

Significance
Thymosin-β4 (Tβ4) sequesters actin monomers to help maintain the high concentrations of unpolymerized actin in higher eukaryotic cells. Despite more than two decades of research investigating the Tβ4–actin interaction, the X-ray structure of the full-length Tβ4:actin complex remained unresolved. Here, we report two X-ray structures of Tβ4:actin complexes. The first structure reveals that Tβ4 has two helices that bind at the barbed and pointed faces of actin, whereas the second structure displays a more open actin nucleotide binding cleft and a disruption of the Tβ4 C-terminal helix interaction. These structures, combined with biochemical assays and molecular dynamics simulations, reveal how Tβ4 prevents monomeric actin from joining actin filaments but participates in the exchange of actin with profilin to ensure controlled actin polymerization.
Keywords: crystallography, protein complex, thermodynamics, molecular dynamics, fluorescence
Abstract
Thymosin-β4 (Tβ4) and profilin are the two major sequestering proteins that maintain the pool of monomeric actin (G-actin) within cells of higher eukaryotes. Tβ4 prevents G-actin from joining a filament, whereas profilin:actin only supports barbed-end elongation. Here, we report two Tβ4:actin structures. The first structure shows that Tβ4 has two helices that bind at the barbed and pointed faces of G-actin, preventing the incorporation of the bound G-actin into a filament. The second structure displays a more open nucleotide binding cleft on G-actin, which is typical of profilin:actin structures, with a concomitant disruption of the Tβ4 C-terminal helix interaction. These structures, combined with biochemical assays and molecular dynamics simulations, show that the exchange of bound actin between Tβ4 and profilin involves both steric and allosteric components. The sensitivity of profilin to the conformational state of actin indicates a similar allosteric mechanism for the dissociation of profilin during filament elongation.
Monomeric actin (G-actin) is maintained at levels far above the critical concentration for polymerization (0.1 μM) (1) in specific areas of a mammalian cell. In lamellipodia, the levels of G-actin and filamentous actin (F-actin) are estimated to be 150 and 500 μM, respectively (2). Generally, actin filaments are thought to be capped to prevent nonproductive polymerization, whereas actively elongating filaments are created through controlled uncapping, severing, or de novo nucleation mechanisms (3) to harness the force of polymerization for a particular biological process. The availability of the pool of G-actin is tightly regulated through the binding of two classes of G-actin sequestering proteins (3, 4). β-Thymosins are short peptides (∼43 aa) that are able to completely sequester G-actin, preventing G-actin from forming a nucleus or joining either end of an existing actin filament. Profilins (molecular mass ∼ 16 kDa), in isolation, also sequester G-actin, precluding nucleation or participation in pointed-end filament elongation. In contrast, they actively participate in barbed-end filament elongation, dissociating from the actin protomer as it is incorporated into the filament. In the presence of actin nucleation and elongation machineries, such as actin-related protein 2/3 complex activators, formins, Wiskott–Aldrich syndrome protein (WASP) homology domain 2 (WH2)-containing nucleators [Spire, protein Cordon-bleu (Cobl), and VopF/L], and vasodilator-stimulated phosphoprotein (VASP) (5, 6), profilin plays an active role in the nucleation and elongation processes.
Cellular concentrations of thymosin-β4 (Tβ4) are in the 100- to 500-μM range, and its association with ATP- and ADP-bound actin is characterized by dissociation constant (Kd) values of 0.1–3.9 and 80–100 μM, respectively (7–11). Profilin I has been reported to be at a concentration of 8.4 μM in CHO cells (12) and can amount from 14% to 100% of the G-actin pool in other cells (13). Profilin I interacts with actin with Kd values of 0.1 and 0.17 μM for ATP- and ADP-bound actin, respectively, and is able to enhance the exchange of ADP for ATP (14). Taken together, these data lead to a model whereby the pool of ATP–G-actin is shared between Tβ4 and profilin. During phases of polymerization, the profilin–actin complex gives up its actin to the elongating filaments. The liberated profilin competes with Tβ4 for its bound ATP–G-actin, effectively restoring the pool of polymerization-competent actin. On filament dissociation, ADP-actin is preferentially sequestered by profiling because of the aforementioned difference in its affinity toward profilin and Tβ4. In the profilin:actin complex, nucleotide exchange is enhanced, and the resulting ATP-actin is subject to competition between profilin and Tβ4, resulting in the restoration of the pool of Tβ4-sequestered actin.
Since the discovery of the actin binding activity of Tβ4 in the early 1990s (15, 16), many efforts have been made to elucidate the atomic details of this interaction, including those by fluorescence (17, 18), NMR (19–23), small-angle X-ray scattering (23), X-ray crystallography [on either part of Tβ4 (24) or its protein homolog (25, 26)], and structure-based modeling (24, 27). Despite these efforts, a detailed picture of the interaction between actin and full-length Tβ4 and in particular, its N-terminal portion remains elusive, which may, in part, be attributed to the dynamic nature of this interaction.
Here, we report two structures of the Tβ4:actin complex, which were realized through the design of hybrid proteins. Comparison of these structures with those of the profilin:actin complex combined with biochemical assays, molecular dynamics (MD) simulations, and principal component analysis (PCA) shows that both steric and allosteric mechanisms are involved in the exchange of bound actin between Tβ4 and profilin. These two proteins have partially overlapping interaction surfaces on actin, and their coexistence in the ternary profilin:actin:Tβ4 complex results in restricted conformational flexibility of the N terminus of Tβ4 (entropy loss). In addition, they also exert allosteric effects onto each other by stabilizing different conformations of the actin monomer through regulating the width of its nucleotide binding cleft.
Results
Two Structures Show Different Conformations of Tβ4:Actin.
To determine the structure of the intrinsically disordered Tβ4 polypeptide in its ordered conformation bound to actin, we created a hybrid protein consisting of full-length human Tβ4 fused through a flexible linker to Pichia (Komagataella) pastoris actin (28). The structure of this chimera was determined to a resolution of 2.3 Å by X-ray crystallography (Fig. 1, Table 1, and Fig. S1A), with two copies of the chimeric protein in the asymmetric unit. The visible residues in the final electron density map include Val5 to Gly36 and Ser52 to Ser365 of actin and Asp5 to Ala40 of Tβ4. There was no interpretable electron density for the N and C termini and the D loop of actin, the linker between actin and Tβ4, or the first four (Ser1 to Pro4) and the last three residues (Gly41 to Ser43) of Tβ4. As had been predicted (27), Tβ4 makes extensive contact with actin, with an N-terminal α-helix (Asp5 to Lys11) and a C-terminal α-helix (Lys31 to Gln39) spanning the nucleotide binding cleft from the barbed to the pointed face of the monomer. This conformation is referred to hereafter as fully bound Tβ4. The occluded interface between Tβ4 and actin is 1,626.8 Å2 according to the Protein interfaces, surfaces and assemblies (PISA) service at the European Bioinformatics Institute (www.ebi.ac.uk/pdbe/prot_int/pistart.html) (29), of which the contribution from the residues after the LKKT motif (Glu21 to Ala40) is 945.1 Å2 and that from the C-terminal helix alone (Lys31 to Gln39) is 391.9 Å2. Comparison with the structure of unbound, unmodified G-actin [native G-actin; Protein Data Bank (PDB) ID code 3HBT] (30) reveals that the nucleotide binding cleft, which is bridged by the Tβ4 C-terminal α-helix, closes, narrowing the distance between subdomains 2 and 4 at the pointed face of actin. This closure is accompanied by a flattening of the monomer structure with respect to that of native G-actin, although to a far lesser extent than observed in the actin filament (31, 32) (Figs. 1D and 3C). These conformational changes on actin clearly indicate that the Tβ4 C-terminal α-helix forms an integral part of the Tβ4:actin interface and is critical in stabilizing actin in a more closed and flattened conformation. Consistent with this observation, previous experimental evidence has shown that (i) removal of the C-terminal portion of Tβ4 results in 4- to 20-fold reduction in its affinity for actin (9, 33), (ii) compared with full-length Tβ4, a 12-fold higher concentration of a truncated version of Tβ4 (Ser1 to Ser30) is needed to inhibit actin polymerization (34), and (iii) NMR analysis showed that the C-terminal α-helix of a chimera containing the N terminus of Ciboulot (Leu8 to Gly24) fused to the C terminus of Tβ4 (residues Phe12-Ser43) maintains stable interactions with actin under physiological conditions (23). Concomitant with the closure of the cleft at the pointed face of the Tβ4-bound actin, there is a slight widening of the groove at the barbed face between subdomains 1 and 3, possibly because of the relative rigidity of the large domain that comprises actin subdomains 3 and 4 (35).
Fig. 1.

Two structures of Tβ4:actin. (A) Domain diagrams of (Upper) the hybrid constructs of P. (K.) pastoris actin–Tβ4 and (Lower) the peptide consisting of Tβ4 and the lysine-rich region of Cobl. (B) Structure of the Pichia actin–Tβ4 hybrid, with Tβ4 capping both the barbed and pointed faces of actin. In both B and C, actin is shown as a surface covering the Cα trace, and Tβ4 is shown as a rainbow-colored cartoon with sticks. Subdomains 1–4 of actin are indicated by numbers. (C) Structure of the Tβ4–Cobl peptide in complex with rabbit skeletal muscle actin. The C-terminal helix of Tβ4 visible in B is disordered in this structure. The visible portion of Tβ4 is referred to as Tβ4N. Actin is in cyan. (D) Significant conformational changes in actin induced by the fully bound Tβ4. The N and C termini of Tβ4 are indicated. The distance between the Cα atoms of Gly63 on subdomain 2 and Pro243 on subdomain 4 of actin reduces from 20.9 (dashed line in E) to 14.5 Å on Tβ4 binding. Actin is colored blue, and the fully bound Tβ4 peptide is in red. The native G-actin (PDB ID code 3HBT) is shown in yellow for comparison. (E) Minor conformational changes on actin induced by Tβ4N. The distance mentioned in D changes from 20.9 to 20.4 Å on Tβ4N binding. Actin is colored cyan, and the Tβ4N peptide is in red. The native G-actin is shown as in D.
Table 1.
Data collection and refinement statistics
| Pichia actin–Tβ4 hybrid* | Tβ4N:muscle actin† | |
| Data collection | ||
| Space group | P 1 21 1 | P 43 21 2 |
| Cell dimension | ||
| a, b, c (Å) | 56.5, 139.5, 56.5 | 93.6, 93.6, 206.3 |
| α, β, γ (°) | 90, 116.2, 90 | 90, 90, 90 |
| Resolution | 20–2.3 (2.38–2.30)‡ | 20–2.0 (2.07–2.00)‡ |
| Rmerge (%) | 10.3 (32.8)‡ | 6.0 (59.5)‡ |
| I/σI | 10.3 (2.4)‡ | 24.8 (4.6)‡ |
| Completeness (%) | 84.2 (54.0)‡,§ | 100.0 (100.0)‡ |
| Redundancy | 3.5 (3.0)‡ | 12.0 (11.8)‡ |
| Refinement | ||
| Resolution (Å) | 19.8–2.3 (2.38–2.30)‡ | 20.0–2.0 (2.07–2.00)‡ |
| No. of reflections | 29,273 (1,870)‡ | 62,729 (6,157)‡ |
| Rwork/Rfree | 19.5/25.2 (26.4/32.4)‡ | 17.1/20.7 (20.1/26.1)‡ |
| No. of atoms | ||
| Protein | 5,976 | 5,703 |
| ATP, Ca2+ | 64 | 64 |
| Water | 227 | 531 |
| B factors | ||
| Protein | 34.1 | 31.3 |
| ATP, Ca2+ | 26.1 | 20.5 |
| Water | 33.0 | 38.4 |
| rmsds | ||
| Bonds (Å) | 0.013 | 0.008 |
| Angles (°) | 1.322 | 1.150 |
| Molprobity statistics | ||
| Clashscore, all atoms | 6.68 | 3.76 |
| Poor rotamers (%) | 0.8 | 0.0 |
| Ramachandran outliers (%) | 0.0 | 0.0 |
| Ramachandran favored (%) | 97.9 | 99.2 |
| MolProbity score | 1.40 | 1.16 |
| Cβ deviations > 0.25 Å (%) | 0.0 | 0.0 |
| Bad backbone bonds (%) | 0.0 | 0.0 |
| Bad backbone angles (%) | 0.0 | 0.0 |
PDB ID code 4PL7.
PDB ID code 4PL8.
Values in parentheses are for the highest resolution shell.
The completeness is 99.4% for the resolution range 20–3.0 Å.
Fig. 3.

PCA of actin from MD simulations. The analysis was performed on the Cα atoms from the subset of residues common to all computational and experimental structures (residues 7–38 and residues 51–370, respectively). (A) Collective motions of actin captured by PC1 and PC2. Motions are illustrated as linear interpolations between the extreme projections of the structures onto the PCs. Each cylinder, therefore, describes the path of each Cα atom between the extremes (on a red–white–blue color scale). (B) Conformational landscape of actin in PC space for the MD ensembles. 2D projections of MD trajectories along PC1 (horizontal axis) and PC2 (vertical axis) were converted into 2D histograms to represent the density of population of each conformational state of actin. Reference structures are 3HBT (native G-actin), 1HLU (profilin-bound actin with widely open nucleotide binding cleft), 2BTF (profilin-bound actin), and 4PL7 (actin with fully bound Tβ4 in the Pichia actin–Tβ4 structure reported in this paper). (C) 2D projections of selected experimental actin structures onto the plane defined by PC1 and PC2. The main functional clusters of structures are highlighted in different colors. The two actin molecules in the newly solved Tβ4N:actin structure reported in this paper have different conformations, which are labeled as 4PL8_A (with bound Tβ4N) and 4PL8_B (Fig. S2A).
In addition to the structure of the complex of Tβ4 with Pichia actin, we solved the structure of a hybrid peptide complexed with rabbit skeletal actin (Fig. 1 and Fig. S1B). The overall sequence identity between Pichia and rabbit skeletal actins is 83%, and 85% (47 of 55) of the residues in the Tβ4 binding interface are identical, with residue substitutions M16L, H28R, T29A, A167E, T199S, S201V, S203T, and G350S from Pichia actin to rabbit skeletal actin. The hybrid peptide comprises the N-terminal portion of human Tβ4 before the LKKT motif (Ser1-Lys16), the lysine-rich region (Phe1095-Asp1106) of Cobl, and full-length human Tβ4 (Ser1-Ser43). This hybrid, in complex with rabbit skeletal actin, was amenable to crystallization and led to an electron density map in which only regions from Tβ4 and actin were visible. There are two copies of actin in the asymmetric unit, forming an antiparallel contact with an interface area of 940.6 Å2 (Fig. S2A). Clear electron density for a polypeptide chain is visible on the surface of one actin molecule, which can be unambiguously assigned to residues Pro4 to Lys25 of Tβ4 (residues 34–55 of the hybrid) (Fig. S2C); for the second actin molecule, there is weak density near its barbed face (Fig. S2D) at a position equivalent to residues Met6 to Ile9 of Tβ4 on the first actin. This density is tentatively assigned to residues 8–11 of the hybrid construct, although we cannot rule out the density arising from a second chain of the hybrid peptide. Subsequent discussion focuses on the first actin molecule with its unambiguously assigned Tβ4 residues. Tβ4 in this structure lacks an ordered C-terminal α-helix (referred to as Tβ4N), and the pointed face of the actin monomer is in a conformation similar to that of native G-actin, which is characterized by subdomains 2 and 4 being relatively separated (Fig. 1 C and E). Otherwise, the ordered Tβ4 residues in contact with actin adopt very similar conformations as those observed in the structure of Tβ4 complexed with Pichia actin. Thus, the Tβ4 C-terminal α-helix acts as a sensor and stabilizer of the closed actin conformation of subdomains 2 and 4, and in the context of the crystal, in which a more open conformation of actin has been stabilized by the formation of antiparallel actin dimers, the Tβ4 C-terminal α-helix has dissociated.
Competition Between Tβ4 and Profilin Affects Actin Polymerization.
Tβ4 and profilin are known to compete with each other for binding actin (36, 37). Because Tβ4 and profilin act oppositely during actin barbed-end elongation (3), with Tβ4 blocking and profilin allowing their bound G-actin to join the barbed end of a filament, the competition between the two proteins creates an effective way of tuning the rate and extent of actin polymerization. To provide additional evidence at the level of single-actin filaments, we visualized, using total internal reflection fluorescence (TIRF) microscopy, the behaviors of Tβ4, profilin, and their combinations in filament elongation (Fig. 2A). Profilin:actin (0.7 and 0.35 μM, respectively) supported elongation of preexisting filaments in a similar manner to uncomplexed actin (0.35 μM), whereas Tβ4, at 3.5 μM (molar ratio 10:1 to actin), completely inhibited barbed-end elongation. Adding profilin to the Tβ4–actin mixture to 0.7 μM (two times the molar concentration of actin) showed no indication of antagonizing Tβ4 sequestration (Fig. S3A, Left), because under these conditions (0.35 μM actin, 0.7 μM profilin, and 3.5 μM Tβ4), the combined concentration of polymerization-competent species (uncomplexed and profilin-complexed actin) was still below 0.1 μM, the critical concentration for actin barbed-end polymerization (Fig. 5 shows the dissociation constants, and Table 3 shows the calculated concentrations). When the total actin concentration was increased to 1.0 μM, while maintaining the previous molar ratios for profilin (2.0 μM) and Tβ4 (10.0 μM), the competition between Tβ4 and profilin became apparent, and barbed-end elongation was observed. As a control, the binary mixture of Tβ4 and actin at these elevated concentrations remains inactive (Fig. S3A, Right).
Fig. 2.

Tβ4/profilin exchange of their bound actin. (A) Competition between profilin and Tβ4 visualized by TIRF microscopy. Preformed actin filaments stained with rhodamine phalloidin (magenta) were immobilized on the surface of the flow cell. Newly formed actin filaments were visualized by BODIPY FL-labeled actin (green; 30% labeling). Because of inefficient incorporation of rhodamine phalloidin at the barbed end of the preformed filaments, dual-color single filaments appear to be disconnected at the magenta–green junction. (B) Model of the profilin:actin:Tβ4 ternary complex. The Pichia actin–Tβ4 structure was superimposed onto the structure of profilin:actin (PDB ID code 2PBD). Actin and profilin from PDB ID code 2PBD are colored gray and green, respectively. The N-terminal helix of Tβ4 causes minimal steric clashes with profilin. (C) Binding of BODIPY-TMR–labeled full-length or N-terminally truncated Tβ4 peptides to (Left) actin or (Right) profilin:actin quantified by fluorescence anisotropy. Data points and error bars represent the averages and the SDs of triplicate measurements. The derived dissociation constants are summarized in Table 2. (D) Binding affinity of profilin to actin quantified by ITC.
Fig. 5.
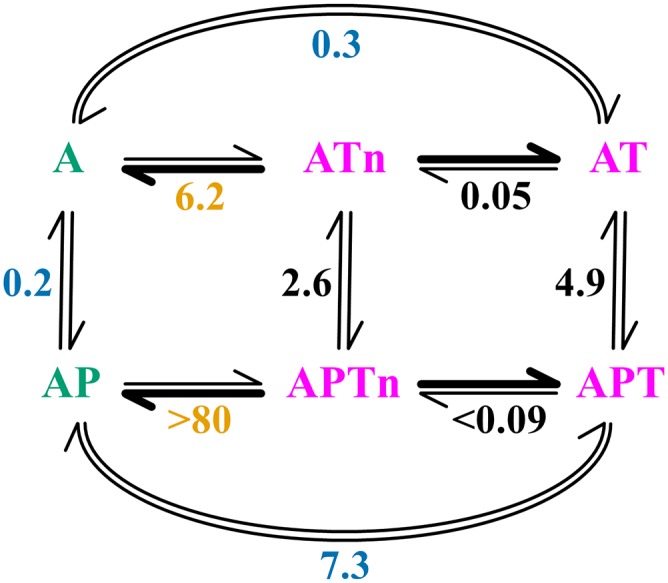
Reaction scheme of actin binding and exchange by Tβ4 and profilin. Species with significant and insignificant contributions to barbed-end elongation are colored green and magenta, respectively. Numbers represent thermodynamic equilibrium constants that are either measured directly [blue, constants in this paper; orange, constants reported by Yarmola and Bubb (9)] or calculated through energy balances (black). A, ATP–G-actin; P, profilin; T, Tβ4; Tn, Tβ4N.
Table 3.
Equilibrium concentrations of systems consisting of Tβ4, profilin, and ATP–G-actin
| Scenario | Total concentration (µM) | Equilibrium concentration (µM) | |||||||||
| At | Tt | Pt | A | AP | AT | APT | ATn | APTn | T | P | |
| 1 | 0.35 | 3.5 | — | 0.03 | — | 0.31 | — | 0.01 | — | 3.18 | — |
| 2 | 0.35 | 3.5 | 0.7 | 0.02 | 0.06 | 0.22 | 0.03 | 0.01 | 0.00 | 3.23 | 0.61 |
| 3 | 1.0 | 10 | — | 0.03 | — | 0.92 | — | 0.04 | — | 9.03 | — |
| 4 | 1.0 | 10 | 2.0 | 0.02 | 0.16 | 0.58 | 0.20 | 0.03 | 0.02 | 9.17 | 1.63 |
| 5 | 90 | 90 | 35 | 1.3 | 11 | 53 | 20 | 2.6 | 1.8 | 13 | 1.8 |
| 6 | 90 | 90 | 35 | 0.7 | 27 | 60 | — | 2.9 | — | 27 | 8.2 |
A, ATP–G-actin; P, profilin; t, total concentration; T, Tβ4; Tn, Tβ4N.
Tβ4:Actin Structures Support the Formation of Profilin:Actin:Tβ4 Complexes.
The competition between Tβ4 and profilin has been proposed to proceed through the formation of a ternary complex of profilin, Tβ4, and actin (38). To gain additional structural insight into such ternary complex formation, the newly solved Tβ4:actin structures were superimposed onto a structure of profilin:actin (PDB ID code 2PBD) (39) to produce models of the profilin:actin:Tβ4 tricomplexes (Fig. 2B and Fig. S3). In these models, the N-terminal helices of Tβ4 (Asp5-Lys11 for the fully bound and Pro4-Lys11 for the Tβ4N) fit into the pocket between profilin and actin, with only the side chain of Pro4 and to an even lesser extent, Asp5 touching the surface of profilin. Thus, the Tβ4:actin structures seem to be compatible with ternary complex formation.
Another prominent feature of the overlaid structures is the conformational difference between the profilin- and Tβ4-bound actins, analogous to the observed difference between the native and Tβ4-bound actins (Fig. S3 C and D). Specifically, binding of the C-terminal helix of Tβ4 reduces the distance between subdomains 2 and 4 and increases the distance between subdomains 1 and 3 of actin. In contrast, profilin binding does not produce a closure of the cleft between subdomains 2 and 4 of actin but slightly reduces the distance between subdomains 1 and 3 (39). This conformational difference in actin is in line with the hypothesis that profilin and Tβ4 mutually influence their interactions with actin through allosteric effects (40). This allosteric mechanism was further explored by fluorescence anisotropy and MD simulations.
Fluorescence Anisotropy Reveals Steric and Allosteric Components of Tβ4/Profilin Exchange.
The newly solved Tβ4–actin structures and their apparent compatibility with profilin binding necessitated a systematic investigation of the contributions of the distal N terminus of Tβ4 and the allosteric mechanism in the exchange of bound actin by Tβ4 and profilin. Fluorescence anisotropy measurements were used with a series of N-terminally truncated Tβ4 peptides. Table 2 summarizes the affinities of the full-length and truncated versions of Tβ4 for actin and the profilin:actin complex. The dissociation constant (Kd) of full-length Tβ4 for actin is in good agreement with reported values (23, 33, 38). In support of the structures, removal of the first three residues, Ser1-Asp2-Lys3, of Tβ4 does not significantly affect the affinity for actin. Truncation beyond Pro4 reduces the binding strength of Tβ4, which may be attributed to disruption of the relatively short N-terminal helix of Tβ4. Exchange of the bound actin between Tβ4 and profilin was measured by the decrease in the fluorescence anisotropy of labeled Tβ4 peptides with increasing concentrations of profilin (Fig. 2C). These data fit well to the model where the exchange proceeds through the ternary complex of profilin:actin:Tβ4 (38) using a single Kd of 0.2 µM for profilin:actin determined by isothermal titration calorimetry (ITC) (Fig. 2D). For both the full-length Tβ4 and the peptide truncated to Pro4, there is an ∼25-fold reduction in their affinity for profilin:actin with respect to actin alone (Table 2), which is comparable with the 10- to 14-fold reductions reported elsewhere (38). The ternary models (Fig. 2B and Fig. S3) predict that truncating the N terminus of Tβ4 to, possibly, Asp5 or more confidently, Met6 would eliminate direct conflicts between Tβ4 and profilin. The difference in affinity of these peptides for profilin:actin narrows compared with actin but is nevertheless apparent. Hence, we conclude that the flexible N-terminal Ser1-Lys3 and the more rigid Pro4 and Asp5 of Tβ4 contribute favorably to the exchange process. There is also an allosteric component clearly shown by the Met6 truncate of Tβ4, which can dissociate profilin in the absence of direct clashes between profilin and Tβ4 in their actin-bound forms. In this situation, exchange proceeds through a pure allosteric mechanism, in which the sequestering proteins stabilize different conformations of actin that are suboptimal for the other sequestering protein.
Table 2.
Dissociation constants of full-length Tβ4 and truncates to actin and profilin:actin
| Tβ4 peptide | Tβ4:actin KdT (µM) | Profilin:actin:Tβ4 KdPT (µM) | Fold reduction KdPT/KdT |
| Full length | 0.32 ± 0.02 (0.27–0.37) | 7.3 ± 1.2 (4.5–10.1) | 23 |
| Pro4 truncate | 0.17 ± 0.02 (0.13–0.21) | 5.0 ± 0.8 (3.2–6.8) | 28 |
| Asp5 truncate | 2.4 ± 0.2 (2.0–2.9) | 13.0 ± 0.9 (10.9–15.1) | 5.4 |
| Met6 truncate | 2.3 ± 0.2 (1.7–2.9) | 5.8 ± 0.4 (4.9–6.7) | 2.5 |
Kd values are expressed as mean values ± SDs, and values in parentheses indicate 95% confidence intervals obtained by fitting the data in Fig. 2C using GraphPad Prism, version 4.00 (GraphPad Software; www.graphpad.com).
MD Simulations Disclose Conformational Landscapes of Tβ4/Profilin Exchange.
To better understand how profilin and Tβ4 modulate the structure of actin, ∼1.0-µs-long MD simulations were carried out on actin, Tβ4:actin, profilin:actin, and profilin:actin:Tβ4 starting from the ATP-bound actin conformation taken from the fully bound Tβ4:actin structure (Table S1). Independent trajectories were generated and analyzed using PCA to extract correlated motions of actin from the matrix of atomic fluctuations. Representative experimental actin structures were included in the PCA to provide quantitative comparison between simulations and experiment (Fig. 3). The first few eigenvectors or principal components (PCs) obtained through this analysis represent the main large-scale functional motions of actin (Fig. S4). The first PC (PC1) corresponds to twisting/flattening of the molecule, whereas the second PC (PC2) describes opening/closing of the nucleotide binding cleft (Fig. 3A).
Fig. 3B shows 2D projections of the first two PCs of actin in different molecular environments, which were captured by MD-generated structural ensembles. Actin alone quickly deviated from its starting structure (Tβ4:actin conformation) to sample a relatively broad energy basin characterized by larger values of PC1 and lower values of PC2, indicating a closed and twisted conformation. This region of the conformational landscape largely corresponds to the one occupied by most experimental unbound G-actin structures (Fig. 3C).
Inclusion of profiling stabilized actin in its more open conformations and increased its flexibility (as judged by the rms fluctuation on Cα positions) (Fig. S5C). The conformational landscape of profilin-bound actin reveals the presence of multiple energy basins corresponding to the widely open (1HLU-like), slightly open (2BTF-like), and G-actin–like conformers, which is in striking contrast with G-actin and consistent with previous reports (41). Interestingly, a plot of the number of atomic contacts between actin and profilin vs. PC2 shows a significant correlation between the strength of the interaction and the opening of the actin nucleotide binding cleft by profilin (Fig. S5E).
Inclusion of Tβ4 restricted the actin conformations to those with narrower distances between subdomains 2 and 4, leading to a landscape with a deep, single-energy minimum. The location of this global minimum is very close to the crystal structure and corresponds to an actin conformer locked in a closed, less twisted state than G-actin. Comparison with experimental structures in which the pointed face is not capped, such as the Tβ4N with skeletal actin (Fig. 3C), indicates that binding of the C-terminal helix from Tβ4 induces a small but significant closure and flattening of actin through direct intermolecular contacts.
Models of the ternary complex were also subjected to MD. These models were stable during the time course of the simulation and showed a reduced overall flexibility of the actin molecule compared with profilin:actin or G-actin but somewhat higher than Tβ4:actin (Fig. S5C). The N terminus of Tβ4 displays a more restricted movement in the ternary complex compared with Tβ4:actin because of the positioning of profilin (Fig. S5 A and B), suggesting an unfavorable entropy loss of the N terminus of Tβ4 in forming the complex. PCA indicates that simultaneous binding of Tβ4 and profilin onto actin results in a relative broadening of the conformational landscape without significantly altering the location of the energy minimum of the actin–Tβ4 complex. Comparison with profilin:actin and Tβ4:actin clearly suggests that profilin and Tβ4 simultaneously exert competing effects on the dynamics of actin and that Tβ4 is able to inhibit profilin-induced cleft opening by maintaining tight contacts with actin through its C-terminal α-helix. This effect is evident in the plot of the number of atomic contacts between actin and profilin (in the ternary complex) vs. PC2, where MD simulated structures cluster in a region of space characterized by low values for PC2 (Fig. S5F). Comparison between the distribution of profilin–actin contacts in profilin:actin and profilin:actin:Tβ4 shows a shift of the main population of structures toward less atomic contacts in the ternary complex, which indicates a slight destabilization of profilin:actin by Tβ4 (Fig. S5 D and F), consistent with experimental affinity measurements (Table 2).
Discussion
Importance of the C-Terminal Helix of Tβ4.
The Tβ4:actin structures described here reveal that the presence and absence of the bound Tβ4 C-terminal helix correlate with significant conformational changes of actin. To further understand these changes, we compared the fully bound Tβ4:actin structure with the structure of actin in complex with a chimera consisting of gelsolin domain 1 (G1) fused to the C-terminal portion of Tβ4 (G1-Tβ4; PDB ID code 1T44) (24). Despite differences in species and isoform, the conformation of cytoplasmic Pichia actin in the Tβ4:actin complex resembles that of rabbit muscle actin complexed with G1-Tβ4 (Fig. S6A), with a closed pointed face cleft. In contrast, G1 alone [in the absence of the Tβ4 C-terminal α-helix; examples are 1P8Z (42), 2FF3 (26), and 1YAG (43)] slightly widens the barbed face groove of bound actin (as seen in 1T44) but fails to induce the closure of the pointed face cleft (Fig. S6B). In good agreement, the PCA shows that, although the structures of G1:actin cluster with the Tβ4N:actin as well as some uncomplexed actins, the fully bound Tβ4:actin and G1-Tβ4:actin structures cluster together in a distinct region (Fig. 3C). Thus, the Tβ4 C-terminal helix is a sensor and stabilizer of the closed pointed face cleft conformation of actin.
Mechanism of G-Actin Sequestration.
To understand the mechanism by which Tβ4 sequesters G-actin, the two structures of Tβ4:actin were overlaid onto the F-actin structure (PDB ID code 3MFP) (32). These structures were analyzed by PISA to locate dual-role surface residues on actin that are involved in both binding Tβ4 in the Tβ4:actin complex and contacting adjacent protomers in the filament. The fully bound C-terminal α-helix of Tβ4 and the first two residues of its N-terminal α-helix sterically prevent the complex from joining the barbed and pointed ends of a filament (Fig. 4), respectively, and there are, indeed, many dual-role residues at both the barbed and pointed faces of actin.
Fig. 4.

Mechanism of G-actin sequestration by Tβ4. The Pichia actin–Tβ4 structure in Fig. 1 was overlaid onto an F-actin model with five protomers generated with PDB ID code 3MFP. (A) Front and side views of superimposed models. Tβ4 residues causing steric hindrance are colored cyan in both A and B. (B) Magnified views at the protomer interface of the filament. Dual-role surface residues on the barbed and pointed face of actin that are involved in both Tβ4 binding and contacting adjacent protomers in the filament are colored green and yellow, respectively.
In addition to the steric exclusion and large competitive binding surface, there also seem to be entropic and allosteric effects that aid Tβ4 sequestration of G-actin. The entropic effect originates from the flexible, distal N terminus of Tβ4, which would become more restricted in movement when the Tβ4:actin complex approaches the pointed end of a filament, analogous to the observations in the MD simulation of the profilin:actin:Tβ4 tricomplex. Evidence for the allosteric effect includes (i) although not as effective as full-length Tβ4, truncated Tβ4s (Ser1-Glu24 or Ser1-Ser30) that lack the C-terminal α-helix reduce the amount of F-actin during salt-induced polymerization (34); (ii) in the absence of a possible steric effect and competitive binding site, profilin and many barbed-end elongating WH2s (see below) reduce the barbed-end association rate of actin (23, 44); and (iii) Tβ4-sequestered actin is in a conformation that is different from the conformations of uncomplexed G- and F-actin (Fig. 3C).
Tβ4 and WH2 Motifs.
To compare the modes of actin interaction between Tβ4 and the WH2 motifs, the available WH2:actin structures were superimposed on the Tβ4:actin structures, with the WH2 from WASP shown as an example (Fig. S6C). The major observation is that none of the WH2s have a helix that makes extensive interactions with the pointed face of actin, and indeed, many WH2s do not even show a stable interaction with actin beyond the central LKKT motif. The Tβ4 C-terminal α-helix is, therefore, the likely major determinant of actin monomer sequestration. Despite being more similar at the barbed face of actin, Tβ4 is distinguished from the WH2s by its short N-terminal α-helix, which is engendered by a helix-breaking proline at position 4, conferring flexibility onto the preceding three residues (Fig. S5A). As discussed earlier, the shorter N-terminal α-helix and the flexible leading residues are functionally significant in that they are compatible with the formation of the profilin:actin:Tβ4 complex.
Many actin polymerization and nucleation factors contain WH2 motifs that are preceded by proline-rich sequences (PRSs), which recruit profilin:actin (3, 5). The currently available crystal structures of WH2s suggest that their comparatively longer N-terminal helices are not compatible with the formation of a relatively stable ternary complex as observed for profilin:actin:Tβ4 (39). Hence, the mechanism by which adjacent PRS and WH2 motifs contribute to polymerization remains open to question. The process may proceed through either (i) an unstable ternary complex, in which profilin and WH2 are bound to the same actin, or alternatively, (ii) a noncanonical WH2:actin interaction in the ternary complex that allows the WH2 to gain access to the actin in the PRS-bound profilin:actin, which was manifested by the WH2-like G-actin–binding (GAB) motif of VASP (39).
Functionally, Tβ4/WH2 motifs have been broadly classified into Tβ4-like peptides that disallow their bound actin to join either end of a filament or barbed-end elongators that participate in barbed- but not pointed-end elongation, albeit at a reduced rate compared with uncomplexed G-actin (23). The barbed-end elongating WH2s are, thus, weak sequesterers for barbed-end elongation, which correlates well with the absence of a C-terminal α-helix in their actin-bound structures. Given the resemblance between the Tβ4N:actin and the WH2:actin structures (Fig. 3C, purple cluster), it seems plausible that Tβ4 may also become a barbed-end elongator on dissociation of its C-terminal α-helix. Indeed, it has been shown that mutations in the C-terminal portion or the linker region of Tβ4 can increase the dynamics of the C-terminal portion of Tβ4 and effectively switch Tβ4 into a barbed-end elongator (23, 25).
Scheme for Actin Binding and Exchange.
The two modes of Tβ4 in complex with actin (fully bound vs. Tβ4N) and the concomitant conformational changes on bound actin suggest that Tβ4 binds to actin through a two-step process: initially, the N-terminal portion interacts followed by the C-terminal portion. This hypothesis is supported in two ways: (i) the isolated N-terminal portion of Tβ4 can bind actin, whereas the isolated C-terminal portion cannot (9, 33), and (ii) for many actin-bound WH2-Tβ4 hybrid peptides, their C-terminal portion is more dynamic than their N-terminal portion (23).
Based on this hypothesis and the Kd values measured here and by others (9), we constructed a reaction scheme for the actin exchange between profilin and Tβ4 (Fig. 5). By comparing the apparent Kd values for the fully bound vs. the Kd values for the Tβ4N (measured with Ser1-Lys25 of Tβ4) (9), the scheme reveals that the C-terminal helix of Tβ4 is critical in stabilizing the association between Tβ4 and actin in both Tβ4:actin and profilin:actin:Tβ4, leading to 10- to 20-fold increase in the binding affinity. The three Kd values (0.2, 2.6, and 4.9 μM) for profilin binding to uncomplexed actin, Tβ4N:actin, and fully bound Tβ4:actin, respectively, show both the steric and the allosteric effects for the Tβ4/profilin exchange.
Using this scheme, we calculated the equilibrium concentrations of all of the species in a system containing Tβ4, profilin, and ATP–G-actin (Table 3). The first four scenarios in Table 3 correspond to the conditions used for the TIRF experiments (Fig. 2A). The concentrations for the polymerization-competent species (A and AP) are below 0.1 μM when the total actin concentration is low (scenarios 1 and 2) or when profilin is absent (scenarios 1 and 3). However, at suitable concentrations of the individual components, profilin can effectively turn the Tβ4-sequestered inactive system into a polymerization-competent system (scenarios 4 vs. 3), which is in good agreement with the TIRF experiments.
Consequences for Filament Elongation.
To assess the in vivo concentrations of all of the species depicted in Fig. 5 and their contributions to actin polymerization, we calculated, in scenario 5 of Table 3, their equilibrium concentrations using the estimated in vivo concentrations for profilin, Tβ4, and ATP–G-actin (45). The benefit of ternary complex formation is illustrated by comparing scenario 5 with scenario 6, in which ternary complex formation was purposely ignored. Clearly, the ternary system is more efficient in lowering the combined concentration of polymerization-competent species (sum of A and AP). Because G-actin is not required to dissociate during Tβ4/profilin exchange, spontaneous nucleation is minimized. Moreover, the ternary complex reduces the concentrations of free profilin, profilin:actin complex, and hence, filament barbed ends associated with profilin, all of which will impact on the actin dynamics regulations in vivo.
Based on this reaction scheme, a small fraction of the Tβ4:actin and profilin:actin:Tβ4 complexes may temporarily lose the Tβ4 C-terminal α-helix interaction with the actin protomer because of the allosteric action of actin (ATn and APTn in Fig. 5). Without the steric hindrance provided by the C-terminal α-helix of Tβ4, the actin in these complexes would be sequestered only by the allosteric mechanism discussed earlier. This loss of steric hindrance seems to impair the capability of Tβ4 as a G-actin sequesterer, turning it into a barbed-end elongator (see above). However, added security is present to counteract such an effect in vivo, in which actin filament elongation is often mediated by polymerization machineries, such as VASP (46) and formins (47). These proteins recruit profilin:actin complexes through their PRS, making Tβ4N:actin (without profilin) a less favorable candidate. Profilin:actin:Tβ4N (APTn in Fig. 5 and Table 3) accounts for only about 14% of the polymerization-competent pool of actin (A and AP). Moreover, this complex does not seem to be compatible with the VASP-mediated elongation process, because Tβ4N would mask the binding site of the GAB motif of VASP (39). Lastly, we speculate that the allosteric sequestration of G-actin by Tβ4N, although quite possibly being attenuated, would not be completely eliminated by the presence of profilin, and consequently, the profilin:actin:Tβ4N complex, like many barbed-end elongating WH2:actin complexes, would not be as competent as profilin:actin in actin polymerization. Taken together, we suggest that, for both Tβ4N:actin and profilin:actin:Tβ4N, their contributions to in vivo actin polymerization will be insignificant.
The facts that Tβ4 and profilin are sensitive to and can alter the conformation of their bound actin suggest that similar allosteric mechanisms will be invoked during filament elongation. G-actin bound to profilin or barbed-end elongating WH2s is able to join the barbed end of a filament. After it is bound, the actin–actin interactions would induce the G- to F-actin structural transition in the newly incorporated protomer away from the optimal conformations that favor binding of profilin or WH2s, leading to the release of bound profilin or WH2 and an uncapped barbed end ready for the incorporation of the next actin protomer. Indeed, there have been continued efforts in the literature aimed at elucidating the interplay between profilin, G-actin, and the barbed end of F-actin (44, 48–52). A recent report detailing kinetic rates at the single-filament level (53) strongly suggests that the conformations of protomers at the barbed end of F-actin differ from those of G-actin. Thus, allosteric effects reduce, in a nucleotide-dependent manner, the affinity of profilin for the barbed end of F-actin relative to G-actin. Additional structural data are needed to elucidate the conformations of the barbed end of F-actin at high resolution to understand these allosteric mechanisms.
Materials and Methods
Proteins and Peptides.
The coding sequence of the P. (K.) pastoris actin (Uniprot accession no. Q9P4D1) was fused to a linker sequence (28) followed by human Tβ4 (Uniprot accession no. P62328). This hybrid was then cloned into a modified pPICZc vector (Invitrogen) that encodes to the C terminus of the insert a human rhinovirus 3C (HRV 3C) protease recognition sequence, a StrepII tag, a tobacco etch virus (TEV) protease recognition sequence, and eight histidines. Protein production was achieved with the X-33 P. pastoris strain (Invitrogen) according to the supplier’s protocols. Cells were lysed with the YeastBuster (Novagen), and the clarified lysate was loaded onto a 5 mL HisTrapFF column attached to an AKTAxpress system (GE Healthcare). After washing the column with 2.0 mM Tris, 0.2 mM ATP, 0.2 mM CaCl2, and 0.2 mM DTT, pH 7.6 (Buffer A), His-tagged HRV 3C protease was injected to release the bound hybrid protein from the column. The hybrid protein was further purified by gel filtration on a Superdex200 column (GE Healthcare) equilibrated with Buffer A and concentrated to 15 mg/mL using a Vivaspin 20 concentrator (Sartorius) with a 30-kDa molecular mass cutoff. Rabbit skeletal muscle actin was purified according to previously published protocols (54) followed by gel filtration. Human profilin I was cloned into the pSY5 vector that features an N-terminal His-tag and an HRV 3C protease recognition sequence (55) and purified by a two-step protocol using HisTrapFF and Superdex75 columns.
Full-length and N-terminal truncated versions of Tβ4, with an extra cysteine introduced at their C terminus, were cloned into the pNIC28-Bsa4 vector (GenBank accession no. EF198106) by ligation-independent cloning (56). The pNIC28-Bsa4 vector features an N-terminal His-tag and a linker that can be completely removed by TEV. An extra glycine is added to both the Ser1 (full-length) and Pro4 to mimic the acetylation in vivo for the former and facilitate TEV protease cleavage for the latter. These constructs were transformed into BL21(DE3) cells (Invitrogen), and protein production was induced using the autoinduction media (57). Cells were lysed by sonication, and the recombinant peptides were purified by a two-step protocol with AKTAxpress using HisTrapFF and Superdex30 columns. In some cases, the His-tag on the peptides was cleaved off by incubating the peptides in solution at 4 °C with His-tagged TEV protease and passing the reaction mixture through the HisTrapFF column again to capture the tag and the TEV protease. The peptides were concentrated to 10 mg/mL using a Vivaspin 15R concentrator with a 2-kDa molecular mass cutoff.
In an effort to characterize the lysine-rich region of Cobl in actin binding, hybrid proteins comprising this lysine-rich region and different WH2/Tβ4 motifs were constructed. One such hybrid protein was made by joining residues S1-K16 of Tβ4, F1095-D1106 of Cobl, and full-length Tβ4 (S1-S43). This hybrid was cloned into the pSY5 vector, produced, and purified as the Tβ4 peptides. The amino acid sequence after HRV 3C protease cleavage is GPSDKPDMAEIEKFDKSKFKPVVQRPVPKDSDKPDMAEIEKFDKSKLKKTETQEKNPLPSKETIEQEKQAGES, with the residues from Tβ4 underlined. The first two extra residues are from the recognition sequence of HRV 3C.
Crystallization, Data Collection, and Structure Solution.
Crystallization of Tβ4:actin was performed using the sitting-drop vapor diffusion method in 1.0-μL drops containing a 1:1 (vol/vol) mixture of protein solution and precipitant. Crystals for the K. pastoris actin–Tβ4 hybrid were obtained by mixing the protein stock at 15 mg/mL in Buffer A with 0.1 M sodium acetate (pH 4.5) and 15% (wt/vol) PEG 4000 at 25 °C. The Tβ4–Cobl hybrid was first incubated at 4 °C in a molar ratio of 1.2:1 with rabbit skeletal muscle actin in Buffer A, concentrated to 10 mg/mL, and mixed with 0.1 M citric acid (pH 3.5) and 15% (wt/vol) PEG 3350 at 15 °C. The crystals were transferred to the precipitant solution supplemented with 20% (vol/vol) glycerol before flash freezing in liquid nitrogen. X-ray diffraction data were collected at beamline BL13B1 on an Area Detection System Corporation Quantum-315 CCD detector at the National Synchrotron Research Center. Data were indexed, scaled, and merged in HKL-2000 (58). Structures were solved by molecular replacement using Saccharomyces cerevisiae actin (PDB ID code 1YAG) (43) or rabbit skeletal muscle actin (PDB ID code 1T44) (24) as models in PHASER (59). The solution was subjected to repetitive rounds of restrained refinement in PHENIX (60) and manual building in COOT (61). TLS parameters generated by the TLSMD web server (62) were included in the final round of refinement. The CCP4 program suite (63) was used for coordinate manipulations. The structures were validated with Molprobity (64). Backbone dihedral angles of the final models that fall within the outliers and the allowed and favored regions of the Ramachandran plots are 0%, 1.7%, and 98.3% for the P. pastoris actin–Tβ4 hybrid structure and 0%, 1.0%, and 99.0% for the Tβ4N:muscle actin structure, respectively. All of the structure-related figures were prepared with the PyMOL Molecular Graphics System (DeLano Scientific LLC).
TIRF Microscopy.
Preparation of sample flow cells was based on published protocols with minor modifications (65). Briefly, glass slides (75 × 25 mm, 1.0 mm thick) and glass coverslips (40 × 24 mm, no. 1, 0.13 to 0.17 mm thick) from Fisher Scientific were sonicated 30 min each in 2% (vol/vol) Hellmanex II solution (Hellma GmbH & Co.), 1.0 M NaOH, and absolute ethanol, with extensive rinses in between with deionized water. After drying with compressed air, the cleaned coverslips were derivatized by coating one side with 80 μL 2 mg/mL mPEG-silane (molecular weight = 2,000; Nanocs Inc.) and 2 μg/mL biotin-PEG-silane (molecular weight = 3,400; Nanocs Inc.) in 80% (vol/vol) ethanol (pH adjusted to 2.0 by HCl) and baked at 60 °C for 16 h. The derivatized coverslips were then rinsed and dried again. Flow cells were assembled by placing three parallel strips of double-sided tape (30 mm × 6 mm × 120 μm) onto a cleaned and dried glass slide with ∼3-mm spacing between the strips. A derivatized coverslip was then positioned over the strips of double-sided tape (with the coated side facing the tape), producing two separate flow chambers per slide.
TIRF images were obtained using a Nikon TE2000-E inverted microscope following published procedures (65, 66). Briefly, the flow cell was first incubated with 1% BSA in Tris-buffered saline (50 mM Tris, 150 mM NaCl, pH 7.6) followed by 100 μg/mL streptavidin in Tris-buffered saline and two washes with 1× TIRF buffer (10 mM imidazole, pH 7.4, 50 mM KCl, 1 mM MgCl2, 1 mM EGTA, 0.2 mM ATP, 10 mM DTT, 15 mM glucose, 20 μg/mL catalase, 100 μg/mL glucose oxidase). Unlabeled rabbit skeletal actin with 0.2% biotinylation was polymerized in the presence of 40 μM rhodamine phalloidin (Molecular Probes) by the addition of equal volume 2× TIRF buffer, and the mixture was introduced into the flow cell. After ∼7 min, the flow cell was washed two times with 1× TIRF buffer so that only the filaments attached to the surface of the coverslip remained. BODIPY FL maleimide (Molecular Probes) -labeled actin (30% fluorophore labeling and no biotinylation in 1× TIRF buffer) was then introduced into the flow cell in the absence or presence of Tβ4 and/or profilin.
Fluorescence Anisotropy Measurements.
His-tagged Tβ4 peptides were exchanged into 20 mM phosphate (pH 7.0) with PD-10 desalting columns (GE Healthcare) and labeled with BODIPY TMR C5-maleimide (Invitrogen) according to the supplier’s protocol. Nickel-Sepharose High Performance Beads (GE Healthcare) were added to the reaction mixture to capture the peptides and facilitate removal of unattached dye by repeated washes. The labeled peptides were eluted with the phosphate buffer containing 250 mM imidazole, and the imidazole concentration was lowered by dilution. Removal of His-tag by TEV protease was conducted as mentioned previously for the unlabeled Tβ4 peptides. The final concentrations of the peptides were quantified by measuring their absorbance at 544 nm and a molar extinction coefficient of 60,000 cm−1 M−1.
Fluorescence anisotropy measurements were conducted using a Tecan Safire II plate reader with excitation and emission wavelengths set at 530 and 570 nm, respectively, and black 96-well nonbinding flat-bottom plates (Greiner) with a total volume of 100 µL in the wells. Buffer A was used throughout the experiments. The peptides were fixed at 0.2 µM. For the profilin competition assays, the actin was at 2.4 µM. Fluorescence anisotropy data for Tβ4 binding to actin were fitted with Eq. 1, and those for the exchange of bound actin between Tβ4 and profilin were fitted with Eqs. 2 and 3 that were originally derived for Tβ4:actin:latrunculin A (67):
| [1] |
| [2] |
| [3] |
where r is the measured fluorescence anisotropy; rf and rb are fluorescence anisotropy values for free Tβ4 and Tβ4 in complex with either actin or profilin:actin, respectively; [A0], [T0], and [P0] are initial concentrations of actin, Tβ4, and profilin, respectively; [A] is free actin concentration; KdT and KdP are dissociation constants for Tβ4:actin and profilin:actin complexes, respectively; and KdPT is the dissociation constant of Tβ4 from the ternary complex.
ITC.
ITC measurements were performed with a MicroCal Auto-iTC200 System (GE Healthcare) in a buffer containing 5 mM Pipes, 0.2 mM ATP, and 0.2 mM CaCl2 (pH 7.5). Profilin at 130 µM was titrated in 10-µL injections into 200 µL 10 µM actin at 25 °C. The duration of each injection was 4 s, with an interval of 2 min between injections. Data analysis was carried out with ORIGIN software, giving a KdP of 0.23 µM.
MD Simulations.
All MD simulations were performed using the GROMACS 4 software package (68) and the AMBER99SB-ILDN* force field (69, 70). At the beginning of each simulation, the protein was immersed in a box of extended simple point charge water. A minimum distance of 1.0 nm was applied between any protein atom and the edges of the box. Sodium ions were added to reach charge neutrality. Long-range electrostatics was treated with the particle mesh Ewald summation (71). Bond lengths were constrained using the P-LINCS algorithm (72). The integration time step was 2 fs. The v-rescale thermostat (73) and the Parrinello–Rahman barostat (74) were used to maintain a temperature of 300 K and a pressure of 1 atm. Each system was energy-minimized using 5,000 steps of steepest descent and equilibrated for 200 ps with restrained protein heavy atoms before the beginning of the production simulation. For each system, three independent production simulations were obtained by using different initial velocities. The aggregated simulation time was ∼4 µs. Calculations of rmsds, root mean square fluctuations, atomic distances, and PCA were carried out using GROMACS routines.
In Silico Calculations.
The procedure for in silico calculations was similar to that reported (45). Briefly, kinetic models based on the reaction scheme (Fig. 5) were constructed in Vcell (75). Reaction rates constants were arbitrarily set to 1.0 for forward reactions and the measured Kd values for reverse reactions. The initial concentrations were set according to the experimental conditions used for TIRF or the estimated in vivo values (45).
Supplementary Material
Acknowledgments
B.X. thanks Drs. Joma K. Joy and Jeffrey Hill from the Experimental Therapeutics Centre of the Agency for Science, Technology and Research (A*STAR) for their help in isothermal titration calorimetry measurements. B.X. and R.C.R. thank A*STAR for support. Portions of this research were carried out at the National Synchrotron Radiation Research Center. The Synchrotron Radiation Protein Crystallographic Facility is supported by the National Research Program for Genomic Medicine. The work presented here made use of the high-performance computing facility provided by the Engineering and Physical Sciences Research Council-funded Centre for Innovation through Grants EP/K000144/1 and EP/K000136/1. The Centre for Innovation is owned and operated by the e-Infrastructure South Consortium formed by the University of Bristol, the University of Oxford, the University of Southampton, and the University College London in partnership with the Science and Technology Facilities Council’s Rutherford Appleton Laboratory. Wellcome Trust Grant 075491/Z/04 is acknowledged for administrative support for C.L. and J.M.G.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
Data deposition: The crystallography, atomic coordinates, and structure factors have been deposited in the Protein Data Bank, www.pdb.org (PDB ID codes 4PL7 and 4PL8).
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1412271111/-/DCSupplemental.
References
- 1.Pollard TD. Rate constants for the reactions of ATP- and ADP-actin with the ends of actin filaments. J Cell Biol. 1986;103(6 Pt 2):2747–2754. doi: 10.1083/jcb.103.6.2747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Koestler SA, et al. F- and G-actin concentrations in lamellipodia of moving cells. PLoS ONE. 2009;4(3):e4810. doi: 10.1371/journal.pone.0004810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pollard TD, Borisy GG. Cellular motility driven by assembly and disassembly of actin filaments. Cell. 2003;112(4):453–465. doi: 10.1016/s0092-8674(03)00120-x. [DOI] [PubMed] [Google Scholar]
- 4.dos Remedios CG, et al. Actin binding proteins: Regulation of cytoskeletal microfilaments. Physiol Rev. 2003;83(2):433–473. doi: 10.1152/physrev.00026.2002. [DOI] [PubMed] [Google Scholar]
- 5.Campellone KG, Welch MD. A nucleator arms race: Cellular control of actin assembly. Nat Rev Mol Cell Biol. 2010;11(4):237–251. doi: 10.1038/nrm2867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dominguez R. Actin filament nucleation and elongation factors—structure-function relationships. Crit Rev Biochem Mol Biol. 2009;44(6):351–366. doi: 10.3109/10409230903277340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kang F, Purich DL, Southwick FS. Profilin promotes barbed-end actin filament assembly without lowering the critical concentration. J Biol Chem. 1999;274(52):36963–36972. doi: 10.1074/jbc.274.52.36963. [DOI] [PubMed] [Google Scholar]
- 8.Carlier MF, Jean C, Rieger KJ, Lenfant M, Pantaloni D. Modulation of the interaction between G-actin and thymosin beta 4 by the ATP/ADP ratio: Possible implication in the regulation of actin dynamics. Proc Natl Acad Sci USA. 1993;90(11):5034–5038. doi: 10.1073/pnas.90.11.5034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yarmola EG, Bubb MR. Effects of profilin and thymosin beta4 on the critical concentration of actin demonstrated in vitro and in cell extracts with a novel direct assay. J Biol Chem. 2004;279(32):33519–33527. doi: 10.1074/jbc.M404392200. [DOI] [PubMed] [Google Scholar]
- 10.Weber A, Nachmias VT, Pennise CR, Pring M, Safer D. Interaction of thymosin beta 4 with muscle and platelet actin: Implications for actin sequestration in resting platelets. Biochemistry. 1992;31(27):6179–6185. doi: 10.1021/bi00142a002. [DOI] [PubMed] [Google Scholar]
- 11.Yu FX, Lin SC, Morrison-Bogorad M, Atkinson MA, Yin HL. Thymosin beta 10 and thymosin beta 4 are both actin monomer sequestering proteins. J Biol Chem. 1993;268(1):502–509. [PubMed] [Google Scholar]
- 12.Finkel T, Theriot JA, Dise KR, Tomaselli GF, Goldschmidt-Clermont PJ. Dynamic actin structures stabilized by profilin. Proc Natl Acad Sci USA. 1994;91(4):1510–1514. doi: 10.1073/pnas.91.4.1510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Southwick FS, Young CL. The actin released from profilin—actin complexes is insufficient to account for the increase in F-actin in chemoattractant-stimulated polymorphonuclear leukocytes. J Cell Biol. 1990;110(6):1965–1973. doi: 10.1083/jcb.110.6.1965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kinosian HJ, Selden LA, Gershman LC, Estes JE. Interdependence of profilin, cation, and nucleotide binding to vertebrate non-muscle actin. Biochemistry. 2000;39(43):13176–13188. doi: 10.1021/bi001520+. [DOI] [PubMed] [Google Scholar]
- 15.Safer D, Golla R, Nachmias VT. Isolation of a 5-kilodalton actin-sequestering peptide from human blood platelets. Proc Natl Acad Sci USA. 1990;87(7):2536–2540. doi: 10.1073/pnas.87.7.2536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Safer D, Elzinga M, Nachmias VT. Thymosin beta 4 and Fx, an actin-sequestering peptide, are indistinguishable. J Biol Chem. 1991;266(7):4029–4032. [PubMed] [Google Scholar]
- 17.De La Cruz EM, et al. Thymosin-beta(4) changes the conformation and dynamics of actin monomers. Biophys J. 2000;78(5):2516–2527. doi: 10.1016/S0006-3495(00)76797-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dedova IV, Nikolaeva OP, Safer D, De La Cruz EM, dos Remedios CG. Thymosin beta4 induces a conformational change in actin monomers. Biophys J. 2006;90(3):985–992. doi: 10.1529/biophysj.105.063081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Domanski M, et al. Coupling of folding and binding of thymosin beta4 upon interaction with monomeric actin monitored by nuclear magnetic resonance. J Biol Chem. 2004;279(22):23637–23645. doi: 10.1074/jbc.M311413200. [DOI] [PubMed] [Google Scholar]
- 20.Czisch M, Schleicher M, Hörger S, Voelter W, Holak TA. Conformation of thymosin beta 4 in water determined by NMR spectroscopy. Eur J Biochem. 1993;218(2):335–344. doi: 10.1111/j.1432-1033.1993.tb18382.x. [DOI] [PubMed] [Google Scholar]
- 21.Safer D, Sosnick TR, Elzinga M. Thymosin beta 4 binds actin in an extended conformation and contacts both the barbed and pointed ends. Biochemistry. 1997;36(19):5806–5816. doi: 10.1021/bi970185v. [DOI] [PubMed] [Google Scholar]
- 22.Simenel C, Van Troys M, Vandekerckhove J, Ampe C, Delepierre M. Structural requirements for thymosin beta4 in its contact with actin. An NMR-analysis of thymosin beta4 mutants in solution and correlation with their biological activity. Eur J Biochem. 2000;267(12):3530–3538. doi: 10.1046/j.1432-1327.2000.01380.x. [DOI] [PubMed] [Google Scholar]
- 23.Didry D, et al. How a single residue in individual β-thymosin/WH2 domains controls their functions in actin assembly. EMBO J. 2012;31(4):1000–1013. doi: 10.1038/emboj.2011.461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Irobi E, et al. Structural basis of actin sequestration by thymosin-beta4: Implications for WH2 proteins. EMBO J. 2004;23(18):3599–3608. doi: 10.1038/sj.emboj.7600372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hertzog M, et al. The beta-thymosin/WH2 domain; structural basis for the switch from inhibition to promotion of actin assembly. Cell. 2004;117(5):611–623. doi: 10.1016/s0092-8674(04)00403-9. [DOI] [PubMed] [Google Scholar]
- 26.Aguda AH, Xue B, Irobi E, Préat T, Robinson RC. The structural basis of actin interaction with multiple WH2/beta-thymosin motif-containing proteins. Structure. 2006;14(3):469–476. doi: 10.1016/j.str.2005.12.011. [DOI] [PubMed] [Google Scholar]
- 27.Xue B, Aguda AH, Robinson RC. Models of the actin-bound forms of the beta-thymosins. Ann N Y Acad Sci. 2007;1112:56–66. doi: 10.1196/annals.1415.010. [DOI] [PubMed] [Google Scholar]
- 28.Noguchi TQ, Kanzaki N, Ueno H, Hirose K, Uyeda TQ. A novel system for expressing toxic actin mutants in Dictyostelium and purification and characterization of a dominant lethal yeast actin mutant. J Biol Chem. 2007;282(38):27721–27727. doi: 10.1074/jbc.M703165200. [DOI] [PubMed] [Google Scholar]
- 29.Krissinel E, Henrick K. Inference of macromolecular assemblies from crystalline state. J Mol Biol. 2007;372(3):774–797. doi: 10.1016/j.jmb.2007.05.022. [DOI] [PubMed] [Google Scholar]
- 30.Wang H, Robinson RC, Burtnick LD. The structure of native G-actin. Cytoskeleton (Hoboken) 2010;67(7):456–465. doi: 10.1002/cm.20458. [DOI] [PubMed] [Google Scholar]
- 31.Oda T, Iwasa M, Aihara T, Maéda Y, Narita A. The nature of the globular- to fibrous-actin transition. Nature. 2009;457(7228):441–445. doi: 10.1038/nature07685. [DOI] [PubMed] [Google Scholar]
- 32.Fujii T, Iwane AH, Yanagida T, Namba K. Direct visualization of secondary structures of F-actin by electron cryomicroscopy. Nature. 2010;467(7316):724–728. doi: 10.1038/nature09372. [DOI] [PubMed] [Google Scholar]
- 33.Chereau D, et al. Actin-bound structures of Wiskott-Aldrich syndrome protein (WASP)-homology domain 2 and the implications for filament assembly. Proc Natl Acad Sci USA. 2005;102(46):16644–16649. doi: 10.1073/pnas.0507021102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Vancompernolle K, Goethals M, Huet C, Louvard D, Vandekerckhove J. G- to F-actin modulation by a single amino acid substitution in the actin binding site of actobindin and thymosin beta 4. EMBO J. 1992;11(13):4739–4746. doi: 10.1002/j.1460-2075.1992.tb05579.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Page R, Lindberg U, Schutt CE. Domain motions in actin. J Mol Biol. 1998;280(3):463–474. doi: 10.1006/jmbi.1998.1879. [DOI] [PubMed] [Google Scholar]
- 36.Goldschmidt-Clermont PJ, et al. The control of actin nucleotide exchange by thymosin beta 4 and profilin. A potential regulatory mechanism for actin polymerization in cells. Mol Biol Cell. 1992;3(9):1015–1024. doi: 10.1091/mbc.3.9.1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pantaloni D, Carlier MF. How profilin promotes actin filament assembly in the presence of thymosin beta 4. Cell. 1993;75(5):1007–1014. doi: 10.1016/0092-8674(93)90544-z. [DOI] [PubMed] [Google Scholar]
- 38.Yarmola EG, Parikh S, Bubb MR. Formation and implications of a ternary complex of profilin, thymosin beta 4, and actin. J Biol Chem. 2001;276(49):45555–45563. doi: 10.1074/jbc.M105723200. [DOI] [PubMed] [Google Scholar]
- 39.Ferron F, Rebowski G, Lee SH, Dominguez R. Structural basis for the recruitment of profilin-actin complexes during filament elongation by Ena/VASP. EMBO J. 2007;26(21):4597–4606. doi: 10.1038/sj.emboj.7601874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yarmola EG, Bubb MR. Profilin: Emerging concepts and lingering misconceptions. Trends Biochem Sci. 2006;31(4):197–205. doi: 10.1016/j.tibs.2006.02.006. [DOI] [PubMed] [Google Scholar]
- 41.Baek K, et al. Modulation of actin structure and function by phosphorylation of Tyr-53 and profilin binding. Proc Natl Acad Sci USA. 2008;105(33):11748–11753. doi: 10.1073/pnas.0805852105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Irobi E, Burtnick LD, Urosev D, Narayan K, Robinson RC. From the first to the second domain of gelsolin: A common path on the surface of actin? FEBS Lett. 2003;552(2-3):86–90. doi: 10.1016/s0014-5793(03)00934-7. [DOI] [PubMed] [Google Scholar]
- 43.Vorobiev S, et al. The structure of nonvertebrate actin: Implications for the ATP hydrolytic mechanism. Proc Natl Acad Sci USA. 2003;100(10):5760–5765. doi: 10.1073/pnas.0832273100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kinosian HJ, Selden LA, Gershman LC, Estes JE. Actin filament barbed end elongation with nonmuscle MgATP-actin and MgADP-actin in the presence of profilin. Biochemistry. 2002;41(21):6734–6743. doi: 10.1021/bi016083t. [DOI] [PubMed] [Google Scholar]
- 45.Xue B, Robinson RC. Guardians of the actin monomer. Eur J Cell Biol. 2013;92(10-11):316–332. doi: 10.1016/j.ejcb.2013.10.012. [DOI] [PubMed] [Google Scholar]
- 46.Krause M, Dent EW, Bear JE, Loureiro JJ, Gertler FB. Ena/VASP proteins: Regulators of the actin cytoskeleton and cell migration. Annu Rev Cell Dev Biol. 2003;19:541–564. doi: 10.1146/annurev.cellbio.19.050103.103356. [DOI] [PubMed] [Google Scholar]
- 47.Schönichen A, Geyer M. Fifteen formins for an actin filament: A molecular view on the regulation of human formins. Biochim Biophys Acta. 2010;1803(2):152–163. doi: 10.1016/j.bbamcr.2010.01.014. [DOI] [PubMed] [Google Scholar]
- 48.Tilney LG, Bonder EM, Coluccio LM, Mooseker MS. Actin from Thyone sperm assembles on only one end of an actin filament: A behavior regulated by profilin. J Cell Biol. 1983;97(1):112–124. doi: 10.1083/jcb.97.1.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pollard TD, Cooper JA. Quantitative analysis of the effect of Acanthamoeba profilin on actin filament nucleation and elongation. Biochemistry. 1984;23(26):6631–6641. doi: 10.1021/bi00321a054. [DOI] [PubMed] [Google Scholar]
- 50.Yarmola EG, Bubb MR. How depolymerization can promote polymerization: The case of actin and profilin. BioEssays. 2009;31(11):1150–1160. doi: 10.1002/bies.200900049. [DOI] [PubMed] [Google Scholar]
- 51.Jégou A, et al. Individual actin filaments in a microfluidic flow reveal the mechanism of ATP hydrolysis and give insight into the properties of profilin. PLoS Biol. 2011;9(9):e1001161. doi: 10.1371/journal.pbio.1001161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Pring M, Weber A, Bubb MR. Profilin-actin complexes directly elongate actin filaments at the barbed end. Biochemistry. 1992;31(6):1827–1836. doi: 10.1021/bi00121a035. [DOI] [PubMed] [Google Scholar]
- 53.Courtemanche N, Pollard TD. Interaction of profilin with the barbed end of actin filaments. Biochemistry. 2013;52(37):6456–6466. doi: 10.1021/bi400682n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Spudich JA, Watt S. The regulation of rabbit skeletal muscle contraction. I. Biochemical studies of the interaction of the tropomyosin-troponin complex with actin and the proteolytic fragments of myosin. J Biol Chem. 1971;246(15):4866–4871. [PubMed] [Google Scholar]
- 55.Chumnarnsilpa S, et al. The crystal structure of the C-terminus of adseverin reveals the actin-binding interface. Proc Natl Acad Sci USA. 2009;106(33):13719–13724. doi: 10.1073/pnas.0812383106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Aslanidis C, de Jong PJ. Ligation-independent cloning of PCR products (LIC-PCR) Nucleic Acids Res. 1990;18(20):6069–6074. doi: 10.1093/nar/18.20.6069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Studier FW. Protein production by auto-induction in high density shaking cultures. Protein Expr Purif. 2005;41(1):207–234. doi: 10.1016/j.pep.2005.01.016. [DOI] [PubMed] [Google Scholar]
- 58.Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. In: Carter CW Jr, Sweet RM, editors. Methods in Enzymology Vol 276: Macromolecular Crystallography, Part A. Academic; New York: 1997. pp. 307–326. [DOI] [PubMed] [Google Scholar]
- 59.McCoy AJ, et al. Phaser crystallographic software. J Appl Cryst. 2007;40(Pt 4):658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Adams PD, et al. PHENIX: A comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr. 2010;66(Pt 2):213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta Crystallogr D Biol Crystallogr. 2010;66(Pt 4):486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Painter J, Merritt EA. TLSMD web server for the generation of multi-group TLS models. J Appl Crystallogr. 2006;39(Pt 1):109–111. [Google Scholar]
- 63.Winn MD, et al. Overview of the CCP4 suite and current developments. Acta Crystallogr D Biol Crystallogr. 2011;67(Pt 4):235–242. doi: 10.1107/S0907444910045749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Chen VB, et al. MolProbity: All-atom structure validation for macromolecular crystallography. Acta Crystallogr D Biol Crystallogr. 2010;66(Pt 1):12–21. doi: 10.1107/S0907444909042073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Breitsprecher D, et al. Rocket launcher mechanism of collaborative actin assembly defined by single-molecule imaging. Science. 2012;336(6085):1164–1168. doi: 10.1126/science.1218062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hernandez-Valladares M, et al. Structural characterization of a capping protein interaction motif defines a family of actin filament regulators. Nat Struct Mol Biol. 2010;17(4):497–503. doi: 10.1038/nsmb.1792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Yarmola EG, Somasundaram T, Boring TA, Spector I, Bubb MR. Actin-latrunculin A structure and function. Differential modulation of actin-binding protein function by latrunculin A. J Biol Chem. 2000;275(36):28120–28127. doi: 10.1074/jbc.M004253200. [DOI] [PubMed] [Google Scholar]
- 68.Hess B, Kutzner C, van der Spoel D, Lindahl E. GROMACS 4: Algorithms for highly efficient, load-balanced, and scalable molecular simulation. J Chem Theory Comput. 2008;4(3):435–447. doi: 10.1021/ct700301q. [DOI] [PubMed] [Google Scholar]
- 69.Best RB, Hummer G. Optimized molecular dynamics force fields applied to the helix-coil transition of polypeptides. J Phys Chem B. 2009;113(26):9004–9015. doi: 10.1021/jp901540t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Lindorff-Larsen K, et al. Improved side-chain torsion potentials for the Amber ff99SB protein force field. Proteins. 2010;78(8):1950–1958. doi: 10.1002/prot.22711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Essmann U, et al. A smooth particle mesh Ewald method. J Chem Phys. 1995;103(19):8577–8593. [Google Scholar]
- 72.Hess B. P-LINCS: A parallel linear constraint solver for molecular simulation. J Chem Theory Comput. 2008;4(1):116–122. doi: 10.1021/ct700200b. [DOI] [PubMed] [Google Scholar]
- 73.Bussi G, Donadio D, Parrinello M. Canonical sampling through velocity rescaling. J Chem Phys. 2007;126(1):014101. doi: 10.1063/1.2408420. [DOI] [PubMed] [Google Scholar]
- 74.Parrinello M, Rahman A. Polymorphic transitions in single crystals: A new molecular dynamics method. J Appl Phys. 1981;52(12):7182–7190. [Google Scholar]
- 75.Loew LM, Schaff JC. The Virtual Cell: A software environment for computational cell biology. Trends Biotechnol. 2001;19(10):401–406. doi: 10.1016/S0167-7799(01)01740-1. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.