

Abstract
Here we report on the structure-based optimization of antibody-recruiting molecules targeting HIV gp120 (ARM-H). These studies have leveraged a combination of medicinal chemistry, biochemical and cellular assay analysis, and computation. Our findings have afforded an optimized analog of ARM-H, which is ~1000 fold more potent in gp120-binding and MT-2 antiviral assays than our previously reported derivative. Furthermore, computational analysis, taken together with experimental data, provides evidence that azaindole- and indole-based attachment inhibitors bind gp120 at an accessory hydrophobic pocket beneath the CD4-binding site and can also adopt multiple unique binding modes in interacting with gp120. These results are likely to prove highly enabling in the development of novel HIV attachment inhibitors, and more broadly, they suggest novel applications for ARMs as probes of conformationally flexible systems.
Introduction
HIV is a global pandemic, which has killed more than 35 million people since its discovery. Although the development of highly active anti-retroviral therapy (HAART) has proven effective in reducing HIV/AIDS-related morbidity and mortality, the virus remains a serious public health threat.Furthermore, the long-term goal of eradicating HIV from the human population is far from being realized. Novel therapeutic agents and strategies for reducing the spread of HIV, and potentially for eradicating the virus altogether, remain highly desirable. To this end, our laboratory has developed a class of anti-HIV compounds termed antibody-recruiting molecules targeting HIV (ARM-Hs).1
Antibody-recruiting molecules, or ARMs, comprise a class of bifunctional small molecules that mediate the formation of ternary complexes between antibodies and disease-causing agents (e.g., cells, viruses, or proteins); ARM-induced antibody opsonization results in the immune-mediated destruction of these targets. Structurally, ARMs consist of three domains: a target binding terminus (TBT), an antibody-binding terminus (ABT), and a chemical linker motif. Variations in TBT and linker functionality have led to development of ARMs to target viruses, bacteria, and cancer cells.2
ARM-Hs are designed to function through three distinct mechanisms of action: (1) recruiting antibodies present in the human blood stream to virus particles and (2) to virus-infected cells, and (3) inhibiting virus attachment to host cells.1 ARMs are bifunctional, and are designed to interact simultaneously with HIV gp120 and anti-dinitrophenyl (anti-DNP) antibodies. HIV gp120 – when partnered with gp41 in the HIV membrane-associated glycoprotein complex – mediates the first step in viral entry into human cells by binding the protein CD4; anti-DNP antibodies are present endogenously in high concentrations in the human bloodstream.3-5 By bringing together these components, ARM-Hs can mediate the formation of a ternary complex, which leads to antibody-mediated immune clearance of Env-expressing cells. Furthermore, ARM-Hs bind competitively with CD4, thus inhibiting entry of virus into human T-cells and interfering with HIV repliciation through multiple complementary mechanisms. In particular, the ability of ARM-H to kill cells displaying the HIV Env protein leaves open the possibility of using these agents for virus eradication following pre-treatment with latency activators, such as prostratin.6 Therefore, development of highly potent, efficacious ARM-H derivatives could prove profoundly beneficial at the societal level.
Here we disclose significantly improved ARM-H derivatives, whose development was enabled by a novel structural model for the interaction between gp120 and azaindole/indole-derived HIV attachment inhibitors. Specifically, our model predicts that both BMS-378806 (1, Figure 1A) and indolo-furan 7 (see Table 2, below) are capable of binding gp120 through at least two distinct orientations, both of which are active in inhibiting its interaction with CD4. This nuanced structural understanding has enabled us to develop a second-generation ARM-H derivative that is about 1,000-fold more potent than our previously published compound.1 Critically, our analysis benefitted from the unique binding properties of ARM-Hs. Structural constraints imposed by simultaneous binding to gp120 and antibodies helped define the ARM-H orientation in computational simulations, while also helping overcome difficulties associated with gp120's high degree of conformational flexibility, which has rendered this target resistant to traditional approaches to “rational” ligand design.7-10 Therefore, the findings in this manuscript are likely to prove useful in the optimization of gp120-binding attachment inhibitors, as well as in illuminating ligand-receptor binding dynamics in a broad range of conformationally flexible systems.
Figure 1.

(A) Images of BMS-378806 (1) and first-generation ARM-H (2). (B) Crystal structure of CD4 bound to HIV gp120 HIV-1 gp120 (PDB 1G9N). CD4 F43 (light blue) engages conserved hydrophobic residues V255, E370, W427, T257 and M475 of gp120 (pink) as well as W112, F382 (orange), which cap a hydrophobic accessory pocket lined by F210, L116, Y435, V255 and V208 (dark blue).
Table 2.
ARM-H analogs derived from indole 2. Viral inhibition was assayed as a function of CD4+ MT-2 T-cell viability after co-incubation with HIV-1 (IIIB) in the presence of increasing concentrations of molecule. IC50 values are obtained in these assays as the dose required to achieve 50% protection of infected cells. CD4 inhibition was measured using a competition ELISA format, in which the binding of soluble CD4 to immobilized HIV-1 gp120 (JRFL) was monitored. IC50 values in these studies are reported as the concentration of ligand required to inhibit 50% of maximal CD4 binding to immoblized HIV-1 gp120. Ab recruiting was determined using an ELISA format, in which binding of monoclonal anti-DNP antibodies to ARM-H complexed with immobilized HIV-1 gp120 (JRFL) was monitored. Here, EC50 values are calculated as the ligand concentration required to obtain 50% maximal anti-DNP binding to immobilized HIV-1 gp120. Values are representative of triplicate data and all assays repeated at least two times for each compound.
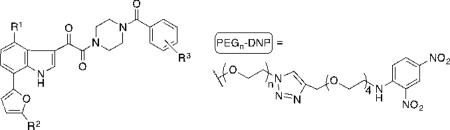
| ||||||
|---|---|---|---|---|---|---|
| Compound | R1 | R2 | R3 | MT-2 IC50 (μM) | CD4 Inhibition IC50 (μM) | Ab recruiting EC50 (μM) |
| 7 | OMe | H | H | 0.00064 | 0.130 | - |
| 8 | PEG6-DNP | H | H | >100 | 70 | >100 |
| 9 | OMe | (CH2NHCOCH2)-PEG5-DNP | H | 0.013 | 0.330 | 1.12 |
| 10 | OMe | H | PEG6-DNP(ortho) | 7.1 | >100 | 1.21 |
| 11 | OMe | H | PEG6-DMP (para) | 100 | >100 | 0.90 |
| 12 | OMe | H | PEG6-DNP (meta) | 0.460 | 0.50 | 1.15 |
Results and Discussion
Our first-generation ARM-H molecule (2, Figure 1A) was constructed by incorporating a PEG linker into the structure of BMS-378806 (1),11 an attachment inhibitor first disclosed by Bristol-Myers Squibb, which advanced through Phase I clinical trials before development was stopped due to failure to achieve desired target exposure levels (Cmin ).12, 13 Our initial choice of the C4 azaindole position as the site to incorporate the linker– DNP functionality was based on previously published modeling studies.14, 15 These reports indicated that BMS-378806 may serve to mimic F43 in CD4 (Figure 1B, light blue residue) – a critical component of the gp120 binding interface – and that the C4 methoxy group of 1 is oriented towards solvent. Although this ARM-H derivative has proven efficacious both as an attachment inhibitor and as a selective antibody-dependent cytotoxic agent, it suffers from a fairly low potency, residing in the mid-micromolar range.1
To identify an optimized analog of 2, and to probe the structural features of ligand-gp120 binding, we initially synthesized derivatives 3–6 (Schemes S1-S3). These compounds were then evaluated in three previously described assays:1 (1) an MT-2 T-cell viability assay to measure ARM-H inhibition of HIV-1 (IIIB) mediated cell lysis, (2) a competition ELISA to measure inhibition of the gp120-CD4 binding interaction, and (3) an antibody-recruiting ELISA to detect the ability of ARM-H analogs to interact with anti-DNP antibody while simultaneously bound to immobilized gp120. Although 3–6 did not exhibit any increases in potency compared to 2, assay results suggested several inconsistencies with structural predictions from previous modeling studies.14, 16, 17 First, the 300-fold loss in potency upon functionalizing 1 with a linker to afford 2 is inconsistent with a picture in which the benzamide motif is exposed to solvent (Table 1).14 Also, the complete abrogation of activity upon substitution of ortho-, para-, but not meta-, positions of the benzamide function suggests a binding mode in which the aromatic ring is at least partially obscured by interactions with the protein.16, 17
Table 1.
Properties of ARM-H analogs derived from BMS-378806. Viral inhibition was assayed as a function of CD4+ MT-2 T-cell viability after co-incubation with HIV-1 (IIIB) in the presence of increasing concentrations of ligand. IC50 values are obtained in these assays as the dose required to achieve 50% protection of infected cells. CD4 inhibition was measured using a competition ELISA format, in which the binding of soluble CD4 to immobilized HIV-1 gp120 (JRFL) was monitored. IC50 values in these studies are reported as the concentration of ligand required to inhibit 50% of maximal CD4 binding to immoblized HIV-1 gp120. Antibody (Ab) recruiting was determined using an ELISA format, in which binding of monoclonal anti-DNP antibodies to ARM-H complexed with immobilized HIV-1 gp120 (JRFL) was monitored. Here, EC50 values are calculated as the ligand concentration required to obtain 50% maximal anti-DNP binding to immobilized HIV-1 gp120. Values are representative of triplicate data and all assays repeated at least two times for each compound. ‘NA’ = no activity; ‘NT’ = not tested.

| ||||||
|---|---|---|---|---|---|---|
| Compound | R1 | R2 | R3 | MT-2 IC50 (μM) | CD4 Inhibition IC50 (μM) | Ab recruiting EC50 (μM) |
| BMS-378806 (1) | OMe | H | H | 0.032 | 8.7 | - |
| 2 | PEG6-DNP | H | H | 12 | 19.9 | 37.9 |
| 3 | OMe | (CH2)2 PEG5-DNP | H | NT | NA | NT |
| 4 | OMe | H | PEG6-DNP(ortho) | >100 | >100 | >100 |
| 5 | OMe | H | PEG6-DMP (para) | 43.0 | 50.3 | 100 |
| 6 | OMe | H | PEG6-DNP (meta) | 26.0 | 28.6 | 28.9 |
To obtain a more accurate model for gp120-ligand binding we next performed detailed computational studies. Careful examination of available gp120 crystal structures revealed a cluster of hydrophobic side-chains (F210, L116, Y435, V255 and V208; Figure 1 and 2, labeled in dark blue), which appears to compose a hydrophobic pocket, separated from the CD4 binding site by two “gate-keeper” residues (W112 and F382, Figure 1 and 2, labeled in orange). Therefore, we speculated that entry of ligands into this non-polar environment, a process enabled by the highly flexible nature of gp120, might afford a stabilizing interaction.
Figure 2.
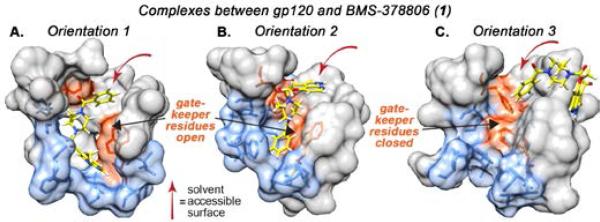
Results of computational studies exploring the interaction between BMS-378806 and gp120. Analysis of rigid-body computational data revealed presence of two “gate-keeper” residues (W112 and F382, orange), which blocked a small accessory hydrophobic pocket (blue). BMS-378806 was docked using “induced-fit” protocol, wherein residues W112 and F382 were mutated to alanine and then re-incorporated after docking. The resulting poses suggested that BMS-378806 could access an accessory hydrophobic pocket in two low-energy orientations (Orientations 1 and 2), and a higher-energy exposed orientation (Orientation 3). Note: some residues omitted for ease of view, see Figure S2 depicting full minimized structures.
To test this hypothesis, we performed induced fit docking calculations (See Supporting Information for more detail). This process involved three steps: (1) compound 1 was docked into a rigid model of gp120 in which residues W112 and F382 were replaced with alanine; (2) side-chains of these residues were restored, and for each ligand pose obtained in Step 1, energetic minimization of the flexible protein was conducted, and (3) the flexible ligand was then re-docked into the optimized protein geometry determined in Step 2. Application of the induced fit protocol yielded two novel ligand binding poses (Figure 2, Orientations 1 and 2), both of which scored significantly more favorable energetically than those arising from rigid-body docking (Orientation 3). Interestingly, while the ligand is buried deep within the hydrophobic accessory pocket in both Orientations 1 and 2, its position is reversed by approximately 180-degrees. Specifically, in Orientation 1, the azaindole core is buried within the hydrophobic pocket and the phenyl ring is exposed to solvent, while in Orientation 2, the phenyl ring is tucked within the protein interior and the azaindole is solvent-exposed.
These computational results provide several useful insights into our experimental observations. For example, the solvent accessibility of only the meta position on the benzamide ring in Orientation 1 provides an explanation for the recovery of potency of compound 6, compared to compounds 3–5, in MT-2, CD4 inhibition ELISA, and antibody-recruiting studies (Figure 1). Notably, the C4-methoxy group is only exposed to solvent in Orientation 3, consistent with the extreme loss in potency observed on functionalization of this group with a linker motif (see above). Taken together, these observations suggest that the core azaindole motif of ARM-Hs 2-6 can bind to CD4 F43 binding pocket on gp120 in such a way that either the alkoxy or the phenyl group can be solvent exposed and thus most likely interacts with gp120 through multiple distinct orientations.
Considering the observed decrease in inhibitory potencies of 3–6 relative to 1, as well as modeling data suggesting a shortage of solvent exposed sites on the azaindole core suitable for linker attachment, we chose a different scaffold upon which to optimize ARM-H derivatives. To this end, we selected known indolo-furan 7,18 a competitive inhibitor of the gp120– CD4 interaction reported to bind gp120 several orders of magnitude more potently than compound 1.
To obtain a more accurate model for gp120-ligand binding we once again performed induced fit modeling, as outlined above. Once again, induced fit modeling gave rise to two nearly-degenerate binding poses for compound 7, which differ in ligand orientation by approximately 180-degrees. To examine the ligand binding stability and side-chain interactions of these orientations, we performed subsequent molecular dynamics (MD) simulations. In Orientation 1, the indole core is situated deep in the accessory pocket, sandwiched between gp120 residues F210 and Y435. The phenyl ring is oriented towards the solvent and only the meta position is partially solvent exposed while the ortho and para positions are blocked.In Orientation 2, on the other hand, the indole engages in π-stacking with W427 and the phenyl group is sandwiched between F210 and F382 suggesting that the phenyl ring is not solvent exposed, but buried within the accessory pocket. Also, in this Orientation, the C4 and C5 substituents on the furan ring project directly toward solvent. In both generated poses, the indole C4 methoxy group is situated beneath the CD4 F43 binding site and therefore predicted to be inaccessible for linker functionalization (8). Furthermore, in order to demonstrate that these modeling results were not an artifact of the specific gp120 starting structure, we repeated calculations for compound 7 starting from two alternate gp120 structures (PDB 3TGS and 3JWD). These simulations yielded ligand poses nearly identical to Orientations 1 and 2 (Figure S4).
Furthermore, comparison of experimentally-determined B-factors with RMSD values for α-carbon atoms relative to the gp120 apostructure, has given rise to a linear association (Figure S1). This correlation – wherein C-alpha atoms with high RMSD values correspond to those with the highest B-factors – supports that major rearrangements of backbone atoms observed in MD simulations is reflective of “real-world” phenomena. The relatively high B-factors and RMSD values observed for “gate-keeper” residues F382 and W112, which are mutated during the induced-fit docking, indicates that they are likely to possess sufficient flexibility to accommodate ligand binding in the proposed Orientations.
Experimental assay data obtained for compounds 7–12 (Schemes S4-S8) proved completely consistent with this computational data. As suggested by binding through Orientation 2, compound 9, which positions the linker at the furan 5-position, is the most potent of the ARM-H derivatives. This compound is only 20-fold less potent than the parent derivative (7), and this slight drop in potency is thought to result from a small degree of steric repulsion between the linker and the residues lining the channel leading to solvent. Furthermore, compounds 10–12 exhibit a similar trend to that seen for BMS-378806-derived ARM-H derivatives, consistent with binding through Orientation 1. Specifically, compounds functionalized at either the ortho (10) or para (11) positions are poor inhibitors of viral cytotoxicity and CD4 binding, while the meta-functionalized derivative (12) retains activity in all three assays. Interestingly, the potency decrease associated with linker attachment in compound 12 is virtually identical to that observed for compound 6 in the azaindole series. Unlike in the azaindole series, however, linker attachment in place of the indole C4 methoxy group almost entirely abrogates compound activity (8). Because this methoxy function is completely buried within the protein interior in both Orientations 1 and 2, this observation might suggest that the energy difference between Orientations 1/2 and Orientation 3 is larger for the indole than the azaindole series. Similar activity trends were observed in recent work by Sato, et al.19 in which 1 and a related analog, BMS-488043, were conjugated to aldolase antibody 38C2 in an effort to enhance HIV fusion inhibition. Consistent with our model, conjugation could occur at multiple positions, with the most significant activity impairmemt occuring through conjugation at the C4 site on 1. Interestingly, the inclusion of anti-DNP antibodies significantly enhances CD4-binding inhibition (~10-fold) of 9 but has little to no effect on its viral neutralization, presumably due to geometric differences between immoblized monomeric gp120 verus membrane bound trimeric gp120 (See Supporting Information and Figure S8 for further discussion).
To confirm that changes in activity are indeed reflective of changes in gp120 ligand binding affinity, we developed a fluorescence polarization (FP) assay using a ARM-H based fluorescent probe (S29), which we used to determine binding affinities of selected compounds (see Supporting Information and Figure S9). Indeed, observed MT-2 IC50 values correlate well with corresponding FP-derived affinities. Taken together, these observations are consistent with computational studies, and support a model in which indole/azaindole attachment inhibitors can bind gp120 through at least two distinct orientations.
We next examined the inhibitory activity of ARM-H 9 against a panel of macrophage- and T cell-tropic HIV-1 strains that use either CCR5 (M-tropic) or CXCR4 (T-tropic) coreceptors to infect cells (Table 3) via a well-established viral pseudotyping assay.20-22 Here, compound 9 possesses a potent inhibitory profile against laboratory pseudotypes from HIV-1 Clade A, B and C. Additionally, in all examined clades, compound 9 proved to be more potent than BMS-378806 (1).
Table 3.
Activity profile of compounds 1, 7, and 9 against a panel of laboratory-derived HIV-1 gp120 pseudotyped viruses in TZM-bl cells (Clade A = green, Clade B = blue, Clade C = red). VSV-G is a non-specific viral glycoprotein control. Value are representative of triplicate data and assay repeated least 2 times for each compound and pseudotype. ‘NT’ = not tested.
| pseudotype | 1 IC50 (nM) | 7 IC50 (nM) | 9 IC50 (nM) |
|---|---|---|---|
| JR-FL | 45 | 0.2 | 1.5 |
| ADA | 246 | 0.14 | 6.3 |
| HXBc2 | 32 | 0.12 | 10.3 |
| BaL | NT | 0.01 | 34.8 |
| LAI | 45 | 0.1 | 4.3 |
| YU2 | 358 | 0.43 | 14.6 |
| 11500 | >1000 | 2.1 | 165 |
| 11502 | >1000 | 0.73 | 2.8 |
| 11506 | >1000 | 0.11 | 8.0 |
| 11528 | >1000 | 0.19 | 5.0 |
| VSV-G | >1000 | >1000 | >1000 |
We also compared the neutralization capacity of 9 to BMS-626529 (13), the active component of prodrug attachemnt inhibitor BMS-663068 (14) currently in clincal development,12, 23 using a panel of clinical envelope clones in a well established cell-cell fusion assay (Table 4).24 Baseline susceptibility of clinical envelopes to BMS-626529 has been shown to be inherently variable, unlike most other anti-retroviral agents. Although the majority of group M envelopes tested are highly susceptible (<10 nM EC50) to BMS-626529, baseline populations from different individuals can exhibit susceptibilities over a range of up to 6 log10.24, 25 An exception are viruses from subtype AE, all of which tested thus far have exhibited resistant phenotypes, due to 375M and 475I polymorphisms found in almost all viruses from this subtype.24 Even within an infected individual, a large variability in susceptibility was observed, as multiple functional envelope clones from single individuals were shown to exhibit a 2-3 log10 range of susceptibility to BMS-626529.24 Thus, in order to further examine the relationship between susceptibility to BMS-626529 and 9, a series of clinical envelopes were examined in a cell-cell fusion assay. Twelve envelopes from 9 infected individuals were chosen so as to cover a wide range of susceptibility to BMS-626529, with EC50s from ~200 pM to > 6 μM. The results are shown in Table 4. As observed with virus infection, the potency of 9 was reduced (from ~10-50-fold) compared to BMS-626529 for each of the 12 functional envelopes tested. However, when results were normalized using fold-change (FC) compared to the LAI envelope control, a good correlation between the activities of the two inhibitors against each individual envelope was observed. Most of the envelopes exhibited less than a 2-fold difference in FC, with the worst being envelope 12N1-13, with a 3.4 fold difference between the two inhibitors. This shows that regardless of the inherent susceptibility of the envelope to BMS-626529, it exhibited a similar degree of reduced susceptibility to 9. Taken together the inhibitory profile of 9 suggests that ARM-H's may be active against a range of clinically relevant HIV-1 subtypes comparabile to that of lead attachment inhibitors.
Table 4.
Activity profile of compounds 9 and clinical lead BMS-626529 against a panel of functional HIV-1 subtype B clinical envelope clones from 9 subjects in cell-cell (transfected HeLa cells) fusion susceptibility assays. Values provided as averages from three replicates.
| Clone ID | EC50 (nM) | Fold Change (vs LAI) | FC Ratio | ||
|---|---|---|---|---|---|
| 9 | 13 | 9 | 13 | 9/13 | |
| 27N4-20 | 7.4 | 0.2 | 0.22 | 0.20 | 1.1 |
| 36N3-5 | 5.5 | 0.4 | 0.16 | 0.40 | 0.40 |
| P15-2-18 | 62 | 1.2 | 1.8 | 1.2 | 1.5 |
| P23-1-13 | 110 | 2.4 | 3.2 | 2.4 | 1.3 |
| P22-2-45 | 73 | 2.5 | 2.1 | 2.5 | 0.84 |
| 66N2-8 | 160 | 8.6 | 4.7 | 8.6 | 0.55 |
| 66N2-16 | 350 | 22 | 10 | 22 | 0.45 |
| 12N1-13 | 550 | 56 | 16 | 56 | 0.29 |
| 21N4-169 | 4300 | 120 | 130 | 120 | 1.1 |
| 21N4-170 | 11000 | 860 | 320 | 860 | 0.38 |
| 16N4-17 | >40000 | 6200 | >1200 | 6200 | |
| 16N4-25 | >40000 | 6400 | >1200 | 6400 | |
| LAI | 34 | 1.0 | 1 | 1 | |
We have previously reported that 2 has the capacity to direct an antibody-mediated, cytotoxic immune response against Env-expressing cells.1 Thus we investigated whether the ternary complex formed between 9, anti-DNP antibodies and HIV-1 Env-expressing HEK293T cells26 could similarly activate complement proteins, thus inducing a specific cytotoxic immune response. To this end, complement preserved serum was added to Env-expressing 293T cells in the presence of a fixed amount of anti-DNP antibodies, and ARMHs 2 (black) or 9 (dark blue), or corresponding analog that lacks the DNP group (S26, turqouise). As shown in Figure 4, cytotoxicity was only observed for target cells expressing gp120 (red versus purple curves), and for ARM-H molecules possessing the DNP group (turquoise curve). Furthermore, removal of anti-DNP antibodies and serum also prevented ARM-H-dependent cytotoxicity (salmon and green curves). Taken together, this data indicates that ARM-H activity depends on the presence of gp120, the DNP group, and anti-DNP antibodies. Perhaps most notably, compound 9 was found to be more potent, and to gave rise to a larger degree of cytotoxicity at high concentrations (i.e., more efficacious), compared to 2 (blue versus black curve). These observations most likely reflect compound 9's enhanced binding affinity for gp120 versus compound 2..
Figure 4.

ARM-H 9 and 2 mediated killing in HIV-1 Env (JRFL)-expressing HEK293T cells. Env-expressing 293T cells (+ gp120) or control cells lacking Env (- gp120) were treated with ARM-H 2 or 9 or control compound S26 at the indicate concentrations in the presence (+Ab) or absence (-Ab) of complement preserved rabbit serum (10%) and rat anti-DNP IgG antibodies (50 μg/mL). Each data point represents the mean ± standard error of at least sextuplicate data. The data trends were reproduced on at least three separate occcasions.
Conclusions
Herein we disclose an intriguing computational model for the binding of HIV gp120 to HIV fusion inhibitors of the azaindole/indole series. Development of this model has exploited the bifunctional nature of ARM-Hs as a molecular tool, which has allowed us to probe ligand binding orientation by virtue of the requirement of the linker to be solvent exposed to bind anti-DNP antibodies while bound to gp120.27-29 This model has led us to hypothesize that the azaindole BMS-378806 (1), and the indole 7, are capable of interacting with gp120 through at least two binding modes within the CD4 F43 binding pocket on gp120. Upon linker attachment, the binding interactions become restricted, resulting in a loss of affinity. Using this model, we have also been able to develop a novel ARM-H scaffold (9) that is close to 1000-fold more potent than our previously published derivative. Evaluation of this new ARM-H analog against a panel of laboratory and clinical HIV pseudotypes has revealed activity against viral subtypes representing Clades A, B, and C. Finally, we have demonstrated that this increase in gp120-binding affinity is accompanied by an enhancement in gp120-targeted immunotoxicity. Overall, our conclusions are likely to provide broad-based insights into the development of novel HIV attachment inhibitors with implications in both fundamental and therapeutically relevant settings.11, 15, 30-34
Experimental
See supplementary materials
Supplementary Material
Figure 3.

Results of computational studies of the complex between 7 and gp120 (PDB 1G9N). Induced-fit docking of the compound in which the “gate-keeper” residues (Trp112 and Phe382, orange) were ”trimmed” (mutated to alanine), resulted in two low-energy ligand orientations (Orientations 1 and 2; panels A and B, respectively), and one high-energy orientation in which the ligand is mostly exposed (Orientation 3, panel C). Subsequent MD simulations indicate that in Orientations 1 and 2, compound 7 accesses an accessory hydrophobic pocket (blue). MD simulation for Orientation 1 was performed for 25 ns with an equilibrium structure observed after 11 ns. For Orientation 2, MD simulation was performed for 214 ns, with equilibrium reached after 178 ns that was stable for the remainder of the simulation. Note: some residues omitted for ease of view, see Figure S3 depicting full minimized structures.
Acknowledgements
This work was funded by the National Institutes of Health through the NIH Director's New Innovator Award Program (Grant DP22OD002913 to D.A.S.), the Alfred P. Sloan Foundation through a Fellowship (to D.A.S.), Bristol-Myers Squibb through an Innovation Award (to D.A.S.), the Camille and Henry Dreyfus Foundation through a New Faculty Award (to D.A.S.), and the Novartis Institute for Biomedical Research through an Early Career Award in Organic Chemistry (to D.A.S.). We would also like to acknowledge generous financial support from NIH grants AI44616 (to W.L.J.), GM32136 (to W.L.J.) and GM49551 (to K.S.A.), as well as NIH/NIAID grant R01AI067034 (R.E.S.) and the NIDA Avant-Garde Award Program (R.E.S.).
Notes and references
- 1.Parker CG, Domaoal RA, Anderson KS, Spiegel DA. J Am Chem Soc. 2009;131:16392–16394. doi: 10.1021/ja9057647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.McEnaney PJ, Parker CG, Zhang AX, Spiegel DA. Acs Chem Biol. 2012;7:1139–1151. doi: 10.1021/cb300119g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ortega E, Kostovetzky M, Larralde C. Molecular immunology. 1984;21:883–888. doi: 10.1016/0161-5890(84)90143-3. [DOI] [PubMed] [Google Scholar]
- 4.Jakobsche CE, Parker CG, Tao RN, Kolesnikova MD, Douglass EF, Jr., Spiegel DA. Acs Chem Biol. 2013;8:2404–2411. doi: 10.1021/cb4004942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Karjalainen K, Makela O. Eur J Immunol. 1976;6:88–93. doi: 10.1002/eji.1830060204. [DOI] [PubMed] [Google Scholar]
- 6.Beans EJ, Fournogerakis D, Gauntlett C, Heumann LV, Kramer R, Marsden MD, Murray D, Chun TW, Zack JA, Wender PA. Proc Natl Acad Sci U S A. 2013;110:11698–11703. doi: 10.1073/pnas.1302634110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Myszka DG, Sweet RW, Hensley P, Brigham-Burke M, Kwong PD, Hendrickson WA, Wyatt R, Sodroski J, Doyle ML. Proc Natl Acad Sci U S A. 2000;97:9026–9031. doi: 10.1073/pnas.97.16.9026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Xiang SH, Kwong PD, Gupta R, Rizzuto CD, Casper DJ, Wyatt R, Wang L, Hendrickson WA, Doyle ML, Sodroski J. J Virol. 2002;76:9888–9899. doi: 10.1128/JVI.76.19.9888-9899.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yuan W, Bazick J, Sodroski J. J Virol. 2006;80:6725–6737. doi: 10.1128/JVI.00118-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kong L, Huang CC, Coales SJ, Molnar KS, Skinner J, Hamuro Y, Kwong PD. J Virol. 2010;84:10311–10321. doi: 10.1128/JVI.00688-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang T, Zhang Z, Wallace OB, Deshpande M, Fang H, Yang Z, Zadjura LM, Tweedie DL, Huang S, Zhao F, Ranadive S, Robinson BS, Gong YF, Ricarrdi K, Spicer TP, Deminie C, Rose R, Wang HG, Blair WS, Shi PY, Lin PF, Colonno RJ, Meanwell NA. J Med Chem. 2003;46:4236–4239. doi: 10.1021/jm034082o. [DOI] [PubMed] [Google Scholar]
- 12.Hanna GJ, Lalezari J, Hellinger JA, Wohl DA, Nettles R, Persson A, Krystal M, Lin P, Colonno R, Grasela DM. Antimicrob Agents Chemother. 2011;55:722–728. doi: 10.1128/AAC.00759-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Xue YJ, Yan JH, Arnold M, Grasela D, Unger S. J Sep Sci. 2007;30:1267–1275. doi: 10.1002/jssc.200600499. [DOI] [PubMed] [Google Scholar]
- 14.Kong R, Tan JJ, Ma XH, Chen WZ, Wang CX. Biochim Biophys Acta. 2006;1764:766–772. doi: 10.1016/j.bbapap.2005.12.017. [DOI] [PubMed] [Google Scholar]
- 15.Wang J, Le N, Heredia A, Song H, Redfield R, Wang LX. Organic & biomolecular chemistry. 2005;3:1781–1786. doi: 10.1039/b415159c. [DOI] [PubMed] [Google Scholar]
- 16.Teixeira C, Serradji N, Maurel F, Barbault F. Eur J Med Chem. 2009;44:3524–3532. doi: 10.1016/j.ejmech.2009.03.028. [DOI] [PubMed] [Google Scholar]
- 17.Da LT, Quan JM, Wu YD. Proteins. 2011;79:1810–1819. doi: 10.1002/prot.23005. [DOI] [PubMed] [Google Scholar]
- 18.Wallace, Tao OBW, Kap-Sun Yeung, Bradley C. Pearce, Nicholas A. Meanwell, Zhilei Qiu, Haiquan Fang, Qiufen May Xue, Zhiwei Yin. PCT. Int. Appl. 2002 WO 2002004440.
- 19.Sato S, Inokuma T, Otsubo N, Burton DR, Barbas CF. Acs Med Chem Lett. 2013;4:460–465. doi: 10.1021/ml400097z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhou T, Georgiev I, Wu X, Yang ZY, Dai K, Finzi A, Kwon YD, Scheid JF, Shi W, Xu L, Yang Y, Zhu J, Nussenzweig MC, Sodroski J, Shapiro L, Nabel GJ, Mascola JR, Kwong PD. Science. 2010;329:811–817. doi: 10.1126/science.1192819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li M, Gao F, Mascola JR, Stamatatos L, Polonis VR, Koutsoukos M, Voss G, Goepfert P, Gilbert P, Greene KM, Bilska M, Kothe DL, Salazar-Gonzalez JF, Wei X, Decker JM, Hahn BH, Montefiori DC. J Virol. 2005;79:10108–10125. doi: 10.1128/JVI.79.16.10108-10125.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li M, Salazar-Gonzalez JF, Derdeyn CA, Morris L, Williamson C, Robinson JE, Decker JM, Li Y, Salazar MG, Polonis VR, Mlisana K, Karim SA, Hong K, Greene KM, Bilska M, Zhou J, Allen S, Chomba E, Mulenga J, Vwalika C, Gao F, Zhang M, Korber BT, Hunter E, Hahn BH, Montefiori DC. J Virol. 2006;80:11776–11790. doi: 10.1128/JVI.01730-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nettles RE, Schurmann D, Zhu L, Stonier M, Huang SP, Chang I, Chien C, Krystal M, Wind-Rotolo M, Ray N, Hanna GJ, Bertz R, Grasela D. The Journal of infectious diseases. 2012;206:1002–1011. doi: 10.1093/infdis/jis432. [DOI] [PubMed] [Google Scholar]
- 24.Zhou N, Nowicka-Sans B, McAuliffe B, Ray N, Eggers B, Fang H, Fan L, Healy M, Langley DR, Hwang C, Lataillade M, Hanna GJ, Krystal M. The Journal of antimicrobial chemotherapy. 2013 doi: 10.1093/jac/dkt412. [DOI] [PubMed] [Google Scholar]
- 25.Nowicka-Sans B, Gong YF, McAuliffe B, Dicker I, Ho HT, Zhou N, Eggers B, Lin PF, Ray N, Wind-Rotolo M, Zhu L, Majumdar A, Stock D, Lataillade M, Hanna GJ, Matiskella JD, Ueda Y, Wang T, Kadow JF, Meanwell NA, Krystal M. Antimicrob Agents Chemother. 2012;56:3498–3507. doi: 10.1128/AAC.00426-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lin PF, Blair W, Wang T, Spicer T, Guo Q, Zhou NN, Gong YF, Wang HGH, Rose R, Yamanaka G, Robinson B, Li CB, Fridell R, Deminie C, Demers G, Yang Z, Zadjura L, Meanwell N, Colonno R. Proc Natl Acad Sci U S A. 2003;100:11013–11018. doi: 10.1073/pnas.1832214100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Madani N, Perdigoto AL, Srinivasan K, Cox JM, Chruma JJ, LaLonde J, Head M, Smith AB, 3rd, Sodroski JG. J Virol. 2004;78:3742–3752. doi: 10.1128/JVI.78.7.3742-3752.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Liu J, Bartesaghi A, Borgnia MJ, Sapiro G, Subramaniam S. Nature. 2008;455:109–113. doi: 10.1038/nature07159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.We cannot rule out that other conduits to the protein surface might exist, such as a “water channel” (see Ref. 31) arising from residues 205, 206, 207, 436, and 437; however, since this channel is narrower than the CD4 binding pocket, and is also more obscured in gp120 trimers (see Ref. 32), we hypothesize that it is not playing a role in observations reported herein.
- 30.Wang T, Yin ZW, Zhang ZX, Bender JA, Yang Z, Johnson G, Yang Z, Zadjura LM, D'Arieno CJ, Parker DD, Gesenberg C, Yamanaka GA, Gong YF, Ho HT, Fang H, Zhou NN, McAuliffe BV, Eggers BJ, Fan L, Nowicka-Sans B, Dicker IB, Gao Q, Colonno RJ, Lin PF, Meanwell NA, Kadow JF. J Med Chem. 2009;52:7778–7787. doi: 10.1021/jm900843g. [DOI] [PubMed] [Google Scholar]
- 31.Wang T, Yin Z, Zhang Z, Bender JA, Yang Z, Johnson G, Zadjura LM, D'Arienzo CJ, DiGiugno Parker D, Gesenberg C, Yamanaka GA, Gong YF, Ho HT, Fang H, Zhou N, McAuliffe BV, Eggers BJ, Fan L, Nowicka-Sans B, Dicker IB, Gao Q, Colonno RJ, Lin PF, Meanwell NA, Kadow JF. J Med Chem. 2009;52:7778–7787. doi: 10.1021/jm900843g. [DOI] [PubMed] [Google Scholar]
- 32.Wang T, Kadow JF, Zhang ZX, Yin ZW, Gao Q, Wu DD, Parker DD, Yang Z, Zadjura L, Robinson BA, Gong YF, Blair WS, Shi PY, Yamanaka G, Lin PF, Meanwell NA. Bioorg Med Chem Lett. 2009;19:5140–5145. doi: 10.1016/j.bmcl.2009.07.076. [DOI] [PubMed] [Google Scholar]
- 33.Meanwell NA, Wallace OB, Fang HQ, Wang H, Deshpande M, Wang T, Yin ZW, Zhang ZX, Pearce BC, James J, Yeung KS, Qiu ZL, Wright JJK, Yang Z, Zadjura L, Tweedie DL, Yeola S, Zhao F, Ranadive S, Robinson BA, Gong YF, Wang HGH, Blair WS, Shi PY, Colonno RJ, Lin PF. Bioorg Med Chem Lett. 2009;19:1977–1981. doi: 10.1016/j.bmcl.2009.02.040. [DOI] [PubMed] [Google Scholar]
- 34.Meanwell NA, Wallace OB, Wang H, Deshpande M, Pearce BC, Trehan A, Yeung KS, Qiu Z, Wright JJ, Robinson BA, Gong YF, Wang HG, Spicer TP, Blair WS, Shi PY, Lin PF. Bioorg Med Chem Lett. 2009;19:5136–5139. doi: 10.1016/j.bmcl.2009.07.027. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.