

Abstract
Analysis of gene sets can implicate activity in signaling pathways that is responsible for cancer initiation and progression, but is not discernible from the analysis of individual genes. Multiple methods and software packages have been developed to infer pathway activity from expression measurements for set of genes targeted by that pathway. Broadly, three major methodologies have been proposed: over-representation, enrichment, and differential variability. Both over-representation and enrichment analyses are effective techniques to infer differentially regulated pathways from gene sets with relatively consistent differentially expressed (DE) genes. Specifically, these algorithms aggregate statistics from each gene in the pathway. However, they overlook multivariate patterns related to gene interactions and variations in expression. Therefore, the analysis of differential variability of multigene expression patterns can be essential to pathway inference in cancers. The corresponding methodologies and software packages for such multivariate variability analysis of pathways are reviewed here. We also introduce a new, computationally efficient algorithm, expression variation analysis (EVA), which has been implemented along with a previously proposed algorithm, Differential Rank Conservation (DIRAC), in an open source R package, gene set regulation (GSReg). EVA inferred similar pathways as DIRAC at reduced computational costs. Moreover, EVA also inferred different dysregulated pathways than those identified by enrichment analysis.
Keywords: gene set analysis, gene expression, variability analysis, multivariate analysis
Introduction
Cellular signaling generates a chain of protein–protein interactions, often terminating in the activation of transcription factors. Such signaling in molecular pathways induces and advances many human cancers. In principle, targeting the specific signaling pathways responsible for individual malignancies would yield an effective treatment. However, identifying the key signaling pathways relies on first inferring the signaling activity in that tumor. Ideally, coordinated changes in the phosphorylation state in network proteins could be measured to directly implicate specific signaling pathways in a malignancy, and the technology to measure such protein states is rapidly advancing. In the meantime, however, many algorithms use the existing transcriptional data to infer differentially regulated pathways. The accuracy of such inference relies in large part on the sets of genes annotated to each pathway (reviewed in Ref. 1–6). In analyses of gene expression data, it is essential to select sets of genes whose expression is altered because of pathway activation. For example, the TRANScription FACtor (TRANSFAC) database2 assembles experimentally validated sets of genes resulting from transcription factor activation. Using these data with set statistics to infer coordinated changes in targets of transcription factors downstream of cell signaling pathways has been an effective substitute for directly inferring differential pathway signaling (eg, Ochs et al.3, and Fertig et al.4). Regardless of the measurement technology, inference of signaling pathways thus requires statistical techniques to be able to account for changes in multiple molecular species.
Historically, analysis of differential pathway regulation from transcriptional data has been divided into two major classes of methodologies (reviewed in Irizzary et al.5): over-representation methods and enrichment methods. Over-representation methods compare sets of genes annotated to pathways to a list of those genes that are significantly differentially expressed (DE) between two phenotypes. Enrichment methods employ a “soft” version of over-representation based on a summary statistic to characterize the level of differential expression of genes in the pathway relative to a null distribution. These methods have been extended to infer pathway members or networks from transcriptional data (eg, Tarca et al.6).
Both over-representation and enrichment methods for detecting differential pathway regulation are robust at inferring consistent up- or down-regulation of pathway genes. However, alterations in cell signaling pathways may be associated with complex changes in gene expression because of pathway interactions.7 Moreover, expression in individual genes is highly variable in human tumors8,9 in part because of the distinct evolution of individual tumors from the same cancer subtype. Thus, individual genes may contribute differently to alterations in the same pathway. As a result, pathways that are dysregulated in human tumors may exhibit complex, multivariate changes in variability that are not captured by the aggregation of statistics of individual genes in over-representation or enrichment analyses. Here, we review more recent methods for detecting differential regulation based directly on multivariate measures of pathway variability. Specifically, we focus on Differential Rank Conservation (DIRAC),10 and a more computationally efficient alternative algorithm, expression variation analysis (EVA). We also introduce a new R package, gene set regulation (GSReg),11 that implements these algorithms to facilitate inference of pathway dysregulation.
Pathway Analysis Methodologies
In this section, we briefly review algorithms for pathway analysis from transcriptional data. Currently, all such algorithms identify significantly perturbed pathways by applying gene set statistics to compare gene expression of pathway targets in one phenotype to gene expression of pathway targets in another phenotype. As a result, they rely critically on the numerous curated databases that annotate genes to pathways.1 Regardless of the pathway targets, algorithms for pathway analysis can be divided into three major classes: over-representation, enrichment, and differential variability analyses. We list software that implements each technique in Table 1 and refer the reader to Irizarry et al.5, Khatri et al.12, and Maciejewski.13 for more reviews and comparisons of over-representation and enrichment analyses. An overview is provided in Figure 1.
Table 1.
Examples of software available for gene set analysis, divided into three major families of algorithms: over-representation, enrichment, and differential variability analyses.
Figure 1.
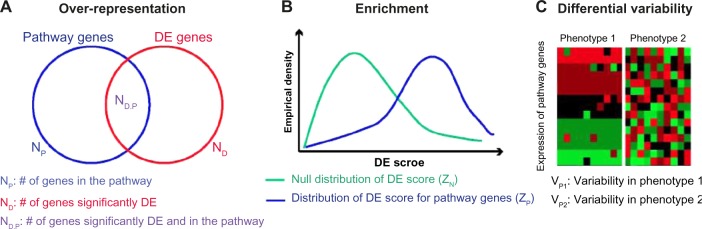
Pathway analysis methodologies from gene expression: (A) Over-representation analysis first performs a statistical test for each gene by comparing expression values in phenotypes to identify a set of significantly DE genes, obtaining a gene count ND. The procedure then counts the number of DE genes that are also annotated to a specified pathway (ND,P) and calculates a P-value for enrichment of that pathway by testing if ND,P is unusually high relative to ND and NP (the number of genes in the pathway). (B) Enrichment analysis first assigns an individual DE score to each of the genes annotated to a pathway, and aggregates these into a pathway score ZP. A similar score is computed for a null distribution, ZN. For example, this null distribution may be defined empirically from the DE score for alternative sets of genes or permuted sample labels. Enrichment analysis forms a pathway statistic by comparing the distribution of DE scores in ZP to that of DE scores in ZN. (C) Differential variability analysis defines a statistic to measure variability of the expression of pathway genes for samples from a given phenotype, denoted by VP1 and VP2 for phenotypes 1 and 2, respectively. If the variability between two phenotypes is significantly high (ie, |VP1 − VP2| >> 0), the pathway is identified as dysregulated.
The first methodology, over-representation analysis, assesses similarity between the set of all DE genes and the set of genes annotated to a pathway (Fig. 1A), and was introduced in Khatri et al.14 First, significantly DE genes between specified phenotypes are identified. For example, a set of DE genes may be defined by computing a Wilcoxon test to compare expression in two phenotypes for each gene measured and selecting significant genes as those having a false discovery rate below a threshold value of 5%. Then a gene set statistic is calculated for each pathway by applying a statistical test (eg, Fisher’s test) that compares each set of pathway genes to the set of DE genes. Pathways whose members are significantly enriched for DE genes are called significant. The methods listed in top of the Table 1 are examples from this family.
Whereas over-representation analysis compares discrete sets of genes, the second methodology – enrichment analysis – formulates set statistics that summarize the overall level of differential expression for the pathway genes between the phenotypes. The first method of this class was gene set enrichment analysis (GSEA).15 Generally, enrichment analysis calculates the differential activity of genes across phenotypes using a differential expression statistic (eg, a t- or Z-statistic). Then the differential activity of a pathway is calculated by applying another statistic (eg, Kolmogrov–Smirnov test, sum, mean, maxmean statistic, etc.) to compare the differential expression statistic for genes in the pathway to a null distribution of differential expression statistics, often defined from alternative sets of genes or permuting sample labels. The algorithms in the middle of Table 1 represent examples from this family accompanied by the software implementing them. A full review of these algorithms is provided in Khatri.12
Although they use different statistics, both over- representation and enrichment methods infer coordinated, average expression changes between phenotypes in sets of genes annotated to a pathway. Because they do not rely on a hard threshold, enrichment methods are more sensitive than over-representation methods at inferring coordinated expression changes in sets of genes. However, they may yield many more false positives. Regardless of their relative advantages, the false-positive rate of both tests may be dependent upon the number of genes in the set.41 Moreover, both methods perform the best when changes in genes annotated to a pathway are consistent and relatively homogeneous in each phenotype; for example, sharply different expression values for a given gene are seen in most samples. However, tumor pathway dysregulation based on interactions among multiple genes may cause differential variability in gene expression between phenotypes. Therefore, the third family of the methods, differential variability analysis, is a multivariate approach that assesses variability within a pathway for a given phenotype and then compares these measures across phenotypes. This emerging methodology, pioneered in Eddy et al.10, and Zhang et al.38, has been extended to a broad set of algorithms summarized in Table 1, and is the focus of the remainder of this paper.
Differential Variability Analysis
Differential variability analysis first measures the level of variability in gene expression in a pathway for a given phenotype and then compares these levels for different phenotypes to determine differential pathway regulation. For example, different pathway genes may have expression outliers in distinct tumor samples relative to normal controls, captured with methods such as Open Grid Services Architecture (OGSA).42 Such distinct alterations in individual tumors may also increase variability of expression in individual genes, motivating approaches that apply over-representation or enrichment analysis to variability statistics for individual genes.43 Nonetheless, in general, alterations in expression may depend strongly on interactions among the genes in the pathway. Consequently, new algorithms employing multivariate statistics are emerging in order to model such complex shifts in variability from one phenotype to another.
Zhang et al.38 developed one of the first methods of this type. Their algorithm calculates correlations between all pairs of genes within a pathway given a phenotype as a measure of pairwise interactions, and then a z-score for the difference in pairwise interactions between two phenotypes. To summarize the change in the correlation pattern, the algorithm applies a “maxmean” statistic to compute the maximum of the mean of positive and negative z-scores corresponding to all gene pairs in the pathway and then ranks pathways by this maxmean statistic. Watkinson et al.40 extended this algorithm by defining synergy between pairs of genes, using an information-theoretic approach. Recently, Liu et al.37 have developed a more sophisticated analysis of variability called gene interaction enrichment and network analysis (GIENA). Instead of correlation, they consider four possible statistics on the expression of two genes: their sum, difference, maximum, and minimum. These operations are assumed to correspond to gene pair cooperation, competition, redundancy, and dependency, respectively. Thereafter, pathway activity is summarized by applying a maxmean statistic over all pairs of interactions within the gene set, similar to Zhang et al.38 In contrast to both Liu et al.37, and Zhang et al.38, Ochs et al.42 provide a formulation for pathway analysis based upon outliers to account for pathway dysregulation and tumor heterogeneity, thereby utilizing a simpler algorithm that does not rely on selecting a variability statistic.
Regrettably, none of the algorithms described above provide a robust software package to facilitate application to new data. They also rely on continuous, normalized gene expression measurements. We have previously shown that rank-based techniques (ie, methods that depend only on the relative ordering of expression values) (i) are more robust to the preprocessing and normalization of data44 than techniques relying on normalized gene expression, (ii) are competitive with the best classification methods in discriminating among phenotypes (eg, Geman et al.45), and (iii) can be far simpler to explain and interpret in biological terms.46,47 Therefore, Eddy et al proposed DIRAC10 as an ordering-based method for differential variability analysis. Given a pathway and a phenotype, DIRAC generates a binary template (one component for each pair of genes) for the ordering of the expression values for the genes in the pathway, and then calculates the average “distance” between training samples and the template as the measure of the pathway variability of the phenotype. The “distance” used in DIRAC involves the Hamming distance over the pairwise comparisons. Permutation tests are used to estimate P-values associated with differences in this variability score between phenotypes, and pathways with significant P-values are identified as perturbed. Consistent with increased complexity in more advanced stages of diseases,10 they found that most dysregulated pathways have higher variability in phenotypes with worse prognosis.
Although DIRAC is effective in inferring dysregulated pathways, the permutation test on which it is based is computationally inefficient, and becomes infeasible when applied to large numbers of pathways and samples. Therefore, we propose an alternative approach called EVA.36 Given a phenotype and pathway, EVA measures the average distance between two randomly chosen expression profiles for the phenotype. More specifically, the EVA variability statistic is the expected Kendall-τ distance48 between the rank vectors corresponding to two independent copies of expression profiles over the pathway. Kendall-τ is a distance that quantifies the difference between the orderings of two vectors. In this case, the permutation distance is defined for the rank vectors of gene expression profiles for pathway genes. The Kendall-τ distance between the two gene expression profiles is essentially the number of disagreeing comparisons between all pairs of genes in the pathway, analogous to the change of rank in DIRAC. To estimate a variability statistic from samples in each phenotype, the EVA algorithm then averages the Kendall-τ distance between each pair of samples from that phenotype. These variability statistics are then compared between two phenotypes for each pathway to estimate pathway dysregulation between phenotypes. The P-values for pathway deregulation are computed analytically from the difference between the empirical Kendall-τ statistics using an approximation for the asymptotic distribution from the theory of U-statistics described in detail in Afsari et al.36 A general description about U-statistics can be found in Van der Vaart.49
GSReg Package
We develop GSReg R package to perform differential variability analysis using DIRAC and EVA, available through Bioconductor. Here, we demonstrate our software by reproducing the results from the DIRAC paper and replicating these results with EVA. Since the original data of DIRAC paper was in Matlab format, we provide the data in a complementary R package, GSBenchMark,50 also available through Bioconductor.
Figure 2 shows the results of variability pathway analysis comparing head and neck squamous cell carcinoma samples to matched normal controls.51 Figure 2 compares variability statistics of pathways in tumors (y-axis) to normal controls (x-axis), revealing that most of the dysregulated pathways have higher variability in tumor samples than normal samples. This was the general trend found for DIRAC10 (Fig. 2A) and persists for EVA (Fig. 2B). In total, DIRAC found 48 dysregulated pathways and EVA discovered 64; there are 45 pathways in common and 68 in total. The general trend that most of the dysregulated pathways have higher variability in the phenotype with poor prognosis remains true for EVA in other datasets compared in DIRAC and provided in GSBenchMark (results not shown).
Figure 2.
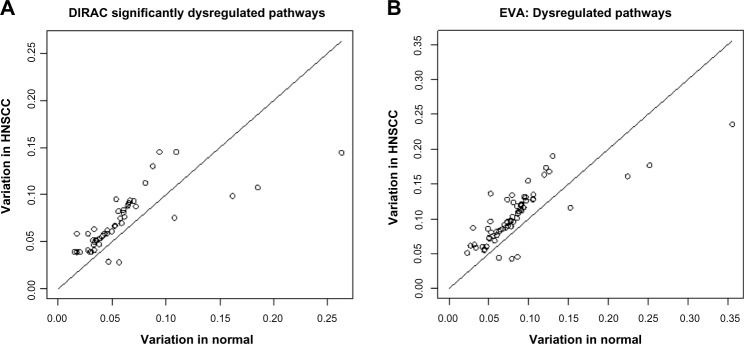
Comparison of dysregulated pathways identified by (A) DIRAC and (B) EVA in comparing head and neck squamous cell carcinoma samples (y-axis) and normal samples (x-axis). Hence, the pathways shown above the line are those with significantly (P-value <0.05) higher variability in tumor than normal samples, and those below the line have significantly higher variability in normal samples.
DIRAC and EVA have been shown mathematically similar.36 The main advantages of the EVA are efficiency in calculation and a more straightforward interpretation that does not involve a “template” but rather is simply the average distance between two samples. To illustrate the computational advantage, for the head and neck cancer data, using a Lenovo Think-Pad with Core™ i7–3720QM Intel CPU at 2.6 GHz and only 1000 permutations of phenotype labels, the DIRAC analysis required 207 seconds while the EVA analytical computation only took 0.3 seconds. Figure 3 compares the corresponding P-values of the differential variability measure generated by DIRAC and EVA. These P-values are highly correlated, with a 0.88 Pearson correlation coefficient (P-value <2 × 10−16). Taken together, these results indicate that EVA can be used as a more efficient alternative for DIRAC analysis.
Figure 3.
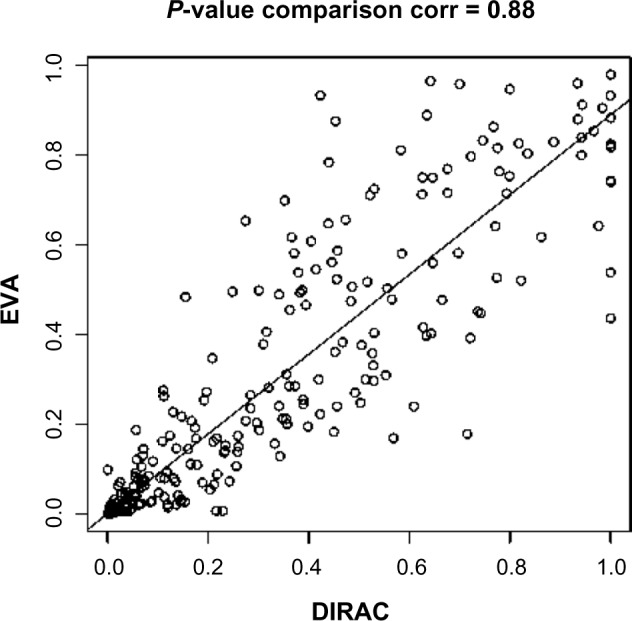
P-value comparison of DIRAC and EVA: Each circle represents a pathway. x-axis and y-axis represent DIRAC and EVA P-values, respectively.
To illustrate the difference between the outcomes of EVA and enrichment analysis, we chose a well-known enrichment method, the Wilcoxon gene set test implemented in Linear Models for Microarray Data (LIMMA).31 For these analyses, we apply the Benjamini–Hotchberg procedure52 to account for multiple hypothesis testing, which was not feasible in the previous comparison with the DIRAC analysis because of the relatively coarse resolution of P-values from the computationally intensive permutation test. In the case of the head and neck squamous tumors, both LIMMA and EVA infer a similar number of differentially regulated pathways (11 and 21, respectively). However, consistent with the test statistic, the significant pathways from EVA have consistently higher variability in the tumor group than those identified with the enrichment statistic. On the other hand, if we apply LIMMA on the univariate F-test statistic for the difference of variances, LIMMA does not identify any pathway as dysregulated. This shows that analyses based on differential variability and enrichment may result in different outcomes.
Conclusion
Cancer is known to be the result of the perturbations in signaling pathways. Many algorithms have been proposed to identify and analyze these perturbations from transcriptional data. We reviewed three major families of pathway analysis methods, each having different criteria for calling a pathway perturbed: over-representation of DE genes, enrichment of large DE statistics in pathway genes, and significant difference in variability of gene expression. This last class of methods is particularly adept at inferring dysregulated pathways with differential variability in multivariate gene expression patterns. Here, we implemented one such variability analysis algorithm, DIRAC,10 and a novel, more efficient alternative EVA in an R package GSReg.
For future work, methods that incorporate more information about biological mechanism may enhance interpretation and reproducibility of learned dysregulated pathways. Also, methods that can assess variability across more than two phenotypes are needed to infer dysregulated pathways in distinct tumor subtypes. Moreover, existing methods for gene set analysis either detect the differential expression or differential variability to identify differential regulation across phenotypes. A more versatile methodology might be a combination of both types of pathway analyses. These combinations may be implemented by using the Kendall-τ distance to compare two independent samples, but from two different phenotypes. Thus, extending the sample comparisons in EVA would provide an algorithm to compare pathway variability within phenotypes with pathway variability between phenotypes.
Figure 4.
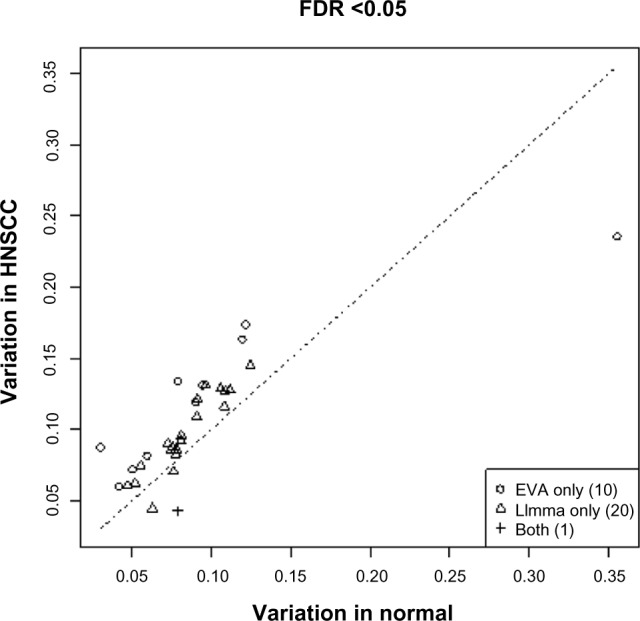
Comparison of dysregulated pathways identified by EVA and LIMMA analysis. Each point represents a pathway, and we compare samples from head and neck squamous cells (y-axis) and normal samples (x-axis). The pathways are detected by at least one of the algorithms significant. This figure shows that LIMMA and EVA may produce different analyses.
Footnotes
ACADEMIC EDITOR: JT Efird, Editor in Chief
FUNDING: EJF and BA acknowledge funding from NCI (CA141053), and EJF further support from the Cleveland Foundation. The work of DG was partially supported by the National Science Foundation under NSF DMS1228248. The authors confirm that the funder had no influence over the study design, content of the article, or selection of this journal.
COMPETING INTERESTS: Authors disclose no potential conflicts of interest.
Paper subject to independent expert blind peer review by minimum of two reviewers. All editorial decisions made by independent academic editor. Prior to publication all authors have given signed confirmation of agreement to article publication and compliance with all applicable ethical and legal requirements, including the accuracy of author and contributor information, disclosure of competing interests and funding sources, compliance with ethical requirements relating to human and animal study participants, and compliance with any copyright requirements of third parties.
Author Contributions
Conceived and designed the experiments: BA, DG, EJF. Analyzed the data: BA. Wrote the first draft of the manuscript: BA. Contributed to the writing of the manuscript: BA, DG, EJF. Agree with manuscript results and conclusions: BA, DG, EJF. Jointly developed the structure and arguments for the paper: BA, DG, EJF. Made critical revisions and approved final version: DG, EJF. All authors reviewed and approved of the final manuscript.
REFERENCES
- 1.Bauer-Mehren A, Furlong LI, Sanz F. Pathway databases and tools for their exploitation: benefits, current limitations and challenges. Mol Syst Biol. 2009;5:290. doi: 10.1038/msb.2009.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Matys V, Fricke E, Geffers R, et al. TRANSFAC: transcriptional regulation, from patterns to profiles. Nucleic Acids Res. 2003;31:374–8. doi: 10.1093/nar/gkg108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ochs MF, Rink L, Tarn C, et al. Detection of treatment-induced changes in signaling pathways in gastrointestinal stromal tumors using transcriptomic data. Cancer Res. 2009;69:9125–32. doi: 10.1158/0008-5472.CAN-09-1709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fertig EJ, Ren Q, Cheng H, et al. Gene expression signatures modulated by epidermal growth factor receptor activation and their relationship to cetuximab resistance in head and neck squamous cell carcinoma. BMC Genomics. 2012;13:160. doi: 10.1186/1471-2164-13-160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Irizarry RA, Wang C, Zhou Y, Speed TP. Gene set enrichment analysis made simple. Stat Methods Med Res. 2009;18(6):565–75. doi: 10.1177/0962280209351908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tarca AL, Draghici S, Khatri P, et al. A novel signaling pathway impact analysis. Bioinformatics. 2009;25:75–82. doi: 10.1093/bioinformatics/btn577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Burrell RA, McGranahan N, Bartek J, Swanton C. The causes and consequences of genetic heterogeneity in cancer evolution. Nature. 2013;501:338–45. doi: 10.1038/nature12625. [DOI] [PubMed] [Google Scholar]
- 8.MacDonald JW Ghosh D. COPA – cancer outlier profile analysis. Bioinformatics. 2006;22:2950–1. doi: 10.1093/bioinformatics/btl433. [DOI] [PubMed] [Google Scholar]
- 9.Bravo HC, Pihur V, McCall M, Irizarry RA, Leek JT. Gene expression anti-profiles as a basis for accurate universal cancer signatures. BMC Bioinformatics. 2012;13:272. doi: 10.1186/1471-2105-13-272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Eddy J, Hood L, Price N, Geman D. Identifying tightly regulated and variably expressed networks by Differential Rank Conservation. PLoS Comput Biol. 2010;6(5):e1000792. doi: 10.1371/journal.pcbi.1000792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Afsari B, Fertig EJ. GSReg: A Package for Gene Set Variability Analysis. R Package Version 0.99 3.
- 12.Khatri P, Sirota M, Butte AJ. Ten years of pathway analysis: current approaches and outstanding challenges. PLoS Comput Biol. 2012;8:e1002375. doi: 10.1371/journal.pcbi.1002375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Maciejewski H. Gene set analysis methods: statistical models and methodological differences. Brief Bioinform. 2013:bbt002. doi: 10.1093/bib/bbt002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Khatri P, Draghici S, Ostermeier GC, Krawetz SA. Profiling gene expression using onto-express. Genomics. 2002;79:266–70. doi: 10.1006/geno.2002.6698. [DOI] [PubMed] [Google Scholar]
- 15.Subramanian A, Tamayo P, Mootha VK, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci USA. 2005;102:15545–50. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dahlquist KD, Salomonis N, Vranizan K, Lawlor SC, Conklin BR. GenMAPP, a new tool for viewing and analyzing microarray data on biological pathways. Nat Genet. 2002;31:19–20. doi: 10.1038/ng0502-19. [DOI] [PubMed] [Google Scholar]
- 17.Doniger SW, Salomonis N, Dahlquist KD, Vranizan K, Lawlor SC, Conklin BR. MAPPFinder: using gene ontology and GenMAPP to create a global gene-expression profile from microarray data. Genome Biol. 2003;4:R7. doi: 10.1186/gb-2003-4-1-r7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zeeberg BR, Feng W, Wang G, et al. GoMiner: a resource for biological interpretation of genomic and proteomic data. Genome Biol. 2003;4:R28. doi: 10.1186/gb-2003-4-4-r28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Al-Shahrour F, Díaz-Uriarte R, Dopazo J. FatiGO: a web tool for finding significant associations of gene ontology terms with groups of genes. Bioinformatics. 2004;20:578–80. doi: 10.1093/bioinformatics/btg455. [DOI] [PubMed] [Google Scholar]
- 20.Beißbarth T Speed TP. GOstat: find statistically overrepresented gene ontologies within a group of genes. Bioinformatics. 2004;20:1464–5. doi: 10.1093/bioinformatics/bth088. [DOI] [PubMed] [Google Scholar]
- 21.Berriz GF, King OD, Bryant B, Sander C, Roth FP. Characterizing gene sets with FuncAssociate. Bioinformatics. 2003;19:2502–4. doi: 10.1093/bioinformatics/btg363. [DOI] [PubMed] [Google Scholar]
- 22.Martin D, Brun C, Remy E, Mouren P, Thieffry D, Jacq B. GOToolBox: functional analysis of gene datasets based on gene ontology. Genome Biol. 2004;5:R101. doi: 10.1186/gb-2004-5-12-r101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Castillo-Davis CI, Hartl DL. GeneMerge – post-genomic analysis, data mining, and hypothesis testing. Bioinformatics. 2003;19:891–2. doi: 10.1093/bioinformatics/btg114. [DOI] [PubMed] [Google Scholar]
- 24.Zheng Q, Wang XJ. GOEAST: a web-based software toolkit for gene ontology enrichment analysis. Nucleic Acids Res. 2008;36:W358–63. doi: 10.1093/nar/gkn276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bindea G, Mlecnik B, Hackl H, et al. ClueGO: a Cytoscape plug-in to decipher functionally grouped gene ontology and pathway annotation networks. Bioinformatics. 2009;25:1091–3. doi: 10.1093/bioinformatics/btp101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Robinson MD, Grigull J, Mohammad N, Hughes TR. FunSpec: a web-based cluster interpreter for yeast. BMC Bioinformatics. 2002;3:35. doi: 10.1186/1471-2105-3-35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Boyle EI, Weng S, Gollub J, et al. GO:TermFinder – open source software for accessing gene ontology information and finding significantly enriched gene ontology terms associated with a list of genes. Bioinformatics. 2004;20:3710–5. doi: 10.1093/bioinformatics/bth456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang B, Kirov S, Snoddy J. WebGestalt: an integrated system for exploring gene sets in various biological contexts. Nucleic Acids Res. 2005;33:W741–8. doi: 10.1093/nar/gki475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Du Z, Zhou X, Ling Y, Zhang Z, Su Z. agriGo: a GO analysis toolkit for the agricultural community. Nucleic Acids Res. 2010;38:W64–70. doi: 10.1093/nar/gkq310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Barry WT, Nobel AB, Wright FA. Significance analysis of functional categories in gene expression studies: a structured permutation approach. Bioinformatics. 2005;21:1943–9. doi: 10.1093/bioinformatics/bti260. [DOI] [PubMed] [Google Scholar]
- 31.Smyth GK. Limma: linear models for microarray data. In: Gentleman R, et al., editors. Bioinformatics and Computational Biology Solutions Using R and Bioconductor. New York, NY: Springer; 2005. pp. 397–420. [Google Scholar]
- 32.Jr GD, Sherman BT, Hosack DA, et al. David: database for annotation, visualization, and integrated discovery. Genome Biol. 2003;4(5):3. [PubMed] [Google Scholar]
- 33.Alexa A, Rahnenfuhrer J. topGO: Enrichment Analysis for Gene Ontology. 2010. R Package Version 28.
- 34.Luo W, Friedman MS, Shedden K, Hankenson KD, Woolf PJ. GAGE: generally applicable gene set enrichment for pathway analysis. BMC Bioinformatics. 2009;10:161. doi: 10.1186/1471-2105-10-161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lai W, Tian L, Park P. sigPathway: Pathway Analysis with Microarray Data. 2013.
- 36.Afsari B. PhD thesis. Baltimore, MD: Johns Hopkins University; 2013. Modeling Cancer Phenotypes with Order Statistics of Transcript Data. [Google Scholar]
- 37.Liu Y, Koyutürk M, Barnholtz-Sloan JS, Chance MR. Gene interaction enrichment and network analysis to identify dysregulated pathways and their interactions in complex diseases. BMC Syst Biol. 2012;6:65. doi: 10.1186/1752-0509-6-65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhang J, Li J, Deng HW. Identifying gene interaction enrichment for gene expression data. PLoS One. 2009;4:e8064. doi: 10.1371/journal.pone.0008064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tsai CA, Chen JJ. Multivariate analysis of variance test for gene set analysis. Bioinformatics. 2009;25:897–903. doi: 10.1093/bioinformatics/btp098. [DOI] [PubMed] [Google Scholar]
- 40.Watkinson J, Wang X, Zheng T, Anastassiou D. Identification of gene interactions associated with disease from gene expression data using synergy networks. BMC Syst Biol. 2008;2:10. doi: 10.1186/1752-0509-2-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Goeman JJ, Bühlmann P. Analyzing gene expression data in terms of gene sets: methodological issues. Bioinformatics. 2007;23:980–7. doi: 10.1093/bioinformatics/btm051. [DOI] [PubMed] [Google Scholar]
- 42.Ochs MF, Farrar JE, Considine M, Wei Y, Meshinchi S, Arceci RJ. Outlier analysis and top scoring pair for integrated data analysis and biomarker discovery. IEEE/ACM Trans Comput Biol Bioinform. 2013:1–1. doi: 10.1109/TCBB.2013.153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ho JW, Stefani M, dos Remedios CG, Charleston MA. Differential variability analysis of gene expression and its application to human diseases. Bioinformatics. 2008;24:i390–8. doi: 10.1093/bioinformatics/btn142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Eddy JA, Sung J, Geman D, Price ND. Relative expression analysis for molecular cancer diagnosis and prognosis. Technol Cancer Res Treat. 2010;9:149. doi: 10.1177/153303461000900204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Geman D, Afsari B, AC Tan DN. Winner, ICMLA Microarray Classification Algorithm Competition. San Diego, CA, USA: 2008. Microarray classification from several two-gene expression comparisons. [Google Scholar]
- 46.Marchionni L, Afsari B, Geman D, Leek JT. A simple and reproducible breast cancer prognostic test. BMC Genomics. 2013;14:336. doi: 10.1186/1471-2164-14-336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Afsari B, Braga-Neto U, Geman D. Rank discriminants for predicting phenotypes from RNA expression. Ann Appl Stat. 2014 [Google Scholar]
- 48.Kendall MG. A new measure of rank correlation. Biometrika. 1938 [Google Scholar]
- 49.Van der Vaart AW. Asymptotic Statistics. Vol. 3. New York, NY: Cambridge University Press; 2000. [Google Scholar]
- 50.Afsari B, Fertig E. Gene Set BenchMark. R Package Version 0.99 3.
- 51.Kuriakose MA, Chen WT, He ZM, et al. Selection and validation of differentially expressed genes in head and neck cancer. Cell Mol Life Sci. 2004;61:1372–83. doi: 10.1007/s00018-004-4069-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc Series B Stat Methodol. 1995;57(1):289–300. [Google Scholar]