

Significance
Specific interactions between proteins control the function of essentially all cellular processes. Despite the importance of interaction specificity, it is unclear how structurally similar proteins achieve their unique recognition preferences. Here, we redesign the specificity of a protein binding domain and quantify the extent to which the designed specificity switch can be transferred to homologous domains. We show that identical mutations in structurally similar domains have a wide range of effects on specificity. We apply a structure-based computational model that recapitulates this context dependence. Our findings show how subtle structural differences between homologous domains contribute to their unique specificities. The differential responses to similar mutation observed here could help explain how families of recognition domains have evolved diverse new interactions.
Keywords: computational design, recognition specificity, promiscuity, protein interaction domains, interface evolution
Abstract
Reengineering protein–protein recognition is an important route to dissecting and controlling complex interaction networks. Experimental approaches have used the strategy of “second-site suppressors,” where a functional interaction is inferred between two proteins if a mutation in one protein can be compensated by a mutation in the second. Mimicking this strategy, computational design has been applied successfully to change protein recognition specificity by predicting such sets of compensatory mutations in protein–protein interfaces. To extend this approach, it would be advantageous to be able to “transplant” existing engineered and experimentally validated specificity changes to other homologous protein–protein complexes. Here, we test this strategy by designing a pair of mutations that modulates peptide recognition specificity in the Syntrophin PDZ domain, confirming the designed interaction biochemically and structurally, and then transplanting the mutations into the context of five related PDZ domain–peptide complexes. We find a wide range of energetic effects of identical mutations in structurally similar positions, revealing a dramatic context dependence (epistasis) of designed mutations in homologous protein–protein interactions. To better understand the structural basis of this context dependence, we apply a structure-based computational model that recapitulates these energetic effects and we use this model to make and validate forward predictions. Although the context dependence of these mutations is captured by computational predictions, our results both highlight the considerable difficulties in designing protein–protein interactions and provide challenging benchmark cases for the development of improved protein modeling and design methods that accurately account for the context.
Many protein–protein interactions are mediated by small modular protein recognition domains (1). These interaction modules, such as PDZ, SH3, and WW domains, generally recognize their protein partners using a structurally conserved binding site, and there are often tens or even hundreds of proteins containing a given type of recognition domain expressed simultaneously in a cell or organism (2). This repeated use of recognition modules with conserved structures poses the following question: how do cells maintain the specificity of binding interactions when so many members of the same domain family are present? Moreover, to what extent do different domain family members in fact have distinct or overlapping preferences for binding their partners? Addressing these questions is of considerable importance, because a significant fraction of protein interactions in a cell is mediated a limited number of protein interaction domain families (1). Despite a large amount of information on the biochemical recognition preferences of many domain members in vitro (3–5), much less is known about the actual extent of specificity and promiscuity of these domains functionally in vivo (6, 7).
One way to dissect the specificity and functional role of a given interaction in the cellular context is the “second-site suppressor” strategy. In this approach, it is inferred that two proteins are involved in the same biological process or even directly interact with each other, if a detrimental mutation in one protein can be functionally compensated for by a mutation in the second. This strategy has been mimicked using computational protein design approaches to reengineer protein–protein interaction specificity. Here, a “computational second-site suppressor” simulation aims to predict mutations in both partners of a protein–protein interface that would be destabilizing when only one of the partners is mutated, but stabilizing if both partners are changed simultaneously (8). This approach has previously been applied to redesign the specificity of a diverse set of interactions, including a DNase–inhibitor pair (8, 9), a small GTPase and its guanine exchange factor (10), as well as the interaction between a ubiquitin ligase and a ubiquitin-conjugating enzyme (11). Despite these success cases, the extent to which a designed specificity switch is transferrable between homologous domains with structurally conserved interaction sites is unknown. Quantifying this transferability is critical for the development of computational and experimental strategies to dissect the biological roles of the large number of proteins that contain structurally similar protein recognition domains.
In this study, we use PDZ domains as a model system for quantifying the extent to which a successfully designed specificity switch can be transferred between homologous domains. PDZ domains are a well-studied family of protein–peptide recognition modules with 364 members in the human genome (2). PDZ domains are typical examples of protein recognition modules that bind to linear peptide motifs using a structurally conserved binding site. Previous work has performed large-scale mutagenesis on an individual PDZ domain to identify mutations that affect interaction specificity (12, 13), but it is unknown if the same mutations have the same effects on specificity in similar PDZ domains. Understanding the context dependence of designed mutations is also important more generally, given the interest in protein-engineering strategies that involve transplanting a functional site from one protein “scaffold” to another (14, 15).
Here, we describe the design of a pair of mutations that modulate peptide recognition specificity of the Syntrophin PDZ domain and confirm the designed interaction biochemically and by solving the crystal structure of the engineered complex. We then quantify the transferability of this specificity switch by transplanting the designed mutations into the context of five related PDZ domain–peptide interfaces. We find that, even though the sites of the sequence changes are structurally superimposable, there is considerable context dependence of the energetic effects of the engineered mutations. To understand the basis of this context dependence, we apply a structure-based computational model to predict the energetic effects of these mutations on each of the homologous PDZ domain–peptide interfaces. Our model recapitulates the experimentally observed energetic effects, suggesting that this context dependence occurs due to subtle structural differences between the homologous domains. Finally, we use this model to make and validate forward predictions that further improve the recognition specificity of our initial design. Taken together, our results address the major challenge of context-dependent effects of designed mutations for engineering protein–protein interactions, provide a valuable benchmark for improved computational design methods that capture these effects, and have implications for the evolution of new interactions.
Results
Computational Redesign of PDZ Specificity.
To create a new PDZ–peptide ligand interaction with altered recognition specificity, we applied a previously described computational second-site suppressor protocol for redesigning protein–protein interactions (8). As input, the computational design method takes the fixed backbones of the PDZ domain and its peptide ligand from a known crystal structure. The design protocol first identifies sequence perturbations in the PDZ ligand that destabilize the complex with the PDZ domain. Second, we apply computational design to search for sequence changes in the PDZ domain that compensate for the perturbation introduced in the PDZ ligand. This approach is illustrated in Fig. 1A.
Fig. 1.

Computational design of a PDZ specificity switch. (A) Cartoon depicting the second-site suppressor design strategy, in which one protein is mutated to destabilize the interaction and the other protein is mutated to rescue the interaction. (B) Computational predictions of the energetic effects for destabilizing mutations at position P−2 on nNOS (gray bars) and for compensating mutations on Syntrophin (black bars). (C) Comparison of the crystal structure of the wild-type interaction (Left) and a model of the designed specificity switch (Right).
We applied this method to the design of a specificity switch in the interface between the PDZ domain of α1-Syntrophin (hereafter referred to as Syntrophin) and the PDZ domain of neuronal nitric oxide synthase (nNOS), which serves as a ligand of Syntrophin. The nNOS PDZ domain forms a pseudopeptide and docks into the PDZ peptide-binding pocket of Syntrophin (16). To design specificity changes, we focused on one of the main specificity-determining residues for PDZ domain ligands, called P−2. The amino acid type present at this position distinguishes two main classes of PDZ domains, with class I recognizing Ser or Thr at this position, and class II recognizing hydrophobic side chains (17). Using computational design, we introduced single amino acid mutations into nNOS, one at the time, at the P−2 position (corresponding to sequence position 1109 in the nNOS complex crystal structure with Syntrophin). For each mutation, we relaxed the area around the site of mutation by optimizing the surrounding side-chain conformations using Monte Carlo-simulated annealing. Then, for each of these mutations, residues in Syntrophin within 6 Å of the nNOS P−2 position were redesigned to find substitutions that compensate for the given residue at the P−2 position by forming low-scoring (favorable) interactions.
This strategy resulted in designed complexes that were then evaluated to maximize the computed predicted energy difference between the destabilized interface (mutant nNOS–Syntrophin complex) and the stabilized interface (mutant nNOS–mutant Syntrophin complex). Fig. 1B shows the computed difference in energy between the destabilized interface when we introduced amino acid substitutions at the P−2 position in nNOS (gray bars), and the difference in energy of the substitution in nNOS with the redesigned Syntrophin (black bars). As expected, essentially all mutations at the P−2 position in nNOS are predicted to destabilize the interaction, except when the P−2 residue is Thr (the original, wild-type residue), or Ser.
Redesign of the interface on the PDZ domain was predicted to stabilize interactions with several modeled residues at the P−2 position in the ligand. These residues included Ser and Thr, as well as the hydrophobic residues Phe, Met, Val, Leu, and Ile. Most of these amino acids are the commonly occurring residues at P−2 in PDZ domain recognition motifs (Ser, Thr for class I PDZ domains and Phe, Tyr, Ile, Leu, and Val for class II PDZ domains). Met, which had the lowest energy (most favorable) compensating interaction, is not commonly observed at P−2 in naturally occurring PDZ domain binding motifs and occurs at P−2 in only 2% of peptides selected for PDZ binding by phage display experiments for 82 human and worm PDZ domains (Fig. S1) (12). We chose several top-ranked predicted specificity-changing (i.e., not Ser or Thr) residues at P−2 (Met, Val, Phe, Leu, Ile, Tyr) for more detailed design simulations (SI Materials and Methods). The top-ranked design was a Syntrophin variant with a single compensating mutation in Syntrophin (H142F) predicted to recognize Met at P−2. Fig. 1C shows a comparison between the crystal structure of the wild-type nNOS–Syntrophin complex and a structural model of the designed complex. In the wild-type complex, His-142 forms a hydrogen bond with Thr at the P−2 position in nNOS. In the designed models, the wild-type polar interaction is replaced by a hydrophobic interaction between the new Met and Phe side chains.
Experimental Characterization of the Specificity Switch.
To experimentally validate the computationally designed specificity switch, we used fluorescence polarization to measure the affinity of wild-type Syntrophin and H142F Syntrophin to a peptide with Ser at the P−2 position (SIESDV) and to a peptide with Met at the P−2 position (SIEMDV) (Materials and Methods). The cognate wild-type and designed complexes had affinities of 8 and 12 µM, respectively, whereas both noncognate interactions had affinities between 60 and 70 µM (Fig. 2A). We therefore designed a Syntrophin–peptide pair with a binding affinity in the same range as the wild-type pair and a moderate but robustly detectable specificity switch, where both of the complexes between a designed and wild-type partner are destabilized.
Fig. 2.

Biochemical and structural characterization of the specificity switch. (A) Fluorescence polarization binding curves of the wild-type and mutant interactions. Error bars show SD for three data points. (B) Comparison of the designed Syntrophin–nNOS crystal structure (pink and lavender) and a model of the designed interface (green). The Cα RMSD between model and crystal structure Syntrophin (entire PDZ) plus nNOS (residues 1106–1111) is 0.45 Å and the heavy-atom RMSD is 0.50 Å.
After in vitro testing of the redesigned interaction and verifying the switch in specificity, we determined the crystal structure of the designed complex between H142F Syntrophin and mutated nNOS (T1109M). The 2.29-Å resolution structure of the redesigned complex (Table S1) showed that the amino acid side-chain conformations of the designed interface residues were nearly identical to those predicted by the model from computational design (Fig. 2B). A structure of the entire complex is shown in Fig. S2, and a close-up of the mutated residues is shown in Fig. S3.
Quantifying the Transferability of the Specificity Switch.
We next investigated to what extent the designed specificity switch in the class I PDZ domain Syntrophin could be transferred to other homologous PDZ domains. We selected five other class I PDZ domains with available cocrystal structures bound to a peptide with Thr at the P−2 position: PDZ2 from tyrosine phosphatase PTP-BL (PTPN13), the PDZ domain from Erbb2-interacting protein (Erbin), PDZ3 from partitioning defective 3 homolog (PAR3), PDZ3 from postsynaptic density protein 95 (PSD95), and PDZ1 from membrane-associated guanylate kinase inverted 1 (MAGI1). A sequence alignment of these PDZ domains is shown in Fig. S4, and the percent identities between each domain and the residues surrounding the specificity switch are shown in Tables S2 and S3, respectively. The positions that were redesigned in the Syntrophin specificity switch are structurally superimposable between the different PDZ domain–peptide complexes (Fig. 3A). The most unique structure is the PDZ domain of PAR3, which has an Asn at the position corresponding to the His-to-Phe mutation in the designed Syntrophin. We purified both the wild type and the His/Asn-to-Phe variant of all five PDZ domains and examined the effects of the mutations on binding for two peptides: either the originally characterized cognate peptide for each PDZ domain (as present in each experimentally determined structure) with Ser/Thr at P−2 or a mutated peptide with a Met at the P−2 position. The different wild-type PDZ domains bound their cognate peptides with affinities between 0.08 and 16.4 μM. All measured binding affinities are shown in Table S4.
Fig. 3.
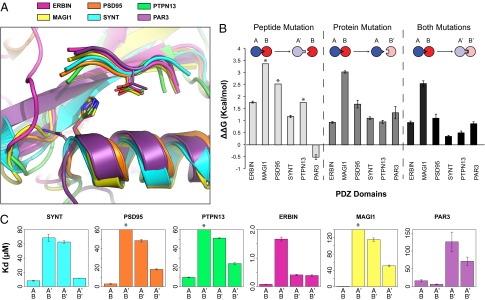
Quantification of the transferability of the specificity switch. (A) Structural alignment of six homologous PDZ domains. The mean overall heavy-atom RMSD between Syntrophin (SYNT) and the other PDZ domains is 1.33 Å, and the mean binding site heavy-atom RMSD is 0.38 Å (the binding site is defined by helix positions 141–151 and strand positions 94–99 in Syntrophin for a total of 15 residues). (B) Experimentally determined energetic effects of specificity switch mutations on binding for the six PDZ domains. Error bars reflect propagated SDs. (C) Binding affinities for the wild-type (A and B) and mutant (A′ and B′) interactions determined by fluorescence polarization. Asterisks denote cases in which affinities could not be accurately quantified because the binding curves did not reach saturation in the concentration range used. In these cases, we estimated the affinity to be greater than 200 µM. A and A′ denote the peptide, and B and B′ denote the protein.
Substituting His/Asn with Phe in the PDZ domain destabilized binding to the original cognate peptide for all six studied PDZ domains (Fig. 3B, dark gray bars). However, the energetic effect of the Phe mutation on peptide binding varied substantially, from moderate (1 kcal/mol for Syntrophin, Erbin, and PTPN13) to large (3 kcal/mol for MAGI1). The substitution of the Thr at P−2 to Met in the peptide (Fig. 3B, light gray bars) also showed remarkable differences among the PDZ domains. The peptide mutation destabilized binding to the PDZ domains to different extents, except for PAR3, which bound with higher affinity to the mutated peptide than to the wild-type peptide. Similarly, the combination of the Phe mutation on the PDZ domain and the Met mutation on the peptide compensated the individual mutations to varying degrees (Fig. 3B, black bars). The double mutations resulted in interactions that were slightly (0.5 kcal/mol) to substantially (2.5 kcal/mol) weaker than the original cognate wild-type complex.
For PSD95 and PTPN13, the interaction had the desired pattern with both cognate pairs forming more stable interactions than both noncognate pairs, as previously seen for Syntrophin (Fig. 3C). In contrast, for Erbin, the mutated PDZ domain binds to both the wild-type and mutated peptides with the same affinity. In the case of MAGI1, the PDZ domain mutation does not compensate the mutation in the peptide well. For PAR3, PDZ domain mutation destabilizes binding to both wild-type and mutated peptides, whereas mutation of the peptide stabilizes the interface. Thus, for some PDZ domains (Syntrophin, PSD95, PTPN13), we created a desired change in specificity by “transplanting” the designed residues, but for others, the simple transplanting of a single residue was not sufficient. These results overall indicate a wide range of energetic effects of identical mutations in structurally similar contexts (Fig. 3A).
Recapitulating the Energetic Effects of the Designed Mutations in Homologous Contexts.
To better understand the basis of the context dependence of designed mutations in PDZ domain interfaces, we first compared the sequence identity and the degree of epistasis for each pair of PDZ domains. However, we did not observe a clear relationship between sequence identities and the effect of the mutations on binding (Fig. S5). Given that subtle structural differences between the PDZ domains may have a significant contribution to the effect of the mutations on binding, we applied a structure-based computational model to predict the energetic effects of mutations on PDZ–peptide binding. This model is based on a computational protocol originally developed for predicting changes in protein stability (18), which we adapted to model changes in binding energy (Materials and Methods). This method differs from the initial second-site suppressor protocol (8) used originally for designing the Syntrophin specificity switch in that the subsequently developed model considers backbone conformational changes in addition to just side-chain rotamer adjustments and that it uses a higher resolution force field with explicit nonpolar hydrogens and a more stringent term for atomic repulsion. Fig. 4A shows that this model captures the observed energetic effects of mutations in both the PDZ domains and the peptide reasonably well. Given this agreement between computational simulations and experimental results, we examined which terms in the energy function were responsible for yielding differences in the predicted energetic effects across the set of six homologous complexes (Fig. S6). We found the main determining factor to be a trade-off between small steric incompatibilities, as captured in small repulsive components of the atomic packing term, and rotamer strain, as captured by an energy term derived from rotamer preferences observed in the Protein Data Bank (PDB) (19). These results suggest that the observed context dependence of designed mutations in PDZ domains occurs due to subtle differences in protein structure and protein interaction geometry among the different PDZ domains.
Fig. 4.
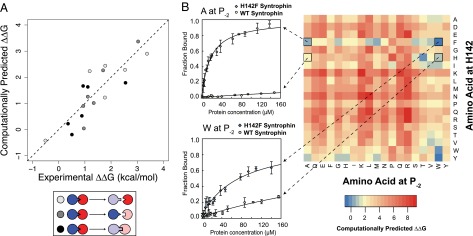
Computational model of energetic effects of mutations and forward predictions. (A) Correlation between experimentally determined and computationally predicted energetic effects of mutations on PDZ–peptide interactions (Pearson correlation coefficient r = 0.85). (B) Computational predictions for all pairs of mutations of position 142 on Syntrophin and position P−2 on the peptide (Right) and characterization of four predictions via fluorescence polarization (Left).
To further test our model, we predicted the mutational effects of all possible pairs of residue changes at the two positions mutated in the Syntrophin specificity switch (Fig. 4B for Syntrophin and Fig. S7 for all of the PDZ domains). These results predicted that the vast majority of mutant pairs are incompatible and would destabilize the interaction; however, there exist a small subset of pairs with predicted near wild-type affinities, including the experimentally characterized designed specificity switch. We experimentally tested two additional predicted specificity switches, both of which involved the H142F mutant on Syntrophin but different mutations on the peptide (Ala at P−2 and Trp at P−2). As predicted, both peptides with the predicted mutations bound to H142F Syntrophin with higher affinity than to wild-type Syntrophin. The affinity of the peptide with Ala at P−2 to H142F and wild-type Syntrophin was 19.9 and >200 µM, respectively; for Trp at P−2 the affinities were 82.4 and >200 µM. In fact, the interaction between H142F Syntrophin and the peptide with Ala at P−2 showed a more significant specificity switch than the initial design. These results both support the computational model (although the interaction with Trp at P−2 was overpredicted) and reveal that H142F Syntrophin is promiscuous and can recognize both Met and Ala at P−2 with high affinity.
Discussion
Our study quantifies the extent to which an engineered switch in protein–protein interaction specificity could be transferred between homologous protein–protein interactions. We found a remarkable context dependence of designed mutations and hypothesized that this context dependence occurs due to subtle structural differences between the related protein–protein interfaces. Supporting this hypothesis, we found that a structure-based computational model recapitulates the differential energetic effects of mutations in homologous contexts. Although encouraging, these results also illustrate the challenges faced by protein-engineering approaches. Improving these approaches will require the ability to successfully capture the context-dependent effects we observed, and our study therefore provides a valuable benchmark for testing the accuracy and sensitivity of protein modeling and design methods.
Context-dependent effects of mutations (epistasis) have been characterized both within single proteins (20–22) and in protein–protein interfaces (23, 24). In general, epistatic effects within proteins, although likely important on evolutionary timescales, have been suggested to be rare individually as the majority of pairwise mutations appear compatible (20, 21). Similarly, in protein–protein interfaces, pairs of mutations have been found to have largely additive energetic effects unless the mutated residues are in direct contact (23) or part of the same tightly interacting cluster (25). In contrast, using PDZ domains as model system, we observe considerable context-dependent effects of mutations even for PDZ domains where the residues directly contacting the engineered mutations are identical (Syntrophin and Erbin) or chemically similar (Val, Leu, and Ile mutations distinguish the contact residues in Syntrophin and Erbin from PTPN13 and MAGI1; Fig. S4 and Table S3).
Our findings of context dependency in PDZ domains are likely relevant for possible evolutionary trajectories to change function (in this case, specificity). As seen in Fig. 3C, the effects of introducing identical single and double mutations on specificity are qualitatively different in different PDZ domains, even in Syntrophin and Erbin that have identical residues contacting the designed positions. For Syntrophin, a single mutation in either partner in the complex destabilizes binding. As a consequence, within this set of mutations there is no “smooth” path to a specificity change, where binding was preserved in at least one of the intermediates. For Erbin, a single mutation in just the protein preserves binding to both peptides, which would allow a specificity-switching path.
Our results also indicate that specificity is not necessarily conferred by the same positions across different members of the PDZ domain family, despite their structural similarity. This observation is of significant interest given that previous studies have conducted exhaustive mutagenesis on an individual PDZ domain to identify the sequence determinants of PDZ domain specificity. Our study suggests that these determinants of specificity may be less generalizable than anticipated as they could differ considerably depending on the particular PDZ domain used in the experiment. Future work that simultaneously applies exhaustive mutagenesis to multiple, homologous PDZ domains will more fully reveal the extent of the context dependence of mutations on PDZ specificity.
Previous studies on the evolution of new protein function have suggested that ancestral proteins might have been functionally promiscuous and subsequently diverged into proteins with specific functions (26). This hypothesis is based on the observation that ancestrally reconstructed proteins have been shown to be more promiscuous than their descendants (27, 28). Although this may be one mechanism to achieve new functions, our initial specificity switch design of Syntrophin PDZ demonstrates that a new function (altered peptide ligand) could be achieved via a single mutation in the peptide recognition domain. This illustrates another mechanism of neofunctionalization that could involve a gene duplication event followed by a specificity altering mutation in one of the resulting gene copies. Considering the abundance of PDZ domains in eukaryotic genomes (2), it is possible that PDZ domain-containing proteins have undergone extensive gene duplication, and the repetition of this process combined with specificity-altering mutations may have allowed PDZ domains to achieve their unique specificities.
Although the effects of mutations on peptide specificity that we observed were highly context dependent between homologous PDZ domains, we were able to recapitulate these effects with a structure-based computational model. Using this model, we predicted and verified that H142F Syntrophin is promiscuous, as it binds with high affinity to peptides with either Ala or Met at the P−2 position. Given that promiscuity is thought to be a precursor for the evolution of new functions, promiscuity-generating mutations like H142F could be extremely useful for protein-engineering efforts where the goal is to redesign naturally occurring proteins to obtain novel functions. Future work could apply the computational models validated in this study to generalized protocols for identifying both specificity and promiscuity-enhancing mutations in protein–protein or protein–ligand interfaces.
Materials and Methods
Computational Protein Design.
The computational interface design protocol for the nNOS–Syntrophin complex was essentially as described (8). Amino acid side chains were modeled as rotamers in an all-atom representation onto a fixed polypeptide template taken from the Syntrophin/nNOS crystal structure, PDB ID code 1QAV (16), with polar hydrogens added. Sequence positions were either designed (allowing rotamers for all 20 naturally occurring amino acids except cysteine and proline using the backbone-dependent library compiled by Dunbrack with additional rotamers for buried residues), repacked (allowing all rotamers of the native amino acid type; this was done for residues directly contacting designed residues), or left unchanged in their native conformation (all other residues). Design simulations used the Rosetta full-atom energy function as described previously (29, 30). In the first step, all 18 possible (20 naturally occurring amino acids except cysteine and proline) single mutations were modeled at the nNOS residue position P−2 (T1109). The predicted binding energy of each peptide with a given amino acid at P−2 was then computed for the complex with (i) wild-type Syntrophin, to estimate the destabilizing effect of the mutation on the wild-type interface (destabilized interface), and (ii) designed Syntrophin, in which three interface residues on Syntrophin (positions 96, 142, and 146) were simultaneously redesigned (compensated interface). In each case, the model of the sequence with the lowest total Rosetta energy was subjected to minimization of the side-chain torsional degrees of freedom before computing the predicted binding energies shown in Fig. 1B. After the initial scan for possible specificity changes, we performed a second round of simulations to generate a large number of possible models with different designed substitutions of PDZ domain residues contacting the residue at P−2 (see SI Materials and Methods for details). We then ranked those models by their predicted binding energy after filtering out models that contained unsatisfied buried hydrogen bond donors and acceptors and steric clashes. The top-ranked design contained a single His-to-Phe mutation in the PDZ domain to compensate for a Thr-to-Met substitution at the peptide residue at P−2, and was chosen as a candidate for a specificity switch, after also considering the destabilization of the His-to-Phe mutation in Syntrophin with the wild-type peptide containing a Thr at P−2.
Protein Expression, Purification, and Structure Determination.
Protein constructs and expression, purification, crystallization, and data collection conditions are given in SI Materials and Methods. The structure was determined at 2.29-Å resolution by molecular replacement using the program Molrep and PDB structure 1QAV (16) as the initial search model. An R factor of 22.6% with an Rfree of 27.3% was obtained for the refined model (PDB ID code 4HOP; Table S1).
Measurement of Binding Affinities and Calculation of Experimental ΔΔG Values.
Affinities for peptide binding to PDZ domains were measured using fluorescence polarization with peptides labeled with fluorescein at the N terminus (SI Materials and Methods). For each PDZ domain, the starting peptide sequence was taken from the publication describing the respective domain–peptide complex structure (Table S4) except for the Syntrophin interaction, where we used a previously characterized peptide (SIESDV), not the protein partner nNOS, to facilitate comparison between all domains using the same peptide-binding assay. Binding measurements were performed in triplicate. Experimentally determined binding affinities (Kd) were converted to ∆G values using ∆G = –RT ln (1/Kd), where T = 298 K. Mutant ∆∆G values were calculated as ∆∆G = ∆Gmut – ∆Gwt. SDs for ∆∆G were calculated by summing the SDs of ∆Gmut and ∆Gwt.
Rosetta Simulations to Estimate Changes in Binding Energy.
Binding energies between PDZ domains and peptides were estimated using a Rosetta protocol originally developed for predicting changes in protein stability in monomeric proteins (18). This protocol consists of two stages: (i) side-chain optimization using a Lennard–Jones potential with a dampened repulsive term and (ii) all-atom energy minimization using harmonic constraints between all Cα atoms within 9 Å. To estimate a binding energy for a given mutation, this protocol was run 50 times for the wild-type complex and 50 times for the mutant complex. The binding energy was computed as the difference in interface score between the lowest scoring wild-type and mutant interfaces. Rosetta command lines and further simulation details are provided in SI Materials and Methods.
Supplementary Material
Acknowledgments
We thank Debbie Jeon for help with experiments. Work on the design of protein interactions in the Kortemme laboratory was supported by awards from the National Science Foundation (NSF) (DBI-1262182) and the National Institutes of Health (R01GM098101). N.O. was supported by an NSF graduate fellowship.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
Data deposition: The atomic coordinates and structure factors have been deposited in the Protein Data Bank (PDB), www.pdb.org (PDB ID code 4HOP).
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1410624111/-/DCSupplemental.
References
- 1.Pawson T. Protein modules and signalling networks. Nature. 1995;373(6515):573–580. doi: 10.1038/373573a0. [DOI] [PubMed] [Google Scholar]
- 2.Letunic I, Doerks T, Bork P. SMART 7: Recent updates to the protein domain annotation resource. Nucleic Acids Res. 2012;40(Database issue):D302–D305. doi: 10.1093/nar/gkr931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Landgraf C, et al. Protein interaction networks by proteome peptide scanning. PLoS Biol. 2004;2(1):E14. doi: 10.1371/journal.pbio.0020014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Stiffler MA, et al. PDZ domain binding selectivity is optimized across the mouse proteome. Science. 2007;317(5836):364–369. doi: 10.1126/science.1144592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yaffe MB, et al. A motif-based profile scanning approach for genome-wide prediction of signaling pathways. Nat Biotechnol. 2001;19(4):348–353. doi: 10.1038/86737. [DOI] [PubMed] [Google Scholar]
- 6.Zarrinpar A, Park S-H, Lim WA. Optimization of specificity in a cellular protein interaction network by negative selection. Nature. 2003;426(6967):676–680. doi: 10.1038/nature02178. [DOI] [PubMed] [Google Scholar]
- 7.Marles JA, Dahesh S, Haynes J, Andrews BJ, Davidson AR. Protein-protein interaction affinity plays a crucial role in controlling the Sho1p-mediated signal transduction pathway in yeast. Mol Cell. 2004;14(6):813–823. doi: 10.1016/j.molcel.2004.05.024. [DOI] [PubMed] [Google Scholar]
- 8.Kortemme T, et al. Computational redesign of protein-protein interaction specificity. Nat Struct Mol Biol. 2004;11(4):371–379. doi: 10.1038/nsmb749. [DOI] [PubMed] [Google Scholar]
- 9.Joachimiak LA, Kortemme T, Stoddard BL, Baker D. Computational design of a new hydrogen bond network and at least a 300-fold specificity switch at a protein-protein interface. J Mol Biol. 2006;361(1):195–208. doi: 10.1016/j.jmb.2006.05.022. [DOI] [PubMed] [Google Scholar]
- 10.Kapp GT, et al. Control of protein signaling using a computationally designed GTPase/GEF orthogonal pair. Proc Natl Acad Sci USA. 2012;109(14):5277–5282. doi: 10.1073/pnas.1114487109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sammond DW, Eletr ZM, Purbeck C, Kuhlman B. Computational design of second-site suppressor mutations at protein-protein interfaces. Proteins. 2010;78(4):1055–1065. doi: 10.1002/prot.22631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tonikian R, et al. A specificity map for the PDZ domain family. PLoS Biol. 2008;6(9):e239. doi: 10.1371/journal.pbio.0060239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.McLaughlin RN, Jr, Poelwijk FJ, Raman A, Gosal WS, Ranganathan R. The spatial architecture of protein function and adaptation. Nature. 2012;491(7422):138–142. doi: 10.1038/nature11500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zanghellini A, et al. New algorithms and an in silico benchmark for computational enzyme design. Protein Sci. 2006;15(12):2785–2794. doi: 10.1110/ps.062353106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Azoitei ML, et al. Computation-guided backbone grafting of a discontinuous motif onto a protein scaffold. Science. 2011;334(6054):373–376. doi: 10.1126/science.1209368. [DOI] [PubMed] [Google Scholar]
- 16.Hillier BJ, Christopherson KS, Prehoda KE, Bredt DS, Lim WA. Unexpected modes of PDZ domain scaffolding revealed by structure of nNOS-syntrophin complex. Science. 1999;284(5415):812–815. [PubMed] [Google Scholar]
- 17.Songyang Z, et al. Recognition of unique carboxyl-terminal motifs by distinct PDZ domains. Science. 1997;275(5296):73–77. doi: 10.1126/science.275.5296.73. [DOI] [PubMed] [Google Scholar]
- 18.Kellogg EH, Leaver-Fay A, Baker D. Role of conformational sampling in computing mutation-induced changes in protein structure and stability. Proteins. 2011;79(3):830–838. doi: 10.1002/prot.22921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shapovalov MV, Dunbrack RL., Jr A smoothed backbone-dependent rotamer library for proteins derived from adaptive kernel density estimates and regressions. Structure. 2011;19(6):844–858. doi: 10.1016/j.str.2011.03.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lunzer M, Golding GB, Dean AM. Pervasive cryptic epistasis in molecular evolution. PLoS Genet. 2010;6(10):e1001162. doi: 10.1371/journal.pgen.1001162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Govindarajan S, et al. Systematic variation of amino acid substitutions for stringent assessment of pairwise covariation. J Mol Biol. 2003;328(5):1061–1069. doi: 10.1016/s0022-2836(03)00357-7. [DOI] [PubMed] [Google Scholar]
- 22.Parera M, Martinez MA. Strong epistatic interactions within a single protein. Mol Biol Evol. 2014;31(6):1546–1553. doi: 10.1093/molbev/msu113. [DOI] [PubMed] [Google Scholar]
- 23.Wells JA. Additivity of mutational effects in proteins. Biochemistry. 1990;29(37):8509–8517. doi: 10.1021/bi00489a001. [DOI] [PubMed] [Google Scholar]
- 24.Schreiber G, Fersht AR. Energetics of protein-protein interactions: Analysis of the barnase-barstar interface by single mutations and double mutant cycles. J Mol Biol. 1995;248(2):478–486. doi: 10.1016/s0022-2836(95)80064-6. [DOI] [PubMed] [Google Scholar]
- 25.Reichmann D, et al. The modular architecture of protein-protein binding interfaces. Proc Natl Acad Sci USA. 2005;102(1):57–62. doi: 10.1073/pnas.0407280102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Khersonsky O, Tawfik DS. Enzyme promiscuity: A mechanistic and evolutionary perspective. Annu Rev Biochem. 2010;79:471–505. doi: 10.1146/annurev-biochem-030409-143718. [DOI] [PubMed] [Google Scholar]
- 27.Wouters MA, Liu K, Riek P, Husain A. A despecialization step underlying evolution of a family of serine proteases. Mol Cell. 2003;12(2):343–354. doi: 10.1016/s1097-2765(03)00308-3. [DOI] [PubMed] [Google Scholar]
- 28.Bridgham JT, Carroll SM, Thornton JW. Evolution of hormone-receptor complexity by molecular exploitation. Science. 2006;312(5770):97–101. doi: 10.1126/science.1123348. [DOI] [PubMed] [Google Scholar]
- 29.Kortemme T, Baker D. A simple physical model for binding energy hot spots in protein-protein complexes. Proc Natl Acad Sci USA. 2002;99(22):14116–14121. doi: 10.1073/pnas.202485799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kortemme T, Morozov AV, Baker D. An orientation-dependent hydrogen bonding potential improves prediction of specificity and structure for proteins and protein-protein complexes. J Mol Biol. 2003;326(4):1239–1259. doi: 10.1016/s0022-2836(03)00021-4. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.