

Summary
The MUC1 transforming protein is overexpressed by most human carcinomas. The present studies demonstrate that the MUC1 C-terminal subunit (MUC1 C-ter) localizes to mitochondria in HCT116/MUC1 colon carcinoma cells and that heregulin stimulates mitochondrial targeting of MUC1 C-ter. We also show that MUC1 attenuates cisplatin-induced (1) release of mitochondrial apoptogenic factors, (2) activation of caspase-3, and (3) induction of apoptosis. Moreover, knockdown of MUC1 expression in A549 lung and ZR-75-1 breast carcinoma cells by MUC1siRNA was associated with increased sensitivity to genotoxic drugs in vitro and in vivo. These findings indicate that MUC1 attenuates the apoptotic response to DNA damage and that this oncoprotein confers resistance to genotoxic anticancer agents.
Introduction
The apoptotic response of cells is induced by extrinsic and intrinsic pathways that activate the caspase family of cysteine proteases. The extrinsic apoptotic pathway is activated by ligand stimulation of the tumor necrosis factor (TNF) family of death receptors. Activation of caspase-8 by death receptor signaling results in cleavage of procaspase-3 (Boldin et al., 1996; Muzio et al., 1996; Stennicke et al., 1998). Caspase-8 also cleaves Bid, a proapoptotic member of the Bcl-2 family, and thereby stimulates release of mitochondrial cytochrome c to the cytosol (Li et al., 1998a; Luo et al., 1998). Activation of the intrinsic pathway by diverse Bid-independent stress signals is also associated with the release of mitochondrial cytochrome c (Kluck et al., 1997; Liu et al., 1996; Yang et al., 1997). In the cytosol, cytochrome c forms a complex with Apaf-1 and activates caspase-9 (Li et al., 1997; Srinivasula et al., 1998). Like caspase-8, caspase-9 can directly activate caspase-3 (Li et al., 1997). In turn, caspase-3 cleaves multiple proteins, which, when inactivated or activated by cleavage, contribute to the induction of apoptosis. Protein kinase Cδ(PKCδ) is one such caspase-3 substrate that is cleaved to a catalytically active fragment, the expression of which is sufficient to induce apoptosis (Emoto et al., 1995). Many genotoxic anticancer drugs induce apoptosis by activation of the intrinsic pathway (Herr and Debatin, 2001; Kroemer and Reed, 2000). Moreover, resistance to cytotoxic anticancer agents is often associated with defects in the intrinsic pathway (Bunz, 2001; Datta et al., 1995).
The human DF3/MUC1 transmembrane glycoprotein is expressed on the apical borders of normal secretory epithelial cells (Kufe et al., 1984). By contrast, transformation of epithelia to carcinomas is associated with marked overexpression of MUC1 throughout the entire cell membrane (Kufe et al., 1984). MUC1 is expressed as a cell surface heterodimer that consists of N-terminal (N-ter) and C-terminal (C-ter) subunits, which form a stable complex following cleavage of a single MUC1 polypeptide in the endoplasmic reticulum (ER) (Ligtenberg et al., 1992). The >250 kDa N-ter ectodomain contains variable numbers of 20 amino acid tandem repeats that are extensively modified by O-linked glycans (Gendler et al., 1988; Siddiqui et al., 1988). The ~20–25 kDa C-ter, which anchors the N-ter to the cell surface, consists of a 58 amino acid extracellular region, a 28 amino acid transmembrane domain, and a 72 amino acid cytoplasmic domain (CD) (Merlo et al., 1989). The MUC1-CD is phosphorylated on Y-46 by the epidermal growth factor receptor (EGFR), c-Src (Li et al., 2001a, 2001b), and Lyn (Li et al., 2003a). Other studies have shown that MUC1-CD is phosphorylated on S-44 by glycogen synthase kinase 3β(GSK3β) (Li et al., 1998b) and on T-41 by PKCδ(Ren et al., 2002). Phosphorylation on Y-46 and T-41 induces binding of MUC1-CD with the Wnt effector β-catenin (Li et al., 2001a, 2001b; Ren et al., 2002). Conversely, GSK3β-mediated phosphorylation of S-44 decreases the interaction of MUC1-CD and β-catenin (Li et al., 1998b). These findings have indicated that MUC1-CD functions in integrating signals from the EGFR and Wnt pathways.
Overexpression of MUC1 confers anchorage-independent growth and tumorigenicity of rodent fibroblasts and human epithelial cells (Li et al., 2003c; Ren et al., 2002). Other work has shown that, in addition to localization at the cell membrane, the MUC1 C-ter is expressed in nuclear complexes with β-catenin (Li et al., 2003a, 2003b, 2003c; Wen et al., 2003). Moreover, treatment of cells with heregulin (HRG), which activates ErbB2-4, is associated with targeting of MUC1 C-ter to the nucleus in a complex with γ-catenin (Li et al., 2003c). These observations have indicated that the function of MUC1 as a transforming protein may be mediated by regulating gene expression. In the present studies, we show that the MUC1 C-ter localizes to mitochondria. Significantly, the results demonstrate that MUC1 blocks activation of the intrinsic apoptotic pathway by DNA damaging agents. Our findings indicate that overexpression of MUC1 by human carcinomas contributes to the resistance of these cells to genotoxic agents.
Results
MUC1 C-ter localizes to mitochondria
MUC1-negative HCT116 cells were transfected to stably ex- press the empty vector, MUC1, or the MUC1(Y46F) mutant. Two clones (A and B) of each were selected from independent transfections. Immunoblot analysis with anti-MUC1 demonstrated no detectable expression of the MUC1 N-ter or C-ter subunits in the vector transfectants (Figure 1A). By contrast, MUC1 N-ter expression was similar in cells transfected with MUC1 or MUC1(Y46F) (Figure 1A). Similar levels of MUC1 C-ter were also found in the MUC1 and MUC1(Y46F) transfectants (Figure 1A). To assess whether MUC1 is expressed at the cell membrane, the transfectants were analyzed by flow cytometry with the anti-MUC1 N-ter antibody. In contrast to HCT116/ vector cells, MUC1 was detectable on the surface of HCT116 cells expressing MUC1 or MUC1(Y46F) (Figure 1B). To further define the distribution of MUC1, confocal microscopy was per- formed with antibodies against the MUC1 N-ter and C-ter. Both subunits were detectable at the cell membrane of the MUC1 transfectants (Figure 1C). Unexpectedly, however, the MUC1 C-ter, and not the N-ter, was also expressed in a pattern that suggested mitochondrial localization (Figure 1C). Indeed, colocalization of the MUC1 C-ter and MitoTracker supported targeting of MUC1 C-ter to mitochondria (Figure 1C). By contrast, there was substantially less mitochondrial localization of the MUC1(Y46F) C-ter (Figure 1C). Higher magnification and focusing of the images within a single HCT116/MUC1 cell showed clear localization of MUC1 C-ter at the cell membrane (Figure 1D, upper panels) and with MitoTracker throughout the mitochondrial network (Figure 1D, lower panels). Of note, detection of MUC1 C-ter at the cell membrane is obscured when focusing the confocal images on mitochondria (Figure 1D, lower panels). Moreover, within the cell, MUC1 C-ter expression is not restricted to mitochondria and, in addition to colocalization with MitoTracker, is detectable in the ER and cytosol (Figure 1D). To confirm these findings, mitochondrial lysates from the transfectants were subjected to immunoblot analysis with anti-MUC1 C-ter. The results demonstrate that the C-ter is detectable in the mitochondrial fraction from HCT116/MUC1, but not from HCT116/vector, cells (Figure 1E). Moreover, in concert with the confocal data, mitochondrial localization of the MUC1(Y46F) C-ter was considerably less than that found for the MUC1 C-ter (Figure 1E). Equal loading of mitochondrial lysates was confirmed by immunoblotting for the mitochondrial HSP60 protein (Figure 1E). Absence of the MUC1 N-ter indicated that the mitochondrial fraction was not contaminated with cell membranes (Figure 1E). Immunoblot mitochondrial lysates with antibodies against the cytosolic IκBα, nuclear PCNA, and ER-associated calreticulin proteins further indicated that the mitochondria are not significantly contaminated with these sub-cellular fractions (Figure 1E).
Figure 1. MUC1 C-ter localizes to mitochondria.

HCT116 cells were transfected to stably express the empty vector, MUC1, or MUC1(Y46F). Clones (A and B) were selected from two independent transfections.
A: Lysates were subjected to immunoblot analysis with anti-MUC1 N-ter (DF3), anti-MUC1 C-ter (Ab5), and anti-β-actin.
B: Cells were incubated with anti-MUC1 N-ter (open patterns) or control mouse IgG (closed patterns) and analyzed for immunofluorescence by flow cytometry.
C: Confocal microscopy of HCT116/Vector-A, HCT116/MUC1-A, and HCT116/MUC1(Y46F)-A cells stained with anti-MUC1 N-ter (DF3) or C-ter (Ab5). Mitochondria were stained with MitoTracker Red. Nuclei were stained with TO-PRO-3. Scale bars represent 20 μm in the upper three panels and 10 μm in the lower panel.
D: Higher magnification of an HCT116/MUC1-A cell stained with anti-MUC1 C-ter (Ab5), Mitotracker Red CMXRos, and TO-PRO-3. Confocal images were focused on cell membrane (upper panels) and mitochondrial (lower panels) staining with anti-MUC1 C-ter. Scale bars represent 10 μm.
E: Mitochondrial fractions from HCT116/vector-A, HCT116/MUC1-A, and HCT116/MUC1(Y46F)-A cells were subjected to SDS-PAGE and immunoblotting with the indicated antibodies. Whole-cell lysate (WCL) was included as a control for detection of the membrane-associated MUC1 N-ter, cytoplasmic IκBα, nuclear PCNA, and endoplasmic reticulum-associated calreticulin proteins.
Targeting of MUC1 C-ter to mitochondria is associated with HRG treatment
To compare MUC1 C-ter expression at the cell membrane with that in mitochondria, lysates from these fractions were subjected to immunoblot analysis with antibodies directed against the extracellular domain (ECD) and cytoplasmic domain (CD) (Figure 2A, upper panel). The results obtained with the Ab5 antibody, which reacts with the C-terminal 17 amino acids of MUC1 CD, demonstrated similar patterns for MUC1 C-ter expressed at the cell membrane and in mitochondria (Figure 2A). Reactivity with Ab5 was observed predominantly at 20–25 kDa (Figure 2A). Reactive bands were also observed at ~17 and 15 kDa (Figure 2A). Immunoblotting with the DF3E antibody, which was generated against the VETQFNQYKTEAAS motif in the MUC1 ECD (Li et al., 2001a), demonstrated reactivity with lysates from both the cell membrane and mitochondria (Figure 2A). Notably, reactivity of the DF3E antibody with the 20–25 kDa MUC1 C-ter and the 17 kDa fragment indicates that the 15 kDa fragment, as detected with Ab5, does not contain the DF3E epitope (Figure 2A). Another antibody generated against the MUC1 ECD, designated ECD1, reacted predominantly with the 20–25 kDa MUC1 C-ter in both the cell membrane and mitochondria. The DF3E and ECD1 antibodies react selectively with the HCT116/MUC1 and not the HCT116/vector cells (data not shown). These results suggest that the 17 and 15 kDa fragments represent MUC1 C-ter cleavage products without certain ECD sequences (Figure 2A). As controls, MUC1 N-ter expression was detectable only in the cell membrane fraction, and HSP60 expression was restricted to the mitochondrial fraction (Figure 2A). Moreover, as shown in Figure 1D, there was no detectable contamination of the mitochondrial fraction with IκBα, PCNA, or calreticulin (data not shown). To further assess localization of MUC1 C-ter in mitochondria, we digested purified mitochondria with trypsin. Treatment with trypsin at concentrations of 50 and 100 μg/ml had little if any effect on MUC1 C-ter as analyzed by immunoblotting with the Ab5, DF3E, and ECD1 antibodies (Figure 2B, left). As a marker protein of the mitochondrial outer membrane, the control and trypsin-treated mitochondria were immunoblotted with an antibody against Tom20, a component of the translocase of the outer membrane of mitochondria (TOM) (Iwahashi et al., 1997). As shown previously (Gotow et al., 2000), Tom20 levels were substantially decreased by trypsin treatment (Figure 2B, left). By contrast, trypsin had no effect on expression of the mitochondrial inner membrane protein Tim23 (Figure 2B, left). To determine whether MUC1 C-ter is integrated into mitochondrial membrane, the purified mitochondria were incubated in alkaline sodium carbonate. The extract was centrifuged, and the supernatant (luminal and peripheral proteins) and the pellet (integral membrane proteins) were analyzed by immunoblotting. The results show that MUC1 C-ter is detectable in the pellet fraction and not the supernatant (Figure 2B, right). Similar results were obtained for Tom20 and Tim23 (Figure 2B, right). As a control, the matrix HSP60 protein was readily solubilized by alkaline sodium carbonate treatment (Figure 2B, right). These results indicate that mitochondrial MUC1 C-ter is an integral membrane protein. To extend these findings, we expressed MUC1 C-ter with a GFP tag at the N terminus and assessed mitochondrial localization. Immunoblot analysis of mitochondrial lysates with anti-GFP and anti-MUC1 C-ter confirmed mitochondrial targeting of the GFP-tagged MUC1 C-ter fusion protein (Figure 2C, upper panels). As controls, expression of the platelet-derived growth factor receptor (PDGFR) and HSP60 was restricted to the cell membrane and mitochondrial fractions, respectively (Figure 2C, upper panels). The results of confocal studies also demonstrate colocalization of GFP-MUC1 C-ter with MitoTracker (Figure 2C, lower panels). The transfection efficiency of HCT116 cells is ~25% under these experimental conditions (Ren et al., 2002). Shown is one HCT116 cell transfected with GFP-MUC1 C-ter surrounded by three cells not expressing the vector (Figure 2C, lower panels). Of note, the GFP-MUC1 C-ter is devoid of a signal sequence and was not detectable at the cell membrane (Figure 2C). As a control, the prominent pattern of mitochondrial localization was not apparent when expressing GFP alone (data not shown). These findings collectively demonstrate that MUC1 C-ter localizes to mitochondria. MUC1 C-ter is targeted to the nucleus with β-catenin in cells stimulated with EGF (Li et al., 2001b, 2003c). Stimulation of HCT116/MUC1 or HCT116/MUC1(Y46F) cells with EGF, however, had little effect on mitochondrial targeting of MUC1 C-ter (Figure 2D, left). In contrast to EGF, HRG activates ErbB2 in the response of epithelial cells to stress (Vermeer et al., 2003) and targets MUC1 C-ter to the nucleolus (Li et al., 2003c). Significantly, HRG treatment for 0.5 hr was associated with a 2.3-fold increase in localization of MUC1 C-ter to mitochondria, and this response persisted through 3 hr (Figure 2D, right). Moreover, HRG had little effect on mitochondrial localization of MUC1(Y46F) C-ter (Figure 2D, right). Similar results were obtained in three separate experiments. In addition, there was no detectable β-catenin or γ-catenin in the mitochondrial fractions from control or HRG-stimulated cells (data not shown). These findings indicate that targeting of MUC1 to mitochondria is regulated, at least in part, by HRG-induced signaling and that the Y46F mutation attenuates this response.
Figure 2. HRG stimulation targets MUC1 C-ter to mitochondria.

A: Schematic representation of MUC1 C-ter with sites recognized by the indicated antibodies (upper panel). Lysates from the cell membrane (CM) and mitochondrial (Mito) fractions of HCT116/MUC1-A cells were subjected to SDS-PAGE and immunoblot analysis with the indicated antibodies (lower panels).
B: Purified mitochondria (0.4 mg protein/ml) were treated with 0, 50, or 100 μg/ml trypsin for 20 min at 4°C (left). The samples were subjected to immunoblot analysis with the indicated antibodies. Purified mitochondria (0.4 mg protein/ml) were resuspended in 0.1 M Na2CO3 (pH 11.5), incubated for 30 min at 0°C, and then centrifuged at 100,000 × μg for 30 min at 4°C (right). The supernatant (S) and pellet (P) were analyzed by immunoblotting with the indicated antibodies.
C: HCT116 cells were transiently transfected with MUC1 C-ter tagged at the N terminus with GFP (GFP-MUC1 C-ter) and harvested at 36 hr. Lysates from cell membrane (CM) and mitochondrial (Mito) fractions were subjected to immunoblot analysis with the indicated antibodies (left). Whole-cell lysate (WCL) was included as a control. Confocal microscopy was performed on the transfected HCT116 cells after staining with MitoTracker Red and TO-PRO-3. Scale bars represent 10 μm.
D: HCT116/MUC1 and HCT116/MUC1(Y46F) cells were stimulated with EGF for 30 min (left) or HRG for the indicated times (right). Mitochondrial lysates were subjected to immunoblot analysis with anti-MUC1 C-ter and anti-HSP60. Intensity of the signals was determined by densitometric scanning.
MUC1 attenuates cytochrome c release and caspase-3 activation
Treatment of cells with DNA-damaging agents is associated with release of mitochondrial cytochrome c and activation of the intrinsic apoptotic pathway. To determine if MUC1 C-ter affects cytochrome c release, the HCT116 transfectants were treated with cisplatin (CDDP). Treatment of HCT116/vector cells with CDDP was associated with increased levels of cytosolic cytochrome c (Figure 3A). Notably, expression of MUC1 significantly attenuated the release of cytochrome c (Figure 3A). By contrast, expression of MUC1(Y46F) was ineffective in blocking CDDP-induced cytochrome c release (Figure 3A). Similar results were obtained in the other separately isolated B clones (data not shown). Release of cytochrome c in the response to genotoxic stress is associated with activation of caspase-3 and cleavage of PKCδ (Emoto et al., 1995). To assess the effects of MUC1 on caspase-3 activation, CDDP-treated cells were analyzed for cleavage of pro-caspase-3. The results demonstrate that, compared to HCT116/vector cells, MUC1 attenuates CDDP-induced activation of caspase-3 (Figure 3B). Cleavage of pro-caspase-3 in CDDP-treated HCT116/MUC1(Y46F) cells was similar to that in HCT116/vector cells (Figure 3B). In concert with these results, caspase-3-mediated cleavage of PKCδ was attenuated in CDDP-treated HCT116/MUC1, as compared to HCT116/vector and HCT116/MUC1(Y46F), cells (Figure 3C). Smac/DIABLO is a mitochondrial protein that induces caspase-dependent cell death by interacting with inhibitor of apoptosis proteins (IAPs) and blocking their caspase inhibitory activity (Du et al., 2000; Verhagen et al., 2000). To determine if MUC1 attenuates release of Smac/DIABLO, HCT116/vector, HCT116/MUC1, and HCT116/ MUC1(Y46F) cells were treated with CDDP for 24, 48, and 72 hr, and cytosolic lysates were subjected to immunoblot analysis. The results demonstrate that, like cytochrome c, release of Smac/DIABLO is attenuated in HCT116/MUC1, as compared to HCT116/vector and HCT116/MUC1(Y46F) cells (Figure 3D). In addition, MUC1 attenuated release of the mitochondrial caspase-independent death effector apoptosis-inducing factor (AIF) (Susin et al., 1999) as compared to that in cells expressing the vector or MUC1(Y46F) (Figure 3D). CDDP treatment of HCT116/vector and HCT116/MUC1(Y46F) cells for 72 hr was associated with >90% cell death and decreases in the β-actin signals used as a control for loading (Figure 3D). By contrast, treatment of HCT116/MUC1 cells with CDDP for 72 hr was associated with cessation of cell growth and <30% cell death. These findings indicate that MUC1 C-ter attenuates DNA damage-induced activation of the intrinsic apoptotic pathway.
Figure 3. MUC1 attenuates CDDP-induced activation of the intrinsic mitochondrial apoptotic pathway.

A: The indicated cells were treated with 100 μM CDDP for 8 and 12 hr. Cytosolic lysates were analyzed by immunoblotting with anti-cytochrome c and anti-β-actin.
B and C: The indicated cells were treated with 100 μM CDDP for 8 and 24 hr. Cell lysates were analyzed by immunoblotting with anti-caspase-3 (B) or anti-PKCδ(C) and anti-β-actin.
D: The indicated cells were treated with 100 μM CDDP for 24, 48, and 72 hr. Cytosolic lysates were analyzed by immunoblotting with the indicated antibodies.
MUC1 blocks DNA damage- and TRAIL-induced apoptosis
To determine if MUC1 affects the induction of apoptosis by CDDP, cells were analyzed for sub-G1 DNA content. Treatment of HCT116/vector cells with CDDP for 24 hr was associated with approximately 40% apoptosis (Figure 4A). Significantly, CDDP-induced apoptosis was attenuated in HCT116/MUC1 but not in HCT116/MUC1(Y46F) cells (Figure 4A). The attenuation of apoptosis by MUC1 as determined from cells with sub-G1 DNA content was confirmed when using TUNEL staining as an alternative method (data not shown). In addition, similar results were obtained in multiple experiments with the separately isolated HCT116 cell clones (Figure 4B). Expression of wild-type MUC1 but not the MUC1(Y46F) mutant also blocked apoptosis induced by the genotoxic agent etoposide (Figure 4C). Stimulation of cell surface death receptors with TNF-α or the TNF-related apoptosis-inducing factor TRAIL is associated with activation of the extrinsic apoptotic pathway. To determine if MUC1 affects death receptor-induced apoptosis, HCT116 cells were treated with TNF-α. In concert with previous work (Tsuchida et al., 1995), TNF-α alone failed to induce apoptosis of HCT116/ vector cells (data not shown). However, treatment with TNF-α in the presence of cycloheximide (CHX) was associated with induction of HCT116/vector cell apoptosis (Figure 4D). Similar results were obtained when HCT116/MUC1 and HCT116/MU-C1(Y46F) cells were treated with TNF-α and CHX (Figure 4D), indicating that MUC1 has no effect on TNF-α+CHX-induced apoptosis. By contrast, TRAIL was effective in inducing apoptosis of HCT116/vector cells without adding CHX and, importantly, MUC1 but not MUC1(Y46F) attenuated this response (Figure 4E). Moreover, when HCT116/MUC1 cells were treated with TRAIL+CHX, MUC1 was ineffective in attenuating TRAIL-induced apoptosis (Figure 4E). Of note, CHX had no apparent effect on expression of MUC1 C-ter (data not shown). These findings indicate that (1) MUC1 attenuates apoptosis induced by activation of the intrinsic pathway, and (2) MUC1 attenuates TRAIL-induced apoptosis by a mechanism that may be mediated by a short-lived protein.
Figure 4. MUC1 attenuates DNA damage- and TRAIL-induced apoptosis.

A: The indicated cells were treated with 100 μM CDDP for 24 hr and then analyzed for sub-G1 DNA.
B: Clones A and B of the indicated cells were left untreated (open bars) or treated with 100 μM CDDP for 24 hr (solid bars).
C: Clones A and B of the indicated cells were left untreated (open bars) or treated with 70 μM etoposide for 48 hr (solid bars).
D: Clones A and B of the indicated cells were left untreated (open bars) or treated with 20 ng/ml TNF-α and 10 μg/ml CHX for 12 hr (closed bars). The results are presented as the percentage apoptosis (mean ± SD of three independent experiments) as determined by analysis of sub-G1 DNA.
E: The indicated cells were left untreated (open bars) or treated with 100 ng/ml TRAIL for 14 hr (closed bars) (left). HCT116/MUC1 cells were treated with 100 ng/ml TRAIL and/or 10 μg/ml CHX as indicated for 14 hr (right). The results are presented as the percentage apoptosis (mean ± SD of three experiments) as determined by analysis of sub-G1 DNA.
Diverse carcinomas express the MUC1 C-ter in mitochondria
To determine if other carcinomas exhibit mitochondrial localization of the MUC1 C-ter, we first performed confocal immuno-fluorescence studies on human SW480 colon carcinoma cells stably transfected to express an empty vector or MUC1. SW480 cells transfected with the empty vector expressed a low level of MUC1 and exhibited little if any MUC1 C-ter in mitochondria (Figure 5A). By comparison, SW480/MUC1 cells exhibited substantially higher levels of MUC1 expression and clear colocalization of MUC1 C-ter with MitoTracker (Figure 5A). To extend these observations to carcinomas that endogenously express MUC1, we performed similar studies on human lung and breast cancer cells. There was no detectable MUC1 N-ter in mitochondria of A549 lung carcinoma cells (data not shown). However, as shown for the MUC1 transfectants, the MUC1 C-ter was detectable in mitochondria (Figure 5B). Similar results were obtained with T-47D (Figure 5C) and ZR-75-1 (Figure 5D) breast carcinoma cells. These findings indicate that the MUC1 C-ter localizes to mitochondria in both MUC1 transfectants and carcinomas that endogenously express MUC1.
Figure 5. MUC1 C-ter localizes to mitochondria of diverse carcinoma cells.

A: SW480 carcinoma cells stably transfected to express an empty vector or MUC1 were incubated with anti-MUC1 C-ter and then a FITC-conjugated secondary antibody. Mitochondria were stained with MitoTracker Red. Nuclei were stained with TO-PRO-3. Scale bars represent 20 μm.
B–D: A549 lung (B), T-47D breast (C), and ZR-75-1 breast (D) carcinoma cells were analyzed by staining with anti-MUC1 C-ter, MitoTracker Red, and TO-PRO-3. Scale bars represent 10 μm (B) or 20 μm (C and D).
Downregulation of MUC1 sensitizes carcinoma cells to apoptosis induced by genotoxic agents
To assess the effects of endogenous MUC1 expression on sensitivity of human carcinoma cells to genotoxic agents, we knocked down MUC1 with small interfering RNA (siRNA) duplexes. Exposure of A549 lung carcinoma cells to MUC1siRNA was associated with downregulation of MUC1 expression (Figure 6A). As a control, there was no detectable downregulation of MUC1 in A549 cells exposed to a control siRNA (CsiRNA) (Figure 6A). A549 cells exposed to mock conditions, MUC1-siRNA or CsiRNA, were then analyzed for annexin V staining and sub-G1 DNA. As determined by annexin V staining, A549 cells exposed to CsiRNA or MUC1siRNA exhibited no significant increase in apoptosis compared to mock conditions (Figure 6B). Importantly, the A549 cells exposed to MUC1siRNA responded to CDDP with an increase in apoptosis compared to that obtained with cells exposed to mock conditions or CsiRNA (Figure 6B). Similar results were obtained in multiple experiments and by analysis for sub-G1 DNA (Figure 6C and data not shown).
Figure 6. Transient downregulation of MUC1 expression sensitizes A549 carcinoma cells to CDDP-induced apoptosis.

A: A549 cells were treated with CsiRNA or MUC1siRNA and harvested 72 hr after transfection. Lysates were analyzed by immunoblotting with anti-MUC1 N-ter and anti-β-actin.
B: A549 cells were left untransfected (MOCK) or transfected with MUC1siRNA or CsiRNA, incubated for 72 hr, and then treated with 10 μM of CDDP for 48 hr. Cells were stained with FITC-conjugated annexin V and analyzed by flow cytometry.
C: The results for MOCK (open bars), MUC1siRNA transfected (solid bars), or CsiRNA transfected (hatched bars) cells are expressed as the percentage apoptosis (mean ± SD of three independent experiments).
To extend these observations to another cell type, we generated a retrovirus expressing MUC1siRNA. Infection of ZR-75-1 breast carcinoma cells with the MUC1siRNA retrovirus and se- lection in G418 was associated with stable downregulation of MUC1 expression (Figure 7A). As a control, stable transduction of ZR-75-1 cells with the empty retrovirus had no effect on MUC1 expression (Figure 7A). Release of mitochondrial apoptogenic factors is associated with loss of the mitochondrial trans-membrane potential (ΔΨm) (Arnoult et al., 2003). CDDP-treated ZR-75-1/vector cells exhibited little if any decrease in ΔΨm (Figure 7B). By contrast, CDDP treatment of ZR-75-1/MUC1s-iRNA cells was associated with a clear loss of ΔΨm (Figure 7B). In concert with these findings, cytochrome c release was attenuated in CDDP-treated ZR-75-1/vector, as compared to ZR-75-1/MUC1siRNA, cells (Figure 7B). In the absence of exposure to a cytotoxic agent, ZR-75-1 cells expressing the empty retroviral vector or MUC1siRNA exhibited less than 5% apoptosis (Figure 7C). Treatment of ZR-75-1/vector cells with CDDP was associated with the induction of ~25% apoptosis (Figure 7C). Significantly, treatment of ZR-75-1/MUC1siRNA cells with CDDP resulted in over 60% apoptosis (Figure 7C). The ZR-75-1 cells were also treated with different concentrations of etoposide. The results show that sensitivity to 10 and 50 μM etoposide was increased substantially by knocking down MUC1 expression (Figure 7D). These findings and those in A549 cells indicate that knocking down MUC1 expression increases sensitivity to genotoxic agents.
Figure 7. Stable downregulation of MUC1 sensitizes ZR-75-1 cells to CDDP- and etoposide-induced apoptosis.
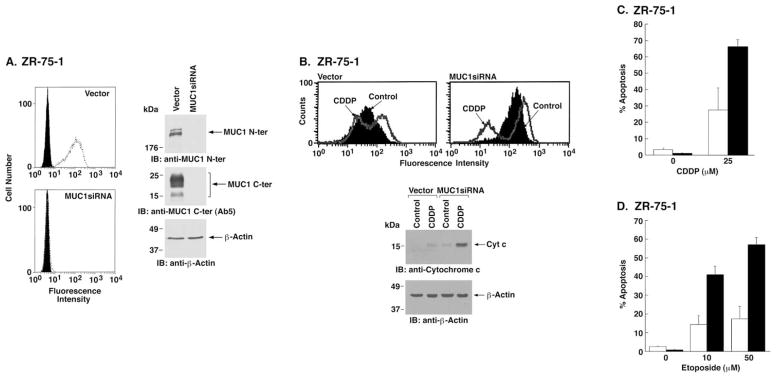
A: ZR-75-1 cells were infected with a control retroviral vector or one expressing MUC1siRNA. Stable transfectants were selected in the presence of G418. Cells were incubated with anti-MUC1 N-ter (open patterns) or control mouse IgG (closed patterns) and analyzed for immunofluorescence by flow cytometry (left). Lysates were analyzed by immunoblotting with the indicated antibodies (right panels).
B: ZR-75-1/vector and ZR-75-1/MUC1siRNA cells were incubated with DiOC6[3] and then left untreated (Control) or exposed to 25 μM CDDP for 48 hr. Mitochondrial transmembrane potential was assessed by flow cytometry (upper panels). Cytosolic lysates were analyzed by immunoblotting with the indicated antibodies (lower panels).
C and D: ZR-75-1/vector (open bars) and ZR-75-1/MUC1siRNA (solid bars) cells were treated with 25 μM CDDP for 72 hr (C) or with 10 and 50 μM etoposide for 72 hr (D). Cells were analyzed for sub-G1 DNA. The results are presented as the percentage apoptosis (mean ± SD of three independent experiments).
MUC1 confers resistance to genotoxic agents in vivo
To determine if MUC1 expression affects chemosensitivity in vivo, HCT116/vector and HCT116/MUC1 cells were injected subcutaneously into the flanks of nude mice. Growth of the HCT116/MUC1 cells was similar to that found for the HCT116/ vector cells (Figure 8A). CDDP treatment of mice injected with the HCT116/vector cells was associated with a significant inhibition of tumor growth (Figure 8A). Notably, however, treatment of the HCT116/MUC1 tumors with CDDP resulted in little if any effect (Figure 8A). To determine if knocking down MUC1 affects chemosensitivity, we injected mice with ZR-75-1 cells stably expressing the empty retroviral vector or MUC1siRNA. Growth of the ZR-75-1/MUC1siRNA tumors was somewhat slowed compared to that found for ZR-75-1/vector cells (Figure 8B). Treatment with CDDP was associated with a partial slowing of ZR-75-1/vector tumor growth (Figure 8B). In contrast, the ZR-75-1/MUC1siRNA tumors were considerably more sensitive to CDDP treatment (Figure 8B). These findings indicate that MUC1 expression contributes to CDDP resistance of carcinoma cells in in vivo tumor models.
Figure 8. MUC1 confers resistance to CDDP treatment in vivo.

A: HCT116/vector (○, ●) or HCT116/MUC1 (□, ■) cells (1 × 106) were injected subcutaneously into the posterior flanks of nude mice.
B: ZR-75-1/vector (○, ●) or ZR-75-1/MUC1siRNA (○, ●) cells (1 × 107) were injected into nude mice that had been pretreated with β-estradiol. The mice were treated as indicated (arrows) with intraperitoneal injections of PBS (□, ○) or 7 mg/kg CDDP (■, ●). Tumor volumes were calculated from bidimensional measurements at the indicated times. The results are expressed as the tumor volume (mean ± SD) of 4–8 mice per group.
Discussion
MUC1 localizes to mitochondria
Recent work has indicated that MUC1 C-ter functions as a transducer of signals from activated ErbB receptor tyrosine kinases and the Wnt pathway to the nucleus (Li et al., 2003c). In the present studies, confocal microscopy showed that MUC1 C-ter also localizes to mitochondria. Subcellular fractionation studies confirmed mitochondrial targeting of MUC1 C-ter and not MUC1 N-ter. Immunoblot analysis of subcellular fractions showed that mitochondrial MUC1 C-ter is similar to MUC1 C-ter at the cell membrane. The results indicate that mitochondrial MUC1 C-ter (20–25 kDa) includes both ECD and CD sequences and that the 17/15 kDa fragments may represent MUC1 C-ter cleavage products. Cell fractionation and confocal microscopy studies of GFP-tagged MUC1 C-ter further confirmed localization of MUC1 C-ter to mitochondria. Our results also indicate that MUC1 C-ter is expressed in mitochondria as an integral membrane protein. MUC1 C-ter most likely localizes to the mitochondrial outer membrane (MOM) based on the lack of a readily identifiable mitochondrial targeting sequence and a functional role in attenuating activation of the intrinsic apoptotic pathway. Little is known, however, about how proteins are targeted to the MOM and then integrated in mitochondrial membranes (Mihara, 2000; Rapaport, 2003). The N terminus of Tom20 is anchored in the mitochondrial outer membrane and, as such, the C terminus is susceptible to protease digestion. Other proteins, such as Bcl-2 and Bcl-xL, are anchored to the MOM by their C termini. Conversely, Tom40 is tightly embedded in the MOM and is not accessible to proteases (Suzuki et al., 2000). The demonstration that MUC1 C-ter is also not susceptible to trypsin digestion indicates that MUC1 C-ter may be embedded in the MOM. Our data, however, do not exclude the possibility that MUC1 C-ter may associate with the mitochondrial inner membrane. In this context, Bcl-2 has been detected in both the mitochondrial outer and inner membranes (Gotow et al., 2000).
The present studies further demonstrate that treatment of cells with HRG but not EGF is associated with increased targeting of MUC1 C-ter to mitochondria. Previous work using ZR-75-1 and HCT116/MUC1 cells has shown that HRG stimulates binding of MUC1 C-ter to γ-catenin and that MUC1 C-ter functions as a shuttle for nuclear targeting of γ-catenin (Li et al., 2003c). The present results indicate that MUC1 C-ter is not complexed with γ-catenin when targeted into mitochondria. In this regard, we have not found γ-catenin in mitochondria of HRG-stimulated ZR-75-1 or HCT116/MUC1 cells (data not shown). These results support a model in which the discrimination between nuclear or mitochondrial localization of MUC1 C-ter is determined by HRG-induced activation of distinct signaling pathways. The results also demonstrate that constitutive and HRG-induced mitochondrial targeting of MUC1 C-ter are attenuated by the Y46F mutation. This finding is consistent, at least in part, with the involvement of a tyrosine kinase that is activated and phosphorylates Y-46 in the HRG response. c-Src is activated by HRG (Belsches-Jablonski et al., 2001; Vadlamudi et al., 2003) and phosphorylates MUC1 on Y-46 (Li et al., 2001a). Thus, c-Src or other tyrosine kinases that are activated by HRG may contribute to mitochondrial targeting of MUC1 C-ter. Additional work has shown that MUC1 C-ter forms a complex with the cytosolic chaperones HSP70 and HSP90 and that these interactions are attenuated by the Y46F mutation (unpublished data). HSP70 and HSP90 function in the delivery of proteins to the mitochondrial import receptor Tom70 and thus may be responsible for targeting MUC1 C-ter to the MOM (Truscott et al., 2003; Young et al., 2003). The GFP-MUC1 C-ter used in our studies was generated without a signal sequence and was not detectable at the cell surface. Thus, the detection of GFP-MUC1 C-ter in mitochondria indicates that MUC1 C-ter can be targeted into mitochondria without prior localization to the cell membrane. MUC1 C-ter (no signal sequence) may be expressed by alternate splicing of MUC1 transcripts and thereby targeted directly to mitochondria. Alternatively, the results obtained with GFP-MUC1 C-ter may reflect a physiologic or stress-induced mechanism in which, after processing in the ER, MUC1 C-ter is released into the cytosol and transported to mitochondria. In support of such mechanisms, the c-Abl kinase localizes to the ER and is trafficked to mitochondria in the response to stress (Ito et al., 2001). Additional studies are ongoing to more precisely define the signals responsible for both the targeting and insertion of MUC1 C-ter as an integral mitochondrial membrane protein.
MUC1 attenuates the intrinsic apoptotic pathway
Genotoxic agents that are used in cancer treatment often induce apoptosis by activation of the mitochondrial pathway (Herr and Debatin, 2001; Kroemer and Reed, 2000). Thus, the identification of antiapoptotic proteins that confer resistance to anticancer agents is of potential importance. The present results further demonstrate that MUC1 expression is associated with attenuation of CDDP-induced release of mitochondrial cytochrome c, Smac/DIABLO, and AIF. In addition, the MUC1(Y46F) mutant was less effective than MUC1 in attenuating CDDP-induced release of these apoptogenic proteins. In concert with these findings, MUC1 but not MUC1(Y46F) was also effective in attenuating CDDP-induced activation of caspase-3 and cleavage of PKCδ. These findings were not restricted to CDDP, since MUC1 also attenuated activation of the intrinsic mitochondrial pathway by etoposide (data not shown). Moreover, the results are not attributable to clonal variation, because independently selected HCT116/MUC1 clones exhibited the same resistant phenotype. Overexpression of MUC1 in rat 3Y1 cells is associated with increases in phospho-Bad and Bcl-xL levels (unpublished data). The finding, however, that HCT116/MUC1 cells do not express increased levels of phospho-Bad or of the Bcl-xL/Bcl-2 proteins (data not shown) indicates that MUC1 C-ter attenuates DNA damage-induced release of apoptogenic mitochondrial effectors by other mechanisms. In this regard, the present results indicate that MUC1 attenuates loss of ΔΨm in the response to DNA damage. Release of mitochondrial apoptogenic factors can occur in part without changes in ΔΨm; however, complete release of cytochrome c and Smac/DIABLO has been associated with loss of ΔΨm (Arnoult et al., 2003). Thus, MUC1 C-ter may attenuate mitochondrial release of apoptogenic factors by stabilizing the ΔΨm in response to stress. Nuclear MUC1 C-ter may also contribute to attenuation of proapoptotic signals that are activated by DNA damage. To further define the mechanistic effects of MUC1 C-ter on the intrinsic pathway, studies are underway to determine whether MUC1 C-ter regulates proapoptotic proteins that induce loss of ΔΨm.
Stimulation of TNF-receptor-1 (TNF-R1) or the TRAIL receptors (R1 and R2) activates caspase-8 and thereby the extrinsic apoptotic pathway. Caspase-8-mediated cleavage of Bid also links death receptor signaling to the mitochondrial pathway by inducing the release of cytochrome c and Smac/DIABLO (Du et al., 2000; Li et al., 1998a; Luo et al., 1998; Verhagen et al., 2000). In this context, death receptor-induced apoptosis of type II cells is dependent on the mitochondrial pathway (Scaffidi et al., 1998). Based on the finding that MUC1 attenuates DNA damage-induced apoptosis by downregulating mitochondrial signaling, we asked if MUC1 also affects stimulation of the extrinsic pathway. Treatment of HCT116/vector cells with TNF-α alone resulted in no detectable apoptosis. However, in concert with reports in other cell types (Tsuchida et al., 1995), HCT116/ vector cells exhibited an apoptotic response when treated with TNF-α and CHX. The results show that MUC1 has no apparent effect on TNF-α+CHX-induced apoptosis. By contrast, apoptosis of HCT116/vector cells was induced by TRAIL in the absence of CHX. Moreover, MUC1 and not MUC1(Y46F) attenuated TRAIL-induced apoptosis, indicating that MUC1 C-ter downregulates mitochondrial amplification of TRAIL death signaling. Importantly, the attenuation of TRAIL-induced apoptosis by MUC1 was reversed by adding CHX. These results indicate that MUC1-mediated downregulation of TRAIL-induced apoptosis is conferred by the synthesis of a short-lived protein (Munshi et al., 2001; Thakkar et al., 2001; Wajant et al., 2000). The use of CHX with TNF-α may also explain why MUC1 had no effect on TNF-α +CHX-induced apoptosis. These findings indicate that attenuation of the intrinsic pathway by mitochondrial localization of MUC1 C-ter may affect death receptor-induced apoptosis. However, the results do not exclude the possibility that MUC1 may downregulate death receptor signaling by other mechanisms.
Have human tumors exploited MUC1 as an otherwise physiologic mechanism to attenuate stress-induced apoptosis?
Stable expression of MUC1 in the MUC1-negative HCT116 cells represents one approach to assess the effects of MUC1 on chemosensitivity. We reasoned, however, that downregulating MUC1 in carcinoma cells that endogenously overexpress this oncoprotein might be equally informative. MUC1siRNA was thus used to knock down MUC1 expression in A549 lung and ZR-75-1 breast cancer cells. The results demonstrate that transient downregulation of MUC1 expression in A549 cells is associated with increased sensitivity to CDDP-induced apoptosis. Stable downregulation of MUC1 in ZR-75-1 cells also resulted in an increased apoptotic response to CDDP treatment in vitro. Moreover, the demonstration that MUC1 downregulation potentiates apoptosis induced by etoposide indicates that MUC1 expression attenuates the apoptotic response to diverse genotoxic agents in vitro. Importantly, stable downregulation of MUC1 increased the sensitivity of ZR-75-1 tumors to CDDP treatment. Conversely, MUC1 expression in HCT116 cells conferred in vivo resistance to CDDP treatment. These findings suggest that MUC1 may contribute to resistance of human tumors to genotoxic anticancer agents.
Mucins represent a defensive physical barrier to environmental stress and may function in transducing signals that protect the epithelium from damage. Mechanical or toxic damage to epithelial cells is associated with disruption of tight junctions between neighboring cells, loss of polarity, and activation of a repair program (Vermeer et al., 2003). Stimulation of ErbB2 by HRG contributes to this repair by promoting cell division and thereby replacement of damaged cells (Vermeer et al., 2003). Our findings that HRG stimulation also induces both nuclear (Li et al., 2003c) and mitochondrial targeting of MUC1 C-ter suggest that MUC1 may play a role in the repair of epithelial integrity. The epithelial stress response is transiently associated with loss of polarity (Vermeer et al., 2003). With transformation and transition to a mesenchymal phenotype, epithelial cells lose the capability to reverse loss of polarity. Thus, in the HRG-induced stress response of normal epithelial cells, nuclear and/or mitochondrial MUC1 could transiently protect against apoptosis following injury. Conversely, irreversible nuclear and/or mitochondrial targeting of MUC1 in carcinoma cells could confer a phenotype that is stably resistant to stress-induced apoptosis. In this regard, our previous studies showed that MUC1 overexpression protects cells against oxidative stress-induced apoptosis (Yin and Kufe, 2003). Moreover, the present finding that MUC1 attenuates the apoptotic response to DNA damaging agents is of potential importance to cancer treatment. Thus, what appears to be a physiologic mechanism to protect normal epithelial cells against apoptosis during stress-induced repair may have been exploited by human tumors to survive under adverse conditions and in response to anticancer agents.
Experimental procedures
Cell culture
Human HCT116 and SW480 colon carcinoma cells (ATCC, Manassas, VA) were cultured in Dulbecco’s modified Eagle’s medium/F12 with 10% heat- inactivated fetal bovine serum, 100 units/ml penicillin, 100 μg/ml streptomycin, and 2 mM L-glutamine. Human A549 lung, T-47D breast, and ZR-75-1 breast carcinoma cells (ATCC) were grown in RPMI 1640 medium containing 10% heat-inactivated fetal bovine serum, antibiotics, and L-glutamine. Insulin (10 ng/ml; Life Technologies, Rockville, MD) was also added to cultures of the T-47D cells. Cells were treated with EGF (10 ng/ml; Calbiochem-Novabiochem, San Diego, CA), HRG (20 ng/ml; Calbiochem-Novabiochem), cisplatin (CDDP; Sigma), etoposide (Sigma), rhTNF-α (Promega, Madison, WI), CHX (Sigma), or rhTRAIL (100 ng/ml; Calbiochem-Novabiochem).
Cell transfections
HCT116 cells were transfected with pIRES-puro2, pIRESpuro2-MUC1, or pIRES-puro2-MUC1(Y46F), as described (Li et al., 2001b). SW480 cells were transfected with pIRES-puro2 or pIRES-puro2-MUC1. Stable transfectants were selected in the presence of 0.4 μg/ml puromycin (Calbiochem-Novabiochem, San Diego, CA). Two independent transfections were performed for each vector. Single cell clones were isolated by limiting dilution and expanded for analysis. In other studies, HCT116 cells were transiently transfected with the pEGFP-C1 vector (Clontech) in which MUC1 C-ter was cloned downstream to sequences encoding the green fluorescence protein (GFP).
Immunoblot analysis
Lysates were prepared from subconfluent cells as described (Li et al., 2001a). Equal amounts of protein were separated by SDS-PAGE and transferred to nitrocellulose membranes. The immunoblots were probed with anti-MUC1 N-ter (DF3) (Kufe et al., 1984), anti-MUC1 C-ter (Ab5; Neomarkers, Fremont, CA), anti-MUC1 C-ter (rabbit polyclonal DF3E) (Li et al., 2001a), anti-MUC1 C-ter (monoclonal ECD1; generated against the MUC1 ECD), anti-β-actin (Sigma), anti-HSP60 (Stressgen Biotechnologies, Victoria, BC, Canada), anti-PCNA (Calbiochem-Novabiochem, San Diego, CA), anti-IκBα(Santa Cruz Biotechnology, Santa Cruz, CA), anti-calreticulin (Stressgen Biotechnologies, Victoria, BC, Canada), anti-PDGFR (Santa Cruz Biotechnology), anti-Tom20 (BD PharMingen, San Diego, CA), anti-Tim23 (BD PharMingen), anti-cytochrome c (BD PharMingen), anti-caspase-3 (BD PharMingen), anti-PKCδ (Santa Cruz Biotechnology), anti-Smac/DIABLO (Medical & Biological Laboratories, Ltd., Japan), or anti-AIP (Santa Cruz Biotechnology). The immunocomplexes were detected with horseradish peroxidase-conjugated secondary antibodies and enhanced chemiluminescence (ECL, Amersham Biosciences, Piscataway, NJ). Intensity of the signals was determined by densitometric scanning.
Flow cytometry
Cells were incubated with anti-MUC1 N-ter or control mouse IgG for 1 hr at 4°C, washed, incubated with goat anti-mouse Ig fluorescein-conjugated antibody (Jackson Immunoresearch Laboratories, West Grove, PA), and then fixed in 2% formaldehyde/PBS. Reactivity was detected by FACScan.
Confocal microscopy
Cells cultured on coverslips were incubated in Dulbecco’s modified Eagle’s/ F12 medium containing 100 nM MitoTracker Red CMXRos (Molecular Probes, Eugene, OR) for 20 min at 37°C. After staining, the cells were washed with fresh growth medium, prefixed in 3.7% formaldehyde/growth medium for 15 min at 37°C, washed with PBS, permeabilized in PBS containing 0.2% Triton X-100 for 5 min at 25°C, washed with PBS, then postfixed in 3.7% formaldehyde/PBS for 5 min at 25°C. After several washes with PBS, the cells were blocked with 10% goat serum for 1 hr at 25°C, stained with anti-MUC1 C-ter antibody for 1.5 hr at 25°C, washed with PBS, incubated with FITC-conjugated secondary antibody (Jackson ImmunoResearch Laboratories, West Grove, PA) for 40 min at 25°C, washed with PBS, and incubated with 2 μM TO-PRO3 (Molecular Probes) for 10 min at 25°C. After mounting the coverslips, images were captured with a LSM510 confocal microscope (ZEISS) at 1024 × 1024 pixel resolution. The excitation wavelength for FITC, MitoTracker Red, and TO-PRO3 were 488 nm, 543 nm, and 633 nm, respectively. Fluorescein fluorescence was captured through a 505–530 nm bandpass filter. MitoTracker Red CMXRos fluorescence was collected through a 560–615 nm band-pass filter. TO-PRO3 staining was visualized through a 650 nm long-pass filter.
Subcellular fractionation
Purified mitochondria and cytoplasmic lysates were prepared as described (Kumar et al., 2003). Cell membranes were purified from supernatants after sedimentation of nuclei and mitochondria as described (Kharbanda et al., 1996). Purified mitochondria (0.4 mg protein/ml) were resuspended in 5 mM HEPES (pH 7.4), 210 mM mannitol, 70 mM sucrose, and 1 mM EGTA before treatment with trypsin (Sigma) at concentrations of 50 and 100 μg/ml for 20 min at 4°C. The samples were then subjected to SDS-PAGE and immunoblotting. Purified mitochondria (0.4 mg protein/ml) were also resuspended in 0.1 M Na2CO3 (pH 11.5), incubated for 30 min at 0°C, and then centrifuged at 100,000 μg for 30 min at 4°C. The supernatant was neutralized with HCl. The supernatant and pellet were then subjected to immunoblot analysis.
Generation of siRNA for transfection
siRNAs were synthesized to target the MUC1 sequence 5′-AAGTTCAGTGC CCAGCTCTAC-3′. The sense and antisense MUC1siRNAs were 5′-GUUCA GUGCCCAGCUCUACdTdT-3′(sense) and 5′-GUAGAGCUGGGCACUGA ACdTdT-3′(antisense) (Dharmacon Research, Inc.). A nonspecific control siRNA (CsiRNA) was also synthesized (5′-GCGCGCUUUGUAGGAUUCG dTdT-3′and 3′-dTdTCGCGCGAAACAUCCUAAGC-5′) (Dharmacon Research, Inc.). Cells were plated in 6-well plates at 1–3 × 105 cells/well, grown in antibiotic-free medium overnight, and then transfected with MUC1siRNA or CsiRNA using Oligofectamine reagent and Opti-MEM I reduced serum medium (Invitrogen Life Technology, Inc.) according to the manufacturer’s instructions. At 72 or 96 hr after transfection, cells were trypsinized, replated, and incubated overnight before treatment.
Generation of retroviral vectors expressing MUC1siRNA
Oligonucleotides of siRNA were designed that contained a sense strand of 19 nucleotide sequences of MUC1 followed by a short spacer (GAGTACTG), the reverse complement of the sense strand, and five thymidines as an RNA polymerase III transcriptional stop signal. Forward oligonucleotides for MUC1 were TCGAG-GGTACCATCAATGTCCACGGAGTACTGCGTGGAC ATTGATGGTACCTTTTT including a XhoI cleavage site and the reverse CTA GAAAAAGGTACCATCAATGTC-CACGCAGTACTCCGTGGACATTGATGG TACCC including a XbaI site. Oligos were annealed and cloned into the XhoI-XbaI site of the pSuppressorRetro vector (Imgenex Co., San Diego, CA). 293T cells were cotransfected with a plasmid expressing MUC1siRNA and pCL-Ampho virus DNA using Fugene (Roche, Indianapolis). The supernatant was collected after 48 hr for infection of target cells.
Analysis of mitochondrial transmembrane potential
Cells were incubated with 0.5 nM 3,3-dihexyloxacarbocyanine iodide (DiOC6[3]; Molecular Probes) for 30 min and analyzed by flow cytometry as described (Shapiro, 2000).
Apoptosis assays
Apoptotic cells were quantified by analysis of sub-G1 DNA, TUNEL staining, and annexin-V staining. At least two methods were used in each experiment. To assess sub-G1 DNA content, cells were harvested, washed with PBS, fixed with 80% ethanol, and incubated in PBS containing 20 ng/ml RNase (Roche) for 60 min at 37°C. Cells were then stained with 40 μg/ml propidium iodide (Sigma) for 30 min at room temperature in the dark. DNA content was analyzed by flow cytometry (EPICS XL-MCL, Coulter Corp.). Apoptotic cells with DNA fragmentation were detected by staining with the In Situ cell death detection kit (TUNEL; Roche Applied Science) and visualized by confocal microscopy (ZEISS LSM510). Apoptosis was also detected by staining cells with annexin V-FITC (BD Biosciences) in annexin V binding buffer (10 mM HEPES, 140 mM NaCl, 2.5 mM CaCl2 [pH 7.4]). After staining, cells were analyzed by flow cytometry.
In vivo treatment models
HCT116/vector or HCT116/MUC1 cells (1 × 106) were injected subcutaneously in the flanks of 4- to 6-week-old female nude (nu/nu) mice. For studies of ZR-75-1 tumors, β-estradiol (1.7 mg, 60 day release; Innovative Research, Toledo, OH) was implanted subcutaneously 3 days prior to subcutaneous injection of 1 × 107 ZR-75-1/control or ZR-75-1/MUC1siRNA cells. Mice were treated intraperitoneally with 7 mg/kg CDDP or, as a control, PBS.
SIGNIFICANCE.
The human MUC1 transmembrane glycoprotein is aberrantly overexpressed in about 800,000 of the 1.3 million tumors diagnosed in the U.S. each year. MUC1 interacts with the ErbB and Wnt signaling pathways and induces transformation. The present studies demonstrate that MUC1 localizes to mitochondria and that MUC1 blocks activation of the intrinsic apoptotic pathway by genotoxic agents. The results also demonstrate that MUC1 confers resistance to treatment in animal tumor models. These findings indicate that overexpression of MUC1 in human tumors could be of importance to the effectiveness of anticancer therapy.
Acknowledgments
This work was supported by grants CA29431 and CA97098 awarded by the National Cancer Institute. The authors acknowledge Kamal Chauhan for excellent technical support. D.K. has a financial interest in ILEX.
References
- Arnoult D, Gaume B, Karbowski M, Sharpe JC, Cecconi F, Youle RJ. Mitochondrial release of AIF and EndoG requires caspase activation downstream of Bax/Bak-mediated permeabilization. EMBO J. 2003;22:4385–4399. doi: 10.1093/emboj/cdg423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belsches-Jablonski AP, Biscardi JS, Peavy DR, Tice DA, Romney DA, Parsons SJ. Src family kinases and HER2 interactions in human breast cancer cell growth and survival. Oncogene. 2001;20:1465–1475. doi: 10.1038/sj.onc.1204205. [DOI] [PubMed] [Google Scholar]
- Boldin M, Goncharov T, Goltsev Y, Wallach D. Involvement of MACH, a novel MORT1/FADD-interacting protease, in Fas/APO-1- and TNF receptor-induced cell-death. Cell. 1996;85:803–815. doi: 10.1016/s0092-8674(00)81265-9. [DOI] [PubMed] [Google Scholar]
- Bunz F. Cell death and cancer therapy. Curr Opin Pharmacol. 2001;1:337–341. doi: 10.1016/s1471-4892(01)00059-5. [DOI] [PubMed] [Google Scholar]
- Datta R, Manome Y, Taneja N, Boise LH, Weichselbaum RR, Thompson CB, Slapak CA, Kufe DW. Overexpression of Bcl-xL by cytotoxic drug exposure confers resistance to ionizing radiation-induced internucleosomal DNA fragmentation. Cell Growth Differ. 1995;6:363–370. [PubMed] [Google Scholar]
- Du C, Fang M, Li Y, Li L, Wang X. Smac, a mitochondrial protein that promotes cytochrome c-dependent caspase activation by eliminating IAP inhibition. Cell. 2000;102:33–42. doi: 10.1016/s0092-8674(00)00008-8. [DOI] [PubMed] [Google Scholar]
- Emoto Y, Manome G, Meinhardt G, Kisaki H, Kharbanda S, Robertson M, Ghayur T, Wong WW, Kamen R, Weichselbaum R, Kufe D. Proteolytic activation of protein kinase C δ by an ICE-like protease in apoptotic cells. EMBO J. 1995;14:6148–6156. doi: 10.1002/j.1460-2075.1995.tb00305.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gendler S, Taylor-Papadimitriou J, Duhig T, Rothbard J, Burchell JA. A highly immunogenic region of a human polymorphic epithelial mucin expressed by carcinomas is made up of tandem repeats. J Biol Chem. 1988;263:12820–12823. [PubMed] [Google Scholar]
- Gotow T, Shibata M, Kanamori S, Tokuno O, Ohsawa Y, Sato N, Isahara K, Yayoi Y, Watanabe T, Leterrier JF, et al. Selective localization of Bcl-2 to the inner mitochondrial and smooth endoplasmic reticulum membranes in mammalian cells. Cell Death Differ. 2000;7:666–674. doi: 10.1038/sj.cdd.4400694. [DOI] [PubMed] [Google Scholar]
- Herr I, Debatin KM. Cellular stress response and apoptosis in cancer therapy. Blood. 2001;98:2603–2614. doi: 10.1182/blood.v98.9.2603. [DOI] [PubMed] [Google Scholar]
- Ito Y, Pandey P, Mishra N, Kumar S, Narula N, Kharbanda S, Saxena S, Kufe D. Targeting of the c-Abl tyrosine kinase to mitochondria in endoplasmic reticulum stress-induced apoptosis. Mol Cell Biol. 2001;21:6233–6242. doi: 10.1128/MCB.21.18.6233-6242.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwahashi J, Yamazaki S, Komiya T, Nomura N, Nishikawa S, Endo T, Mihara K. Analysis of the functional domain of the rat liver mitochondrial import receptor Tom20. J Biol Chem. 1997;272:18467–18472. doi: 10.1074/jbc.272.29.18467. [DOI] [PubMed] [Google Scholar]
- Kharbanda S, Saleem A, Yuan ZM, Kraeft S, Weichselbaum R, Chen LB, Kufe D. Nuclear signaling induced by ionizing radiation involves colocalization of the activated p56/p53lyn tyrosine kinase with p34cdc2. Cancer Res. 1996;56:3617–3621. [PubMed] [Google Scholar]
- Kluck RM, Bossy-Wetzel E, Green DR, Newmeyer DD. The release of cytochrome c from mitochondria: a primary site for Bcl-2 regulation of apoptosis. Science. 1997;275:1132–1136. doi: 10.1126/science.275.5303.1132. [DOI] [PubMed] [Google Scholar]
- Kroemer G, Reed JC. Mitochondrial control of cell death. Nat Med. 2000;6:513–519. doi: 10.1038/74994. [DOI] [PubMed] [Google Scholar]
- Kufe D, Inghirami G, Abe M, Hayes D, Justi-Wheeler H, Schlom J. Differential reactivity of a novel monoclonal antibody (DF3) with human malignant versus benign breast tumors. Hybridoma. 1984;3:223–232. doi: 10.1089/hyb.1984.3.223. [DOI] [PubMed] [Google Scholar]
- Kumar S, Mishra N, Raina D, Kharbanda S, Saxena S, Kufe D. Abrogation of the apoptotic response to oxidative stress by the c-Abl tyrosine kinase inhibitor STI571. Mol Pharmacol. 2003;63:276–282. doi: 10.1124/mol.63.2.276. [DOI] [PubMed] [Google Scholar]
- Li P, Nijhawan D, Budihardjo I, Srinivasula SM, Ahmad M, Alnemri ES, Wang X. Cytochrome c and dATP-dependent formation of Apaf-1/caspase-9 complex initiates an apoptotic protease cascade. Cell. 1997;91:479–489. doi: 10.1016/s0092-8674(00)80434-1. [DOI] [PubMed] [Google Scholar]
- Li H, Zhu H, Xu CJ, Yuan J. Cleavage of BID by caspase 8 mediates the mitochondrial damage in the fas pathway of apotosis. Cell. 1998a;94:491–501. doi: 10.1016/s0092-8674(00)81590-1. [DOI] [PubMed] [Google Scholar]
- Li Y, Bharti A, Chen D, Gong J, Kufe D. Interaction of glycogen synthase kinase 3β with the DF3/MUC1 carcinoma-associated antigen and β-catenin. Mol Cell Biol. 1998b;18:7216–7224. doi: 10.1128/mcb.18.12.7216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Kuwahara H, Ren J, Wen G, Kufe D. The c-Src tyrosine kinase regulates signaling of the human DF3/MUC1 carcinoma-associated antigen with GSK3β and β-catenin. J Biol Chem. 2001a;276:6061–6064. doi: 10.1074/jbc.C000754200. [DOI] [PubMed] [Google Scholar]
- Li Y, Ren J, Yu WH, Li G, Kuwahara H, Yin L, Carraway KL, Kufe D. The EGF receptor regulates interaction of the human DF3/ MUC1 carcinoma antigen with c-Src and β-catenin. J Biol Chem. 2001b;276:35239–35242. doi: 10.1074/jbc.C100359200. [DOI] [PubMed] [Google Scholar]
- Li Y, Chen W, Ren J, Yu W, Li Q, Yoshida K, Kufe D. DF3/MUC1 signaling in multiple myeloma cells is regulated by interleukin-7. Cancer Biol Ther. 2003a;2:187–193. doi: 10.4161/cbt.2.2.282. [DOI] [PubMed] [Google Scholar]
- Li Y, Liu D, Chen D, Kharbanda S, Kufe D. Human DF3/ MUC1 carcinoma-associated protein functions as an oncogene. Oncogene. 2003b;22:6107–6110. doi: 10.1038/sj.onc.1206732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Yu WH, Ren J, Huang L, Kharbanda S, Loda M, Kufe D. Heregulin targets γ-catenin to the nucleolus by a mechanism dependent on the DF3/MUC1 protein. Mol Cancer Res. 2003c;1:765–775. [PubMed] [Google Scholar]
- Ligtenberg MJ, Kruijshaar L, Buijs F, van Meijer M, Litvinov SV, Hilkens J. Cell-associated episialin is a complex containing two proteins derived from a common precursor. J Biol Chem. 1992;267:6171–6177. [PubMed] [Google Scholar]
- Liu X, Kim C, Yang J, Jemmerson R, Wang X. Induction of apoptotic program in cell-free extracts: requirement for dATP and cyto-chrome c. Cell. 1996;86:147–157. doi: 10.1016/s0092-8674(00)80085-9. [DOI] [PubMed] [Google Scholar]
- Luo X, Budihardjo H, Zou H, Slaughter C, Wang X. Bid, a Bcl2 interacting protein, mediates cytochrome c release from mitochondria in response to activation of cell surface death receptors. Cell. 1998;94:481–490. doi: 10.1016/s0092-8674(00)81589-5. [DOI] [PubMed] [Google Scholar]
- Merlo G, Siddiqui J, Cropp C, Liscia DS, Lidereau R, Callahan R, Kufe D. DF3 tumor-associated antigen gene is located in a region on chromosome 1q frequently altered in primary human breast cancer. Cancer Res. 1989;49:6966–6971. [PubMed] [Google Scholar]
- Mihara K. Targeting and insertion of nuclear-encoded preproteins into the mitochondrial outer membrane. Bioessays. 2000;22:364–371. doi: 10.1002/(SICI)1521-1878(200004)22:4<364::AID-BIES6>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- Munshi A, Pappas G, Honda T, McDonnell TJ, Younes A, Li Y, Meyn RE. TRAIL (APO-2L) induces apoptosis in human prostate cancer cells that is inhibitable by Bcl-2. Oncogene. 2001;20:3757–3765. doi: 10.1038/sj.onc.1204504. [DOI] [PubMed] [Google Scholar]
- Muzio M, Chinnaiyan AM, Kischkel FC, O”Rourke K, Shevchenko A, Ni J, Scaffidi C, Bretz JD, Zhang M, Gentz R, et al. FLICE, a novel FADD-homologous ICE/CED-3-like protease, is recruited to the CD95 (Fas/APO-1) death-inducing signaling complex. Cell. 1996;85:817–827. doi: 10.1016/s0092-8674(00)81266-0. [DOI] [PubMed] [Google Scholar]
- Rapaport D. Finding the right organelle. Targeting signals in mito-chondrial outer-membrane proteins. EMBO Rep. 2003;4:948–952. doi: 10.1038/sj.embor.embor937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren J, Li Y, Kufe D. Protein kinase C δ regulates function of the DF3/MUC1 carcinoma antigen in β-catenin signaling. J Biol Chem. 2002;277:17616–17622. doi: 10.1074/jbc.M200436200. [DOI] [PubMed] [Google Scholar]
- Scaffidi C, Fulda S, Srinivasan A, Friesen C, Li F, Tomaselli KJ, Debatin KM, Krammer PH, Peter ME. Two CD95 (APO-1/ Fas) signaling pathways. EMBO J. 1998;17:1675–1687. doi: 10.1093/emboj/17.6.1675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shapiro HM. Membrane potential estimation by flow cytometry. Methods. 2000;21:271–279. doi: 10.1006/meth.2000.1007. [DOI] [PubMed] [Google Scholar]
- Siddiqui J, Abe M, Hayes D, Shani E, Yunis E, Kufe D. Isolation and sequencing of a cDNA coding for the human DF3 breast carcinoma-associated antigen. Proc Natl Acad Sci USA. 1988;85:2320–2323. doi: 10.1073/pnas.85.7.2320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srinivasula S, Ahmad M, Alnemri T, Alnemri E. Autoactivation of procaspase-9 by Apaf-1-mediated oligomerization. Mol Cell. 1998;1:949–957. doi: 10.1016/s1097-2765(00)80095-7. [DOI] [PubMed] [Google Scholar]
- Stennicke H, Jurgensmeier J, Shin H, Deveraux Q, Wolf BB, Yang X, Zhou Q, Ellerby H, Ellerby L, Bredesen D, et al. Pro-caspase-3 is a major physiologic target of caspase-8. J Biol Chem. 1998;273:27084–27090. doi: 10.1074/jbc.273.42.27084. [DOI] [PubMed] [Google Scholar]
- Susin SA, Lorenzo HK, Zamzami N, Marzo I, Snow BE, Brothers GM, Mangion J, Jacotot E, Costantini P, Loeffler M, et al. Molecular characterization of mitochondrial apoptosis-inducing factor. Nature. 1999;397:441–446. doi: 10.1038/17135. [DOI] [PubMed] [Google Scholar]
- Suzuki H, Okazawa Y, Komiya T, Saeki K, Mekada E, Kitada S, Ito A, Mihara K. Characterization of rat TOM40, a central component of the preprotein translocase of the mitochondrial outer membrane. J Biol Chem. 2000;275:37930–37936. doi: 10.1074/jbc.M006558200. [DOI] [PubMed] [Google Scholar]
- Thakkar H, Chen X, Tyan F, Gim S, Robinson H, Lee C, Pandey SK, Nwokorie C, Onwudiwe N, Srivastava RK. Pro-survival function of Akt/protein kinase B in prostate cancer cells. Relationship with TRAIL resistance. J Biol Chem. 2001;276:38361–38369. doi: 10.1074/jbc.M103321200. [DOI] [PubMed] [Google Scholar]
- Truscott KN, Brandner K, Pfanner N. Mechanisms of protein import into mitochondria. Curr Biol. 2003;13:R326–R337. doi: 10.1016/s0960-9822(03)00239-2. [DOI] [PubMed] [Google Scholar]
- Tsuchida H, Takeda Y, Takei H, Shinzawa H, Takahashi T, Sendo F. In vivo regulation of rat neutrophil apoptosis occurring spontaneously or induced with TNF-alpha or cycloheximide. J Immunol. 1995;154:2403–2412. [PubMed] [Google Scholar]
- Vadlamudi RK, Sahin AA, Adam L, Wang RA, Kumar R. Heregulin and HER2 signaling selectively activates c-Src phosphorylation at tyrosine 215. FEBS Lett. 2003;543:76–80. doi: 10.1016/s0014-5793(03)00404-6. [DOI] [PubMed] [Google Scholar]
- Verhagen AM, Ekert PG, Pakusch M, Silke J, Connolly LM, Reid GE, Moritz RL, Simpson RJ, Vaux DL. Identification of DIABLO, a mammalian protein that promotes apoptosis by binding to and antagonizing IAP proteins. Cell. 2000;102:43–53. doi: 10.1016/s0092-8674(00)00009-x. [DOI] [PubMed] [Google Scholar]
- Vermeer PD, Einwalter LA, Moninger TO, Rokhlina T, Kern JA, Zabner J, Welsh MJ. Segregation of receptor and ligand regulates activation of epithelial growth factor receptor. Nature. 2003;422:322–326. doi: 10.1038/nature01440. [DOI] [PubMed] [Google Scholar]
- Wajant H, Haas E, Schwenzer R, Muhlenbeck F, Kreuz S, Schubert G, Grell M, Smith C, Scheurich P. Inhibition of death receptor-mediated gene induction by a cycloheximide-sensitive factor occurs at the level of or upstream of Fas-associated death domain protein (FADD) J Biol Chem. 2000;275:24357–24366. doi: 10.1074/jbc.M000811200. [DOI] [PubMed] [Google Scholar]
- Wen Y, Caffrey T, Wheelock M, Johnson K, Hollingsworth M. Nuclear association of the cytoplasmic tail of MUC1 and β-catenin. J Biol Chem. 2003;278:38029–38039. doi: 10.1074/jbc.M304333200. [DOI] [PubMed] [Google Scholar]
- Yang J, Liu X, Bhalla K, Kaekyung Kim C, Ibrado AM, Cai J, Peng TI, Jones DP, Wang X. Prevention of apoptosis by Bcl-2: release of cytochrome c from mitochondria blocked. Science. 1997;275:1129–1132. doi: 10.1126/science.275.5303.1129. [DOI] [PubMed] [Google Scholar]
- Yin L, Kufe D. Human MUC1 carcinoma antigen regulates intracellular oxidant levels and the apoptotic response to oxidative stress. J Biol Chem. 2003;278:35458–35464. doi: 10.1074/jbc.M301987200. [DOI] [PubMed] [Google Scholar]
- Young JC, Hoogenraad NJ, Hartl FU. Molecular chaperones Hsp90 and Hsp70 deliver preproteins to the mitochondrial import receptor Tom70. Cell. 2003;112:41–50. doi: 10.1016/s0092-8674(02)01250-3. [DOI] [PubMed] [Google Scholar]