Ther. 2007;14:1287–1297. doi: 10.1038/sj.gt.3302958. [DOI] [PubMed] [Google Scholar]
- Fabian MR, Sonenberg N. The mechanics of miRNA-mediated gene silencing: a look under the hood of miRISC. Nat Struct Mol Biol. 2012;19:586–593. doi: 10.1038/nsmb.2296. [DOI] [PubMed] [Google Scholar]
- Hrecka K, Hao C, Gierszewska M, Swanson SK, Kesik-Brodacka M, Srivastava S, et al. Vpx relieves inhibition of HIV-1 infection of macrophages mediated by the SAMHD1 protein. Nature. 2011;474:658–661. doi: 10.1038/nature10195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Towers GJ, Noursadeghi M. Interactions between HIV-1 and the cell-autonomous innate immune system. Cell Host Microbe. 2014;16:10–18. doi: 10.1016/j.chom.2014.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paddison PJ, Caudy AA, Bernstein E, Hannon GJ, Conklin DS. Short hairpin RNAs (shRNAs) induce sequence-specific silencing in mammalian cells. Genes Dev. 2002;16:948–958. doi: 10.1101/gad.981002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paddison PJ, Cleary M, Silva JM, Chang K, Sheth N, Sachidanandam R, et al. Cloning of short hairpin RNAs for gene knockdown in mammalian cells. Nat Methods. 2004;1:163–167. doi: 10.1038/nmeth1104-163. [DOI] [PubMed] [Google Scholar]
- He L, Hannon GJ. MicroRNAs: small RNAs with a big role in gene regulation. Nat Rev Genet. 2004;5:522–531. doi: 10.1038/nrg1379. [DOI] [PubMed] [Google Scholar]
- Matveeva OV, Kang Y, Spiridonov AN, Saetrom P, Nemtsov VA, Ogurtsov AY, et al. Optimization of duplex stability and terminal asymmetry for shRNA design. PLoS One. 2010;5:e10180. doi: 10.1371/journal.pone.0010180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ringpis GE, Shimizu S, Arokium H, Camba-Colón J, Carroll MV, Cortado R, et al. Engineering HIV-1-resistant T-cells from short-hairpin RNA-expressing hematopoietic stem/progenitor cells in humanized BLT mice. PLoS One. 2012;7:e53492. doi: 10.1371/journal.pone.0053492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mottet-Osman G, Iseni F, Pelet T, Wiznerowicz M, Garcin D, Roux L. Suppression of the Sendai virus M protein through a novel short interfering RNA approach inhibits viral particle production but does not affect viral RNA synthesis. J Virol. 2007;81:2861–2868. doi: 10.1128/JVI.02291-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giry-Laterrière M, Cherpin O, Kim YS, Jensen J, Salmon P. Polyswitch lentivectors: “all-in-one” lentiviral vectors for drug-inducible gene expression, live selection, and recombination cloning. Hum Gene Ther. 2011;22:1255–1267. doi: 10.1089/hum.2010.179. [DOI] [PubMed] [Google Scholar]
- Giry-Laterrière M, Verhoeyen E, Salmon P. Lentiviral vectors. Methods Mol Biol. 2011;737:183–209. doi: 10.1007/978-1-61779-095-9_8. [DOI] [PubMed] [Google Scholar]
- McDougal JS, Cort SP, Kennedy MS, Cabridilla CD, Feorino PM, Francis DP, et al. Immunoassay for the detection and quantitation of infectious human retrovirus, lymphadenopathy-associated virus (LAV) J Immunol Methods. 1985;76:171–183. doi: 10.1016/0022-1759(85)90489-2. [DOI] [PubMed] [Google Scholar]
- Moore JP, McKeating JA, Weiss RA, Sattentau QJ. Dissociation of gp120 from HIV-1 virions induced by soluble CD4. Science. 1990;250:1139–1142. doi: 10.1126/science.2251501. [DOI] [PubMed] [Google Scholar]
- Nègre D, Mangeot PE, Duisit G, Blanchard S, Vidalain PO, Leissner P, et al. Characterization of novel safe lentiviral vectors derived from simian immunodeficiency virus (SIVmac251) that efficiently transduce mature human dendritic cells. Gene Ther. 2000;7:1613–1623. doi: 10.1038/sj.gt.3301292. [DOI] [PubMed] [Google Scholar]
- Matveeva OV, Nazipova NN, Ogurtsov AY, Shabalina SA. Optimized models for design of efficient miR30-based shRNAs. Front Genet. 2012;3:163. doi: 10.3389/fgene.2012.00163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matveeva O. What parameters to consider and which software tools to use for target selection and molecular design of small interfering RNAs. Methods Mol Biol. 2013;942:1–16. doi: 10.1007/978-1-62703-119-6_1. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.