

Significance
Inhibitory serpin plasminogen activator inhibitor 1 (PAI-1) helps regulate blood clot clearance, and high levels of active PAI-1 are associated with poor cancer prognoses and cardiovascular diseases. PAI-1 transitions from an active form to an inactive, latent form via large structural rearrangements. Our simulations provide the first realistic atomistic characterization, to our knowledge, of this transition. They correctly predict the kinetic effects of mutations and explain why PAI-1 binding to specific molecules accelerates the transition. Structures from these simulations provide templates to design drugs that speed up the latency transition, with potentially important biomedical applications. Serpins are large proteins, and the first-passage times of the simulated conformational transitions are many orders of magnitude longer than the time intervals accessible using conventional molecular dynamics.
Keywords: molecular simulations, conformational change, plasminogen activator inhibitor-1
Abstract
Protease inhibition by serpins requires a large conformational transition from an active, metastable state to an inactive, stable state. Similar reactions can also occur in the absence of proteases, and these latency transitions take hours, making their time scales many orders of magnitude larger than are currently accessible using conventional molecular dynamics simulations. Using a variational path sampling algorithm, we simulated the entire serpin active-to-latent transition in all-atom detail with a physically realistic force field using a standard computing cluster. These simulations provide a unifying picture explaining existing experimental data for the latency transition of the serpin plasminogen activator inhibitor-1 (PAI-1). They predict a long-lived intermediate that resembles a previously proposed, partially loop-inserted, prelatent state; correctly predict the effects of PAI-1 mutations on the kinetics; and provide a potential means to identify ligands able to accelerate the latency transition. Interestingly, although all of the simulated PAI-1 variants readily access the prelatent intermediate, this conformation is not populated in the active-to-latent transition of another serpin, α1-antitrypsin, which does not readily go latent. Thus, these simulations also help elucidate why some inhibitory serpin families are more conformationally labile than others.
The serpin plasminogen activator inhibitor 1 (PAI-1) negatively regulates blood clot clearance (fibrinolysis) by mechanically inhibiting important serine proteases, including tissue type plasminogen activator and urokinase type plasminogen activator (1). Suicide inhibition, initiated by proteolytic cleavage of the PAI-1 reactive center loop (RCL), requires insertion of the cleaved RCL into the central β-sheet (sheet A). This process expands sheet A, inhibits the covalently attached protease by mechanical disruption of the active site (2), and results in a thermodynamically stable serpin conformation. Alternatively, PAI-1 can spontaneously deactivate by inserting its intact, uncleaved RCL into sheet A, resulting in the more stable but inactive latent conformation (1) (Fig. 1).
Fig. 1.

PAI-1 latency transition. (Left) In the active state, the RCL is solvent-exposed and s1C is hydrogen-bonded to β-sheet C. (Right) The active state contrasts with the latent state, where s1C is released from sheet C and the RCL is inserted into β-sheet A as the fourth strand in a now six-stranded, fully antiparallel β-sheet. The sequence of the RCL and s1C is shown below the structures. h, helix; s, strand.
The latency transition provides a facile way to regulate PAI-1 activity. Physiologically, this regulation is achieved by binding to the cell adhesion factor vitronectin, leading to an ∼50% increase in the active state t1/2 (3). Because high levels of active PAI-1 are associated both with poor prognoses for some cancers, presumably due to interactions with vitronectin, and with cardiovascular diseases (1), PAI-1 inhibitors that accelerate the latency transition are under development (1, 4, 5). However, drug design efforts are hampered by the lack of detailed molecular mechanisms for PAI-1 conformational changes. Numerous studies have identified mutations that either accelerate or retard the conformational transition, as well as antibodies that can accelerate latency. Despite these efforts, the molecular details of the latency transition and the residues involved in the key interactions that drive it are still unclear.
In principle, molecular dynamics (MD) simulations can aptly complement these experiments and provide an atomistic description of protein structural transitions. Unfortunately, even using the most powerful special-purpose supercomputer, MD simulations can only cover time intervals up to about a millisecond for polypeptide chains consisting of nearly 100 amino acids (6), which are considerably smaller than PAI-1. Additionally, the PAI-1 active state has a t1/2 of 1–2 h at 37 °C (3), a time scale clearly beyond the reach of any present or foreseen conventional all-atom MD simulation.
In view of the computational limitations of MD, more sophisticated alternative algorithms have been developed and then applied to investigate rare biological transitions (an incomplete list is provided in refs. 7–11). To date, the only attempt to simulate the active-to-latent transition in PAI-1 in atomic detail used the targeted MD algorithm (12). Unfortunately, even application of a strong steering force to drive the protein toward the latent state did not result in a transition. To overcome this problem, an ad hoc intermediate structure was postulated and used as the target for the steered simulation. This procedure, however, resulted in trajectories that disagree with the results of kinetic experiments.
In this paper, we show that using the so-called “dominant reaction pathways” (DRP) (13–15) approach, it is finally possible to overcome the existing computational limitations and perform atomistic simulations of the active-to-latent transitions of several PAI-1 variants and of human α1-antitrypsin (A1AT), with mean first-passage times as long as several days. The DRP method is a path integral-based approach that yields the reaction pathways with the highest probability of being realized within Langevin dynamics. This scheme has been extensively tested against the results of MD protein folding simulations using both reduced (16, 17) and realistic atomistic force fields (15). It was then successfully applied to much more complex processes, such as the folding of a natively knotted protein (14).
Simulating the serpin active-to-latent transition only involved a few tens of central processing units (CPUs) on a regular personal computing (PC) cluster, running for about 2 d. By analyzing the resulting pathways, we have characterized the reaction mechanism and validated it against available experimental data. Our simulations reveal the likely structure of the metastable prelatent intermediate whose existence has been inferred from experimental results (18–20). They also predict the effects of mutations known to speed up or slow down the PAI-1 latency transition (21, 22) and demonstrate that the energy barriers between the active and latent states are very different for PAI-1, which readily goes latent, and A1AT, which rarely goes latent (23, 24). Finally, DRP simulations also allow us to identify a small subset of amino acid residues involved in key favorable and unfavorable interactions during the transition.
Results
DRP-based simulations of the latency transition were performed for three PAI-1 variants: WT, a stabilized PAI-1 variant; PAI-1stab (N150H, K154T, Q319L, and M354I), a stable variant that goes latent more slowly than WT (21); and a destabilized variant, PAI-1destab (G38S and Q322H), with a faster latency transition (22) (Fig. S1). To gain further insight into what drives the latency transition, simulations were also performed for WT A1AT, which does not readily go latent (23, 24). Despite the very low probability of the WT A1AT latency transition, the ratchet-and-pawl biasing potential used in the DRP method allowed this transition to be simulated within a few thousands of CPU hours on a regular PC cluster.
Structural Intermediate Between the PAI-1 Active and Latent States.
Selected snapshots of the latency transition for WT PAI-1 are shown in Fig. 2A. The boxed structure is a long-lived intermediate that occupies a local minimum in the potential energy along the transition. In Fig. 3A, we plot the potential energy profile as a function of the Euclidean distance covered by the Cα atoms along the reaction pathway. This distance was proposed as a natural reaction coordinate for conformational transitions (25). In all of the PAI-1 simulations, the protein is trapped for an extended period in a local energy minimum located at ∼2,000 nm before proceeding to the latent state.
Fig. 2.

Conformations sampled along the PAI-1 active-to-latent pathway. (A) Snapshots along the DRP generated active → latent pathway. The boxed structure is the proposed prelatent state. (B) Superimposed frames from 200 ns of an unbiased MD simulation starting from the boxed structure in A. The RCL and gate regions are colored red (0 ns), orange (20 ns), yellow (100 ns), and blue (200 ns).
Fig. 3.

PAI-1 energy landscape shows pronounced minima for the prelatent and latent states. (A) Potential energy evaluated along the dominant pathways for PAI-1 WT (black curve), the PAI-1 variants (red and blue curves), and A1AT (brown curve) as a function of the total Euclidean distance covered by the Cα atoms during the latency transition. The structure in the upper right corner corresponds to the PAI-1 prelatent state. To resolve the energy landscapes better, the plotted potential energies were averaged over 15 consecutive frames in the DRP trajectories. The Euclidean distance is defined as where Ns is the number of frames in the dominant trajectory, NCα is the number of residues in the protein, and is the position of the kth Cα atom in the ith frame. Upot, potential energy. (B) rmsd to the latent state as a function of the number of rMD steps for all three PAI-1 variants. The prelatent state is revealed by a clear plateau in all three curves. (C) Mutations and active t1/2s for the PAI-1 variants.
In the prelatent state detected in our simulations, strand 1C (s1C) has detached from sheet C and moved around the gate formed by β-strands 3C and 4C, sheet A is open between strands 3 and 5, and the RCL is inserted into sheet A up to P11 (Ser336) (Movies S1–S3). The transition from the active state to the prelatent state also seems to require changing the bend (intrahelical angle) in helix A (Fig. S2). These DRP results agree with a number of experiments suggesting that in solution, PAI-1 exists in equilibrium between a state resembling the active form seen in crystal structures and a prelatent state, whose exact structure remains a matter of conjecture (18–20).
When the DRP-generated prelatent structure shown in Fig. 2A is used as the starting point for conventional MD simulations, the RCL tends to move up toward the gate region, whereas the gate appears to move down (Fig. 2B). This finding suggests that the exact conformation of the DRP-generated prelatent state is influenced by the biasing force used to generate the initial trial trajectories, and that alternate conformations may be obtained when the system is allowed to explore a larger configuration space. Such alternate conformations derived from equilibrium simulations may be more physiologically relevant.
Experimentally, incubation of active PAI-1 with the antibody H4B3 has been shown to accelerate the latency transition in a concentration-dependent manner (20). However, the epitope recognized by H4B3 is only accessible when s1C has detached from sheet C. Although this epitope is accessible in the latent state, in the prelatent state predicted by the DRP and in the prelatent conformations extracted from the equilibrium simulations described above, it is not accessible in the active conformation, suggesting that active PAI-1 is in equilibrium with a form in which s1C is detached. In this model, H4B3 binding prevents s1C reattachment, pushing this equilibrium away from the active form, and thereby facilitating latency.
In addition to antibodies, a number of small molecules are known to inhibit PAI-1 (26). One of these molecules, AZ3976, binds to latent PAI-1 in a pocket between helices E and F (5) (Fig. 4). It has been suggested that AZ3976 binding to the prelatent state prevents a return to the active conformation. Our simulations support this mechanism. Indeed, the top hit from randomly docking AZ3976 to the DRP-generated prelatent structure places AZ3976 in the experimentally identified pocket (Fig. 4). By contrast, AZ3976 could not be docked to active PAI-1. Thus, AZ3976 and the antibody H4B3 likely use similar mechanisms to accelerate the latency transition: binding to epitopes revealed in the prelatent conformation and biasing the equilibrium toward the prelatent conformation.
Fig. 4.

Drug candidate AZ3976 binds to the DRP prelatent state structure. The DRP prelatent structure for WT PAI-1 (green) is superimposed on the X-ray crystal structure of latent PAI-1 (gray) bound to the small-molecule inhibitor AZ3976 (blue) [PDB ID code 4AQH (5)]. AZ3976 (red) is shown in the top-scoring position found by docking to the DRP prelatent structure using Autodock (47). (Right) Region containing AZ3976 is enlarged.
We observe no long-lived partially RCL-inserted state in simulations of the WT A1AT latency transition, in agreement with the fact that there is no experimental evidence for the existence of an A1AT prelatent state. We emphasize that our simulation protocol included no assumptions regarding the existence of any metastable intermediate states and that the hypothetical prelatent state described here occurs spontaneously in the DRP simulations.
Overview of the Active-to-Latent Transition.
In all four serpins, we observe that the conformational transition is initiated by overcoming a marginally activated enthalpy barrier of about 200 in units of thermal energy (kBT) associated with peeling s1C away from sheet C and moving the RCL around the gate. Notably, in the PAI-1 simulations, the removal of s1C, the opening of sheet A, and a large movement of the gate region all occur in a strikingly concerted manner (Movies S1–S3). By contrast, conformational changes for A1AT are much less concerted (Movie S4).
In PAI-1, after the RCL transits the gate region, the reaction proceeds downhill until the reaction coordinate reaches a value of ∼2,000 nm, corresponding to the prelatent state discussed above. This configuration is separated from the final latent state by a high-energy barrier. The structure of PAI-1 in the high-energy region is remarkably close to the final latent structure, with the RCL almost fully inserted. Progress at this point is blocked by steric clashes between the RCL and the loop from helix F to s3A. The final stage of the transition involves displacing this loop and helix F and inserting the final residues of the RCL (P6–P4) into sheet A. There is a notable twist in the bottom of s4A (the inserted RCL) immediately before formation of the final latent structure, with several unfavorable dihedral angles in the P6–P4 region (Fig. 5A). In the final stage of the transition, this twist is relaxed and helix F simultaneously shifts its position to form closer contacts with sheet A (Fig. 5B). These main chain motions are coupled to significant side-chain reorientations of Val341 and Val343 in s4A, which require significant repacking of the PAI-1 core. These processes are accompanied by a significant drop in energy, indicating that relatively subtle structural changes relieve a large amount of the PAI-1 conformational strain. Consistent with our results, recent studies of the A1AT inhibitory transition detected a long-lived intermediate state in which the RCL was almost fully inserted and concluded that the final stage of the transition involved movement of helix F (27).
Fig. 5.
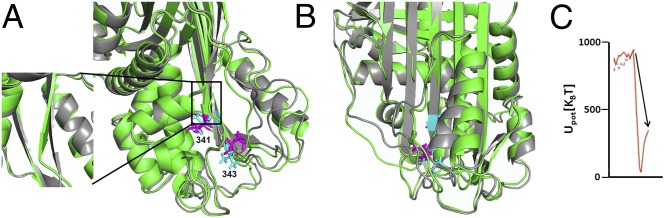
Final conformational changes in the latency transition involve twisting of s4A (the RCL) and movement of helix F. (A) Superimposed DRP structures of WT PAI-1 in the high-energy form immediately before completion of the latency transition (green) and in the final low-energy latent state (gray). Val341 and Val343, which undergo large rotamer changes, are shown in magenta for the high-energy form and in cyan sticks for the latent conformation. The expanded box highlights the relaxation of the twist in s4A (RCL) that accompanies the final stages of the latency transition. (B) As in A, but rotated to highlight the change in position of helix F. (C) Change in energy upon moving from the high-energy structure to the latent structure.
Reaction Kinetics.
The experimentally determined t1/2s for the three PAI variants are summarized in the table reported in Fig. 3C. These data show that point mutations on PAI-1 can significantly affect the reaction kinetics by slowing or accelerating the rate of latency by nearly 10-fold. In contrast, without proteolytic cleavage, WT A1AT can remain in the active state for weeks (23, 24). We now address whether our numerical simulations are compatible with these experimental t1/2s. The DRP approach focuses on the nonequilibrium reactive dynamics and is not directly applicable to compute free-energy barriers and reaction rates. However, it can still be used to infer qualitative predictions on the effects of mutations on the latency transition. In particular, we note that the behavior of the rmsd to native forms of the three variants shown in Fig. 3B correctly reproduces the ordering of lifetimes observed in experiments, predicting that the stab (or destab) mutations lead to a longer (or shorter) prelatent state lifetime. This feature is also reflected by the structure of the molecular energy profile along the Euclidean distance reaction coordinate l (Fig. 3A). Indeed the barrier from the prelatent state to the latent state in the stab (or destab) mutant is higher (or lower) than that in WT PAI-1.
Let us now compare the energy profiles evaluated along the reaction pathway of PAI-1 and A1AT, which share 29% sequence identity. We recall that for A1AT, the transition to latency is highly disfavored (23, 24) compared with PAI-1. Consistently, we find that for A1AT, the overall energy change remains unfavorable for almost the entire reaction, long after moving the RCL around the gate region. In addition, the energy landscape does not display a deep minimum compatible with an on-pathway prelatent intermediate (Fig. 3A). Interestingly, to our knowledge, the prelatent state has never been observed for A1AT. These differences in the A1AT and PAI-1 DRP trajectories provide insight into the physical origin of the different kinetic stabilities of these two proteins. In PAI-1, a large downward bending of s3C and s4C occurs before passage of the RCL around the gate. We note that similar downward bending motions are observed in our conventional MD simulations of the prelatent state, indicating that this gate bending is not an artifact of the DRP method (Fig. 2B). In contrast, for A1AT, movement of the gate is concurrent with passage of the RCL, and the gate appears to be physically pushed out of its normal position by the RCL. Previous hydrogen/deuterium exchange measurements support the notion that the gate, including s3C and s4C, is more flexible in PAI-1 than in A1AT (28, 29).
Our results on PAI-1 and A1AT indicate that the DRP method is able to distinguish between serpins that spontaneously undergo the transition to the latent state and those serpins that do not. To test this capability further, we have computed DRP trajectories for an A1AT destabilized variant, (A1ATdestab), which, unlike WT A1AT, readily goes latent and for which an X-ray crystal structure of the latent state is available [Protein Data Bank (PDB) ID code 1iz2] (24). Relative to WT A1AT, this variant contains 10 mutations: (i) the multi-seven mutations (F51L, T59A, T68A, A70G, M374I, S381A, and K387R) that stabilize active A1AT without compromising function (30); (ii) two naturally occurring variations (R101H and E376D); and (iii) the mutation in s1C, V364A, that increases the probability of the latency transition. Note that the variant containing just the multi-seven mutations and the two naturally occurring mutations is similar to WT A1AT and does not readily go latent (30). In Fig. 6, we compare the energy profiles of WT A1AT, A1ATdestab, and WT PAI-1. We see that the mutations strongly affect the energy landscape of A1AT, making it qualitatively similar to the energy landscape of PAI-1, thereby explaining why A1ATdestab goes latent spontaneously.
Fig. 6.

Energy landscape of A1ATdestab is qualitatively similar to that of PA1-1. The molecular energy profile evaluated along the reaction coordinate l for WT A1AT (solid brown line), WT PA1-1 (solid black line), and A1ATdestab (dashed blue line) is shown.
We analyzed the evolution of the rmsd relative to the latent state of several secondary structures during the reaction. For almost all of the secondary structures considered, the evolution of this parameter is quantitatively similar in the two proteins. In contrast, a significant difference emerges for helix A. Indeed, whereas in A1AT, this helix remains close to its native configuration throughout the entire reaction, in PAI-1, helix A is unfolded in the early part of the reaction. This prediction is supported hydrogen/deuterium exchange MS experiments for PAI-1, where vitronectin binding both increases the active state t1/2 and decreases exchange in helix A (29). Similarly, for A1AT, the process of inserting the proteolytically cleaved RCL into sheet A is associated with increased exchange in this helix (31). These results support the importance of helix A flexibility for serpin conformational changes. Even more notable is a recent hydrogen/deuterium exchange MS study that directly monitored the transition of PAI-1 from the active state to the latent state (32). This study found evidence of cooperative unfolding of helix A and s5A during the transition. Further, although unfolding in helix A and s5A was slow (t1/2 of ∼20 min), it was nevertheless much faster than the complete latency transition under the same conditions (t1/2 of ∼450 min), indicating that disruption of these regions did not correspond to the rate-limiting step of the transition. This result is consistent with our simulations, where disruption of both helix A and s5A occur during the active → prelatent transition, whereas a significant additional barrier exists for the prelatent → latent transition.
Significant Interactions During the Conformational Reaction.
We now ask what interactions shape the underlying energy landscape and drive the transition. Decomposing the potential energy into Coulomb and van der Waals terms, we find that the initial transition from the active to the prelatent state is driven primarily by the formation of favorable Coulomb-type interactions, amounting to a total favorable ΔECoulomb of −6.8 kcal/mol. These interactions in the prelatent state do not appear to be concentrated in any one region of the molecule. Interestingly, nearly 40% of the favorable Coulomb energy (−2.7 kcal/mol) is accounted for by only 16 residues, which are uniformly distributed throughout the sequence. We note that the favorable Coulomb energy is almost entirely lost during the transition from the prelatent state to the high-energy plateau region immediately preceding the final transition to the latent state (ΔECoulomb = +6.8 kcal/mol), and that a large portion is regained during the final transition to the latent state (ΔECoulomb = −6.2 kcal/mol). A similar trend is seen for the van der Waals energy, although the magnitudes of these changes are ∼50% smaller.
At each stage of the transition, the same subset of residues makes the dominant contribution (Fig. S3 and Table S1). It is somewhat unexpected that the same subset of residues showing the largest favorable energy changes going from the active state to the prelatent state also show the largest unfavorable energy changes going from the prelatent state to the high-energy plateau region. These residues are not clustered in particular regions but are widely distributed, consistent with the hypothesis of “distributed strain” proposed by Seo et al. (33). A statistically significant number of these residues (nine of 16) have previously been shown to play a significant role in function and/or disease (details and statistical significance are provided in SI Methods, Fig. S3, and Table S1). This correspondence suggests that our simulations have identified residues that play important roles in mediating the latency transition. We note that Li et al. (34) recently identified a pocket at the interface of sheets B and C that appears to be important for PAI-1 function, and a number of this subset of energetically important residues (K176, F207, E226, G264, and T267) are clustered in this region.
Discussion
In the DRP PAI-1 simulations, a prelatent state appears to facilitate the active-to-latent transition. This result is consistent with several lines of experimental evidence, including binding of the H4B3 antibody to an epitope that is only revealed after s1C detaches from sheet C (20); cross-linking studies using double-cysteine mutants in the RCL and β-sheet A, indicating that PAI-1 samples conformations in which the RCL is inserted into sheet A up to at least P11 (Ser336) and perhaps as far as P8 (Thr339) (19); and FRET measurements indicating that the distance between P1 (Arg346) or P3 (Ser344) and the bottom of the molecule (∼Glu313) is considerably smaller than distances seen in crystal structures of the active form (19). All of these experiments are consistent with the prelatent structure revealed in the DRP simulations (Fig. 2). Binding of either the H4B3 antibody or the small-molecule AZ3976 appears to increase the rate of latency by altering this equilibrium and increasing the probability of occupying the prelatent state. The docking results show that AZ3976 can bind to the prelatent state and suggest that DRP-based structures of the prelatent state will be useful for computational drug screening.
As noted above, in PAI-1, s1C detachment is accompanied by the separation of s3A and s5A along their entire length. Although the simultaneity of s1C detachment and s3A-s5A separation may be exaggerated slightly by the nonequilibrium nature of the simulations, the concerted opening of sheet A is supported by a number of observations. It has been argued from crystal structures and mutagenesis data that vitronectin blocks the latency transition by sterically hindering the sliding of the lower portions of β-strands 1A and 2A into the gap between helices D and E (35). In our simulations, this sliding occurs as s1C detaches and moves around the gate, whereas the RCL inserts between s3A and s5A up to P11. In none of our simulations do we see evidence of PAI-1 behaving in a manner similar to antithrombin III, where s3A and s5A separate at the top to accommodate partial RCL insertion, whereas the bottom of sheet A remains intact. If passage of s1C and partial RCL insertion require opening of the entire length of sheet A, then vitronectin should block not just the latency transition but also the active-to-prelatent transition. Experiments have shown that when PAI-1 is immobilized on vitronectin, the antibody H4B3 (which is thought to bind to the prelatent state) cannot bind (20), suggesting that vitronectin does, in fact, prevent the transition to the prelatent state. Hence, this effect of vitronectin is rationalized by the concerted nature of the initial transition seen in our simulations.
Facile opening of sheet A in PAI-1 is also supported by the fact that it lacks a highly conserved hydrogen bond network at the center of the shutter region (22, 36). The effects of this network are apparent if we compare the latency transitions of PAI-1 and A1AT (Fig. S4). Early in the A1AT transition, it can be seen that although the upper and lower ends of s3A and s5A are separating, the center of these strands, the conserved hydrogen bond network, resists separation. In contrast, similar behavior is not seen in PAI-1, where the entire length of the strands readily separates (a more detailed discussion is provided in SI Methods). This full-sheet A opening is also consistent with the observation that, compared with A1AT, the amide hydrogens in sheet A in PAI-1 are more labile to exchange with deuterium, indicating weaker amide hydrogen bonds (28, 29). Further support is also provided by the ability of an RCL-derived peptide to bind between s3A and s5A at both the upper and lower ends, inhibiting PAI-1 activity (37). These results suggest both a role for the conserved network in stabilizing sheet A and that the concerted opening seen in PAI-1 is not an artifact of the DRP algorithm, because the algorithm does not find similarly concerted behavior in A1AT, despite structural similarities.
In conclusion, in this work, we have presented a theoretical/computational study of the dynamical mechanisms involved in the conformational transitions of serpins from the biologically active, metastable state to the thermodynamically stable, latent state. Simulations using realistic force fields and all-atom models of PAI-1 have resulted in order of magnitude estimates for the relative acceleration and delay of the latency transition for different PAI-1 variants. The relative ordering of the time scales for the latency transition agrees well with the experimental results, where PAI-1stab has a significantly increased active-state lifetime, whereas the active-state lifetime of PAI-1destab is significantly decreased. The simulations also identify residues throughout the PAI-1 structure that are energetically important for the latency transition, and mutagenesis of the majority of these residues is associated with dysfunction and/or disease. Finally, the DRP simulations also provide strong support for the existence of an on-pathway intermediate in the PAI-1 transition, which had been proposed in the past based on the analysis of FRET, disulfide cross-linking, and antibody binding experiments. The computational efficiency of these simulations and the size of the serpins suggest that the DRP method may be applied to study even larger systems and/or much slower reactions, thereby significantly decreasing the gap between protein processes that are computationally accessible and those protein processes that are biologically relevant.
Methods
Atomistic Force Field.
Our atomistic DRP simulations were performed using the AMBER ff99SB force field in implicit solvent (38), with generalized Born formalism implemented in GROMACS 4.5.2 (39). The Born radii were calculated according to the Onufriev–Bashford–Case algorithm (40).
Preparation of the PDB Files.
The active and latent structures of the WT PAI-1 and A1AT were downloaded from the PDB. The corresponding PDB ID codes are as follows: 1oc0 [active PAI-1 (35)], 1dvn [latent PAI-1 (41)], 1qlp [active A1AT (42)], and 1iz2 [latent A1AT (24)]. The missing residues in the active structure of PAI-1 were modeled using the ModLoop package (43) and refined by RosettaBackrub (44). Appropriate mutations were introduced to make sure that the corresponding sequences of the active and latent serpins were identical.
The PAI-1destab and PAI-1stab mutants were defined by the following residue substitutions: G38S/Q322H [PAI-1destab (22)] and N150H/K154T/Q319L/M354I [PAI-1stab (21)]. These mutations were introduced into the active and latent WT structures. The coordinates of the mutated residues were provided by ModLoop (43) and refined by RosettaBackrub (44), and the new structures were energy-minimized using GROMACS (39).
DRP Algorithm.
The DRP approach applies to systems that can be described by the Langevin equation, which, in the overdamped limit and discretized (Ito) form, reads
| [1] |
In this equation, xk and Dk are the position and diffusion constants of the kth atom, respectively. X(ti) = [x1(ti)…,xN(ti)] is the position in configuration space at time step ti, U(X) is the potential energy, kBT is the thermal energy, and ηk(ti) is a memoryless white Gaussian noise with unitary variance acting on the kth atom. The probability density that a specific stochastic trajectory connecting a given initial state and final state in Nt time steps is realized in the Langevin dynamics defined in Eq. 1 and is given by
| [2] |
The DRP algorithm is defined by the following two-step procedure. In the first step, a set of trial paths connecting the given initial and final configurations is generated using the ratchet-and-pawl MD (rMD) algorithm, introduced by Paci and Karplus (45) and Camilloni et al. (46). Next, these trial paths are scored according to their probabilities to be realized in an unbiased Langevin simulation, using Eq. 2. This procedure returns the most probable path among the set of trial rMD reactive trajectories. The second step of the DRP procedure is very important, because it serves to minimize the effects of the bias. We emphasize that Eq. 2 provides a way to estimate the relative likelihood of the trial trajectories with the same boundary conditions (i.e., of paths that leave from the same initial conformation and are bound to reach the final conformation within the same time interval).
Supplementary Material
Acknowledgments
Simulations were performed mostly on the high-performance computing cluster at the University of Maryland Computer Aided Drug Design center. G.C., S.a.B., and P.F. are members of the Interdisciplinary Laboratory for Computational Science. This work was partially supported by NIH Awards R01HL085469 (to P.L.W.) and R01GM094848 (to A.G.).
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1407528111/-/DCSupplemental.
References
- 1.Van De Craen B, Declerck PJ, Gils A. The biochemistry, physiology and pathological roles of PAI-1 and the requirements for PAI-1 inhibition in vivo. Thromb Res. 2012;130(4):576–585. doi: 10.1016/j.thromres.2012.06.023. [DOI] [PubMed] [Google Scholar]
- 2.Huntington JA, Read RJ, Carrell RW. Structure of a serpin-protease complex shows inhibition by deformation. Nature. 2000;407(6806):923–926. doi: 10.1038/35038119. [DOI] [PubMed] [Google Scholar]
- 3.Lindahl TL, Sigurdardottir O, Wiman B. Stability of plasminogen activator inhibitor 1 (PAI-1) Thromb Haemost. 1989;62(2):748–751. [PubMed] [Google Scholar]
- 4.Lin Z, et al. Structural insight into inactivation of plasminogen activator inhibitor-1 by a small-molecule antagonist. Chem Biol. 2013;20(2):253–261. doi: 10.1016/j.chembiol.2013.01.002. [DOI] [PubMed] [Google Scholar]
- 5.Fjellström O, et al. Characterization of a small molecule inhibitor of plasminogen activator inhibitor type 1 that accelerates the transition into the latent conformation. J Biol Chem. 2013;288(2):873–885. doi: 10.1074/jbc.M112.371732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shaw DE, et al. Atomic-level characterization of the structural dynamics of proteins. Science. 2010;330(6002):341–346. doi: 10.1126/science.1187409. [DOI] [PubMed] [Google Scholar]
- 7.Elber R, West A. Atomically detailed simulation of the recovery stroke in myosin by Milestoning. Proc Natl Acad Sci USA. 2010;107(11):5001–5005. doi: 10.1073/pnas.0909636107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shimamura T, et al. Molecular basis of alternating access membrane transport by the sodium-hydantoin transporter Mhp1. Science. 2010;328(5977):470–473. doi: 10.1126/science.1186303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Biarnés X, Bongarzone S, Vargiu AV, Carloni P, Ruggerone P. Molecular motions in drug design: The coming age of the metadynamics method. J Comput Aided Mol Des. 2011;25(5):395–402. doi: 10.1007/s10822-011-9415-3. [DOI] [PubMed] [Google Scholar]
- 10.Ovchinnikov V, Karplus M, Vanden-Eijnden E. Free energy of conformational transition paths in biomolecules: The string method and its application to myosin VI. J Chem Phys. 2011;134(8):085103. doi: 10.1063/1.3544209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Moradi M, Tajkhorshid E. Mechanistic picture for conformational transition of a membrane transporter at atomic resolution. Proc Natl Acad Sci USA. 2013;110(47):18916–18921. doi: 10.1073/pnas.1313202110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Krüger P, Verheyden S, Declerck PJ, Engelborghs Y. Extending the capabilities of targeted molecular dynamics: Simulation of a large conformational transition in plasminogen activator inhibitor 1. Protein Sci. 2001;10(4):798–808. doi: 10.1110/ps.40401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Faccioli P, Sega M, Pederiva F, Orland H. Dominant pathways in protein folding. Phys Rev Lett. 2006;97(10):108101. doi: 10.1103/PhysRevLett.97.108101. [DOI] [PubMed] [Google Scholar]
- 14.a Beccara S, Škrbić T, Covino R, Micheletti C, Faccioli P. Folding pathways of a knotted protein with a realistic atomistic force field. PLoS Comput Biol. 2013;9(3):e1003002. doi: 10.1371/journal.pcbi.1003002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.a Beccara S, Škrbić T, Covino R, Faccioli P. Dominant folding pathways of a WW domain. Proc Natl Acad Sci USA. 2012;109(7):2330–2335. doi: 10.1073/pnas.1111796109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Faccioli P. Characterization of protein folding by dominant reaction pathways. J Phys Chem B. 2008;112(44):13756–13764. doi: 10.1021/jp805762d. [DOI] [PubMed] [Google Scholar]
- 17.Faccioli P, Lonardi A, Orland H. Dominant reaction pathways in protein folding: A direct validation against molecular dynamics simulations. J Chem Phys. 2010;133(4):045104. doi: 10.1063/1.3459097. [DOI] [PubMed] [Google Scholar]
- 18.Gorlatova NV, Elokdah H, Fan K, Crandall DL, Lawrence DA. Mapping of a conformational epitope on plasminogen activator inhibitor-1 by random mutagenesis. Implications for serpin function. J Biol Chem. 2003;278(18):16329–16335. doi: 10.1074/jbc.M208420200. [DOI] [PubMed] [Google Scholar]
- 19.Hägglöf P, Bergström F, Wilczynska M, Johansson LB-Å, Ny T. The reactive-center loop of active PAI-1 is folded close to the protein core and can be partially inserted. J Mol Biol. 2004;335(3):823–832. doi: 10.1016/j.jmb.2003.11.005. [DOI] [PubMed] [Google Scholar]
- 20.Dupont DM, et al. Evidence for a pre-latent form of the serpin plasminogen activator inhibitor-1 with a detached β-strand 1C. J Biol Chem. 2006;281(47):36071–36081. doi: 10.1074/jbc.M606851200. [DOI] [PubMed] [Google Scholar]
- 21.Berkenpas MB, Lawrence DA, Ginsburg D. Molecular evolution of plasminogen activator inhibitor-1 functional stability. EMBO J. 1995;14(13):2969–2977. doi: 10.1002/j.1460-2075.1995.tb07299.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hansen M, Busse MN, Andreasen PA. Importance of the amino-acid composition of the shutter region of plasminogen activator inhibitor-1 for its transitions to latent and substrate forms. Eur J Biochem. 2001;268(23):6274–6283. doi: 10.1046/j.0014-2956.2001.02582.x. [DOI] [PubMed] [Google Scholar]
- 23.Lomas DA, Elliott PR, Chang W-SW, Wardell MR, Carrell RW. Preparation and characterization of latent α1-antitrypsin. J Biol Chem. 1995;270(10):5282–5288. doi: 10.1074/jbc.270.10.5282. [DOI] [PubMed] [Google Scholar]
- 24.Im H, Woo M-S, Hwang KY, Yu M-H. Interactions causing the kinetic trap in serpin protein folding. J Biol Chem. 2002;277(48):46347–46354. doi: 10.1074/jbc.M207682200. [DOI] [PubMed] [Google Scholar]
- 25.Elber R, Shalloway D. Temperature dependent reaction coordinates. J Chem Phys. 2000;112:5539. [Google Scholar]
- 26.Brown NJ. Therapeutic potential of plasminogen activator inhibitor-1 inhibitors. Ther Adv Cardiovasc Dis. 2010;4(5):315–324. doi: 10.1177/1753944710379126. [DOI] [PubMed] [Google Scholar]
- 27.Maddur AA, Swanson R, Izaguirre G, Gettins PGW, Olson ST. Kinetic intermediates en route to the final serpin-protease complex: Studies of complexes of α1-protease inhibitor with trypsin. J Biol Chem. 2013;288(44):32020–32035. doi: 10.1074/jbc.M113.510990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tsutsui Y, Liu L, Gershenson A, Wintrode PL. The conformational dynamics of a metastable serpin studied by hydrogen exchange and mass spectrometry. Biochemistry. 2006;45(21):6561–6569. doi: 10.1021/bi060431f. [DOI] [PubMed] [Google Scholar]
- 29.Trelle MB, et al. Hydrogen/deuterium exchange mass spectrometry reveals specific changes in the local flexibility of plasminogen activator inhibitor 1 upon binding to the somatomedin B domain of vitronectin. Biochemistry. 2012;51(41):8256–8266. doi: 10.1021/bi3008998. [DOI] [PubMed] [Google Scholar]
- 30.Lee KN, Im H, Kang SW, Yu MH. Characterization of a human α1-antitrypsin variant that is as stable as ovalbumin. J Biol Chem. 1998;273(5):2509–2516. doi: 10.1074/jbc.273.5.2509. [DOI] [PubMed] [Google Scholar]
- 31.Baek J-H, Yang WS, Lee C, Yu M-H. Functional unfolding of α1-antitrypsin probed by hydrogen-deuterium exchange coupled with mass spectrometry. Mol Cell Proteomics. 2009;8(5):1072–1081. doi: 10.1074/mcp.M800365-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Trelle MB, Madsen JB, Andreasen PA, Jørgensen TJD. Local transient unfolding of native state PAI-1 associated with serpin metastability. Angew Chem Int Ed Engl. 2014;53(37):9751–9754. doi: 10.1002/anie.201402796. [DOI] [PubMed] [Google Scholar]
- 33.Seo EJ, Lee C, Yu M-H. Concerted regulation of inhibitory activity of α1-antitrypsin by the native strain distributed throughout the molecule. J Biol Chem. 2002;277(16):14216–14220. doi: 10.1074/jbc.M110272200. [DOI] [PubMed] [Google Scholar]
- 34.Li S-H, et al. Mechanistic characterization and crystal structure of a small molecule inactivator bound to plasminogen activator inhibitor-1. Proc Natl Acad Sci USA. 2013;110(51):E4941–E4949. doi: 10.1073/pnas.1216499110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhou A, Huntington JA, Pannu NS, Carrell RW, Read RJ. How vitronectin binds PAI-1 to modulate fibrinolysis and cell migration. Nat Struct Biol. 2003;10(7):541–544. doi: 10.1038/nsb943. [DOI] [PubMed] [Google Scholar]
- 36.Irving JA, Pike RN, Lesk AM, Whisstock JC. Phylogeny of the serpin superfamily: Implications of patterns of amino acid conservation for structure and function. Genome Res. 2000;10(12):1845–1864. doi: 10.1101/gr.gr-1478r. [DOI] [PubMed] [Google Scholar]
- 37.Xue Y, et al. Interfering with the inhibitory mechanism of serpins: Crystal structure of a complex formed between cleaved plasminogen activator inhibitor type 1 and a reactive-centre loop peptide. Structure. 1998;6(5):627–636. doi: 10.1016/s0969-2126(98)00064-1. [DOI] [PubMed] [Google Scholar]
- 38.Wang J, Cieplak P, Kollman PA. How well does a restrained electrostatic potential (RESP) model perform in calculating conformational energies of organic and biological molecules? J Comput Chem. 2000;21:1049–1074. [Google Scholar]
- 39.Hess B, Kutzner C, van der Spoel D, Lindahl E. GROMACS 4: Algorithms for highly efficient, load-balanced, and scalable molecular simulation. J Chem Theory Comput. 2008;4:435–447. doi: 10.1021/ct700301q. [DOI] [PubMed] [Google Scholar]
- 40.Onufriev A, Bashford D, Case DA. Exploring protein native states and large-scale conformational changes with a modified generalized Born model. Proteins. 2004;55(2):383–394. doi: 10.1002/prot.20033. [DOI] [PubMed] [Google Scholar]
- 41.Stout TJ, Graham H, Buckley DI, Matthews DJ. Structures of active and latent PAI-1: A possible stabilizing role for chloride ions. Biochemistry. 2000;39(29):8460–8469. doi: 10.1021/bi000290w. [DOI] [PubMed] [Google Scholar]
- 42.Elliott PR, Pei XY, Dafforn TR, Lomas DA. Topography of a 2.0 A structure of α1-antitrypsin reveals targets for rational drug design to prevent conformational disease. Protein Sci. 2000;9(7):1274–1281. doi: 10.1110/ps.9.7.1274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fiser A, Sali A. ModLoop: Automated modeling of loops in protein structures. Bioinformatics. 2003;19(18):2500–2501. doi: 10.1093/bioinformatics/btg362. [DOI] [PubMed] [Google Scholar]
- 44.Lauck F, Smith CA, Friedland GF, Humphris EL, Kortemme T. RosettaBackrub—A web server for flexible backbone protein structure modeling and design. Nucleic Acids Res. 2010;38(Web Server issue):W569–W75. doi: 10.1093/nar/gkq369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Paci E, Karplus M. Forced unfolding of fibronectin type 3 modules: An analysis by biased molecular dynamics simulations. J Mol Biol. 1999;288(3):441–459. doi: 10.1006/jmbi.1999.2670. [DOI] [PubMed] [Google Scholar]
- 46.Camilloni C, Broglia RA, Tiana G. Hierarchy of folding and unfolding events of protein G, CI2, and ACBP from explicit-solvent simulations. J Chem Phys. 2011;134(4):045105. doi: 10.1063/1.3523345. [DOI] [PubMed] [Google Scholar]
- 47.Trott O, Olson AJ. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem. 2010;31(2):455–461. doi: 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.