

Abstract
Tumor growth is associated with the inhibition of host antitumor immune responses that can impose serious obstacles to cancer immunotherapy. To define the potential contribution of Qa-1-restricted CD8 regulatory T cells (Treg) to the development of tumor immunity, we studied B6.Qa-1 D227K mice that harbor a point mutation in the MHC class Ib molecule Qa-1 that impairs the CD8 Treg suppressive activity. Here, we report that growth of B16 melanoma is substantially delayed in these Qa-1 mutant mice after therapeutic immunization with B16 melanoma cells engineered to express GM-CSF compared to Qa-1 B6.WT controls. Reduced tumor growth is associated with the enhanced expansion of follicular T helper cells, germinal center B cells, and high titers of antitumor autoantibodies, which provoke robust antitumor immune responses in concert with tumor-specific cytolytic T cells. Analysis of tumor-infiltrating T-cells revealed that the Qa-1 DK mutation was associated with an increase in the ratio of CD8+ T effectors compared with CD8 Treg cells. These data suggest that the CD8+ T effector–Treg ratio may provide a useful prognostic index for cancer development and raise the possibility that depletion or inactivation of CD8 Treg represents a potentially effective strategy to enhance antitumor immunity.
Keywords: melanoma, autoimmune disease, autoantibody, regulatory T cells
Introduction
Recent advances in cancer immunology have generated considerable interest in approaches that interrupt inhibitory pathways which can constrain antitumor immunity (1, 2). For example, definition and analysis of CTLA-4 expression by regulatory and other T cells led to the generation of a CTLA-4 monoclonal antibody (ipilimumab) that substantially extends survival in patients with metastatic melanoma (3, 4) and shows promise for achieving durable remissions of other types of cancer. Although ipilimumab has been approved as a standard-of-care therapy for advanced melanoma since 2011, a relatively small fraction (approximately 1/5) of patients treated with this drug achieved long-term clinical responses, suggesting that targeting additional inhibitory pathways might increase the efficacy of this general therapeutic strategy.
Most current approaches directed at regulatory T cells (Treg) have targeted cells within the CD4 lineage. However, there is increasing evidence that CD8 T cells also include a regulatory cell lineage that inhibits T cell responses (5). These regulatory CD8 T cells (CD8 Treg) eliminate activated T follicular helper (TFH) cells through the recognition of the MHC class Ib molecule, Qa-1 (HLA-E in human) expressed at their surface. This regulatory subset represents about 5% of CD8 T cells and expresses a triad of surface receptors – CD44, CD122 and Ly49 – that can be used to purify them (6, 7). Qa-1 knock-in mice (B6.Qa-1 D227K) have been generated to express a point mutation (position 227, D→K) that disrupts the binding of Qa-1 to the T cell receptor (TCR)–CD8 complex of CD8 Treg (8). These mutant mice develop increased germinal centers, high titers of autoantibodies and a lethal SLE-like autoimmune disease (6) highlighting the contribution of this Qa-1-restricted inhibitory interaction to the regulation of immune response and self-tolerance.
These considerations led us to examine the potential contribution of Qa-1-restricted inhibitory pathways to antitumor immunity. Analysis of mice that had been inoculated with GM-CSF-expressing B16 melanoma cells revealed robust upregulation of Qa-1 on lymphocytes and tumor infiltration of CD8 Treg. We reasoned that increased targeting of Qa-1+ TFH cells by CD8 Treg might inhibit tumor immunity and disruption of this inhibitory interaction might enhance the protective immune response. We therefore analyzed the response of B6.Qa-1 D227K (B6-DK) mice challenged with B16 melanoma after vaccination with irradiated GM-CSF-transduced tumor cells. We find that genetic disruption of CD8 Treg activity results in enhanced antitumor immunity that is associated with a robust antibody response to tumor-associated antigens (TAA) that cooperates with CD8 effector T cells to constrain tumor growth.
Methods
Mice
C57BL/6J (B6) (Jackson Laboratory), B6.Rag2−/− (Taconic), and B6.Qa-1 D227K mice (N11; 99% congenic according to SNP analysis), OT-I [C57BL/6-Tg(TcraTcrb)1100Mjb/J] and OT-II [B6.Cg-Tg(TcraTcrb)425Cbn/J] were housed in pathogen-free conditions. All experiments were performed in compliance with federal laws and institutional guidelines, as approved by Dana-Faber Cancer Institute's Animal Care and Use Committee.
Cell lines and tissue culture
B16F10 (ATCC) and GVAX (B16-GM-CSF) were cultured in complete DMEM medium with 10% FCS. B16-OVA was cultured in complete DMEM medium with 10% FCS supplemented with 250µg/ml G418 (Invitrogen). All lines were maintained at 37°C and 5% CO2. All cell lines used were checked for mycoplasma. B16-OVA was authenticated by PCR for OVA expression. No additional authentication was performed.
Reagents and flow cytometry
Single-cell suspensions were stained with target antibodies for 30 min in the dark at 4°C in ice-cold FACS buffer (2% FCS, 0.1% NaN3 in PBS), washed and analyzed using a FACSCalibur or LSR Fortessa (BD Biosciences) and FlowJo software (TriStar). Tumors were digested with collagenase/dispase for 2h at 37°C with agitation and fractionated on Nyco-Prep 1.007 separation medium (PROGEN) with centrifugation at 2000 rpm, RT. Lymphoid fraction was collected, washed extensively with ice-cold FACS buffer and analyzed. Tumor infiltrates were defined as CD45+ cells. Anti-B220, anti-CD44, anti-CD45, anti-CD122, anti-Ly49 C/I/F/H, anti-NK1.1, anti-CD11b, anti-CD25, anti-CXCR5, anti-ICOS, IgG1 isotype, anti-Qa-1b, anti-CD69, anti-Fas, anti-IgM (BD Bioscience) or anti-CD4, anti-CD8, anti-CD3, anti-CD200, anti-BTLA4, anti-PD-1, anti-FoxP3 (eBioscience), CD1d tetramer (NIH Tetramer Core Facility at Emory U) were used for cell analysis.
Tumor challenge and treatment
Female 6–12 weeks old B6-WT or B6-DK mice were challenged subcutaneously (s.c.) with 2–5×105 B16 (or B16-OVA) tumor cells and immunized with irradiated 1×105 GVAX (B16 retrovirally transduced with GM-CSF) s.c. on the opposite flank on day 0. Mice were treated by immunization with 5×105 irradiated (150 Gy) B16 (or B16-OVA) and 5×105 GVAX s.c. on days 7 and 14 after challenge on alternating sides. Tumors were measured 2–3 times per week using calipers and tumor volume was presented as (x×y×z)/2 mm3. Mice exhibiting signs of distress or with tumors reaching 2 cm on the longest axis were sacrificed according to IACUC guidelines. Survival plot was created using Prizm 6.0 (GraphPad Software, Inc.). In some experiments mice were vaccinated with irradiated 5×105 B16-OVA and 5×105 GVAX s.c. one week prior to challenge with 1×106 B16-OVA, and tumor incidence was recorded. In serum transfer experiments, Rag2−/− hosts were challenged with 1×106 B16-OVA and treated with 400 µl serum i.p. on days 0, 8, and 15 after challenge. Mice also received 2×106 naïve OT-I Tg CD8 T cells i.v. on day 2 and 1 µg IL15–IL15Rα complex i.p. on days 2, 7, and 14 after challenge. Serum was prepared from either B6-WT or B6-DK mice vaccinated on days 0, 7, 10, and 17 with irradiated 1×106 B16-OVA and 5×105 GVAX. Serum was collected beginning on day 13 after the first immunization and equal volumes from each collection date were pooled before treatment. Serum was prepared as described (9), total IgG was obtained by precipitation with ammonium sulfate (Sigma), diluted in PBS and desalted on 50,000 MW Amicon filters (EMD Millipore).
Enzyme-linked immunosorbent assay (ELISA)
Detection of NP-specific antibodies by ELISA was performed as described (6). Serum harvested 14 days after immunization with NP19-KLH in CFA and re-immunization with NP19-KLH in IFA was used as a standard. A 1:1000 dilution of this immune serum was defined as 100 Units/ml. OVA-specific antibodies were detected on ELISA plates coated with 10 µg/ml OVA (Pierce) overnight at 4°C. Total IgG1 was detected by incubating plates with biotinylated anti-mouse IgG1 conjugated to streptavidin peroxidase. Anti-OVA IgG1 standard was from Abcam. Anti-double-stranded-DNA (dsDNA) and anti-nuclear antibodies (ANA) in mouse sera were determined by ELISA according to the manufacturer’s protocol (Alpha Diagnostic International). Anti-macrophage migration inhibitory factor (MIF) and anti-Angiopoietin-2 (Ang-2) IgG antibodies were measured as described (10). Recombinant mouse MIF and Ang-2 were from R&D Systems. IFNγ in mouse sera was measured using an OptEIA ELISA kit (BD Biosciences); IL-21 was measured using an ELISA Duo set (R&D Systems).
Immunohistochemistry
Sections from frozen spleens and tumors, 7 µm and 10 µm respectively, were fixed in cold acetone, air-dried, blocked with 3% BSA in PBS and 1:1000 Fc block (BD Biosciences) for 30 min at RT, and stained with antibodies overnight in blocking buffer in the dark at 4°C using a moist chamber. Images were acquired on Nikon inverted wide-field fluorescence microscope with 100× magnification. Germinal centers (GC) in spleen were defined by labeling with phycoerythrin (PE)-conjugated anti-B220 antibodies (BD, RA3-6B2) and fluorescein isothiocyanate (FITC)-conjugated anti-GL-7 (BD, clone GL7) antibodies. IgG depositions in tumor were detected with FITC-conjugated anti-mouse IgG antibody (Sigma), complement membrane activated complex (MAC) with biotinylated C5b-9 antibody (Bioss), macrophages with F4/80 antibody (eBioscience), desmin with anti-desmin Ab (Abcam) and fibroblast with ant-ERTR7 Ab (Santa Cruz). Data were quantified using ImageJ software and presented as fluorescent area (pixels2) per field of view (FOV) or using NIS Elements software (Nikon Instruments Inc.) and presented as fluorescent area (pixels2) per region of interest (ROI). Vasculature was visualized using PE-conjugated anti-CD31 antibody (eBioscience) and vessel diameter was measured on the longest axis by NIS Elements software.
Adoptive transfer experiments
Naïve CD4+CD25− cells were purified from spleens of OT-II Tg WT or B6-DK mice using a CD4 Cell Enrichment Set and biotinylated anti-CD25 antibody (BD Biosciences). CD4 cells were in vitro polarized to TFH cells with 50 ng/ml IL-21, 20 ng/ml IL-6, 10 µg/ml anti-IL-4, 10 µg/ml anti-IFNγ, 20 µg/ml anti-TGFβ and cultured for 5d in the presence of 1 µg/ml OT-II peptide and APC. Naïve B cells were isolated from the spleens of B6-WT mice using a B Lymphocyte Enrichment Set (BD Biosciences). B cell and CD4 cell purity was >95%. CD8 Treg were isolated from the spleens of B6-WT mice immunized i.p. with 100 µg of KLH in CFA, and re-immunized 7 days later with 100 µg of KLH in IFA. CD8 Treg were first enriched with a CD8 Cell Enrichment Set (BD Bioscience) followed by sorting for CD3+CD8+CD44+CD122+Ly49+ T cells. Rag2−/− mice were adoptively transferred with 1.5×106 OT-II TFH cells, 3.5×106 naïve B cells, and 1×105 CD8 Treg. Immediately after transfer, mice were immunized i.p. with 100 µg of NP13-OVA in CFA, after 10 days mice were re-immunized i.p. with 50 µg of NP13-OVA in IFA, and challenged with 1×106 B16-OVA s.c. 4 days later. Tumor size was monitored as above.
Statistical analyses
Data are presented as mean ± SEM. Statistical analyses were performed using Prizm 6.0 software, the Wilcoxon–Mann–Whitney rank sum test for comparison of two groups or conditions, unless otherwise noted. P < 0.05 was considered to be statistically significant (P * <0.05, P ** <0.01, P *** <0.001).
Results
Genetic disruption of CD8 Treg activity is associated with reduced melanoma growth and enhanced TFH cell responses
We utilized the B16 melanoma model to investigate the contribution of CD8 Treg to antitumor immunity (11). Tumor-bearing B6 mice were vaccinated with irradiated B16 melanoma cells engineered to express GM-CSF (GVAX) to induce an immune response to the tumor (12). Following the completion of GVAX, we noted substantial upregulation of Qa-1 expression by splenocytes and by tumor-infiltrating lymphocytes (TILs), but not by tumor cells (Fig. 1A). Since B16 tumor cells do not express detectable Qa-1, host cells in the spleen and in tumor infiltrates represent potential targets of Qa-1-restricted CD8 Treg cells. Upregulation of Qa-1 expression by immune cells was associated with the infiltration of B16 tumors by cells expressing markers of the CD8 Treg phenotype (Fig. 1B). Increased proportions of CD8 Treg within tumor-infiltrating CD8 T cells correlated with rapid tumor growth (~day 20) (Fig. 1B, right). We directly tested the contribution of Qa-1-restricted CD8 T cells to tumor rejection using Qa-1 knock-in mice (B6.Qa-1 D227K; B6-DK) that harbor defective Qa-1-restricted CD8 Treg activity secondary to a Qa-1 point mutation that disrupts the TCR recognition of Qa-1–peptide ligands (8). We inoculated B6.Qa-1 WT (B6-WT) and B6-DK mice with B16 tumor cells and immunized them with irradiated GM-CSF-transduced B16 cells on day 0, 7 and 14. B6-DK mice showed significantly extended survival and reduced tumor growth compared to B6-WT mice (Fig. 1C).
Figure 1. GVAX immunization combined with disruption of CD8 Treg activity in B6-DK mice significantly potentiates antitumor immunity.

A) B6-WT mice were challenged with 2×105 B16 cells s.c. and immunized with irradiated 1×106 GVAX (grey box) or not (black box) on days 5, 8 and 11. Qa-1 expression was determined on gated subsets of splenocytes and TILs 20–24h after the last immunization. Data are representative of 2 independent experiments and presented as ratio of mean fluorescence intensity (MFI) of specific Qa-1 staining and MFI of background (bg) staining with streptavidin only. B) CD8 Treg efficiently infiltrate into tumors. Mice were challenged with 5×105 B16 cells. The lymphoid fraction of collagenase/dispase digested tumors was collected by gradient centrifugation on days 10, 20, and 30 after challenge. The proportions of CD8 Treg (CD122+Ly49+) were determined by FACS as percent (%) of CD8+CD3+ cells. A representative FACS plot from day 20 after challenge is shown on the left. Tumor growth is shown on the right. C) B6-WT (■) or B6-DK (□) mice were challenged with 2×105 B16 and immunized on the same day with irradiated 1×105 GVAX followed by immunization with irradiated 5×105 B16 and 5×105 GVAX s.c. on day 7 and 14, n=15 mice per group. Data are presented as cumulative survival (left panel) or tumor growth (right panel) of B16 challenged mice from two independent experiments. D) B6-WT (■) or B6-DK (grey □) mice were treated as in B). Percent (%; left panel) or absolute number (right panel) of CD4 TFH cells (CD200+ICOS+PD-1hi gated from CD4+CD3+) from either spleens or tumors (Tm) of treated animals was determined 3 days after the last immunization. ICOS expression by tumor-infiltrating CD3+CD4+ lymphocytes was determined by FACS 3 days after the last immunization. Data are representative of at least 3 independent experiments. E) Cellular analysis of tumor-bearing B6-WT (■) or B6-DK (grey □) mice immunized with GVAX. The proportion and absolute number of CD8, Ly49+ CD8, NK (NK1.1+CD3−) or CD4 Treg (CD25+FoxP3+ gated from CD3+CD4+) lymphocytes from the spleens, tumor draining lymph nodes (dLN) or tumors (Tm) was determined by FACS 3 days after the last immunization. Data are representative of at least 2 independent experiments.
Further analysis of B6-DK mice revealed increased numbers of TFH cells (CD4+ ICOS+ CD200+) compared to B6-WT mice (Fig. 1D), consistent with previous findings that immunization of B6-DK mice with foreign antigens results in increased expansion of TFH cells and high titers of autoantibodies (6). Moreover, tumor-infiltrating CD4 T cells in Qa-1-mutant mice displayed a more activated phenotype, as judged by levels of ICOS expression (Fig. 1D) (3). No significant difference was noted in the numbers of CD4 Treg or NK cells (Fig. 1E). Interestingly, increased intra-tumoral expansion of effector CD8 T cells and reduced numbers of CD8 Treg were detected in B6-DK mice compared to B6-WT controls (Fig. 1E). This resulted in a substantial increase in the intra-tumoral CD8+ Teff/Treg ratio that was associated with the Qa-1 DK mutation.
Vaccination of B6-DK mice results in enhanced antibody responses to tumor-associated antigens
In view of the increased numbers of TFH cells (e.g., Fig. 1D) and GC B cells (see below) in tumor-bearing B6-DK mice, we asked whether these Qa-1 mutant mice developed antibody responses to surrogate tumor-associated antigens (TAA). We immunized B6-WT and B6-DK mice with irradiated GM-CSF-transduced B16 tumor cells that expressed an ovalbumin transgene (B16-OVA) (13). We detected high titers of OVA-specific antibody in the sera from B6-DK, but not B6-WT mice (Fig. 2A). Increased anti-TAA antibody formation was associated with enhanced GC formation in B6-DK mice (Fig. 2B). Vaccination of B6-DK mice with irradiated B16-OVA and GVAX 7 days before inoculation with B16-OVA tumor cells also induced high titers of anti-OVA antibodies. This antibody response was maintained during the course of B16 tumor growth and correlated with increased tumor protection (Fig. S1), suggesting that antibodies induced by vaccination might have durable antitumor activity. Moreover, higher titers of anti-OVA antibodies in B6-DK mice were also detected when mice were first injected with B16-OVA tumor cells 7 days before immunization with irradiated B16-OVA and GVAX, supporting the idea that the enhanced response to TAA in B6-DK mice might have therapeutic value (Fig. 2C).
Figure 2. GVAX immunization induces robust antibody response to tumor-associated antigens in B6-DK mice.

A) B6-WT (■) or B6-DK (grey □) mice were vaccinated with irradiated 1×106 B16-OVA and 5×105 GVAX on day 7 and 14. Sera were collected 14, 21, and 28 days after vaccination and analyzed for anti-OVA IgG1 antibodies by ELISA, n=3 mice per group. Data are representative of two independent experiments. B) Histology of spleens from A) on day 28. Green, GL7; red, B220. Germinal center (GC) area, pixel2 per field of view (FOV) and GC numbers per FOV were quantified with ImageJ. C) Mice were challenged with 5×105 B16-OVA and immunized with irradiated 1×105 GVAX followed by immunization with irradiated 1×106 B16-OVA and 5×105 GVAX s.c. on day 7 and 14, n=5 mice per group. Sera were collected on day 25 after challenge and analyzed for anti-OVA IgG1 antibodies by ELISA. Data are representative of 2 independent experiments.
To evaluate the contribution of TFH cells to antitumor antibody responses in Qa-1 mutant mice, we adoptively transferred in vitro polarized TFH cells from B6-WT or B6-DK × OT-II TCR Tg mice into Rag2−/− hosts along with naive B6-WT B cells and CD8 Treg. Following inoculation with B16-OVA cells and challenge with NP-OVA, recipients of TFH cells from B6-DK mice produced substantially higher titers of both total anti-NP (anti-NP23 IgG1) and high affinity (anti-NP4 IgG1) Abs and anti-OVA antibodies than recipients of TFH cells from B6.Qa-1 B6-WT donors, and this response was associated with reduced tumor growth (Fig. 3A, B). We again observed enhanced expansion of TFH cells (but not CD8 Treg) in recipients of B6-DK TFH cells (Fig. 3C). The expansion of TFH cells was accompanied by increased levels of IL-21, but not IFNγ in the sera of B6-DK mice (Fig. 3D), indicating that the expanded T cells were producing a canonical TFH cytokine.
Figure 3. Disrupted CD8 Treg activity in B6-DK mice is associated with decreased tumor formation and increased antibody response to tumor-associated antigens (TAA).

Rag2−/− mice were adoptively transferred with OT-II WT or B6-DK polarized in vitro to TFH cells, naïve B cells, and CD8 Treg. Mice were immunized with NP-OVA in CFA, boosted on day 10 with NP-OVA in IFA, and challenged with 5×105 B16-OVA 4 days later. A) Tumor volume was recorded every 2–3 days B) Sera were collected on day 15–16 after challenge and analyzed for anti-OVA IgG1 or anti-NP IgG1 antibodies by ELISA. C) Absolute numbers of TFH, GC B (B220+Fas+IgM−) or CD8 Treg (CD3+CD8+ CD122+Ly49+) cells from B6-WT (■) or B6-DK (grey □) mice were determined in the spleens, tumor draining lymph nodes (dLN) or tumors (Tm). D) IL-21 and IFNγ levels were quantified by ELISA in serum collected from B6-WT (■) or B6-DK (grey □) mice on day 15–16 after challenge. Data are representative of at least 3 independent experiments, n=5 mice per group.
Next, we analyzed the repertoire of antibodies present in the serum of GVAX-vaccinated B6-WT and B6-DK mice. We found increased titers of autoantibodies in the sera of B6-DK mice, including anti-dsDNA antibodies (Fig. 4A). The same trend was observed after treatment of tumor-bearing mice with GVAX (Fig. 4B). In addition, we noted several types of autoantibodies that have been reported to have anti-angiogenic properties (10, 14, 15) in a portion of the B6-DK mice, but not B6-WT mice (Fig. 4C). These Abs were induced upon GVAX vaccination and were not detectable in naïve B6-WT or B6-DK mice. To determine whether the tumor vasculature was affected by the therapeutic antibodies in B6-DK mice, we performed histology on tumor sections and immunostaining with CD31 antibodies. Although the number of vessels was similar, the tumor vessels appeared smaller in tumors of B6-DK than in B6-WT controls (Fig. 4D), suggesting that increased vasculature pathology was associated with increased Ab responses of B6-DK mice, which may contribute to delayed tumor growth.
Figure 4. High autoantibody titers induced after GVAX immunization in the absence of CD8 Treg activity.
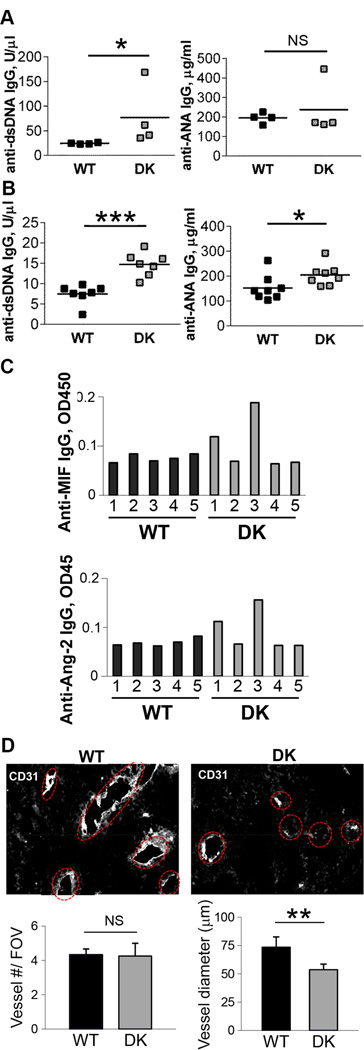
A) Mice were vaccinated with irradiated 5×105 B16-OVA and 5×105 GVAX on day 7 and 14. Sera were collected 4 weeks later and analyzed for anti-dsDNA IgG or anti-ANA IgG antibodies by ELISA B) Mice were challenged with 5×105 B16-OVA and immunized with irradiated 1×105 GVAX followed by immunization with irradiated 5×105 B16-OVA and 5×105 GVAX s.c. on day 7 and 14. Sera were analyzed as above. C) Mice were challenged and treated as in B). Sera were analyzed for anti-MIF or anti-Angiopoietin-2 (Ang-2) IgG antibodies by ELISA. Data are representative of at least two independent experiments. D) Remodeling of the tumor microenvironment with therapeutic antibodies results in decreased tumor vasculature. Histology of B16-OVA tumors that were collected on day 20 from either B6-WT or B6-DK mice. Mice were immunized with irradiated 5×105 B16-OVA and 5×105 GVAX on day 7 and 14 after challenge. Vessel numbers were counted manually per slide, and vessel diameter was measured on the longest axis by NIS Elements software. Data are representative of three independent experiments.
Most analyses of protective tumor immunity have focused on cell-mediated immunity, including the host cytotoxic response. Our analysis of enhanced antitumor immunity in animals with defective CD8 Treg activity suggested a role for autoantibody responses, including antibodies to TAA. To determine whether antibodies produced after GVAX immunization of B6-DK mice contributed to antitumor immunity, Ig was prepared from serum of Qa-1 WT or B6-DK mice after GVAX immunization. Rag2−/− mice were inoculated with B16-OVA and treated with 400 µl of serum-derived Ig on days 0, 8, and 15. Mice also received 2×106 naïve OT-I cells on day 2 and 1µg IL15/IL15Rα complex on days 2, 7, and 14 after tumor inoculation as a source of activated CD8 T cells. We found that 60% of the mice that received antibodies from B6-DK mice remained tumor-free, while all mice that received serum Ig from WT controls developed tumors (Fig. 5A). This antitumor response did not depend on the contribution of anti-OVA antibody alone, since the provision of high titers of anti-OVA Ig (500 µg anti-OVA IgG1) to Rag2−/− mice without T cells had no detectable effect on tumor growth (Fig. S2). Further analysis of B16 tumors revealed increased IgG deposition in the tumors of B6-DK mice after treatment with GVAX (Fig. 5B), which co-localized with the C5b-C9 complement membrane-activated complexes (MAC) (Fig. 5B) and was associated with macrophage infiltrates (Fig. 5C); Ig did not co-localize with other tumor stromal components, including CD31+ and ERTR-7+ cells (Fig. 5D). We also used a polyclonal antibody to the desmin stromal protein that cross-reacts with single-stranded and double-stranded DNA (16) for additional histological analysis (Fig. S3). Immunofluorescence analysis using anti-desmin Ab showed co-localization with IgG deposits in addition to the presence of filamentous structures normally detected by this Ab (Fig. 5D). Areas containing co-localized anti-desmin Ab accounted for a substantial fraction of the intra-tumoral IgG foci (~30–50%), suggesting that a significant portion of intra-tumoral Ig may represent anti-DNA antibodies derived from sera of tumor-bearing B6-DK mice.
Figure 5. Antitumor antibodies produced in B6-DK mice enhance tumor rejection and remodel tumor microenvironment.

A) Serum was prepared from either B6-WT (■) or B6-DK (grey □) mice serially vaccinated with irradiated 1×106 B16-OVA and 5×105 GVAX. Rag2−/− hosts were challenged with 1×106 B16-OVA and treated with concentrated serum i.p. on days 0, 8, and 15 after challenge. Mice also received naïve OT-I Tg CD8 T cells on day 2 and 1 µg IL15/IL15Rα complex on days 2, 7, and 14 after challenge (mice n=4). B) Histology of B16-OVA tumors that were collected on day 20 from either B6-WT (■) or B6-DK (grey □) mice. Mice were either untreated or immunized with irradiated 5×105 B16-OVA and 5×105 GVAX on day 7 and 14 after challenge. Green, anti-mouse IgG; red, complement membrane-activated complex (MAC), blue, DAPI. Data were quantified using ImageJ and are presented as fluorescent area, pixels2 per FOV (right panel). Data are representative of at least two independent experiments. C) Histology of B16-OVA tumors that were collected on day 20 from either B6-WT (■) or B6-DK (grey □) mice. Mice were untreated or immunized with irradiated 5×105 B16-OVA and 5×105 GVAX on day 7 and 14 after challenge. Green, anti-mouse IgG; red, F4/80. Data were quantified using NIS Elements software and are presented as fluorescent area, pixels2 per region of interest (ROI) or using ImageJ and presented as fluorescent area, pixels2 per FOV (right panel). Data are representative of at least two independent experiments. P values were calculated to compare F4/80+ fluorescent area within IgG+ and IgG− regions. D) Tissue sections obtained from above were analyzed for IgG deposition (green) in relation to stromal components by staining with anti-CD31 (red, 100×), anti-ERTR7 (red, 100×) and anti-desmin (red, 200×). Nuclei were stained with DAPI (blue). Data shown are representative of IgG deposit area within tumors from B6-DK mice containing 5–10 distinct foci.
Discussion
We report here that antitumor immunity against melanoma is enhanced by the genetic disruption of CD8 Treg activity that was associated with the expansion of TFH cells and GC B cells, the generation of anti-TAA antibodies and delayed tumor growth. These findings concerning the antitumor response are consistent with previous findings that targeting of TFH cells by CD8 Treg constrains the expansion of TFH cell and the development of high affinity Ab and autoantibody responses. Excessive production of IL-21 by activated TFH cells in B6-DK mice may also stimulate a CD8 T-cell response to exert antitumor activity (17).
The therapeutic activity of the Ab response mounted by B6-DK mice was confirmed from studies of tumor-bearing hosts infused with IgG from immunized tumor-bearing B6-DK, but not from B6-WT mice. Transfer of IgG from the former, but not the latter donors resulted in significant protection from lethal tumor growth in the adoptive hosts (Fig. 5A). The Ab response in B6-DK mice included anti-dsDNA, anti-MIF and anti-Ang-2 specificities after GVAX treatment and tumor challenge (Fig. 4). Increased IgG deposition in tumors of B6-DK mice was associated with tumor tissue destruction and often co-localized with membrane-activating complexes (C5b-C9) that have the potential to lyse tumor cells. Whether antibodies generated in this setting directly target B16 melanoma or act through the activation of tumor-infiltrating immune cells, such as macrophages, is not clear. NK mediated cytolysis of B16 melanoma does not appear to be involved in this process, since NK cells did not co-localize with intra-tumoral IgG and we did not observe antibody-dependent cell-mediated cytotoxicity (ADCC) against B16 cells after incubation with B6-DK-derived Ab.
Considerable attention has been given to the association of inflammatory and autoimmune responses and the development of cancers (18–22). However, the influence of chronic autoimmunity on the natural history of solid tumors is not well understood. For example, although SLE patients have decreased risk of breast cancer and melanoma (23), the underlying mechanisms and biological significance of these observations are unclear. It may be relevant that longitudinal analyses of sera from long-term surviving melanoma patients after vaccination with GM-CSF-secreting autologous melanoma cells have revealed that the generation of autoantibodies was associated with increased tumor destruction (15).
These findings raise the intriguing possibility that interruption of CD8 Treg activity may act in part through enhanced antibody responses to “classical” autoimmune target antigens, including dsDNA. While the full repertoire of Abs generated in B6-DK mice upon GVAX treatment and their relative contribution to antitumor immunity has not been determined, the production of high levels of anti-dsDNA Ab in tumor-bearing B6-DK mice after GVAX treatment was associated with protective antitumor immunity. Although the development of anti-dsDNA antibody is considered a pathogenic sign in the context of SLE, the appearance of these autoantibodies in cancer patients has been associated with improved clinical outcome (24). Anti-dsDNA antibodies may affect tumor growth through direct binding and induction of apoptosis (25, 26). Cell-penetrating anti-DNA antibodies, for example, inhibit DNA single strand and double strand DNA-repair in tumor cells, and sensitize them to DNA-damaging therapy (27). Anti-dsDNA antibody complexed to dsDNA can also stimulate B cells and dendritic cells in a TLR9-dependent manner to produce increased complement-fixing antibodies and IFNα, respectively, which can have inhibitory effects on malignancy (28, 29). These considerations support the notion that generation of a broad-based range of autoantibodies, including dsDNA, in the absence of CD8 Treg activity may contribute to the enhanced anti-melanoma responses noted here as well as to the response against a wide range of tumors.
The ability of antibodies specific for intracellular rather than cell surface molecules to mediate antitumor immunity is not well understood. The pathogenic effects of antibodies to the cytoplasmic enzyme glucose-6-phosphate isomerase (GPI) in the K/BXN arthritis mouse model may be instructive. Here, anti-GPI antibodies initiate local inflammatory changes leading to increased extracellular GPI and enhanced vascular permeability to serum autoantibodies and inflammatory cells (30, 31). In the present study Ab-mediated antitumor effects depended on the cooperative action of tumor-specific CD8 T cells (Fig. 5A). Induction of inflammatory foci in the tumor environment may enhance the release of intracellular antigens (e.g., dsDNA), resulting in increased permeability of the vasculature, and mobilization of antigen-specific CD8+ cytolytic T cells into the tumor sites. This coordinated humoral and cellular response may be essential for optimal antitumor activity. Indeed, a combined antibody and CD8+ antitumor immune response to the intracellular melanoma antigen NY-ESO-1 has been correlated with clinical benefit in advanced melanoma patients treated with anti-CTLA4 (ipilimumab) (32). Increased ICOS+ CD4 cells within the tumor and enhanced IL-21 production in the serum of DK mice are consistent with previous findings that TFH cells are preferentially expanded in DK mice. Additional experiments, e.g., with ICOS−/− mice, are needed to document the contribution of elevated ICOS+ CD4 cells to antitumor responses and delineate the potential link between ICOS+ CD4 cells, IL-21 production and enhanced antitumor responses. Recent analysis of spatiotemporal dynamics of the tumor-immune interaction during tumor progression revealed that CXCL13 and IL-21 constitute the pivotal antitumor players and increased numbers of TFH and B cells are strongly correlated with a positive prognosis (33). Promotion of effector T cells by IL-21 in a mouse melanoma model, and the efficacy of IL-21 treatment of metastatic melanoma patients suggest a multifaceted role for IL-21, in addition to its potential contribution to Ab-mediated antitumor responses noted here (34, 35).
Here we find that an increase in the ratio of CD8 Teff to CD8 Treg (and no change in CD4 Treg) within tumors of B6-DK mice compared to those in B6-WT mice is associated with enhanced antitumor activity after tumor vaccination. Our observations are congruent with previous findings that increased ratios of CD8 T cells to FoxP3+ Treg is associated with favorable prognosis in the context of human and mouse cancers (36–38). Blockade of CD8 Treg-dependent inhibition may tip the balance within the tumor microenvironment in favor of the Teff compartment, resulting in enhanced antitumor immunity.
We also detected anti-MIF and anti-Ang2 autoanibodies in some GVAX-treated tumor-bearing B6-DK mice, but not in B6-WT controls (Fig. 4C). These antibodies may exert antitumor effects through the inhibition of the Tie-2 pathway in macrophages and MMP-9 production (10). Interestingly, increases in both anti-MIF and anti-Ang-2 antibodies were also noted in response to treatment with GVAX and anti-CTLA4 antibody therapy and correlated with increased tumor vasculopathy (10). Whether Ab activity present in IgG-enriched sera from B6-DK mice directly contributes to the antitumor response requires further experiments, including evaluation of the effects of absorption of purified IgG by tumor lysate. These considerations suggest that the disruption of normal immunoregulatory T cells may favor increased autoantibody responses that can target molecular elements that contribute to tumor growth and spread.
Here we identify a new immune phenotype of Qa-1 mutant mice. Genetic disruption of the inhibitory interaction between CD8 Treg and Qa-1+ TFH cells results in the induction of robust autoimmune antitumor responses driven by enhanced TFH cell helper activity and the upregulation of therapeutic antibody production by B cells. Our data indicate that the CD8+ Teff–Treg ratio in tumor lymphocyte infiltrates may represent a useful prognostic index for cancer development. These data also suggest that specific depletion of CD8 Treg represents a novel and potentially effective strategy for cancer treatment.
Supplementary Material
Acknowledgements
We thank Dr. Lisa Cameron (DFCI Microscopy Core) for help with image preparation and analysis, A. Angel for manuscript and figure preparation and V. Garcia for technical assistance.
Grant Support
This work was supported in part by NIH Research Grant AI 037562 and a gift from the LeRoy Schecter Research Foundation to H. Cantor; NRSA Fellowships (T32AI07386) to D. Alvarez Arias and (T32CA070083) to H-J. Kim; T.A.W. Holderried is supported by the University of Düsseldorf.
Footnotes
Author Contributions: D. Alvarez Arias, H-J. Kim, G. Dranoff and H. Cantor conceived and planned experiments; D. Alvarez Arias, H-J. Kim, T.A.W. Holderried and X. Wang performed experiments; D. Alvarez Aias, H-J. Kim, G. Dranoff and H. Cantor analyzed data and wrote the paper.
References
- 1.Kantoff PW, Schuetz TJ, Blumenstein BA, Glode M, Bilhartz DL, Wyand M, et al. Overall survival analysis of a phase II randomized controlled trial of a Poxviral-based PSA-targeted immunotherapy in metastatic castration-resistant prostate cancer. J Clin Oncol. 2010;28(7):1099–1105. doi: 10.1200/JCO.2009.25.0597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Vanneman M, Dranoff G. Combining immunotherapy and targeted therapies in cancer treatment. Nat Rev Cancer. 2012;12(4):237–251. doi: 10.1038/nrc3237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sharma P, Wagner K, Wolchok JD, Allison JP. Novel cancer immunotherapy agents with survival benefit: recent successes and next steps. Nat Rev Cancer. 2011;11(11):805–812. doi: 10.1038/nrc3153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hodi FS, O'Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, Gonzalez R, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N.Engl.J.Med. 2010;363:711–723. doi: 10.1056/NEJMoa1003466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kim HJ, Cantor H. Regulation of self tolerance by Qa-1-restricted CD8+ regulatory T cells. Semin.Immunol. 2011;23:446–452. doi: 10.1016/j.smim.2011.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kim HJ, Verbinnen B, Tang X, Lu L, Cantor H. Inhibition of follicular T helper cells by CD8 + Treg is essential for self tolerance. Nature. 2010;467:328–332. doi: 10.1038/nature09370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kim HJ, Wang X, Radfar S, Sproule TJ, Roopenian DC, Cantor H. CD8+ T regulatory cells express the Ly49 class I MHC receptor and are defective in autoimmune-prone B6-Yaa mice. Proc.Natl.Acad.Sci.U.S.A. 2011;108:2010–2015. doi: 10.1073/pnas.1018974108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lu L, Kim HJ, Werneck MB, Cantor H. Regulation of CD8+ regulatory T cells: Interruption of the NKG2A-Qa-1 interaction allows robust suppressive activity and resolution of autoimmune disease. Proc.Natl.Acad.Sci.U.S.A. 2008;105(49):19420–19425. doi: 10.1073/pnas.0810383105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Reilly RT, Machiels JP, Emens LA, Ercolini AM, Okoye FI, Lei RY, et al. The collaboration of both humoral and cellular HER-2/neu-targeted immune responses is required for the complete eradication of HER-2/neu-expressing tumors. Cancer Res. 2001;61(3):880–883. [PubMed] [Google Scholar]
- 10.Schoenfeld J, Jinushi M, Nakazaki Y, Wiener D, Park J, Soiffer R, et al. Active immunotherapy induces antibody responses that target tumor angiogenesis. Cancer Res. 2010;70(24):10150–10160. doi: 10.1158/0008-5472.CAN-10-1852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Klein C, Bueler H, Mulligan RC. Comparative analysis of genetically modified dendritic cells and tumor cells as therapeutic cancer vaccines. J Exp.Med. 2000;191(10):1699–1708. doi: 10.1084/jem.191.10.1699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dranoff G, Jaffee E, Lazenby A, Golumbek P, Levitsky H, Brose K, et al. Vacciniation with irradiated tumor cells engineered to secrete murine granulocyte-macrophage colony-stimulating factor stimulates potent, specific, and long-lasting anti-tumor immunity. Proc Natl Acad Sci USA. 1993;90:3539–3543. doi: 10.1073/pnas.90.8.3539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bellone M, Cantarella D, Castiglioni P, Crosti MC, Ronchetti A, Moro M, et al. Relevance of the tumor antigen in the validation of three vaccination strategies for melanoma. J Immunol. 2000;165(5):2651–2656. doi: 10.4049/jimmunol.165.5.2651. [DOI] [PubMed] [Google Scholar]
- 14.Leow CC, Coffman K, Inigo I, Breen S, Czapiga M, Soukharev S, et al. MEDI3617, a human anti-angiopoietin 2 monoclonal antibody, inhibits angiogenesis and tumor growth in human tumor xenograft models. Int J Oncol. 2012;40(5):1321–1330. doi: 10.3892/ijo.2012.1366. [DOI] [PubMed] [Google Scholar]
- 15.Sittler T, Zhou J, Park J, Yuen NK, Sarantopoulos S, Mollick J, et al. Concerted potent humoral immune responses to autoantigens are associated with tumor destruction and favorable clinical outcomes without autoimmunity. Clin Cancer Res. 2008;14(12):3896–3905. doi: 10.1158/1078-0432.CCR-07-4782. [DOI] [PubMed] [Google Scholar]
- 16.Kamei H. Nuclear antigens, as DNA, of DSB389 MAb and some other anti-desmin monoclonal antibodies. Cell Biol Int. 1996;20(11):769–775. doi: 10.1006/cbir.1996.0099. [DOI] [PubMed] [Google Scholar]
- 17.Zeng R, Spolski R, Finkelstein SE, Oh S, Kovanen PE, Hinrichs CS, et al. Synergy of IL-21 and IL-15 in regulating CD8+ T cell expansion and function. Journal of Experimental Medicine. 2005;201(1):139–148. doi: 10.1084/jem.20041057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Grivennikov SI, Greten FR, Karin M. Immunity, inflammation, and cancer. Cell. 2010;140(6):883–899. doi: 10.1016/j.cell.2010.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Coussens LM, Zitvogel L, Palucka AK. Neutralizing tumor-promoting chronic inflammation: a magic bullet? Science. 2013;339(6117):286–291. doi: 10.1126/science.1232227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bernatsky S, Kale M, Ramsey-Goldman R, Gordon C, Clarke AE. Systemic lupus and malignancies. Curr Opin Rheumatol. 2012;24(2):177–181. doi: 10.1097/BOR.0b013e32834ff258. [DOI] [PubMed] [Google Scholar]
- 21.Ekstrom K, Hjalgrim H, Brandt L, Baechlund E, Klareskog L, Ekbom A, Askling J. Risk of malignant lymphomas in patients with rheumatoid arthritis and in their first-degree relatives. Arthritis Rheum. 2003;48(4):963–970. doi: 10.1002/art.10939. [DOI] [PubMed] [Google Scholar]
- 22.Ngalamika O, Zhang Y, Yin H, Zhao M, Gershwin ME, Lu Q. Epigenetics, autoimmunity and hematologic malignancies: a comprehensive review. J Autoimmun. 2012;39(4):451–465. doi: 10.1016/j.jaut.2012.09.002. [DOI] [PubMed] [Google Scholar]
- 23.Bernatsky S, Easton DF, Dunning A, Michailidou K, Ramsey-Goldman R, Gordon C, et al. Decreased breast cancer risk in systemic lupus erythematosus: the search for a genetic basis continues. Lupus. 2012;21(8):896–899. doi: 10.1177/0961203312443992. [DOI] [PubMed] [Google Scholar]
- 24.Syrigos KN, Charalambopoulos A, Pliarchopoulou K, Varsamidakis N, Machairas A, Mandrekas D. The prognostic significance of autoantibodies against dsDNA in patients with colorectal adenocarcinoma. Anticancer Res. 2000;20(6B):4351–4353. [PubMed] [Google Scholar]
- 25.Torchilin VP, Iakoubov LZ, Estrov Z. Antinuclear autoantibodies as potential antineoplastic agents. Trends Immunol. 2001;22(8):424–427. doi: 10.1016/s1471-4906(01)01984-6. [DOI] [PubMed] [Google Scholar]
- 26.Cao Q, Xu W, Wen Z, Xu L, Li K, Chu Y, Xiong S. An anti-double-stranded DNA monoclonal antibody induced by tumor cell-derived DNA inhibits the growth of tumor in vitro and in vivo via triggering apoptosis. DNA Cell Biol. 2008;27(2):91–100. doi: 10.1089/dna.2007.0633. [DOI] [PubMed] [Google Scholar]
- 27.Hansen JE, Chan G, Liu Y, Hegan DC, Dalal S, Dray E, et al. Targeting cancer with a lupus autoantibody. Sci Transl Med. 2012;4(157):157ra42. doi: 10.1126/scitranslmed.3004385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tarhini AA, Gogas H, Kirkwood JM. IFN-alpha in the treatment of melanoma. J Immunol. 2012;189(8):3789–3793. doi: 10.4049/jimmunol.1290060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kis-Toth K, Szanto A, Thai TH, Tsokos GC. Cytosolic DNA-activated human dendritic cells are potent activators of the adaptive immune response. J Immunol. 2011;187(3):1222–1234. doi: 10.4049/jimmunol.1100469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Matsumoto I, Maccioni M, Lee DM, Maurice M, Simmons B, Brenner M, et al. How antibodies to a ubiquitous cytoplasmic enzyme may provoke joint-specific autoimmune disease. Nat Immunol. 2002;3(4):360–365. doi: 10.1038/ni772. [DOI] [PubMed] [Google Scholar]
- 31.Binstadt BA, Patel PR, Alencar H, Nigrovic PA, Lee DM, Mahmood U, et al. Particularities of the vasculature can promote the organ specificity of autoimmune attack. Nat Immunol. 2006;7(3):284–292. doi: 10.1038/ni1306. [DOI] [PubMed] [Google Scholar]
- 32.Yuan J, Adamow M, Ginsberg BA, Rasalan TS, Ritter E, Gallardo HF, et al. Integrated NY-ESO-1 antibody and CD8+ T-cell responses correlate with clinical benefit in advanced melanoma patients treated with ipilimumab. Proc Natl Acad Sci U S A. 2011;108(40):16723–16728. doi: 10.1073/pnas.1110814108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bindea G, Mlecnik B, Tosolini M, Kirilovsky A, Waldner M, Obenauf AC, et al. Spatiotemporal dynamics of intratumoral immune cells reveal the immune landscape in human cancer. Immunity. 2013;39(4):782–795. doi: 10.1016/j.immuni.2013.10.003. [DOI] [PubMed] [Google Scholar]
- 34.Kim-Schulze S, Kim HS, Fan Q, Kim DW, Kaufman HL. Local IL-21 promotes the therapeutic activity of effector T cells by decreasing regulatory T cells within the tumor microenvironment. Mol Ther. 2009;17(2):380–388. doi: 10.1038/mt.2008.249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Petrella TM, Tozer R, Belanger K, Savage KJ, Wong R, Smylie M, et al. Interleukin-21 has activity in patients with metastatic melanoma: a phase II study. J Clin Oncol. 2012;30(27):3396–3401. doi: 10.1200/JCO.2011.40.0655. [DOI] [PubMed] [Google Scholar]
- 36.Sato E, Olson SH, Ahn J, Bundy B, Nishikawa H, Qian F, et al. Intraepithelial CD8+ tumor-infiltrating lymphocytes and a high CD8+/regulatory T cell ratio are associated with favorable prognosis in ovarian cancer. Proc Natl Acad Sci U S A. 2005;102(51):18538–18543. doi: 10.1073/pnas.0509182102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Quezada SA, Peggs KS, Curran MA, Allison JP. CTLA4 blockade and GM-CSF combination immunotherapy alters the intratumor balance of effector and regulatory T cells. J Clin.Invest. 2006;116:1935–1945. doi: 10.1172/JCI27745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hodi FS, Butler M, Oble DA, Seiden MV, Haluska FG, Kruse A, et al. Immunologic and clinical effects of antibody blockade of cytotoxic T lymphocyte-associated antigen 4 in previously vaccinated cancer patients. Proc Natl Acad Sci USA. 2008;105(8):3005–3010. doi: 10.1073/pnas.0712237105. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.