

Significance
Mycobacterium tuberculosis infects more than one-third of humans yet no effective vaccine exists. This study shows how a subset of αβ T cells targets M. tuberculosis lipid antigens that are presented by the MHC molecule CD1c. In contrast to many T cells that recognize CD1d, these αβ T cells express diverse T-cell receptors and have differing footprints on CD1c during lipid recognition. This study also shows that some CD1c-specific αβ T cells are exquisitely specific for the lipid presented, whereas others have a more promiscuous reactivity, demonstrating that the αβ T-cell response to CD1c lipid presentation is diverse and adaptable. These data may provide additional resources for development of MHC-independent vaccines against M. tuberculosis.
Keywords: group 1 CD1, Mycobacterium tuberculosis, T-cell reactivity
Abstract
CD1c is a member of the group 1 CD1 family of proteins that are specialized for lipid antigen presentation. Despite high cell surface expression of CD1c on key antigen-presenting cells and the discovery of its mycobacterial lipid antigen presentation capability, the molecular basis of CD1c recognition by T cells is unknown. Here we present a comprehensive functional and molecular analysis of αβ T-cell receptor (TCR) recognition of CD1c presenting mycobacterial phosphomycoketide antigens. Our structure of CD1c with the mycobacterial phosphomycoketide (PM) shows similarities to that of CD1c-mannosyl-β1-phosphomycoketide in that the A' pocket accommodates the mycoketide alkyl chain; however, the phosphate head-group of PM is shifted ∼6 Å in relation to that of mannosyl-β1-PM. We also demonstrate a bona fide interaction between six human TCRs and CD1c-mycoketide complexes, measuring high to moderate affinities. The crystal structure of the DN6 TCR and mutagenic studies reveal a requirement of five complementarity determining region (CDR) loops for CD1c recognition. Furthermore, mutagenesis of CD1c reveals residues in both the α1 and α2 helices involved in TCR recognition, yet not entirely overlapping among the examined TCRs. Unlike patterns for MHC I, no archetypical binding footprint is predicted to be shared by CD1c-reactive TCRs, even when recognizing the same or similar antigens.
Human CD1 molecules are antigen-presenting proteins that capture and display lipid antigens to T cells (1). CD1 proteins are categorized based on their genomic organization, sequence homology, and cellular function into group 1 (CD1a, CD1b, and CD1c), group 2 (CD1d), and group 3 (CD1e) (2, 3). Our understanding of CD1 antigen presentation and recognition by a T-cell receptor (TCR) is largely based on CD1d/natural killer T (NKT) cell receptor interactions (4). However, the five human CD1 molecules are markedly different in the architecture of their antigen binding pockets, which in turn gives rise to distinct antigen specificities. Furthermore, each CD1 family member exhibits different tissue expression profiles and intracellular trafficking patterns, demonstrating that CD1 molecules perform specific, nonredundant immunological tasks (5). CD1c, in particular, is a common and broadly expressed antigen-presenting molecule in the human immune system, where it is found at high surface density on most myeloid dendritic cells and marginal zone B cells.
A number of Mycobacterium tuberculosis lipid antigens have been identified that are presented by group 1 CD1 molecules (6). CD1c molecules present several mycobacterial or synthetic lipid antigens that share a key chemical motif: methylated alkyl chains. These antigens include synthetic mannosyl phosphodolichols that are structurally related to mycobacterial mannosyl-β1-phosphomycoketide (MPM) (7–9) and recently described phosphomycoketide (PM) (10). The unusual methyl branches associated with these mycoketide antigens, synthesized by polyketide synthase 12 (pks12), serve as a molecular signature for M. tuberculosis and are required for loading into the CD1c antigen binding groove (8, 10). MPM binds in the A′ pocket of CD1c and the mannose head group extends out of ligand binding groove to become accessible to a TCR for recognition (11). Intracellular processing pathways might fine-tune lipid antigen processing as CD1c expressing antigen-presenting cells (APCs) are thought to process glycosylated antigens such as MPM and present deglycosylated, neoepitope PM to T cells (10). In addition, human CD1c has been reported to present lipopeptide antigens (12) and sulfatide to human αβ T cells (13). CD1c was also the first CD1 molecule implicated as a ligand for human Vδ1+ γδ T cells (14). CD1c-reactive cells have been detected at high frequency in human blood (15), expand in numbers during human tuberculosis infection (9), and infiltrate organs during autoimmune disease (16). Despite accumulating evidence for an important, natural function of this broadly expressed protein in the human immune response, TCR binding to CD1c has not been measured, and the structural basis of antigen recognition remains unknown.
Studies with human cell lines showed that CD1c interacts with both αβ and γδ T cells (9, 12, 14, 17, 18), and recently it has been demonstrated that CD1c-PM tetramers detect polyclonal T-cell populations from the blood of M. tuberculosis-infected patients (10). Together, these findings suggest that there are specific, selective expansions of T-cell populations in response to CD1c recognition, and these serve as evidence for in vivo immune responses. However, despite an abundance of studies on NKT cell recognition of CD1d, there is no information on the molecular basis of TCR recognition of mycobacterial antigens presented by CD1c.
We sought to understand how diverse αβ T cells specifically interact with two structurally related mycobacterial antigens, PM and MPM, when presented by CD1c molecules. Whereas it was previously thought that CD1c-reactive TCRs show absolute specificity of glycosylated (MPM) or unglycosylated forms of phosphomycoketide (PM), here we report a new T-cell response that is cross-reactive to both types of antigens. We cloned six prototypical CD1c-reactive TCRs representing two of the observed mycoketide reactivities (PM specific and MPM/PM cross-reactive) and made the first measurements of a TCR-CD1c interaction. Four of these TCRs use the TRVB7-9 segment, which was overrepresented in the CD1c-reactive T-cell pool. Using mutational analysis, we defined the critical amino acid residues and energetic hotspots on the surface of CD1c that are responsible for interaction with these diverse TCRs. These six TCRs show differences in the degree to which they recognize the protruding phosphate or carbohydrate moieties, and the mutational analysis maps three distinct footprint patterns. Due to these differences, we sought to determine the structure of the CD1c-PM complex to compare it with that of our previously determined CD1c-MPM complex (11). Furthermore, we determined the structure of the CD1c-PM reactive DN6 TCR in its unliganded state. Peptide-reactive TCRs and invariant NKT TCRs show highly stereotyped orientation on MHC I and CD1d. In contrast, our study suggests that even structurally related antigens are recognized by differing approaches of their respective TCRs, which sample different facets of the CD1c surface.
Results
Three Distinct Patterns of T-Cell Recognition of Mycoketides.
PM and MPM antigens are useful tools for probing the TCR recognition of the distal surface of CD1c-lipid complexes because both antigens have identical phospholipid anchors, but carry head groups of differing size. Activation of the CD8-1 T-cell clone requires the larger β-linked mannose for recognition (9), whereas the clone DN6 responds to the smaller primary phosphate epitope (10). To expand the number of TCRs available for structural analysis, we isolated PM reactive T-cell clones by sorting peripheral blood mononuclear cells (PBMCs) with PM-loaded CD1c tetramers and cloned cells at limiting dilution. We rescreened a panel of clones for CD1c tetramer-PM staining (Fig. 1A) and IFN-γ response (Fig. 1B) using K562 cells that did or did not express CD1c. The resulting CD1c and PM reactive T cells from donor 1 (clones 1.6 and 1.22), donor 2 (clone 2.4), and donor 22 (clones 22.2 and 22.5) were confirmed as clones based on the homogenous expression of coreceptors such as CD4 (Fig. 1A), as well detection of only one TCR α and one TCR β chain sequence.
Fig. 1.

CD1c reactive T-cell clones secrete IFN-γ in response to CD1c-PM stimulation. (A) After sorting PBMCs based on CD1c-PM tetramer staining, cells were cloned at limiting dilution and then screened for CD1c-PM tetramer positivity, CD4/CD8 coreceptor expression and (B) IFNγ release after stimulation with C32-PM or C32-MPM presented by CD1-expressing K562 cells. In all cases, cell lines were screened for antigen reactivity two or more times.
Each clone’s TCR incorporates unique pairings of Vα and Vβ domains in their rearrangements: DN6, TRAV36 and TRBV6-6, 22.2, TRAV17 and TRBV7-9, 22.5, TRAV26-2 and TRBV7-9, 1.6, TRAV9-2 and TRBV20-1, 1.22, TRAV22 and TRBV7-9; 2.4, TRAV26 and TRBV7-9 (Fig. S1A). The CD1c-MPM-specific CD8-1 clone TCR sequence, using TRAV8-6 and TRBV20-1, is shown for comparison. The α chains use different joining segments (TRAJ52, TRAJ47, TRAJ36, TRAJ35, TRAJ49, and TRAJ17) but the β chains include TRBJ1 and similar TRBJ2 and TRBD2 segments. The four TCRs using TRBV7-9 all had diverse complementarity determining region 3 (CDR3) loop sequences (Fig. S1B). These seven (including CD8-1) distinct TCRs have three patterns of mycoketide antigen recognition: CD8-1 recognizes MPM but not PM; DN6, 1.6, 22.2, 1.22, and 2.4 recognize PM presented by CD1c, and surprisingly, the clone 22.5 showed apparent cross-reactivity for PM and MPM when presented by K562 cells. However, a recent study indicated that MPM can be processed through a de-glycosylation reaction to create PM within cells (10). Therefore, we could not distinguish with these experiments whether the 22.5 TCR showed a true cross-reactivity to CD1c-MPM complexes or whether MPM was processed by cells to create a CD1c-PM epitope. We therefore extended our approach to using recombinant protein interaction studies to verify these specificities.
Direct Recognition of CD1c/Mycoketide by Diverse αβ TCRs.
To determine first whether these TCRs recognize CD1c-lipid complexes directly, we expressed soluble versions of the TCR heterodimeric ectodomains of the DN6, 22.2, 22.5, 1.6, 1.22, and 2.4 clones and the extracellular region of CD1c in insect cells. Despite considerable effort, we were unable to produce a recombinant version of the CD8-1 TCR, so it is not included in this study. To test the antigenic specificity of these TCRs with CD1c/lipid, we loaded CD1c with PM, MPM, phosphatidic acid (PA), or phosphatidylcholine (PC), whose chemical structures of these lipids are shown in Fig. 2A. We measured their association with each TCR by either surface plasmon resonance (SPR) (Fig. 2B) or biolayer interferometry (BLI) (Fig. 2 C and D). PM and MPM are related, both having a phosphate moiety in their head group linked to a single alkyl chain of C32 with repeating methyl branches at C4 and every fourth carbon thereafter, all of which are in the S-configuration (all-S). MPM differs from PM based in its β-anomerially linked mannose residue on the phosphate moiety. Similar to PM, PA has a phosphate moiety as a head group yet has two alkyl chains as a tail. Although PC has a choline group attached to the phosphate moiety of PA, it is a common endogenous lipid and thus was included as an additional control (Fig. 2A). We also included the lyso form of phosphatidic acid [lysophosphatidic acid (LPA)] to determine whether the number of lipid chains was a factor in recognition (Fig. S2). Unloaded CD1c was used throughout as a control for background reactivity; this protein is loaded with endogenous lipid molecules from insect cells (19).
Fig. 2.
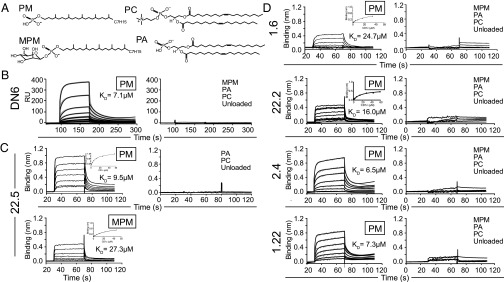
Binding analysis of human αβ-TCRs to CD1c loaded with mycobacterial lipid antigens using surface plasmon resonance and blitz bio-layer interferometry. (A) Chemical structures of lipids used in the analysis: phosphomycoketide (PM), β-mannosyl- phosphomycoketide (MPM), 1, 2-dioleoyl-sn-glycero-3-phosphocholine (PC), and 1, 2-dioleoyl-sn-glycero-3-phosphate (PA). (B) SPR sensogram showing reference-subtracted binding [response units (RUs)] of the DN6 TCR with increasing concentrations of CD1c-PM (shown in gray), associated association (Ka) and dissociation (Kd) rate fits (black lines) and calculated dissociation constants (KD) (Left). (Right) Binding response of the DN6 TCR with CD1c loaded with the lipids: MPM, PA, and PC and unloaded CD1c. (C) BLI sensogram showing reference-subtracted binding (binding in nm) of 22.5 TCR with increasing concentrations of CD1c-PM (Upper Left) and CD1c-MPM (Lower Left). Associated equilibrium analysis fits and calculated dissociation constants (KD) are shown in the Upper Right of each sensogram panel. (Right) Binding response of 22.5 TCR with PC and PA loaded CD1c and CD1c unloaded. (D) BLI sensograms showing the reference-subtracted binding response (binding in nm) of the 1.6, 22.2, 2.4, and 1.22 TCRs with increasing concentrations of CD1c-PM (Left). For the 2.4 and 1.22 TCRs, experimentally measured binding curves are colored in gray. Equilibrium analysis fits for the 1.6 and 22.2 TCRs are shown in the Upper Right of each sensogram panel; Ka and Kd fits for the 2.4 and 1.22 sensograms are shown in black lines. Calculated dissociation constants (KD) are shown for each sensogram. (Right) Binding response of these TCRs with PC, PA loaded CD1c, and CD1c unloaded. Data are representative of three or more experiments.
Each TCR exhibited direct binding to CD1c-mycoketide complexes but not control CD1c complexes (Fig. 2 and Fig. S2), although clear differences were observed in their antigen specificities. The DN6 TCR specifically interacts with CD1c loaded with PM in a concentration-dependent manner; we calculated the affinity in terms of equilibrium dissociation constant (KD) to be 7.1 ± 0.8 μM (Fig. 2B). Consistent with functional results (10), the DN6 TCR had no detectable binding to CD1c loaded with MPM. In contrast, the 22.5 TCR recognized both CD1c loaded with PM and CD1c loaded with MPM with KDs of 9.5 ± 1.2 and 27.3 ± 3.5 μM, respectively (Fig. 2C). The additional mannose residue of MPM resulted in an ∼2.7-fold reduction in the measured affinity. Similar to DN6, the 1.6, 1.22, 2.4, and 22.2 TCRs specifically recognized only CD1c-PM in a concentration-dependent manner with KDs of 24.7 ± 3.4, 7.3 ± 2.0, 6.5 ± 2.0, and 16.0 ± 2.4 μM, respectively (Fig. 2D). These results are summarized in table format in Fig. S3.
Such quantitative measurements of TCRs binding to CD1c-lipid resolve questions that arise from differing patterns of antigen recognition observed in cellular assays. First, although DN6 recognizes the MPM antigen when presented by live APCs (10), the lack of detectable binding to CD1c-MPM complexes by its TCR directly confirms the model that the true epitope of DN6 is CD1c-PM complexes that are generated during cellular processing. In contrast, the apparent cross-reactivity of 22.5 to MPM and PM in cellular assays (Fig. 1B) is validated by our binding measurements showing interactions between both CD1c-PM and CD1c-MPM with the 22.5 TCR. Thus, these results validate three patterns of TCR recognition of CD1c-mycoketide antigen: absolute specificity for MPM (CD8-1), absolute specificity for PM (DN6, 1.6, 1.22, 2.4, and 22.2), and true cross-reactivity to both antigens (22.5). More generally, these affinities are within the range of those measured for CD1d/iNKT cells and on the high end of those for TCR/MHCp binding (20, 21). In theory, the phosphate anion might have dominated reactivity, but all of the TCRs exhibited remarkable specificity to PM as contrasted with PA, or even the lyso-version of PA, LPA. This specific recognition suggests that different positioning or stability of the phosphate head groups, mediated by their chemically distinct lipid chains, plays an important role in TCR reactivity. This conclusion is further supported by our results demonstrating that TCRs do not recognize PC loaded CD1c or unloaded CD1c, indicating they are not autoreactive and instead are exquisitely specific to mycobacteria-derived antigens (Fig. 2).
Structural Determination of CD1c in Complex with PM Reveals a Shifted Head Group.
The CD1c crystal structure was first solved in complex with MPM (11). PM has now emerged as a biologically relevant antigen, which is both a processed product of MPM (10) and a de novo precursor to MPM production within M. tuberculosis. The majority of existing mycoketide clones recognize PM. To understand the molecular basis of PM presentation, we determined the CD1c-PM complex crystal structure. We used the previously developed CD1copt construct (11) and loaded this with chemically synthesized PM. As PM exhibited a much lower solubility in buffer compared with MPM, we devoted considerable effort to optimizing loading, as discussed in detail in Materials and Methods. CD1c-PM crystallized in the same condition as CD1c-MPM and occupied the same space group. The structure was determined via molecular replacement, and the previously solved structure of CD1c (PDB: 3OV6), minus ligand, was used as a search model. A single CD1c-PM molecule was found in the asymmetric unit. The structure was refined to 2.7-Å resolution with final Rwork and Rfree of 23.2% and 27.1%, respectively, and more than 90% of residues distributed in the favored region in the Ramachandran plot (Table 1). Occupancies were set to zero for amino acid residues with weak or absent electron density. In this structure, CD1c adopts an architecture highly similar to CD1c-MPM [root mean square (RMS) deviation = 0.319 Å] with an MHC-like fold with the two hydrophobic pockets called A′ and F′ shared by all human CD1 molecules. The A′ pocket accommodated the alkyl chain of PM (Fig. 3 A and B), whereas the F′ pocket exhibited an open groove-like structure, similar to that in the CD1c/MPM structure.
Table 1.
Data collection and refinement statistics (molecular replacement) for CD1c-PM and the DN6 TCR
| Data collection and refinement | DN6 | CD1c-PM |
| Data collection | ||
| Space group | C 1 2 1 | P 21 21 21 |
| Cell dimensions | ||
| a, b, c (Å) | 143.8, 64.1, 69.5 | 54.3, 87.0, 90.0 |
| α, β, γ (°) | 90.00, 115.88, 90.00 | 90.00, 90.00, 90.00 |
| Resolution (Å) | 50.00–3.00 (3.05–3.00) | 46.0–2.70 (2.75–2.70) |
| Rmerge | 12.2 (56.1) | 9.3 (39.4) |
| I/σI | 10.7 (2.6) | 18.8 (2.4) |
| Completeness (%) | 98.8 (93.4) | 96.4 (76.5) |
| Redundancy | 3.8 (2.8) | 5.5 (3.1) |
| Refinement | ||
| No. reflections | 10,895 | 11,212 |
| Rwork/Rfree | 26.6/32.0 | 23.2/27.1 |
| No. atoms | ||
| Protein | 3,168 | 5,786 |
| Ligand/ion | 38 | 173 |
| Water | 5 | 6 |
| B-factors | ||
| Protein | 103 | 65 |
| Ligand/ion | 110 | 68 |
| Water | 55 | 44 |
| RMSD | ||
| Bonds (Å) | 0.007 | 0.007 |
| Angles (°) | 1.133 | 1.573 |
Values in parentheses are for highest-resolution shell.
Fig. 3.

Structure of CD1c in complex with PM. (A) Side view of CD1c-PM complex shown as ribbon diagram with CD1c and β2 microglobulin colored as deep purple and cyan, respectively. PM is colored as yellow. Omit map (light blue) densities at a σ =1 are shown for PM and an N-linked glycosylation at position N20 (NAG-NAG). The PM is bound in the A′ pocket. (B) Top view of CD1c-PM complex showing an omit map (light blue) and 2Fo-Fc map (green) for the PM lipid ligand. (C) A sectioned top view of the complex shows the packing of the mycoketide alkyl chains of PM and MPM in the A′ pocket. (D) Superimposition of CD1c-PM and CD1c-MPM complexes colored as purple and white, respectively. PM is also colored purple; MPM is in white. The alkyl chains of PM and MPM are similarly bound in the A′ pocket, traversing underneath the α1 helix, and exiting through the D′/E′ portal. (Inset) Both PM and MPM head groups are exposed and accessible for TCR recognition but are positioned ∼6 Å away from each other (as measured from phosphates). (E) Hydrogen bonds between PM head group and CD1c. Ribbon diagram of CD1c with residues involved in hydrogen bonding with PM are shown as stick. PM is colored as follows: carbon, yellow; oxygen, red; phosphorus, orange. Dotted lines represent probable hydrogen bonds.
Omit and 2Fo-Fc maps show clear electron density for the PM alkyl tail and phosphate head group bound in CD1c (Fig. 3 A and B). The methylated lipid chain of PM traverses through the A′ pocket, bent in a clockwise manner around the A′ pole to fit within the tunnel, exiting through the D′ portal (Fig. 3C). The C32-PM alkyl chain contains five methyl branches in an all-S configuration, which are necessary for T-cell activation in biological assays (7, 10). From the crystal structure, it is evident that the downward clockwise spiral of the PM backbone around the A′ pole positions the S-methyl branches on the outer surface of the toroid. This mode of binding specifically positions all methyl branches so that they increase hydrophobic interactions with the A′ pocket, similar to that described for MPM (11). Overall, the PM alkyl tail superimposed with that of MPM with a RMS deviation of 1.6 Å, confirming that methylated mycoketide tails mediate CD1c binding and thereby serve as a molecular pattern for mycobacterial detection (Fig. 3C). In contrast to the similarities in alkyl chain positioning, the phosphate head group of PM, lacking the mannose moiety found in MPM, is positioned ∼6 Å from its equivalent position in the CD1c/MPM structure (Fig. 3D), adopting a more extended conformation toward the F′ pocket. The phosphate head group of PM is stabilized by several possible H-bonds with main chain atoms of Tyr73 and Leu77 of the α1 helix of CD1c (Fig. 3E and Table S1).
DN6 TCR Crystal Structure.
To understand the overall structure and general conformations of the CDR loops of the PM-specific TCRs, we pursued crystallization trials using standard methodologies. Diffracting crystals were only obtained for the DN6 TCR; data were collected to 3.0 Å, and the DN6 TCR structure was solved via molecular replacement. One TCR heterodimer was present in the asymmetric unit. The DN6 structure was refined to 3.0-Å resolution with and Rfree and Rwork of 26.6% and 32.0%, respectively, and ∼90% amino acid residues were distributed in the favorable region in the Ramachandran plot (Table 1).
The overall structure of the DN6 TCR adopts a canonical αβ TCR structure (Fig. S4A) composed of four Ig-like domains with a variable and a constant domain per chain. The two variable domains of TRAV36 and TRBV6-6 together form the six hypervariable CDR loops, forming the potential ligand-binding site (Fig. S4A). In this structure, clear and distinct electron density was observed for all CDR loops of the DN6 TCR, in particular the CDR3α and CDR3β (Fig. S4 B and C). The CD1d-specific iNKT TCR [Protein Data Bank (PDB) ID code 2CDE] and CD1b-specific TCR clone 18 (PDB ID code 4G8E) were used for comparative structural analyses with DN6. The RMS values for pairwise superimposition between the Cα backbone of the complete DN6 TCR, iNKT TCR and the clone 18 TCR were 1.08 (2,318 atoms) and 1.37 Å (2,411 atoms), respectively, similar to values for comparisons of the variable domains alone [1.3 (1,226 atoms) and 1.25 Å (1,202 atoms), respectively]. These low RMS values reflect a conserved overall structural architecture of the TCR Ig domains, although structural differences in some of the CDR loops are evident from the superposition of the variable domains via Cα backbone comparison (Fig. S4D). The CDR1β and CDR2β loops exhibited striking structural similarities as DN6, clone 18 TCRs are comprised of similar TRBV6 chains (Fig. S1), and the iNKT TCR uses a TRBV25-1 chain with sequence similarities in those CDRs. The conformation of the CDR3β and all three α chain CDR loops of DN6 adopted unique conformational states compared with that of those of the iNKT and clone 18 TCRs (Fig. S4D).
The DN6 TCR Uses Five of the Six CDRs Loops to Recognize CD1c-PM.
The crystal structure of the DN6 TCR elucidated the organization of the hypervariable CDR loops and the surface exposed amino acid residues that could be involved in the antigen recognition. This information was used to perform extensive alanine-scanning mutagenesis of the CDR loops of DN6 to determine the CDR loops, as well as the specific residues possibly involved in recognition of CD1c-PM. Alanine-scanning mutagenesis is useful to determine the energetic contribution of an amino acid residue past the β-carbon and has been widely used in protein–protein interaction studies, although some caveats are associated with it (21–25). Based on the structure, we selected 30 solvent-exposed residues, 14 from TRAV36 chain and 16 from TRBV6-6 chain, to mutate to alanine. These mutant TCRs were cloned and expressed in insect cells; expression levels, reducing and nonreducing SDS/PAGE mobility, and size exclusion elution profiles of these mutants were comparable to the WT TCR. To rapidly screen these mutants, an ELISA-based assay (26) involving PM-CD1c tetramers was used to determine the effect of mutation on binding; WT DN6 TCR was included in all assays as a comparative control.
Fig. 4 presents a summary of the DN6 TCR scanning results; residues were colored based on the extent to which mutation to alanine affected binding: residues with relative binding ranges 0–50% were colored as red, 50–100% were as orange, 100% were blue, and more than 100% were as green. This analysis revealed a clear bias toward the α chain in CD1c/PM recognition. All CDR loops of the α chain are affected by alanine mutations (Fig. 4A); six residues in CDR1α (V25, T26, N27, F28, R29, and S30), three residues in CDR2α (T48, S50, and I52) and three residues in CDR3α (S94, Y95, and K97) resulted in reduced binding when mutated to alanine, thus reflecting a potential role in CD1c/PM interaction (Fig. 4A). Recognition by the β chain appeared skewed toward the CDR2β and CDR3β loops, as mutation of several residues within these loops (Y48, V50, I54, and T55 in CDR2β and H95, L97, S99, and E101 in CDR3β) affected CD1c-PM tetramer binding (Fig. 4B). Interestingly, alanine substitution of two hydrophobic residues on the CDR2β, V50 and I54, had the greatest overall effect on binding. Two hydrophobic residues in the CDR2β of NKT TCRs also make important contacts for recognition of CD1d-αGalcer (27). We also noted that several key residues were hydrophobic in nature and thus may interact with hydrophobic residues of CD1c. These TCR CDR loop residues used in CD1c-PM recognition involves both germ-line (CDR1 and CDR2 loops) and residues encoded by recombined CDR3 sequences, but N-encoded residues least affected binding when mutated.
Fig. 4.

Energetically important amino acid residues in α and β chains of DN6 TCR for interaction with CD1c-PM. ELISA results using CD1c-PM tetramers to probe reactivity to alanine-scanned CDR1, CDR2, and CDR3 loops of the DN6 α chain (A) and DN6 β chain (B). Results are shown as relative binding: CD1c-PM binding to mutant TCR divided by WT DN6 TCR binding. Columns in the graph are colored based on the level of relative binding: RB < 50%, red; 50% < RB < 100%, orange; RB = 100%, blue; RB > 100%, green. Mutant positions are shown on the x axis. BSA was used as control, and measurements were performed three times for each mutant.
Important residues were mapped on the DN6 surface based on the relative binding (RB) to visualize CDR hotspots for binding CD1c (Fig. S5). Residues were colored according to the mutational analysis shown in Fig. 4A. The alanine mutations that markedly affect binding derive from the CDR1α (V25, T26, N27, F28, R29, and S30), CDR3α (S94, Y95, and K97), and CDR2β (Y48, V50, I54, and T55), forming a continuous stretch along the lower half of the TCR as visualized in Fig. S5. Several residues in the CDR3β (notably H95 and L97) provide important contributions that could represent a stabilizing anchor for TCR binding (Fig. S5). These critical residues are different from those identified in the iNKT TCR, which are biased toward the CDR1α, CDR3α, and CDR2β loops (27), and together comprise a larger surface area. Thus, DN6 recognition of CD1c is unlike that of iNKT TCR recognition of CD1d, but is reminiscent of type II NKT TCR recognition of CD1d-sulfatide where all of the CDR loops engaged in contacts (28, 29).
TCR Footprint Mapping of CD1c Reveals Diverse Docking Modes for Each TCR.
To identify side chain residues of CD1c that contribute to TCR recognition, we used the CD1c-PM and CD1c-MPM structures to select 13 solvent exposed residues located in the α1 and α2 helices that were available for TCR contact but not involved in PM or MPM presentation (Fig. S6). Alanine mutants were expressed in insect cells, loaded with PM and/or MPM, and equilibrium dissociation constants (KD) were measured using SPR (DN6) or BLI (22.2, 22.5, 1.6, 2.4, and 1.22) (Fig. 5). Measurements of WT interactions were included with every mutant measurement to control for minor differences between measurement runs. The ratio of mutant KD to WT KD was calculated for all of the TCRs to determine the effect of mutation. The results from this analysis grouped the TCRs into three general categories: α1 biased (Fig. 5, Top: DN6, 2.4, and 1.22), α1/α2 distributed (Fig. 5, Middle: 22.5 and 22.2), and α2 biased (Fig. 5, Bottom: 1.6). CD1c residues affecting DN6, 2.4, and 1.22 binding were concentrated on the α1 helix, in particular residues D65, L68, F72, F75, and R79, where mutation to alanine resulted in undetectable binding in at least one of these TCRs. Mutation of F72 and F75 abrogated binding in all three TCRs, suggesting this is a focal point of binding. Mutation of residues in the α2 helix only had a moderate effect: L147A, Y152A, and S165A mutants were the only positions to reduce binding (one- to threefold reduction). The remaining residues tested either had no effect or an increased affinity (Fig. 5, Top). The 1.6 TCR, also specific for PM, showed a very different pattern of recognition. Important CD1c contract residues were found in both the α1 and α2 helices, with intact L68, L147, Y152, and R169 being required for recognition (Fig. 5, Bottom). Mutation of D65 and E157 also had a severe impact on binding, resulting in more than a threefold decrease in binding affinity. Thus, the predicted footprint of the 1.6 TCR spans almost the entire surface of CD1c/PM with a bias toward the α2 helix.
Fig. 5.
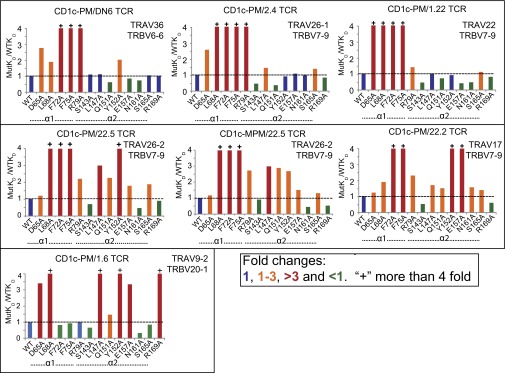
Energetically important amino acid residues in α1 and α2 helices of CD1c for interaction with αβ TCRs. Ratio of mutant KD to WT CD1c indicates the fold changes in the binding to CD1c reactive TCRs. (Top) DN6, 2.4, 1.22 TCRs. (Middle) 22.5 (PM and MPM), 22.2 TCRs. (Bottom) 1.6 TCR; as determined by SPR and BLI. Mutations with more than threefold reduction in the binding are colored as red, one to threefold reduction are colored as orange, no change is colored as blue, and less than one fold are colored as green. +, substitutions resulting in binding over fourfold reduction. Positions of mutant residues are also indicated below the horizontal axis of the curve. Data are representative of one experiment.
The 22.5 TCR differs from DN6 and 1.6 in that it can recognize both PM and MPM when presented by CD1c, so we took the advantage of this dual recognition and measured the binding of mutants with CD1c loaded with PM or MPM. The two footprints were very similar to each other, indicating that the 22.5 TCR likely docks in a similar footprint when recognizing PM and MPM (Fig. 5, Middle). The 22.2 TCR had a similar footprint to 22.5; both TCRs required the same two critical α1 helix residues, F72 and F75 (similar to DN6, 2.4, and 1.22), with similar moderate involvement of D65, L68, and R69. However, 22.5 and 22.2 clearly use residues in the α2 helix: in particular, L147, Q151, Y152, and E157. As noted with the DN6 TCR mutagenesis, there appeared to be a considerable involvement of hydrophobic residues in TCR docking, most notably L68, F72, F75, L147, and Y152, suggesting that hydrophobic interactions may play an important role in CD1c recognition by this TCR repertoire.
To visualize the location of the residues on CD1c involved in TCR binding as estimated from our alanine scanning mutagenesis, we mapped the mutations onto a surface representation of the corresponding CD1c-antigen structure for all six TCRs and two antigens (PM and MPM) (Fig. 6). In the case of the DN6, 2.4, and 1.22 TCRs, the interaction was mostly dominated by residues that were present in the α1 helix with a hotspot formed by F72 and F75 residues, with only one residue in the α2 helix, Y152, moderately involved in DN6 binding (Fig. 6A). Therefore, the footprints of these TCRs all have an extreme α1 helical bias, with a potential focus over the F′ pocket of CD1c, positioned over the PM phosphate head group. The positioning of these TCRs in this way may explain, in part, the exquisite specificity to PM and not MPM (10).
Fig. 6.

Energetic hotspots on CD1c for recognition of PM and MPM mediated by αβ TCRs. Surface representation of α1 and α2 domain on CD1c shown in white. Mutant residues are mapped on the surface, labeled, and colored on the basis of fold changes in binding (as in Fig. 5) with TCRs in respect to WT CD1c: (A) CD1c-PM with DN6, 2.4, 1.22 TCRs; (B) CD1c-PM, MPM with 22.5 TCR, CD1c-PM with 22.2; (C) 1.6 TCR/CD1c-PM. Critical residues for type I NKT TCR/CD1d-α-Galcer recognition are shown in D, mutations resulted in more than fourfold reduction in binding are colored as red as reported by ref. 27. (E) Type II NKT TCR footprint for CD1d-lysosulfatide recognition is shown, and the figure is constructed based on CD1d and TCR contact (PDB ID code 4ELM) (28).
The contact residues for the 22.5 TCRs were similar between PM and MPM and suggested a broad contact surface, spanning almost the entirety of both α1 and α2 helices. This pattern was similar to that of the 22.2 TCR, which was even more distributed across these helices (Fig. 6B). The antigenic head group would be thus centrally positioned in the 22.2 and 22.5 TCR docking; however, differences in the head group orientation only moderately affect 22.5 TCR binding. Therefore, the extensive contact surface with CD1c likely compensates for changes in TCR contacts with antigen. Finally, mapping the 1.6 TCR interaction onto CD1c revealed an additional unique docking footprint. Whereas the majority of the critical residues were located across the α2 helix (L147, Q151, Y152, E157, and R169), two residues at the N-terminal end of the α1 helix (D65 and L68) were also noted in our mutagenesis scan. This docking mode positions the TCR instead over the A′ pocket but also suggests the involvement of the PM head group (Fig. 6C). The docking orientations observed in our study contrast with those observed in CD1d-restricted type I and type II NKT TCRs binding (Fig. 6 D and E). The type I NKT TCR footprint has a much less central footprint, as it is focused almost entirely over the F′ pocket and extends to the margin of the CD1d surface, whereas the type II NKT TCR footprint is located on the other edge of the platform, mostly positioned over the A′ pocket (Fig. 6 D and E) (27–29). CD1c recognition not only involves TCRs of diverse genetic makeup, but also differing patterns of antigen fine specificity and flexibility in how these TCRs dock onto CD1c during antigen recognition.
Discussion
CD1c molecules are abundantly expressed on three key APCs in humans: thymocytes, dendritic cells (DCs), and B cells. In addition, group 1 CD1 isoforms are well suited to present mycobacterial lipid antigens to T cells because they are up-regulated in vitro and in vivo in response to M. leprae or M. tuberculosis infections (30–32). Also tuberculosis patients have CD1c-dependent polyclonal responses in vivo during natural infection (9). In particular, PM is suspected as a major antigen for a polyclonal T-cell response in humans as T-cell populations are readily stained with CD1c-PM tetramers from TB patient blood (10), although MPM reactive T-cell clones have also been isolated (33). Approximately 2% of all circulating αβ T cells are reactive to CD1c presenting endogenous lipids, which suggests a broader role of CD1c in human immunity (15, 34). However, the molecular features dictating TCR recognition of group I CD1 molecules are the least well understood of any CD1 isoform. Our studies show direct binding of human TCRs to CD1c, providing a molecular and structural foundation for understanding M. tuberculosis mycoketide presentation by CD1c, one that distinguishes it from the stereotyped response of germ-line encoded mycolyl (GEM)-reactive T cells to CD1b or invariant NKT cells to CD1d.
The CD1c-reactive T-cell repertoire has not been characterized in detail, but initial reports suggest diverse T-cell receptors respond to CD1c (6). The DN6, 22.5, 22.2, 1.6 1.22, and 2.4 T-cell clones examined here are representatives of this diverse repertoire of CD1c-restricted T cells. All of these clones recognize PM, a lipid that occurs through de novo production by M. tuberculosis, as well as through cellular antigen processing of MPM to its deglycosylated epitope. Previously, antigen processing for MPM to PM was inferred based on the observation that DN6 can recognize MPM but only when displayed by intact cells with antigen processing capabilities (10). Direct binding of the DN6 TCR to PM but not MPM bound to CD1c directly establishes PM as the true TCR epitope. Unexpectedly, the apparent cross-reactivity of clone 22.5 to PM and MPM results from the TCR’s binding to either lipid in complex with CD1c.
All seven TCR reactivities studied here show affinities within the range reported for conventional and invariant T-cell populations (20, 35) and generate IFN-γ as a functional response. In contrast to many other group I CD1 studies, these TCRs do not respond to endogenous lipid antigens and are thus unlikely to be autoreactive. Our structural studies clearly show that mycoketide antigens are bound in the A′ pocket with a conserved architecture of alkyl chain placement. Methyl groups in the mycoketide chain are essential for loading into CD1c, and the crystal structures of CD1c with MPM and PM explain their role in anchoring the lipid within the CD1c A′ pocket. This feature reinforces the mycoketide alkyl chain, with a length between C30 and C34, as a key molecular recognition determinant for M. tuberculosis infection (7). However, our structure of CD1c/PM demonstrates that head group positioning in CD1c differs between PM and MPM (11), with a shift of ∼6 Å between the phosphate groups of the two head groups. This difference suggests that specificity to these antigens might involve both the positioning of the antigen in CD1c and the chemical makeup of the head group. Our data also demonstrate the underlying molecular features of the polyclonal immune response against mycoketides; some TCRs have intrinsic plasticity in mycoketide recognition with the ability to respond to both MPM and PM, such as the 22.5 TCR, which might impart improved recognition of cells, which are expected to display both antigens simultaneously. However, the CD8-1, DN6, 22.2, 1.6, 1.22, and 2.4 TCRs are nonpermissive for these particular changes and instead are specific to either MPM or PM (10, 33). Therefore, three classes of reactivity exist to this particular antigen pair: specificity to MPM, specificity to PM, or cross-reactivity to both.
The DN6 T-cell clone has been widely used for M. tuberculosis antigen presentation assays and has recently been shown to specifically recognize PM when presented by CD1c (10). Our crystal structure of the DN6 TCR provided important information on the conformation of the CDR loops, and importantly, the solvent-exposed residues that are available for CD1c/antigen recognition. The superimposition of the DN6 TCR structure with those the iNKT TCR and the CD1b-specific TCR clone 18 showed surprising similarities in CDR1β and CDR2β loop structure; however, the loop architecture of the DN6 CDR3β loop and all three CDR loops of the α chain differ, likely due to the sequence divergence between these TCRs. Most of the surface-exposed amino acid residues in the CDR3 loops of DN6 originate from the germ-line encoded TRAJ and TRBJ segments; the N residues (W in the CDR3α and RH in the CDR3β) are mostly buried inside the structure. Indeed, alanine scanning of the DN6 CDR loops demonstrate that the majority of the energetically important residues are encoded by germ-line templates. Interestingly, the TCRs examined in this study used different TRAJ segments, but five of the six TCRs incorporated similar TRBJ2 and TRBD2 segments (2.4 used TRBJ1), perhaps because residues encoded in these segments are used in CD1c recognition. Similar germ line-mediated recognition enables NKT cells to tolerate considerable structural variation among glycolipid antigens (36); however, whether this strategy is ubiquitously used among CD1c-reactive T cells awaits a broader characterization of this T-cell repertoire.
Alanine-scanning of the DN6 CDR loops revealed involvement of five of the six CDR loops in recognition of PM-loaded CD1c. All of the CDR loops of the α chain were affected by mutation to alanine, suggesting a dominant role for the α chain in CD1c antigen engagement. This α-chain dominance is reminiscent of type I NKT recognition where the invariant α chain overall makes more energetically important contacts than β chain (27); however, DN6 recognition of PM-loaded CD1c appears distributed almost equally across the three CDR loops instead of being concentrated predominantly in the CDR3α, as is the case with iNKT TCR recognition of CD1d. The CDR2 and CDR3 of the β chain also were affected by mutagenesis and indicate the important contribution to binding made by these loops. The broad involvement of these five CDR loops and the TCR diversity we have thus far documented for CD1c-reactive TCRs suggests a molecular recognition strategy unlike that of the type I NKT cells, with a concentrated binding footprint on CD1d mostly involving the CDR3α and CDR2β loops. Instead, CD1c-reactive T cells appear to use a recognition strategy like that of type II NKT cells, expressing a more diverse TCR repertoire and using all six CDR loops in contacting CD1d-lysosulphatide (28, 29). However, the footprint of these TCRs on CD1c suggest that the particular docking modes seen here are unlike those described for type II NKT TCRs.
Our alanine scanning of the CD1c surface for six representative CD1c-reactive TCRs specific for the same antigen has revealed new and potentially important insight into the strategy used by this T-cell population in CD1c recognition. Conventional αβ TCRs recognize classical class I MHC molecules with a relatively conserved diagonal orientation (37); NKT (38) and mucosal associated invariant T (MAIT) TCRs (39–41) also dock with a conserved orientation on their CD1d and MR1 ligands, respectively. In contrast, our results reveal diverse docking strategies to one kind of CD1c-phospholipid complex, even though two structurally related antigen complexes are studied. One mode of binding focuses on residues of the α1 helix (DN6, 2.4, and 1.22), one focused predominantly on the α2 helix (1.6) and the last with contacts distributed across the α1 and α2 helices (22.5 and 22.2). At a minimum, this indicates that CD1c TCRs do not have an invariant docking strategy, as is known to occur in invariant NKT cells and expected for the conserved T cells in the CD1b system known as GEM T cells (42). Also, functional mapping of the footprint suggests that the centrally located footprint of these CD1c-reactive TCRs is distinct from type I (focused on the F′ pocket) or type II (focused on the A′ pocket) NKT populations. The semiconserved footprint of the 22.5 TCR for PM-loaded CD1c and MPM-loaded CD1c suggest that variation in antigen structure or positioning does not dramatically affect the footprint of this TCR on CD1c, so it is possible that there may be a defined set of docking modes that are used, dictated by which TRAV/TRAJ and TRBV/TRBJ gene segments are used in the TCR. Further characterization of the CD1c restricted TCR repertoire will contribute significantly to answering this question.
Materials and Methods
T-Cell Cloning.
The cloning of C32-PM loaded CD1c tetramer+ cells were as previously described (10). Briefly, PBMCs were collected after informed consent from leukapheresis of asymptomatic tuberculin-positive subjects and overseen by the institutional review boards of the Lemuel Shattuck Hospital (00000786) and Partners Healthcare (2002-P-000061) or from blood collars from the Kraft Family Blood Donor clinic. PBMCs were separated by Ficoll density gradient and enriched for T cells using the Dynabeads Untouched Human T Cells Primary negative selection kit (Invitrogen). After resting overnight, T cells were stained with C32-PM loaded-CD1c tetramers and sorted using a FACSAria flow cytometer. C32-PM loaded-CD1c tetramer binding cells were plated at limiting dilution (∼1 cell per well) in the presence of an expansion mixture, consisting of irradiated PBMCs, EBV-transformed B cells, and 50 ng/mL anti-CD3 (clone OKT3) and IL-2. After an initial round of expansion, clones were screened for C32-PM CD1c tetramer binding and coreceptor expression with phycoerythrin-labeled anti-CD8 (clone HIT8a) and allophycocyanin-labeled anti-CD4 (clone RPA-T4). An aliquot of tetramer-reactive clones were collected for cDNA, with the rest further expanded for functional studies. Clones were restimulated every other week with expansion mixture. For functional studies, T-cell clones were stimulated for 24 h [37 °C, 5% (vol/vol) CO2] in the presence of 15 µM of antigen presented by 5 × 104 K562 expressing CD1c or control vector, and IFN-γ release was measured in Elispot (Mabtech).
Protein Expression and Purification.
We used our previously engineered CD1c construct (11) for many of the studies reported here. In addition, WT CD1c fused with β2m was cloned into a pAcGP67A vector (BD Biosciences) containing a C-terminal BirA biotinylation sequence followed by a six-Histidine tag. All constructs were expressed in High Five cells with the baculovirus expression system as previously described (11). Alanine-scanning mutagenesis was performed through overlapping PCR with specific primers containing the desired mutations with glycosylated CD1copt (11) as a template; the resulting mutants were cloned into the pAcGP67A vector (BD Biosciences) with a deca-His tag at the C terminus. Baculovirus was produced and used to transduce High Five cells; mutant proteins were purified as previously described (11).
We used standard RT-PCR with immunogenetics (IMGT) degenerate primer sets (IPS000029 for TCRα chains and IPS000003 for TCRβ chains) to determine the sequence of the 22.5 and 1.6 TCRs (10). The TCR ectodomains of CD1c-PM—reactive T-cell clones DN6, 2.4, 1.22, 22.5, and 1.6 were cloned from cDNA. The sequence of the variable domain of TRAV9 chain of the 1.6 TCR was codon-optimized for insect cell expression and synthesized through overlapping PCR from a reverse-translated sequence. Chains were modified to favor proper αβ chain heterodimer formation as previously described (43) by inserting T48C and S57C mutations in the α and β constant domains, respectively, and elimination of the WT interchain disulphide cysteines. TCRs were expressed and purified as previously described (39).
CD1c and TCR Biophysical Interaction Analysis.
All interaction measurements for DN6 TCR with CD1c-ligands were performed using surface plasmon resonance on a Biacore 3000 with streptavidin chips (SA chip; GE Healthcare) used for immobilization of biotinylated DN6 TCR. Hepes buffer saline (HBS) buffer (10 mM Hepes, pH 7.4, and 150 mM NaCl) was used for all measurements. Binding signals were reference subtracted, and traces were plotted in the BIAevaluation software. All interaction analyses of the 2.4, 1.22, 22.2, 22.5, and 1.6 TCRs were carried out in real time by BLI in a Blitz System Package (Fortebio; Pal Lifescience) using a similar strategy to that of SPR. GraphPad Prism was used to determine the affinity constants for these interactions. The CD1copt protein was expressed in insect cells as described above and loaded with MPM, PM, and the control lipids PA (cat no. 840875C), PC (cat no. 850375C), and lysophosphatidylcholine (LPC) (cat no. 845875C) from Avanti Polar Lipids and LPA (Cat no. 62215) from Cayman Chemicals.
CD1c Loading with PM, Purification, and Crystallization.
The CD1copt protein was expressed in insect cells as previously described (11). As PM was less soluble in HBS than MPM due to the absence of a mannose in the head group, we optimized the PM loading into CD1c and the purification method for crystallization. CD1copt was briefly treated with 0.005% Tween20 to remove partially endogenously loaded lipid, and the protein sample was again purified using a Mono-Q anion exchange (GE Healthcare) column. PM was partially solubilized into HBS by sonicating in a 50 °C water bath cleaner (Branson) for 30 min. For loading into CD1c, PM was incubated with CD1copt at a 10 molar excess at 37 °C overnight. The protein sample was then centrifuged for 30 min at 16,000 × g, and the samples were subjected to anion exchange chromatography to separate the fraction of CD1copt loaded with PM from unloaded CD1c and free lipids. The purified CD1c-PM peak fractions were assessed by SPR to confirm TCR binding. CD1copt-PM fractions were concentrated to 10 mg/mL and crystallized via a sitting-drop method at 22 °C by combining 0.5 μL protein solution and 0.5 μL mother liquor solution containing 1.05 M sodium citrate, 100 mM 2-(cyclohexylamino) ethanesulfonic acid (pH 9.4), and 25 mM triglycine.
Crystallization of DN6 TCR.
The WT DN6 TCR was expressed in insect cells and purified as described above. The protein was treated with endoglycosidase F3 at 37 °C for 2 h to minimize heterogeneity present by the N-linked glycosylation sites. The glycosylation-minimized DN6 TCR was purified by anion exchange chromatography. Protein was concentrated to 10 mg/mL and crystallized using the sitting drop method, combining 0.5 μL of protein solution and 0.5 μL of reservoir solution containing 0.1 M Mes, pH 6.5, 20% PEG 4000, and 0.6 M NaCl at 22 °C.
Crystallographic Data Collection, Structure Determination, Refinement, and Analysis.
CD1copt-PM crystals were cryo-protected with 50% sodium malonate, and DN6 crystals were cryo-cooled in mother liquor solution supplemented with 20% (vol/vol) glycerol before cooling to 100° K with liquid nitrogen. X-ray datasets were collected on a MAR300 CCD at beamline 23 ID-D at the Advanced Photon Source (APS) at Argonne National Laboratory. HKL2000 was used to index, integrate, and scale the data (44). Coordinates of the ligandless CD1copt (PDB ID code 3OV6) was used as a search model to solve the CD1copt-PM complex structure by molecular replacement with the program Phaser (45). The DN6 structure was solved using coordinates of Tk3 (chain D; PDB ID code 3MV7) and A6 TCR (chain E, PDB ID code 1A07) as search models. Initially, rigid body and then restrained refinement was performed with the Phenix software suite (46). A PM coordinate file was generated from CD1copt-MPM coordinates in Coot (47). Subsequent cycles of manual building in Coot and refinement were carried out, and the lipid or covalently bound sugar moiety were introduced into the model guided by Fo-Fc and SA omit map. Translation/libration/screw (TLS) refinement was performed with Phenix for the DN6 structure. All refinement was performed by taking a random 5% of reflections and excluding them for statistical validation purposes (Rfree). Hydrogen bonding contacts between PM and CD1copt were calculated using the program Contacts in the CCP4 package (48). All structural figures were generated with the program Pymol (Schrödinger Scientific).
Alanine-Scanning Mutagenesis Analysis.
CD1c tetramers were prepared by adding one-fourth molar ratio of streptavidin-HRP (Pierce) over 10 time intervals to ensure maximum tetramerization. Wells of an ELISA plate were coated with WT and mutant DN6 TCRs in triplicate at a concentration of 10 μg/mL in HBS and incubated overnight at 4 °C. Wells were washed with PBS and blocked with BSA for 2 h. CD1c-PM tetramer was diluted in HBS containing 1 mg/mL purified BSA and added to the TCR-coated wells. Plates were then incubated for 30 min at 22 °C and washed 10 times with PBS. TMB (3,3′,5,5′-tetramethylbenzidine) ELISA (Pierce) substrate was added to wells and color development was stopped with 2 M sulfuric acid. The optical density of each well was measured at 450 nm with a reference wavelength of 490 nm. The optical density for each mutant TCR was compared with WT, in triplicate, to estimate the effect of each mutation. CD1c mutant constructs were cloned and expressed as described above and were analyzed for binding with the Blitz system, as described above for the WT TCR association studies.
Supplementary Material
Acknowledgments
We thank the staff of the Advanced Proton Source at The General Medical Sciences and Cancer Institutes Structural Biology Facility–Collaborative Access Team (23ID) for use and assistance with X-ray beamlines and Ruslan Sanishvili, Steven Corcoran, and Michael Becker in particular for help and advice during data collection. A. De Jong generously provided the K562 transfectants, and Prof. Steve Porcelli generously provided the DN6 T-cell line. This study was supported by National Institutes of Health (NIH) Grants R56_AI097386 and R01AI073922 (to E.J.A.); R01GM102489 (to J.A.P.); R01AI049313, R01AR048632, and the Burroughs Wellcome Fund Program in Translational Medicine (to D.B.M.); and NIH Contract HHSN272201300006C and NIH Grant P51 RR000165 (to J.D.A.).
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
Data deposition: The atomic coordinates and structure factors have been deposited in the Protein Data Bank, www.pdb.org (PDB ID code 4ONO and 4ONH).
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1408549111/-/DCSupplemental.
References
- 1.Porcelli SA, Modlin RL. The CD1 system: Antigen-presenting molecules for T cell recognition of lipids and glycolipids. Annu Rev Immunol. 1999;17:297–329. doi: 10.1146/annurev.immunol.17.1.297. [DOI] [PubMed] [Google Scholar]
- 2.Brigl M, Brenner MB. CD1: Antigen presentation and T cell function. Annu Rev Immunol. 2004;22:817–890. doi: 10.1146/annurev.immunol.22.012703.104608. [DOI] [PubMed] [Google Scholar]
- 3.Adams EJ, Luoma AM. The adaptable major histocompatibility complex (MHC) fold: Structure and function of nonclassical and MHC class I-like molecules. Annu Rev Immunol. 2013;31:529–561. doi: 10.1146/annurev-immunol-032712-095912. [DOI] [PubMed] [Google Scholar]
- 4.Young MH, Gapin L. Group 1 CD1-restricted T cells take center stage. European Journal of Immunology. 2011;41(3):592–594. doi: 10.1002/eji.201141408. [DOI] [PubMed] [Google Scholar]
- 5.Kasmar A, Van Rhijn I, Moody DB. The evolved functions of CD1 during infection. Curr Opin Immunol. 2009;21(4):397–403. doi: 10.1016/j.coi.2009.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Van Rhijn I, Ly D, Moody DB. CD1a, CD1b, and CD1c in immunity against mycobacteria. Adv Exp Med Biol. 2013;783:181–197. doi: 10.1007/978-1-4614-6111-1_10. [DOI] [PubMed] [Google Scholar]
- 7.de Jong A, et al. CD1c presentation of synthetic glycolipid antigens with foreign alkyl branching motifs. Chem Biol. 2007;14(11):1232–1242. doi: 10.1016/j.chembiol.2007.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Matsunaga I, et al. Mycobacterium tuberculosis pks12 produces a novel polyketide presented by CD1c to T cells. J Exp Med. 2004;200(12):1559–1569. doi: 10.1084/jem.20041429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Moody DB, et al. CD1c-mediated T-cell recognition of isoprenoid glycolipids in Mycobacterium tuberculosis infection. Nature. 2000;404(6780):884–888. doi: 10.1038/35009119. [DOI] [PubMed] [Google Scholar]
- 10.Ly D, et al. CD1c tetramers detect ex vivo T cell responses to processed phosphomycoketide antigens. J Exp Med. 2013;210(4):729–741. doi: 10.1084/jem.20120624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Scharf L, et al. The 2.5 Å structure of CD1c in complex with a mycobacterial lipid reveals an open groove ideally suited for diverse antigen presentation. Immunity. 2010;33(6):853–862. doi: 10.1016/j.immuni.2010.11.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Van Rhijn I, et al. CD1c bypasses lysosomes to present a lipopeptide antigen with 12 amino acids. J Exp Med. 2009;206(6):1409–1422. doi: 10.1084/jem.20082480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shamshiev A, et al. Presentation of the same glycolipid by different CD1 molecules. J Exp Med. 2002;195(8):1013–1021. doi: 10.1084/jem.20011963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Spada FM, et al. Self-recognition of CD1 by gamma/delta T cells: Implications for innate immunity. J Exp Med. 2000;191(6):937–948. doi: 10.1084/jem.191.6.937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.de Lalla C, et al. High-frequency and adaptive-like dynamics of human CD1 self-reactive T cells. Eur J Immunol. 2011;41(3):602–610. doi: 10.1002/eji.201041211. [DOI] [PubMed] [Google Scholar]
- 16.Roura-Mir C, et al. CD1a and CD1c activate intrathyroidal T cells during Graves’ disease and Hashimoto’s thyroiditis. J Immunol. 2005;174(6):3773–3780. doi: 10.4049/jimmunol.174.6.3773. [DOI] [PubMed] [Google Scholar]
- 17.Beckman EM, et al. CD1c restricts responses of mycobacteria-specific T cells. Evidence for antigen presentation by a second member of the human CD1 family. J Immunol. 1996;157(7):2795–2803. [PubMed] [Google Scholar]
- 18.Porcelli S, et al. Recognition of cluster of differentiation 1 antigens by human CD4-CD8-cytolytic T lymphocytes. Nature. 1989;341(6241):447–450. doi: 10.1038/341447a0. [DOI] [PubMed] [Google Scholar]
- 19.Haig NA, et al. Identification of self-lipids presented by CD1c and CD1d proteins. J Biol Chem. 2011;286(43):37692–37701. doi: 10.1074/jbc.M111.267948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gadola SD, et al. Structure and binding kinetics of three different human CD1d-alpha-galactosylceramide-specific T cell receptors. J Exp Med. 2006;203(3):699–710. doi: 10.1084/jem.20052369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Huseby ES, Crawford F, White J, Marrack P, Kappler JW. Interface-disrupting amino acids establish specificity between T cell receptors and complexes of major histocompatibility complex and peptide. Nat Immunol. 2006;7(11):1191–1199. doi: 10.1038/ni1401. [DOI] [PubMed] [Google Scholar]
- 22.Baker BM, Turner RV, Gagnon SJ, Wiley DC, Biddison WE. Identification of a crucial energetic footprint on the alpha1 helix of human histocompatibility leukocyte antigen (HLA)-A2 that provides functional interactions for recognition by tax peptide/HLA-A2-specific T cell receptors. J Exp Med. 2001;193(5):551–562. doi: 10.1084/jem.193.5.551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.DeLano WL. Unraveling hot spots in binding interfaces: Progress and challenges. Curr Opin Struct Biol. 2002;12(1):14–20. doi: 10.1016/s0959-440x(02)00283-x. [DOI] [PubMed] [Google Scholar]
- 24.Feng D, Bond CJ, Ely LK, Maynard J, Garcia KC. Structural evidence for a germline-encoded T cell receptor-major histocompatibility complex interaction ‘codon’. Nat Immunol. 2007;8(9):975–983. doi: 10.1038/ni1502. [DOI] [PubMed] [Google Scholar]
- 25.Manning TC, et al. Alanine scanning mutagenesis of an alphabeta T cell receptor: Mapping the energy of antigen recognition. Immunity. 1998;8(4):413–425. doi: 10.1016/s1074-7613(00)80547-6. [DOI] [PubMed] [Google Scholar]
- 26.Luoma AM, et al. Crystal structure of Vδ1 T cell receptor in complex with CD1d-sulfatide shows MHC-like recognition of a self-lipid by human γδ T cells. Immunity. 2013;39(6):1032–1042. doi: 10.1016/j.immuni.2013.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wun KS, et al. A minimal binding footprint on CD1d-glycolipid is a basis for selection of the unique human NKT TCR. J Exp Med. 2008;205(4):939–949. doi: 10.1084/jem.20072141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Girardi E, et al. Type II natural killer T cells use features of both innate-like and conventional T cells to recognize sulfatide self antigens. Nat Immunol. 2012;13(9):851–856. doi: 10.1038/ni.2371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Patel O, et al. Recognition of CD1d-sulfatide mediated by a type II natural killer T cell antigen receptor. Nat Immunol. 2012;13(9):857–863. doi: 10.1038/ni.2372. [DOI] [PubMed] [Google Scholar]
- 30.Krutzik SR, et al. TLR activation triggers the rapid differentiation of monocytes into macrophages and dendritic cells. Nat Med. 2005;11(6):653–660. doi: 10.1038/nm1246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Roura-Mir C, et al. Mycobacterium tuberculosis regulates CD1 antigen presentation pathways through TLR-2. J Immunol. 2005;175(3):1758–1766. doi: 10.4049/jimmunol.175.3.1758. [DOI] [PubMed] [Google Scholar]
- 32.Sieling PA, et al. CD1 expression by dendritic cells in human leprosy lesions: Correlation with effective host immunity. J Immunol. 1999;162(3):1851–1858. [PubMed] [Google Scholar]
- 33.Rosat JP, et al. CD1-restricted microbial lipid antigen-specific recognition found in the CD8+ alpha beta T cell pool. J Immunol. 1999;162(1):366–371. [PubMed] [Google Scholar]
- 34.Adams EJ. Diverse antigen presentation by the Group 1 CD1 molecule, CD1c. Mol Immunol. 2013;55(2):182–185. doi: 10.1016/j.molimm.2012.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.van der Merwe PA, Davis SJ. Molecular interactions mediating T cell antigen recognition. Annu Rev Immunol. 2003;21:659–684. doi: 10.1146/annurev.immunol.21.120601.141036. [DOI] [PubMed] [Google Scholar]
- 36.Scott-Browne JP, et al. Germline-encoded recognition of diverse glycolipids by natural killer T cells. Nat Immunol. 2007;8(10):1105–1113. doi: 10.1038/ni1510. [DOI] [PubMed] [Google Scholar]
- 37.Garcia KC, Adams EJ. How the T cell receptor sees antigen—a structural view. Cell. 2005;122(3):333–336. doi: 10.1016/j.cell.2005.07.015. [DOI] [PubMed] [Google Scholar]
- 38.Rossjohn J, Pellicci DG, Patel O, Gapin L, Godfrey DI. Recognition of CD1d-restricted antigens by natural killer T cells. Nat Rev Immunol. 2012;12(12):845–857. doi: 10.1038/nri3328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.López-Sagaseta J, et al. The molecular basis for Mucosal-Associated Invariant T cell recognition of MR1 proteins. Proc Natl Acad Sci USA. 2013;110(19):E1771–E1778. doi: 10.1073/pnas.1222678110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.López-Sagaseta J, et al. MAIT recognition of a stimulatory bacterial antigen bound to MR1. J Immunol. 2013;191(10):5268–5277. doi: 10.4049/jimmunol.1301958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Patel O, et al. Recognition of vitamin B metabolites by mucosal-associated invariant T cells. Nat Commun. 2013;4:2142. doi: 10.1038/ncomms3142. [DOI] [PubMed] [Google Scholar]
- 42.Van Rhijn I, et al. A conserved human T cell population targets mycobacterial antigens presented by CD1b. Nat Immunol. 2013;14(7):706–713. doi: 10.1038/ni.2630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Boulter JM, et al. Stable, soluble T-cell receptor molecules for crystallization and therapeutics. Protein Eng. 2003;16(9):707–711. doi: 10.1093/protein/gzg087. [DOI] [PubMed] [Google Scholar]
- 44.Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 45.McCoy AJ, et al. Phaser crystallographic software. J Appl Cryst. 2007;40(Pt 4):658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Adams PD, et al. PHENIX: A comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr. 2010;66(Pt 2):213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Emsley P, Cowtan K. Coot: Model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60(Pt 12, Pt 1):2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 48.Collaborative Computational Project, Number 4 The CCP4 suite: Programs for protein crystallography. Acta Crystallogr D Biol Crystallogr. 1994;50(Pt 5):760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.