

Abstract
Although green tea is considered to be a healthy beverage, hepatotoxicity associated with the consumption of green tea extract has been reported. In the present study, we characterized the hepatotoxicity of green tea extract in rats and explored the responsible mechanism. Six-week-old IGS rats received a single intraperitoneal (ip) injection of 200 mg/kg green tea extract (THEA-FLAN 90S). At 8, 24, 48 and 72 hrs and 1 and 3 months after exposure, liver damage was assessed by using blood-chemistry, histopathology, and immunohistochemistry to detect cell death (TUNEL and caspase-3) and proliferative activity (PCNA). Analyses of malondialdehyde (MDA) in serum and the liver and of MDA and thymidine glycol (TG) by immunohistochemistry, as oxidative stress markers, were performed. Placental glutathione S-transferase (GST-P), which is a marker of hepatocarcinogenesis, was also immunohistochemically stained. To examine toxicity at older ages, 200 mg/kg green tea extract was administered to 18-wk-old female rats. In 6-wk-old rats, 12% of males and 50% of females died within 72 hrs. In 18-wk-old rats, 88% died within 72 hrs. The serum levels of aspartate aminotransferase, alanine aminotransferase and/or total bilirubin increased in both males and females. Single-cell necrosis with positive signs of TUNEL and caspase-3 was seen in perilobular hepatocytes from 8 hrs onward in all lobular areas. PCNA-positive hepatocytes increased at 48 hrs. MDA levels in the serum and liver tended to increase, and MDA- and TG-positive hepatocytes were seen immunohistochemically. GST-P–positive hepatocellular altered foci were detected in one female rat at the 3-month time point. In conclusion, a single injection of green tea extract induced acute and severe hepatotoxicity, which might be associated with lipid peroxidation and DNA oxidative stress in hepatocytes.
Keywords: apoptosis, green tea, hepatotoxicity, oxidative stress, rats
Introduction
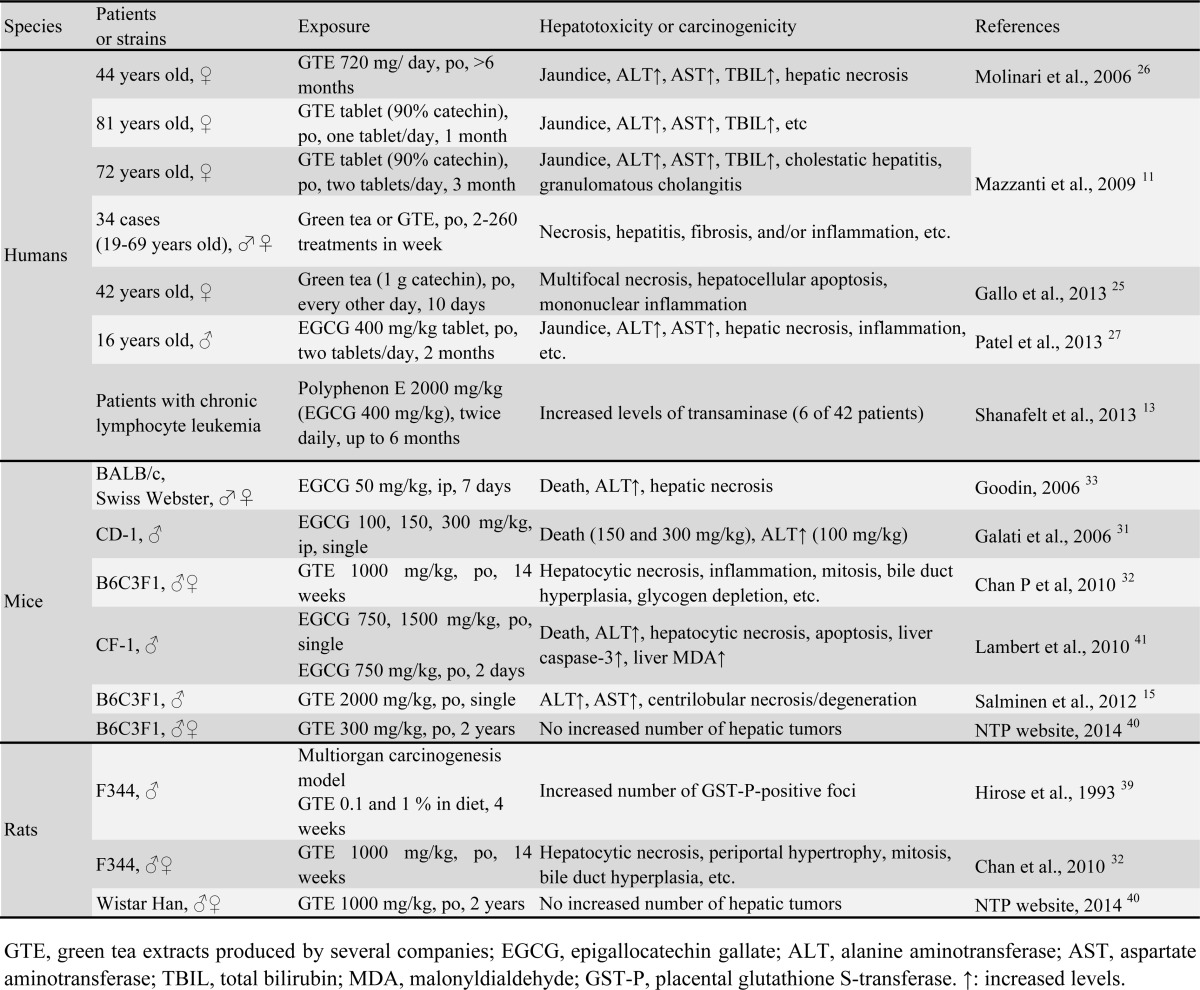
Tea (Camellia sinensis) is a widely consumed beverage worldwide. Green tea, which is popular in Japan and China, accounts for about 20% of worldwide tea consumption. About 78% of tea consumed is black tea, which is the main tea beverage in the United States, Europe and Western Asia. The remaining 2% of tea consumed is oolong tea, which is mainly consumed in southeastern China and Taiwan1. Green tea includes polyphenolic catechins, such as epicatechin, epicatechin gallate, epigallocatechin and epigallocatechin gallate (EGCG), the most abundant and probably the most pharmacologically active polyphenolic catechin2, 3. There is wide interest in the medical benefits of green tea, including the prevention and treatment of cancers such as upper gastrointestinal tract, breast and prostate cancers and oral leukoplakia1, 2, 4, and non-cancer health benefits, such as improved cardiac function, decreased atherosclerosis, cholesterol-lowering activity, and obesity prevention5-7. Although normal consumption of green tea seems safe, the consumption of concentrated green tea extract (GTE) has been associated with clinical cases of hepatotoxicity, and it can induce hepatocellular damage in rodents (Table 1). Since 1966, at least 216 cases of toxicities attributed to GTE have been reported; most of these cases presented with an acute hepatocellular injury pattern, and most patients recovered after stopping the use of GTE8, 9. In 2003, regulatory agencies in France and Spain suspended market authorization for a weight-loss product containing a hydroalcoholic GTE because of hepatotoxicity concerns10-12. In a recent clinical study of chronic lymphocyte leukemia, Polyphenon E including 400 mg/kg EGCG increased the levels of liver transaminase, suggesting severe liver damage13. However, green tea or GTE effectively inhibited the hepatotoxicities induced by 2-nitropropane, galactosamine, carbon tetrachloride, pentachlorophenol and acetaminophen when administered prior to chemical exposure in rats or mice14, 15. In our preliminary pharmacological studies of a rat model of retinal degeneration, GTE induced death due to acute hepatotoxicity. The purpose of this study was to characterize and clarify the mechanism of GTE hepatotoxicity in rats following a single intraperitoneal (ip) exposure.
Table 1. Selected Literature Information about Hepatotoxicity and Carcinogenicity Induced by Green Tea, Green Tea Extract or Catechin in Humans and Rodents.
Materials and Methods
Chemical and dose formulation
The GTE used was THEA-FLAN 90S (Ito En, Ltd., Tokyo, Japan; GTE), a decaffeinated product sold as a commercial food additive. GTE is mainly comprised of polyphenols (about 90%) that include tea catechins with a galloyl moiety (70.0%)6; epigallocatechin gallate (54%, EGCG), epicatechin (12.4%), gallocatechin gallate (2.8%), catechin gallate (0.4%), epigallocatechin (0.3%), gallocatechin (0.1%), caffeine (0.0%) and other catechins (0.3%). (THEA-FLAN 90S content information was kindly provided by Ito En, Ltd.) GTE was dissolved in physiologic saline just prior to use.
Animal procedures
The study protocol and all animal procedures were approved by the Animal Care and Use Committee of Kansai Medical University and were in accordance with the guidelines for animal experimentation at Kansai Medical University. Male and female SPF rats [Crl:CD(SD)] were purchased from Charles River Laboratories Japan (Yokohama, Japan). To examine the sequential changes in hepatotoxicity, at six weeks of age, 33 males and 42 females received a single intraperitoneal (ip) injection of 200 mg/kg GTE dissolved in physiologic saline. Three to six surviving rats of each sex were selected for analysis at 8, 24, 48, and 72 hrs, and 1 and 3 months after GTE injections (Fig. 1). In addition, to confirm the susceptibility to toxicity in older animals, eight female rats were injected with GTE at 18 wks of age and analyzed 7 days after treatment. In our preliminary study, ip injection of 200 mg/kg GTE once or twice induced severe degrees of hepatocellular necrosis one day later, as shown by a histopathological examination; however 100 mg/kg GTE did not induce lesions in the liver. Therefore, in this study, we selected 200 mg/kg as the dosage that can induce hepatotoxicity by single ip exposure. Rats were maintained under specific pathogen-free conditions and had free access to water and a CMF diet (Oriental Yeast, Chiba, Japan). They were housed in plastic cages with paper-chip bedding (Paper Clean, SLC, Hamamatsu, Japan) in an air-conditioned room at 22°C ± 2°C and 60% ± 10% relative humidity with a 12-hr light/dark cycle. The age-matched control groups consisted of 4 to 5 male and/or female rats that were treated with vehicle (physiological saline) only. Each day, the rats were weighed and observed for clinical signs of toxicity. On the day of sacrifice, GTE- and saline-treated rats were anesthetized with isoflurane (ForaneR; Abbott Japan, Tokyo, Japan) and sacrificed by exsanguination from aortic transection.
Fig. 1.

Experimental protocol. Solid arrows, injection of green tea extract (GTE); broken arrows, time points of necropsy.
Blood chemical analysis to detect hepatotoxicity
At the time of sacrifice, blood samples of all surviving rats were withdrawn from the abdominal aorta under anesthesia. The serum concentrations of aspartate aminotransferase (AST), alanine aminotransferase (ALT), total bilirubin (TBIL) and alkaline phosphatase (ALP) were detected with a Fuji DRI- CHEM 3500V analyzer (Fujifilm Corporation., Tokyo, Japan). AST and ALT levels reflect damage to hepatocytes and are therefore valuable biomarkers of hepatotoxicity induced by chemicals16, while TBIL and ALP are markers of bile procession alterations and bile duct injury15.
EGCG concentrations in serum and the liver
Serum and liver samples were collected from male and female rats, two control rats and two GTE-treated rats 48 hrs after the administration of GTE. The serum and hepatic levels of EGCG were determined by HPLC with an electrochemical detector (Coulochem III electrochemical detector, ESA Inc., USA) (HPLC-EC). To determine the total amount of EGCG, including free and conjugated forms, a plasma sample (200 µL) was incubated with a mixture of β-glucuronidase (500 units) at 37°C for 45 min. Immediately after incubation, 400 µL of acetonitrile and 100 µL of ethyl gallate solution (1 µg/mL) in methanol as internal standard were added to the reaction mixture and vigorously mixed for 5 min, followed by centrifugation at 10,000 ×g for 10 min at 4°C to precipitate protein. The resulting supernatant was transferred to another tube and then evaporated to dryness in a vacuum. Then a liver sample (0.2 g) was homogenized with 0.8 mL of saline and centrifuged at 10,000 ×g for 10 min at 4°C. The resulting supernatant (400 µL) was treated in the same manner as the plasma sample. The residues were redissolved in 1 mL of 5% methanol aqueous solution and then applied to a Bond Elute cartridge (C18, size 50 mg/1 mL). EGCG in the cartridge was eluted with 1 mL of 50% methanol-50% acetonitrile. The resulting eluents were evaporated to dryness in a vacuum. The residue was redissolved in 200 µL of mobile phase for HPLC (10% methanol, 8% acetonitrile, 0.1% folic acid), filtered through a 0.45-µm filter and then applied to the HPLC-EC system. The analytical conditions were as follows: HPLC column, Inertsil ODS-3 (3 x 150 mm, 5 µm, GL Sciences Inc., Tokyo, Japan); mobile phase, 10% methanol, 8% acetonitrile and 0.1% folic acid; flow rate, 0.4 ml/min; column temperature, 40°C; detector, ESA Coulochem III, 150 mV.
Malondialdehyde analyses in serum and the liver
The malondialdehyde (MDA) levels, an indicator of free-radical generation that increases at the end of the lipid peroxides17, were measured in serum and liver tissue of the control and 8, 24, 48 and 72 hrs GTE groups by using an NWLSS kit (Northwest Life Sciences Specialties, Vancouver, Canada). Centrifugation of 10% w/v liver homogenates in cold assay buffer was conducted at 1600 g for 10 minutes at 4°C, and the assay was performed according to the manufacturer’s instructions. This assay is based on the reaction of MDA with thiobarbituric acid to form MDA-TBA adducts. The absorbance of reaction mixtures was recorded as spectra from 400-700 nm by using a spectrophotometer, and a 3rd derivative analysis was performed. The results were expressed as micromoles of MDA per gram of wet liver. Serum MDA levels were expressed as micromoles of MDA per liter of the sample.
Histopathological examination of liver
Liver tissues were fixed overnight in 10% neutral buffered formalin, and the right, medial and left lobes were embedded in paraffin, sectioned at a thickness of 4 μm, and stained with hematoxylin and eosin (H&E). Partial left lobes were fixed in Bouin fixative, and paraffin blocks were prepared for immunohistochemical staining of MDA. The histopathological terminology for nonneoplastic hepatic lesions is in accordance with the International Harmonization Nomenclature and Diagnostic Criteria for Lesions in Rats and Mice Project18.
Immunohistochemical examination of the liver
Sequential sections of livers of the control, 8, 24, 48 and 72 hrs GTE groups were used for immunohistochemical analyses. Cell death was observed by terminal deoxynucleotidyl transferase (TdT)-mediated dUTP digoxigenin nick end-labeling (TUNEL) by using an in situ apoptosis detection kit (Apop-Tag; Millipore, Billerica, MA, USA) according to a previous report19. Sequential sections were also immunohistochemically stained with anti-active caspase 3 polyclonal antibody (×200; Promega Corporation, Madison, USA) by the modified NIEHS method20. To detect the proliferative activity of hepatocytes in all groups including the 1- and 3-month groups, the monoclonal antibody to proliferating cell nuclear antigen (PCNA) (clone PC10, ×100; Leica Biosystems, Newcastle Upon Tyne, UK) was used21. In the control and 8, 24, 48 and 72 hrs GTE groups, immunohistochemistry for malondialdehyde (MDA) (clone 1F83, ×40; Japan Institute for the Control of Aging, Shizuoka, Japan) and thymidine glycol (TG) (clone 2E8, ×20; Japan Institute for the Control of Aging) was conducted to detect the oxidative stress of cytoplasmic lipids and DNA, respectively22, 23. Placental glutathione S-transferase (GST-P) is specifically expressed during rat hepatocarcinogenesis, and it has been used as a reliable tumor marker for experimental rat carcinogenesis24. The liver sections of the control and 1 and 3 months GTE groups were stained with anti-GST-P polyclonal antibody (prediluted; Medical & Biological Laboratories, Tokyo, Japan). Liver sections fixed in Bouin solution were used for MDA, and liver sections fixed in formalin were used for the other immunostainings. In brief, the staining protocols were as follows: sections were deparaffinized, hydrated and blocked for endogenous peroxidase. Heat-induced epitope retrieval was performed for caspase 3 and PCNA. All primary antibodies were reacted at room temperature for one hour. The antigen-antibody complexes were identified by using a streptavidin-biotin (LSAB) staining kit (Dako, Carpinteria, CA, USA) according to the manufacturer’s instructions. The reaction products were visualized with 3-3’-diaminobenzidine tetrahydrochloride. Histopathological and immunohistochemical examinations were conducted by two toxicologic pathologists certified by the Japanese Society of Toxicologic Pathology (K.Y., A.T.).
Statistical analysis
All discrete values except for histopathological and immunohistochemical results were expressed as the mean ± standard error (SE) and analyzed by using the two-tailed independent Student’s t-test for unpaired samples after confirming the homogeneity of variances. The results presented below include comparisons between GTE-treated rats and saline-treated controls. P-values < 0.05 were considered statistically significant.
Results
General remarks
During the experimental period, the mortality after administration of 200 mg/kg GTE was higher in female rats than in male rats. Acute death within 72 hrs occurred in 4 of 33 males (12% in mortality) and in 21 of 42 females (50% in mortality) that received GTE at the age of 6 weeks (Fig. 1). A 1 week GTE exposure group could not set up due to the large number of dead animals at the age of 6 wks. Because so many 6-wk-old rats died soon after GTE exposure, there were not enough surviving animals to study one week after exposure. Similarly, 7 of 8 females that received GTE at the age of 18 wks died within 72 hrs (88% mortality). The cause of death in these rats was severe liver toxicity, as confirmed by histopathological analysis (data not shown). Susceptibility to GTE-induced toxicity seemed to be higher in older rats.
The body weights in the GTE groups significantly decreased at maximum 12 g and 8.4 g within 24 hrs in males and females, respectively (data not shown). Thereafter, the body weights returned to control levels. At 1 and 3 months after GTE exposure, their weights were comparable to those of the age-matched control rats.
Blood chemistry
The AST levels in the GTE-treated male rats tended to increase with time (Fig. 2). They peaked at 72 hrs in male rats (1268 U/L) and at 48 hrs in female rats (1570 U/L), while they remained low in control rats (male control, 86 U/L; female control, 69 U/L). The AST levels in male rats were significantly increased at 8 and 48 hrs. The ALT levels exhibited the same increasing trend as the AST levels (Fig. 2); the ALT levels peaked at 72 hrs in male rats (818 U/L) and at 48 hrs in female rats (1315 U/L), while the levels remained low in control rats (male control, 35 U/L; female control, 29 U/L). The ALT levels were significantly increased at 8 hrs in the male GTE-treated rats.
Fig. 2.

Blood chemistry. The serum concentrations of aspartate aminotransferase (AST), alanine aminotransferase (ALT), alkaline phosphatase (ALP) and total bilirubin (TBIL) were analyzed. *: p<0.05; **: p<0.01.
As a marker of bile production alterations and bile duct injury, there was a trend for increased TBIL levels with time in male and female rats (Fig. 2), and both sexes reached a peak level of 1.4 mg/dL at 48 hrs (male control, 0.2 mg/ dL; female control, 0.2 mg/ dL). In contrast, the ALP levels decreased to 1530 U/L in males at 24 hrs and 1002 U/L in females at 8 hrs (male control, 2698 U/L; female control, 1970 U/L). The ALP levels returned to control levels in the female rats at 72 hrs, but the decreased levels persisted in males even at 72 hrs (Fig. 2).
At 1 and 3 months after GTE exposure, none of the blood chemistry parameters were different from the levels in the age-matched control rats (data not shown).
EGCG concentrations in serum and liver
EGCG was not detected in the serum or liver of male and female control rats. In the serum of GTE-exposed rats, the EGCG levels reached 0.388 μg/mL (0.880 μM) in males and 0.276 μg/mL (0.602 μM) in females at 48 hrs after GTE exposure. The EGCG levels in the livers of GTE-exposed rats were lower than in the serum; the hepatic levels were 0.107 μg/g tissue (0.232 μM) in males and 0.084 μg/g tissue (0.182 μM) in females.
MDA values in serum and the liver
The serum MDA levels increased beginning a 24 hrs (Fig. 3) and peaked at 72 hrs in males (1.85 μmol/L) and at 48 hrs (1.87 μmol/L) in females (0.44 μmol/L in both male and female controls). There was a trend for increased hepatic MDA levels beginning at 8 hrs, and the levels peaked at 0.33 and 0.22 μmol/ g tissue at 72 hrs in male and female rats, respectively (0.14 and 0.03 μmol/ g tissue in male and female controls, respectively). The hepatic MDA levels were significantly increased at 8 to 72 hrs in female rats (Fig. 3).
Fig. 3.

Analysis of malondialdehyde in serum and the liver. *: p<0.05; **: p<0.01.
Liver morphology
The absolute and relative liver weights decreased in males and females at 8, 24 and 48 hrs after exposure (data not shown). The change might be related to the severely decreased body weights. At 1 and 3 months after exposure, the weights in the male and female GTE groups were similar to those in the age-matched control groups.
The histopathological changes induced by GTE exposure are summarized in Table 2. Beginning at 8 hrs after exposure, the levels of glycogen in the hepatocellular cytoplasm of males and females were decreased as compared with the controls (Fig. 4a and b). Single-cell necrosis of hepatocytes appeared in perilobular areas 8 hrs after exposure (Fig. 4b, c and d) and thereafter progressed to all lobular areas as massive necrosis (Fig. 4e). At 72 hrs, the average score of necrosis was 3.0 in both males and females. Inflammatory reactions in the parenchyma and capsules of livers were detected beginning at 24 hrs and 8 hrs after exposure, respectively (Fig. 4c, d, and e). The inflammatory reactions, which were composed mainly of mononuclear cells, suggested acute inflammation. At 72 hrs, the average inflammation scores in the parenchyma were 2.4 and 1.2 in males and females, respectively. Hepatocellular mitosis, hypertrophy (Fig. 4c) and bile duct proliferation (Fig. 4e) were also seen at mainly 48 and 72 hrs after exposure. In some rats of the 1 and 3 months GTE groups, although lobular adhesion related to capsular inflammation was seen, the liver structure was similar to that of the age-matched controls (Fig. 4f).
Table 2. Histopathological Scoring of Liver.
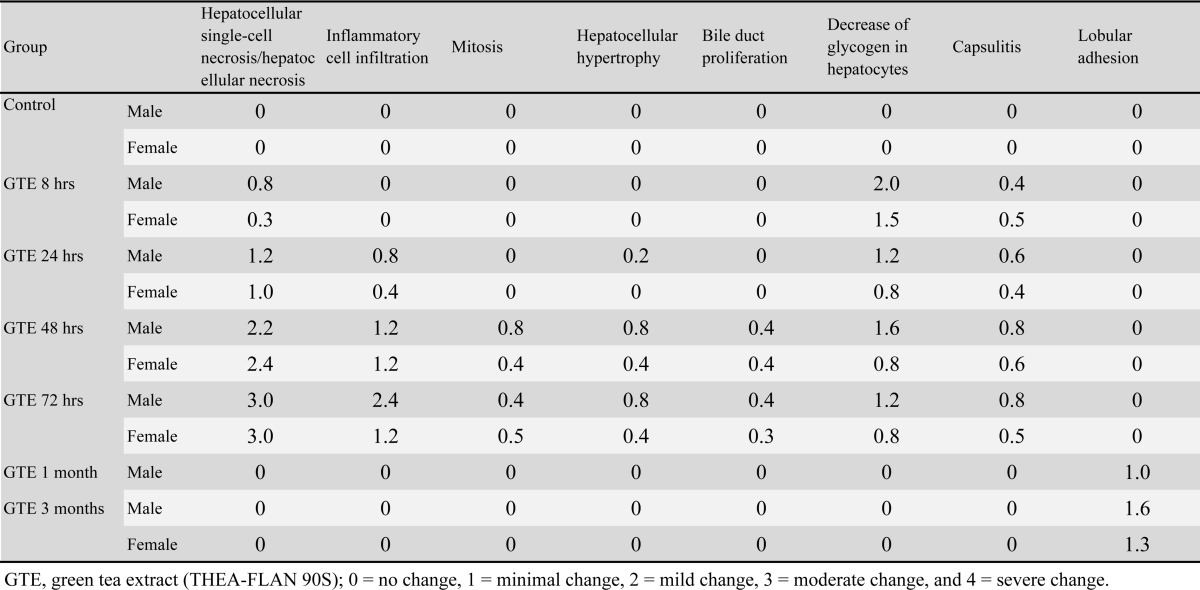
Fig. 4.

Histopathological findings. a, Control; b–f, 8 hrs, 24 hrs, 48 hrs, 72 hrs and 1 month after green tea extract (GTE) exposure, respectively. Note that hepatocellular damage progresses gradually from the perilobular (c) to centrilobular area after GTE exposure (e). C: central vein. *Glisson’s sheath. H & E, ×200.
Immunohistochemistry of the liver
Cell death expression
In control and 8 hrs groups, there were few positive hepatocytes for TUNEL and caspase 3 antibodies (Fig. 5a, b, e, and f). Twenty-four hours after exposure, TUNEL- and caspase 3-positive hepatocytes were seen in the perilobular area of males and females (Fig. 5c and g). Positive cells were scattered to all lobular areas at 48 and 72 hrs (Fig. 5d and h). Single-cell necrosis was correlated with positive results from TUNEL and caspase-3 immunohistochemistry. One and 3 months after exposure, there were a few TUNEL- and caspase 3-positive cells in the GTE-treated rats, which was similar to the age-matched controls.
Fig. 5.

Immunohistochemistry for cell death in the liver. The signals of terminal deoxynucleotidyl transferase-mediated d UTP digoxigenin nick and end-labeling (TUNEL) can be in the nuclei of hepatocytes of green tea extract (GTE)-exposed rats. a, Control; b–d, 8 hrs, 24 hrs and 48 hrs after GTE exposures, respectively. Caspase 3 signals can also be seen in the nuclei of hepatocytes of GTE-exposed rats. e, Control; f–h, 8 hrs, 24 hrs and 48 hrs after GTE exposure, respectively. C: central vein. *Glisson’s sheath. ×200.
Proliferative activity
PCNA-positive cells were scattered in the perilobular area of male and female control rats (Fig. 6a). In contrast, at 48 and 72 hrs after GTE exposure, more positive cells were seen in all lobular areas of the male and female GTE-treated rats as compared with the controls (Fig. 6b). In the 1- and 3-month groups, PCNA-positive cells were scattered in the liver of GTE-exposed rats, which was similar to the age-matched controls.
Fig. 6.

Immunohistochemistry for proliferative activity and oxidative stress in the liver. Proliferating cell nuclear antigen (PCNA) signals can be seen in the nuclei of hepatocytes of control (a) and green tea extract (GTE)-exposed rats (b). Note that more positive cells can be seen in the rats 48 hrs after GTE exposure (b). No thymidine glycol (TG) signals can be seen in control rats (c), while TG signals in the nuclei of hepatocytes were detected in the rats 48 hrs after GTE exposure (d). No malondialdehyde (MDA) signals can be seen in control rats (e), while positivity for MDA in the hepatocellular cytoplasm was detected in the rats 48 hrs after GTE exposure (f). C: central vein. *Glisson’s sheath. ×200.
Oxidative stress
In the control group, hepatocytes were negative for TG (Fig. 6c). In the GTE groups, at 48 and 72 hrs after exposure, TG signals were seen in the nuclei of hepatocytes of males and females (Fig. 6d). There was a negative reaction with MDA in control hepatocytes (Fig. 6e), while a positive reaction was detected in the hepatocellular cytoplasm of all lobular areas of males and females at 48 and 72 hrs after exposure (Fig. 6f).
GST-P positivity
In the control female rats at three months, only a few small GST-P–positive foci were seen in GST-P–immunostained slides, and the foci were composed of a small number of hepatocytes (Fig. 7a). In one of three female rats at three months after exposure, large positive foci were scattered throughout the slides and were composed of more than one hundred hepatocytes (Fig. 7b). Positive foci were not seen in any rats one month after exposure, in males three months after exposure, or in the control group at 3 months.
Fig. 7.
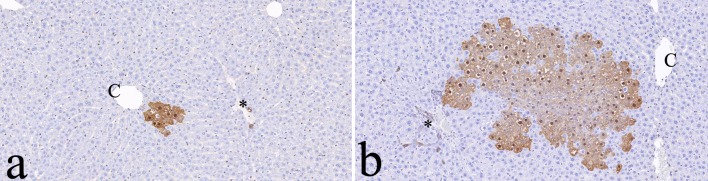
Immunohistochemistry for placental glutathione S-transferase (GST-P). Note that the larger GST-P-positive foci are found 3 months after green tea extract (GTE) exposure (b) compared with the age-matched controls (a). Bile duct epithelial cells are normally positive for GST-P in both groups. C: central vein. *Glisson’s sheath. ×100.
Discussion
In the present study, the toxicological effects of GTE in rats included death, weight loss, and blood chemical and histopathological changes related to hepatocellular damage. Histopathologically, single-cell necrosis with positive results for TUNEL and caspase-3 was seen in perilobular hepatocytes in the early phase and thereafter in all lobular areas. Cellular hypertrophy and mitoses of hepatocytes, inflammation and bile duct hyperplasia were observed.
Information about hepatotoxicity and carcinogenicity induced by green tea, GTE or catechins in humans and rodents is summarized in Table 1. In some human cases, hepatic necrosis, apoptosis and inflammation were detected histopathologically, as well as elevated levels of transaminases11, 25-27. In rodents, oral or ip exposure to green tea, GTE or catechins also induced severe acute hepatotoxicity as evaluated by blood chemistry and histopathology. General toxicity studies of several types of GTE in rodents have been conducted. In 4 and 26 wk oral studies of GTE-exposed rats conducted by Kao Corporation (Tokyo, Japan), the no-observed-adverse-effect doses were 1000 mg/kg and 400 mg/kg, respectively28, 29, and no hepatic damage could be detected clinicopathologically or histopathologically. Intraperitoneal injection of 82 mg/kg EGCG for 7 days into SD rats did not change biochemical markers of liver damage, such as AST, ALT or ALP30. However, a single ip injection of 150 mg/kg EGCG induced death in mice within one day, while 100 mg/kg elevated the ALP level to 170 U/L, as compared with 49 U/L in controls31. Recently, in 14-wk oral toxicity studies of GTE (Amax NutraSource, Inc., Eugene, OR, USA) in rats and mice that were conducted by the National Toxicology Program (NTP)32, 1000 mg/kg GTE induced hepatotoxicity, such as hepatocyte necrosis, hypertrophy, mitosis, glycogen deposition, and bile duct hyperplasia. These changes are similar to our results obtained after a single ip exposure to GTE.
In the present study, mortality after administration of 200 mg/kg GTE was higher in female rats than in male rats. The susceptibility to GTE-induced toxicity seemed to be higher in older rats. From the results of previous studies, female rodents seem to have higher susceptibility to hepatotoxicity induced by GTE or EGCG; ip exposure to 50 mg/kg EGCG for 7 days induced higher mortality due to hepatotoxicity in female BALB/c mice33, and oral exposure to 1000 mg/kg GTE for 14 weeks induced hepatotoxicity only in female rats. The reasons for the sex- and age-based differences in susceptibility in the present study are not clear, and further in vivo research regarding GTE metabolism in the case of ip exposure is required.
Catechins are chemical antioxidants that quench free-radical species. There is evidence that some effects of catechins, such as EGCG, are related to the induction of reactive oxidative stress; these prooxidant effects appear to be responsible for the induction of apoptosis in tumor cells3, 34. GTEs were negative for toxicity in in vivo genotoxic studies, such as a bone marrow micronucleus assay, although in vitro studies (chromosomal aberration assay or mouse lymphoma assay) revealed a positive trend35. The in vitro genotoxic effects of green tea are likely due to H2O2, while its genoprotective effects could be due to the direct antioxidant action of green tea or to cellular adaptations to subtle changes in the cellular redox balance induced by the low concentrations of catechins achieved in vivo36. In vitro, GTE promotes the prooxidant property of catechins to generate active oxygen species that cause DNA damage35, 37.
In the present study, GTE induced hepatocellular apoptosis, as reflected by TUNEL and caspase-3 positivity. Moreover, 48 hrs after treatment, serum and hepatic MDA levels tended to increase and MDA- and TG-positive hepatocytes increased. The most widely used index of lipid peroxidation is MDA formation, often assayed with the thiobarbituric acid (TBA) assay, and TG is one of the DNA hydroxylation products that acts as an in vivo biomarker for oxidative DNA damage17. Oxidative base modifications may result in mutations, whereas oxidation of deoxyribose moieties may induce base release or DNA strand breaks. Our results suggest that GTE induced oxidative stress in the liver, followed by DNA double-strand breaks and apoptosis in cells.
Seven GTEs significantly enhanced resazurin reduction in an alamarBlue assay at 1-3 mg/ml medium in primary culture of rat hepatocytes, and among EGCG, (-)-EC, caffeine and theanine, cytotoxicity was found with EGCG only, calculated 400–1200 μM of EGCG in medium12. Among four catechins, EGCG, epicatechin-3-gallate, epigallocatechin and epicatechin, EGCG was the most cytotoxic toward isolated rat hepatocytes as it collapsed the mitochondrial membrane and induced reactive oxidative species formation31. The LD50 of EGCG cytotoxicity was 200 μM. There is little information in vivo about the blood concentration of catechins in the case of intraperitoneal exposure as compared with oral exposures38. After a single ip injection of 100 mg/kg EGCG in SD rats, the plasma EGCG levels were 24, 2, 4, 1 and 1 μM at 0.5, 1, 2, 5 and 24 hrs, respectively. A plasma EGCG concentration of 1 μM would be similar to the levels in a 70-kg human 1 hr after drinking 6–12 cups (200 mL/cup) of tea30. In the present study, the serum and hepatic EGCG concentrations 48 hrs after exposure were 0.880 μM and 0.232 μM in males and 0.602 μM and 0.182 μM in females, respectively. The data revealed high concentrations of EGCG distributed in the livers of rats in the present study. The serum EGCG levels in the early phase after GTE exposure in our study, such as after a few hours, would be much higher than human exposure levels. Further studies are necessary to explain the discrepancy in EGCG concentrations that induce hepatocellular damage between in vitro and in vivo studies.
In rats, GST-P has been shown to be the most effective marker for altered hepatocellular foci. Single hepatocytes immunoreactive to GST-P that appear early in hepatocarcinogenesis may sometimes represent initiated cells24. The foci may regress by remodeling of the liver. In a rat multiorgan carcinogenesis model, 1% green tea catechins in the diet increased the numbers of altered hepatocellular foci positive for GST-P, suggesting slightly enhanced hepatocarcinogenesis39. However, in the recent NTP rodent carcinogenesis studies, there was no increased occurrence of liver tumors even at 2 years after treatment with 1000 mg/kg GTE administered orally40. In the present study, an increased number and area of GST-P–positive foci were seen in one of the three female rats 3 months after exposure to 200 mg/kg ip GTE. The hepatocarcinogenesis of GTE might not occur in humans, as 200 mg/kg GTE is a lethal dose in rats, and ip injection is not the route of exposure in humans.
In conclusion, a single injection of GTE induced severe acute hepatotoxicity in rats via oxidative stress to hepatocellular lipids and DNA. Although GTEs are widely used for their supposed health benefits, their use is associated with adverse events, particularly hepatotoxic reactions. Further studies may be required to elucidate the dose-dependent hepatotoxic effects and underlying molecular mechanism of GTE in our rat model.
Acknowledgments
We thank Dr. Y. Sagesaka of Ito En, Ltd. (Makinohara, Shizuoka, Japan), for providing THEA-FLAN 90S and for providing information on the chemical content. We also thank Dr. Takayasu Moroki of Maruho Co., Ltd. (Kyoto, Japan), for blood chemistry analysis and Dr. Kazuo Igarashi, Department of Research Development, Association of Medicinal Analysis, Amagasaki, Japan (http://www.median-lab.jp/), for EGCG analysis. The authors have no competing financial interests. This work was supported in part by a Grant-in-Aid for Scientific Research (C) from the Japan Society for the Promotion of Science (JSPC 25462740).
Footnotes
This is an open-access article distributed under the terms of the Creative Commons Attribution Non-Commercial No Derivatives (by-nc-nd) License <http://creativecommons.org/licenses/by-nc-nd/3.0/>.
References
- 1.Yuan JM, Sun C, and Butler LM. Tea and cancer prevention: epidemiological studies. Pharmacol Res. 64: 123–135. 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chow HHS, and Hakim IA. Pharmacokinetic and chemoprevention studies on tea in humans. Pharmacol Res. 64: 105–112. 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lambert JD, and Elias RJ. The antioxidant and pro-oxidant activities of green tea polyphenols: a role in cancer prevention. Arch Biochem Biophys. 501: 65–72. 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cooper R, Morré DJ, and Morré DM. Medicinal benefits of green tea: part II. review of anticancer properties. J Altern Complement Med. 11: 639–652. 2005. [DOI] [PubMed] [Google Scholar]
- 5.Cooper R, Morré DJ, and Morré DM. Medicinal benefits of green tea: Part I. Review of noncancer health benefits. J Altern Complement Med. 11: 521–528. 2005. [DOI] [PubMed] [Google Scholar]
- 6.Kobayashi M, Unno T, Suzuki Y, Nozawa A, Sagesaka Y, Kakuda T, and Ikeda I. Heat-epimerized tea catechins have the same cholesterol-lowering activity as green tea catechins in cholesterol-fed rats. Biosci Biotechnol Biochem. 69: 2455–2458. 2005. [DOI] [PubMed] [Google Scholar]
- 7.Suzuki Y, Miyoshi N, and Isemura M. Health-promoting effects of green tea. Proc Jpn Acad, Ser B, Phys Biol Sci. 88: 88–101. 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Navarro VJ, Bonkovsky HL, Hwang SI, Vega M, Barnhart H, and Serrano J. Catechins in dietary supplements and hepatotoxicity. Dig Dis Sci. 58: 2682–2690. 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sarma DN, Barrett ML, Chavez ML, Gardiner P, Ko R, Mahady GB, Marles RJ, Pellicore LS, Giancaspro GI, and Low Dog T. Safety of green tea extracts : a systematic review by the US Pharmacopeia. Drug Saf. 31: 469–484. 2008. [DOI] [PubMed] [Google Scholar]
- 10.Bun SS, Bun H, Guédon D, Rosier C, and Ollivier E. Effect of green tea extracts on liver functions in Wistar rats. Food Chem Toxicol. 44: 1108–1113. 2006. [DOI] [PubMed] [Google Scholar]
- 11.Mazzanti G, Menniti-Ippolito F, Moro PA, Cassetti F, Raschetti R, Santuccio C, and Mastrangelo S. Hepatotoxicity from green tea: a review of the literature and two unpublished cases. Eur J Clin Pharmacol. 65: 331–341. 2009. [DOI] [PubMed] [Google Scholar]
- 12.Schmidt M, Schmitz HJ, Baumgart A, Guédon D, Netsch MI, Kreuter MH, Schmidlin CB, and Schrenk D. Toxicity of green tea extracts and their constituents in rat hepatocytes in primary culture. Food Chem Toxicol. 43: 307–314. 2005. [DOI] [PubMed] [Google Scholar]
- 13.Shanafelt TD, Call TG, Zent CS, Leis JF, LaPlant B, Bowen DA, Roos M, Laumann K, Ghosh AK, Lesnick C, Lee MJ, Yang CS, Jelinek DF, Erlichman C, and Kay NE. Phase 2 trial of daily, oral Polyphenon E in patients with asymptomatic, Rai stage 0 to II chronic lymphocytic leukemia. Cancer. 119: 363–370. 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hasegawa R, Takekida K, Sai K, Umemura T, Tanimura A, Inoue T, and Kurokawa Y. [Inhibitory effect of green tea infusion of hepatotoxicity]. Kokuritsu Iyakuhin Shokuhin Eisei Kenkyusho Hokoku. 116: 82–91. 1998; (Abstract in English, text in Japanese). [PubMed] [Google Scholar]
- 15.Salminen WF, Yang X, Shi Q, Greenhaw J, Davis K, and Ali AA. Green tea extract can potentiate acetaminophen-induced hepatotoxicity in mice. Food Chem Toxicol. 50: 1439–1446. 2012. [DOI] [PubMed] [Google Scholar]
- 16.Ozer J, Ratner M, Shaw M, Bailey W, and Schomaker S. The current state of serum biomarkers of hepatotoxicity. Toxicology. 245: 194–205. 2008. [DOI] [PubMed] [Google Scholar]
- 17.de Zwart LL, Meerman JH, Commandeur JNM, and Vermeulen NP. Biomarkers of free radical damage applications in experimental animals and in humans. Free Radic Biol Med. 26: 202–226. 1999. [DOI] [PubMed] [Google Scholar]
- 18.Thoolen B, Maronpot RR, Harada T, Nyska A, Rousseaux C, Nolte T, Malarkey DE, Kaufmann W, Küttler K, Deschl U, Nakae D, Gregson R, Vinlove MP, Brix AE, Singh B, Belpoggi F, and Ward JM. Proliferative and nonproliferative lesions of the rat and mouse hepatobiliary system. Toxicol Pathol. 38(Suppl): 5S–81S. 2010. [DOI] [PubMed] [Google Scholar]
- 19.Yoshizawa K, Sasaki T, Uehara N, Kuro M, Kimura A, Kinoshita Y, Miki H, Yuri T, and Tsubura A. N -ethyl- N -nitrosourea induces retinal photoreceptor damage in adult rats. J Toxicol Pathol. 25: 27–35. 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.National Institute of Environmental Health Sciences (NIEHS) website. Detection of cleaved caspase-3 in formalin-fixed, paraffin-embedded rat tissue. 2014. http://www.niehs.nih.gov/research/atniehs/labs/assets/docs/a_d/caspase3rat.pdf.
- 21.Yoshizawa K, Emoto Y, Kinoshita Y, Kimura A, Uehara N, Yuri T, Shikata N, and Tsubura A. Histopathological and immunohistochemical characterization of spontaneously occurring uterine deciduomas in young adult rats. J Toxicol Pathol. 26: 61–66. 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ito K, Yano T, Morodomi Y, Yoshida T, Kohno M, Haro A, Shikada Y, Okamoto T, Maruyama R, and Maehara Y. Serum antioxidant capacity and oxidative injury to pulmonary DNA in never-smokers with primary lung cancer. Anticancer Res. 32: 1063–1067. 2012. [PubMed] [Google Scholar]
- 23.Yamada S, Kumazawa S, Ishii T, Nakayama T, Itakura K, Shibata N, Kobayashi M, Sakai K, Osawa T, and Uchida K. Immunochemical detection of a lipofuscin-like fluorophore derived from malondialdehyde and lysine. J Lipid Res. 42: 1187–1196. 2001. [PubMed] [Google Scholar]
- 24.Ito N, Hasegawa R, Imaida K, Hirose M, Shirai T, Tamano S, and Hagiwara A. Medium-term rat liver bioassay for rapid detection of hepatocarcinogenic substances. J Toxicol Pathol. 10: 1–11. 1997. [Google Scholar]
- 25.Gallo E, Maggini V, Berardi M, Pugi A, Notaro R, Talini G, Vannozzi G, Bagnoli S, Forte P, Mugelli A, Annese V, Firenzuoli F, and Vannacci A. Is green tea a potential trigger for autoimmune hepatitis? Phytomedicine. 20: 1186–1189. 2013. [DOI] [PubMed] [Google Scholar]
- 26.Molinari M, Watt KDS, Kruszyna T, Nelson R, Walsh M, Huang WY, Nashan B, and Peltekian K. Acute liver failure induced by green tea extracts: case report and review of the literature. Liver Transpl. 12: 1892–1895. 2006. [DOI] [PubMed] [Google Scholar]
- 27.Patel SS, Beer S, Kearney DL, Phillips G, and Carter BA. Green tea extract: a potential cause of acute liver failure. World J Gastroenterol. 19: 5174–5177. 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chengelis CP, Kirkpatrick JB, Regan KS, Radovsky AE, Beck MJ, Morita O, Tamaki Y, and Suzuki H. 28-Day oral (gavage) toxicity studies of green tea catechins prepared for beverages in rats. Food Chem Toxicol. 46: 978–989. 2008. [DOI] [PubMed] [Google Scholar]
- 29.Morita O, Kirkpatrick JB, Tamaki Y, Chengelis CP, Beck MJ, and Bruner RH. Safety assessment of heat-sterilized green tea catechin preparation: a 6-month repeat-dose study in rats. Food Chem Toxicol. 47: 1760–1770. 2009. [DOI] [PubMed] [Google Scholar]
- 30.Kao YH, Hiipakka RA, and Liao S. Modulation of endocrine systems and food intake by green tea epigallocatechin gallate. Endocrinology. 141: 980–987. 2000. [DOI] [PubMed] [Google Scholar]
- 31.Galati G, Lin A, Sultan AM, and O’Brien PJ. Cellular and in vivo hepatotoxicity caused by green tea phenolic acids and catechins. Free Radic Biol Med. 40: 570–580. 2006. [DOI] [PubMed] [Google Scholar]
- 32.Chan PC, Ramot Y, Malarkey DE, Blackshear P, Kissling GE, Travlos G, and Nyska A. Fourteen-week toxicity study of green tea extract in rats and mice. Toxicol Pathol. 38: 1070–1084. 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Goodin MG, Bray BJ, and Rosengren RJ. Sex- and strain-dependent effects of epigallocatechin gallate (EGCG) and epicatechin gallate (ECG) in the mouse. Food Chem Toxicol. 44: 1496–1504. 2006. [DOI] [PubMed] [Google Scholar]
- 34.Lambert JD, Sang S, and Yang CS. Possible controversy over dietary polyphenols: benefits vs risks. Chem Res Toxicol. 20: 583–585. 2007. [DOI] [PubMed] [Google Scholar]
- 35.Ogura R, Ikeda N, Yuki K, Morita O, Saigo K, Blackstock C, Nishiyama N, and Kasamatsu T. Genotoxicity studies on green tea catechin. Food Chem Toxicol. 46: 2190–2200. 2008. [DOI] [PubMed] [Google Scholar]
- 36.Ho CK, Siu-wai C, Siu PM, and Benzie IF. Genoprotection and genotoxicity of green tea (Camellia sinensis): Are they two sides of the same redox coin? Redox Rep. 18: 150–154. 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Elbling L, Weiss RM, Teufelhofer O, Uhl M, Knasmueller S, Schulte-Hermann R, Berger W, and Micksche M. Green tea extract and (-)-epigallocatechin-3-gallate, the major tea catechin, exert oxidant but lack antioxidant activities. FASEB J. 19: 807–809. 2005. [DOI] [PubMed] [Google Scholar]
- 38.Feng WY. Metabolism of green tea catechins: an overview. Curr Drug Metab. 7: 755–809. 2006. [DOI] [PubMed] [Google Scholar]
- 39.Hirose M, Hoshiya T, Akagi K, Takahashi S, Hara Y, and Ito N. Effects of green tea catechins in a rat multi-organ carcinogenesis model. Carcinogenesis. 14: 1549–1553. 1993. [DOI] [PubMed] [Google Scholar]
- 40.National Toxicology Program (NTP) website. TR-585: Technical Report Pathology Tables and Curves (Green Tea Extract). 2014. http://ntp.niehs.nih.gov/?objectid=76498EC3-EDBD-3E87-BFD9C2662A40C368.
- 41.Lambert JD, Kennett MJ, Sang S, Reuhl KR, Ju J, and Yang CS. Hepatotoxicity of high oral dose (-)-epigallocatechin-3-gallate in mice. Food Chem Toxicol. 48: 409–416. 2010b. [DOI] [PMC free article] [PubMed] [Google Scholar]