

Abstract
In a tumor cell, the development of acquired therapeutic resistance and the ability to survive in extracellular environments that differ from the primary site are the result of molecular adaptations in potentially highly plastic molecular networks. The accurate prediction of intracellular networks in a tumor remains a difficult problem in cancer informatics. In order to make truly rational patient-driven therapeutic decisions, it will be critical to develop methodologies that can accurately infer the molecular circuitry in the cells of a specific tumor. Despite enormous heterogeneity, cellular networks elicit deterministic digital-like responses. We discuss the use and limitations of methodologies that model molecular networks in cancer cells as a digital circuit. We also develop a network model of Notch signaling in colon cancer using a novel reverse engineering logic-based method and published western blot data to elucidate the interactions likely present in the circuits of the SW480 colon cancer cell line. Within this framework, we make predictions related to the role that honokiol may be playing as an anti-cancer drug.
Keywords: Notch, honokiol, reverse engineering, signaling pathways, logic networks
Background
The targeted inhibition of specific molecules in signaling networks has emerged as a leading anti-cancer strategy. The large number of off-target effects associated with targeted therapies in cancer has been termed as the “whack a mole problem”1 because modulation of one molecular target often results in the activation of another non-targeted molecule. In the cancer research community, off-target effects are generally attributed to non-specific drug interactions or nonlinear feedback connections within an intracellular network, but may also be due to retroactive signaling.2 Moreover, as a consequence of the varied ways an individual cell may adapt to changes in its local environment, the connectivity and inter-functional dependence of molecular networks is potentially unique to each sub-clonal population within a tumor. Determination of precisely how key dysregulated networks are wired as an integrated network circuit in tumor cells as well as in surrounding normal tissue will greatly enhance the search for more rational and patient-specific therapeutic interventions.
The Notch pathway plays a complex role in the tumorigenesis of both hematologic and solid tissues.3 Notch signaling is an evolutionarily conserved pathway that plays a critical role in embryonic development by tightly regulating cellular differentiation, angiogenesis, proliferation, and apoptosis.3–6 There is increasing evidence that tumor-initiating cells (also known as cancer stem cells) are dependent on Notch signaling.4,5 Notch pathway crosstalk is not well understood, and a paradoxical relationship between RAS and Notch signaling has been reported in the literature.7 Unraveling the complex circuitry of Notch signaling in both normal and malignant tissue is of critical importance in cancer.
The accurate prediction of intracellular networks from data remains a difficult problem in molecular systems biology and bioinformatics. A number of reverse engineering methods have been developed8–11 to attempt to infer intracellular networks from experimental steady state or time course protein concentration data, which are often collected from a series of perturbation experiments. While some of these methods have proven highly predictive, one limitation is that they often require prohibitive amounts of experimental data to be predictive. The dialogue on reverse-engineering assessment and methods (DREAM)12 was developed to encourage the innovation of reverse engineering approaches for the prediction of gene regulatory networks from high throughput datasets, and a number of networks generated by teams participating in DREAM challenges have proven highly predictive. While the networks predicted as part of these challenges are directional (indicating causal relationships), they are unsigned (which means they do not provide a prediction of whether an interaction is activating or inhibiting). The methodology we rely on in this work attempts to reverse engineer not only the sign of the interactions but also the logical hier archy when multiple nodes regulate the same target.
Are cellular networks digital circuits?
Individual cells exist in diverse states depending on environmental cues; and, in spite of their heterogeneity, they often exhibit deterministic responses to stimuli. Is it, therefore, reasonable to model intracellular molecular networks as digital circuits? A number of switch-like all-or-nothing type cellular responses have been observed experimentally. For example, signaling cascades often exhibit an ultrasensitive and highly switch-like response.13,14 Ferrell et al.15 showed the all-or-nothing switch-like character of the MAPK cascade in Xenopus oocyte maturation in response to a continuously variable stimulus. Cytokine secretion in T-cells has also been characterized as digital.16,17 Notch ligand regulation can generate all-or-nothing responses that induce cellular phenotypes in a variety of animal models.18,19 The endoplasmic reticulum has sophisticated stress signaling pathways, which switches off protein synthesis and activates key chaperones to improve effective protein processing.20,21 Stem cells convert graded signaling to digital outputs that promote or repress proliferation in an all-or-nothing manner.22
Lahav et al.23 showed the digital-like response of p53 oscillations in response to ionizing radiation (IR). In their experiments, damped p53 oscillations were measured in cells after exposure to radiation. They observed p53 and MDM2 oscillations that strongly resembled digital behavior. DNA damage from the IR lead to lower MDM2 levels that stabilized p53 levels, which, in turn, allowed p53 activity to increase and, consequently, MDM2 expression to also increase. The mean height and width of the pulses observed corresponded to the oscillatory p53 activity peaks but did not vary with the irradiation dose, indicating that the strength of output did not correlate with the strength of the input.23
Recently, Tamsir et al.24 molecularly engineered simple logic gates linked by diffusible chemical signals in bacteria. This was accomplished by compartmentalizing logic gates between strains of bacteria and using quorum sensing to allow for communication between strains, combining two methods of digital computation. In this work, promoters served as the inputs for logic gates, offering modularity through the ability to switch inputs and induce the corresponding responses by arranging genetic promoters and repressors of specific genetic targets according to a desired Boolean logic rules.
Building logic-based network models of cancer
All these examples support the notion that cellular molecular networks behave similar to digital electronic circuits. In an analog circuit, an increase in signal strength monotonically corresponds to the strength of the output, while in a binary digital system the same output, regardless of the strength of input, is observed.
We view logic networks as models of the signal transfer in a digital circuit that hold great promise for inferring biological networks in cancer cells. A number of predictive logic-based network models of cancer exist in the literature.25–27 Two-state logic-based models (which simulate the functional activation and deactivation of each molecule represented as a node in the network) can be powerful tools for investigating complex intracellular molecular networks because they are qualitatively predictive and require little to no prior knowledge of the parameter values and mechanistic details that are essential for deterministic and quantitatively precise kinetic methods.28–31 Fuzzy logic and multistate logic-based models (which permit nodes in the network to be in more than two states) are also powerful but require parameter estimates that are not always easy to correlate with biophysical chemical theory.31
Molecular perturbation experiments that provide readouts of key nodes in a network reveal important clues about the structure of a network. Logic methods and perturbation experiments can be leveraged to infer the underlying structure of a molecular network. In this work, we use a logic-based network modeling framework to simulate Notch signaling in the SW480 colon cancer cell line. We relied on data from perturbation experiments recently published by Ponnurangam et al.5 and develop and employ a novel reverse engineering approach to predict the interactions most likely at work in these cells. We also make predictions of direct and indirect interactions of honokiol and ionizing radiation in SW480 cells.
Results
Notch is a transmembrane receptor protein consisting of two primary subunits. Upon ligand binding, Notch is cleaved by the γ-secretase complex, and the Notch intracellular domain (NICD) is released in the cytoplasm where it translocates to the nucleus and promotes the transcription of a number of target genes. In mammalian cells there are five primary ligands for Notch: DLL1, DLL3, DLL4, Jagged 1, and Jagged 2.3–6,18,19 Through transmembrane receptor ligand binging, Notch can also play a role in synchronizing signaling between cells (Fig. 1A).
Figure 1.

Honokiol and ionizing radiation (IR) affect Notch signaling. (A) Upon binding to a ligand, the transmembrane Notch receptor is cleaved by γ-secretase, allowing the intracellular domain to translocate to the nucleus, where it can activate a variety of transcription programs, including that of its own ligand. Notch signaling can, therefore, play a role in synchronizing signaling across cells in a population. (B) Western blot readouts of proteins affected by honokiol and/or IR from Ponnurangam et al.5 are represented as discrete digital values of activate and inactive states. (C) The initial proposed interaction network describing the effect honokiol and IR on cellular signaling with cross talk to the Notch pathway. Black edges with arrows indicate activating regulations and red edges with blunt ends indicate inhibiting regulations.
Model development and data discretization
In a recent study, Ponnurangam et al.5 investigated the effects of combined therapy of honokiol and IR in colon cancer. Part of their study involved perturbations of the SW480 cell line treated with or without honokiol and/or IR. Honokiol, which is isolated from Magnolia officinalis, has anti-inflammatory and antioxidant activity, and can induce apoptosis or inhibit proliferation in several cancer cell lines.5,32 The four primary perturbation conditions used in their study were (1) control (no honokiol and no IR), (2) honokiol only, (3) IR only, and (4) both honokiol and IR. The authors reported readouts of proliferation and apoptosis under the four perturbation conditions as well as western blot (WB) data for a number of key proteins involved in Notch signaling. We converted the data for CDK1, MYC, BAX, Jagged, NICD, and HES (along with the reported readouts of proliferation and apoptosis) to discrete digital readouts (Fig. 1B, Supplementary Table S1) of either ON or OFF. It is important to note what ON and OFF values represent in a logic network. A node state of ON implicitly assumes that the molecule represented by the node is at an expression or activity level that is above the threshold needed to induce a regulatory change in the molecules it regulates. Similarly, a node state of OFF implicitly assumes the node is at an expression or activity level that is below the threshold.31 Because the data reported5 provide qualitative readouts of protein levels but do not include activation readouts (eg, from phospho- WB data), the model we constructed implicitly assumes that the presence of the protein corresponds to the active state of the protein.
Construction of an initial hypothesized interacting network
We first constructed a hypothesized interaction network using knowledge from the literature (Fig. 1C). Cyclin D1, c-MYC, and P27 are involved in the regulation of cell cycle progression and proliferation,33–35 while Bax plays a key regulatory role in controlling apoptosis.36 In our model, Jagged represents the ligand for Notch because it was the only ligand measured in the experiment and, along with HES, is one of several transcription targets downstream of NICD signaling. For simplicity we did not include a separate node for Notch in the model and instead allowed the NICD node to represent both the expressed receptor as well as the cleaved protein domain. We also included activation of AKT and RAS by serum in our model. The data were collected from experiments performed in rich media containing serum, which is known to activate AKT and ERK (a kinase downstream of RAS).37,38 RAS is mutated in SW480 cells, which is positive for H-RAS, K-RAS, and N-RAS expression. In a separate study, a WB performed in SW480 in rich media showed RAS to be in the active GTP bound state under control conditions.39 We therefore assumed the presence of serum is sufficient to activate both AKT and RAS.
A significant amount of data indicates that honokiol has a variety of functions in the cell (including anti-inflammatory, anti-angiogenic, anti-viral, and anti-tumor functions).40,41 While the exact pharmacological mechanisms of honokiol are still being elucidated, they are thought to include interactions with several targets including the inhibition of AKT.41 Given its reported role in modulating proliferation, we initially hypothesized that honokiol may also directly or indirectly alter the RAS pathway (a hypothesis that is supported by data).42 Moreover, in developmental systems, Notch signaling involves the negative repression of a downstream gene target by its protein product, such as the her1/7 genes in zebrafish.43,44 We, therefore, hypothesized that it is possible for the HES protein (a mammalian ortholog represented as HES-P in our model) to exert regulatory control on the transcription of the hes gene (represented as hes-g in our model). We also included possible cross talk between RAS and Notch signaling.3,6
An interaction network diagram provides information about the directionality of an interaction (eg, which nodes regulate other nodes) and ideally, as in our case, it also includes signed interactions defining whether a regulation is activating or inhibiting. An interaction network, however, is not designed to provide information about the expected response when multiple molecules acting on the same node are present. In contrast, a logic network, which is similar to a circuit diagram, explicitly shows directionality as well as how multiple signals acting on a single target should be integrated31 by incorporating logic gates made of logic operators describing the interaction in a rule-based manner.
Construction of initial hypothesized logic network
Using the interaction network as a guide as well as literature knowledge, we constructed an initial hypothesized logic network of Notch signaling in SW480 cells (Fig. 2A) and compared the steady state output (Fig. 2B and C) of this model to the observed output (Fig. 1B). A penalty was assessed for any differences between simulation and experiment. A penalty of 0.0 indicates an exact qualitative match with the observed data. A penalty of 1.0 indicates the simulated values were wrong for all nodes in all test conditions. The initial hypothesized network was assessed a penalty of 0.44, indicating that this network does not fit the data well (Fig. 2C). Much of the difference between the simulated and experimental values can be attributed to predicted oscillations in the hypothesized network (Fig. 2B and C). An important limitation of the current approach is that we are relying on WB data collected at a single time point that does not reveal whether a molecule is oscillating. Additional time course data would be required to elucidate any oscillations.
Figure 2.

Initial proposed logic circuitry controlling honokiol and IR effects on Notch signaling in SW480 cells. (A) Logic regulations were carefully considered for each interaction in the network. The graphical representation of the logical circuit is equivalent to the logic rules listed to right. Black edges with arrows indicate activating regulations and red edges with blunt ends indicate inhibiting regulations. The * after a node on the left-hand side of an expression indicates the value the node will assume at the next time step. (B) Asynchronous simulation of the logic network in (A) was performed under the four experimental conditions. The y-axis represents the probability a node is ON. Probabilities between 0.0 and 1.0 after the first 5–10 time steps indicate a node is oscillating in the steady state. (C) The predicted steady state values for the network are represented in discrete from. The red Xs indicate differences between predicted values and the experimental values in Figure 1B. The raw penalty of this network is 14. After scaling by the maximum raw penalty of 32, a scaled penalty of 0.44 was generated. Much of the difference between the simulated and experimental values can be attributed to predicted oscillations.
We next ran a series of simulations where we perturbed the network by sequentially dropping each edge in the logic network and then swapping each logic operator for its opposite operator. The smallest penalty observed after a single network perturbation was 0.34, which suggests that the network as constructed remains incomplete.
Logic network search space generation
We next enumerated all possible ways each of the nodes in the interaction network could be logically controlled by their regulatory nodes. A list of each node and its potential regulators is summarized in Supplementary Figure S1A. As an example, CDK1 is inhibited by P53 and P27 in the interaction network (Fig. 1C). When we generated all possible ways P53 and P27 could potentially combine to regulate CDK1, however, we did not restrict the logical control to inhibition only, which allows for the possibility of non-canonical adaptations in these cells (Supplementary Fig. S1A). This led to a total of 135 potential logic rules that could describe the regulation of all nodes in the network. Together these rules combined to produce a total of 2.6 × 1012 possible unique logic networks.
Constraint-based network inference search
As a consequence, it was not possible to exhaustively test each of these networks via direct simulation. Therefore, we used a genetic algorithm (GA)45 as a constraint-based search to look for networks that best fit the data by minimizing the difference between the simulated and observed data using a fitness function to assign a penalty score for incorrect predictions (see Methods section). After several repeated runs, we did not find any network that produced penalty values less than 0.0625 (there were, however, tens of thousands of networks that produced this penalty value). Therefore, we concluded that critical regulations were missing from the hypothesized network model. Given the marked reduction in proliferation and increase in caspase 3 cleavage reported by Ponnurangam et al.5 when IR and honokiol are used in combination, we reasoned that honokiol may interact directly with MYC and/or BAX via interactions not previously included in our model. Adding honokiol as a potential regulator of MYC and BAX added an additional 164 possible logic regulations to the existing set of 135 logic regulations and further increased the set of possible logic networks in the search space to 1.4 × 1014. We repeated the constraint-based network search as before and this time found far fewer networks (1,850) with a penalty of 0.0625, again the smallest penalty found. We observed that each of these networks were wrong in the same two conditions: (1) the Jagged value in the presence of both IR and honokiol and (2) the MYC value when only honokiol is present. In the former case, all tested networks predicted that Jagged was ON (when it was expected OFF), and in the latter case, all tested networks predicted MYC OFF (when it was expected ON).
Development of truth tables to predict the likely regulation of Jagged and MYC
If we consider the digital representation of both Jagged and MYC in Figure 1B, we can construct a set of truth tables to test the possible ways that IR and honokiol can be combined to produce the expected readouts (Fig. 3). We found that the only logical combination of honokiol and IR that will produce the readout expected for Jagged or MYC is a rule that contains “NOT Honokiol or NOT IR”. Because our hypothesized network does not allow for any effects from IR stimulation to modulate proteins involved the Notch pathway, we next allowed the network search to proceed by permitting IR and honokiol to be more promiscuous in their potential interactions in the network. Specifically, all but the following three nodes were allowed to potentially be regulated by honokiol: proliferation, apoptosis, and hes-g. In addition, four Notch pathway nodes were permitted to be potentially regulated by IR: Jagged, NICD, hes-g, and the HES-P (Supplemental Fig. S1B). These additional potential regulations increased the number of logic networks that can be formed to 1.8 × 1024.
Figure 3.
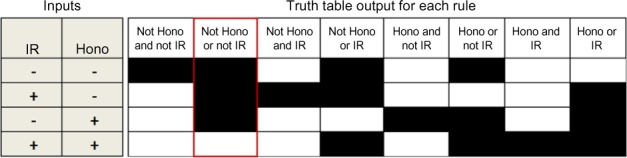
Truth tables for honokiol and IR regulation. If honokiol and IR are the only inputs to a regulation, eight unique logical expression can be generated using the AND, OR, and NOT operators. The logical output of each rule is presented in truth table form. Black indicates a target node (eg, Jagged) is ON and white indicates a target node is OFF for the given inputs. The only logical regulation of honokiol and IR that produces the readout expected for Jagged or MYC in Figure 2B is indicated in red in the truth table.
When we ran the constraint-based network search, hundreds of thousands of networks were found with a penalty of 0.0, indicating that a very large number of networks can generate output that matches SW480 expected values. To attempt to pare this list to a more reasonable number, filters were added to further constrain the network search so that unlikely interactions were not permitted. The filters selected removed interactions we assumed would be detrimental to the tumor cells’ survival and, therefore, represent unlikely adaptations. The regulations not permitted were
honokiol activates AKT
honokiol activates RAS
honokiol activates MYC
honokiol inhibits p27
serum inhibits AKT
serum inhibits RAS
AKT inhibits MYC
RAS inhibits MYC
IR activates MYC
AKT activates BAX
P27 activates CDK1
MYC activates P27
We again ran the constraint-based network search with these filters and found 30,347 unique network configurations with a penalty score of 0.0.
Regulatory frequencies in the candidate networks
Without performing additional experiments (which ideally would include readouts for several more proteins as well as activation data in the form of phospho-WB readouts), it is not easy to rationally cull the list further. Therefore, we next calculated the frequency of distinct regulations present for each node in the set of 30,347 candidate networks (Table 1).
Table 1.
Frequency of each regulation type in 30,347 candidate networks that fit the SW480 data.
| TARGET NODE | % OF CANDIDATE NETWORKS WITH REGULATION | |
|---|---|---|
| CDK1 | honokiol NOT honokiol NOT P27 NOT P53 P53 |
10.64% 74.65% 94.24% 81.33% 16.99% |
| MYC |
AKT NOT honokiol NOT IR RAS |
84.42% 93.45% 100.00% 76.35% |
| BAX |
honokiol NOT AKT NOT honokiol NOT P53 P53 |
73.32% 86.25% 1.11% 21.95% 78.05% |
| JAGGED | HES-P honokiol NOT HES-P NOT honokiol NOT IR NOT RAS RAS |
29.57% 0.08% 28.41% 99.00% 100.00% 10.88% 42.27% |
| NICD |
honokiol IR JAGGED NOT honokiol NOT IR\ NOT JAGGED |
96.98% 77.36% 99.36% 1.33% 5.03% 0.64% |
| HES-P | hes1-g | 100.00% |
| hes1g | HES-P IR NICD NOT HES-P NOT IR |
48.77% 59.43% 100.00% 27.82% 16.92% |
| RAS |
NOT honokiol RAS serum |
87.24% 4.34% 50.27% |
| AKT | AKT NOT honokiol serum |
0.77% 92.80% 63.07% |
| P27 | HES-P honokiol NOT HES-P NOT MYC P27 |
41.69% 73.84% 33.09% 92.58% 0.05% |
| P53 | honokiol IR MDM2 NOT honokiol NOT IR NOT MDM2 |
21.52% 78.05% 1.39% 0.13% 21.95% 16.54% |
| MDM2 | honokiol MDM2 NOT H NOT P53 P53 |
47.70% 5.96% 30.16% 39.52% 28.63% |
| PROLIF | CDK1 | 100.00% |
| APOP | BAX | 100.00% |
Note: Regulations in bold were included in at least 70% of the networks. The presence of the auto-regulation of a node (eg, AKT by AKT) represents the logic rule that allows for constitutive activation of the node. Regulatory nodes preceded by “NOT” indicate inhibitory regulations of the target node, and all others indicate activating regulations of the target node.
MYC regulations
There were four distinct types of regulations of MYC found in the 30,347 candidate networks: “AKT,” “not honokiol,” “not IR,” and “RAS”, each of which occurred in more than 76% of the networks, while “not IR” occurred in 100% of networks. It is worth noting that this analysis only tells us the sign (activating or inhibiting) of an interaction and does not tell us how, for example, “AKT” and “not IR” (which must occur together in 84% of the networks; Table 1) are logically integrated to regulate MYC. More experimental data will be required to unravel the nuanced details of the logic circuitry in the cells.
NICD regulations
The regulation of NICD by “IR” or “not IR” occurs in 77% and 5%, respectively, of the candidate networks. If there was strong reason to believe that IR induced signaling plays no role in Notch expression and/or NICD cleavage, the removal of both activating and inhibiting IR regulations of the NCID node would eliminate the majority of the candidate networks (leaving a much smaller list of candidate networks to evaluate). Doing so, in this case, however, is not recommended without performing additional validation experiments given published data on the potential cross talk between P53 and Notch signaling.46
In the case of NICD regulation by honokiol, activation by honokiol occurs in 96% and repression occurs in just over 1% of networks. This result strongly suggests that honokiol is likely to play some role regulating either the expression of Notch or cleavage of the intracellular domain. The role of honokiol and IR in these regulations may represent an effect on the γ-secretase complex, which is involved in NICD cleavage. Alternatively, it could represent a direct effect on Notch transcription. In any case, these results provide a specific and tangible set of tests to consider in the laboratory. Honokiol is generally associated with disruption of cancer causing events.5 One reasonable next set of experiments to perform in the laboratory would be to directly test the role of honokiol and IR on Notch and NICD cleavage. If either IR or honokiol are discovered to destabilize NICD in some way, this will allow paring the candidate network down to ~5% of the total networks.
Possible antagonistic regulation of Notch signaling by ionizing radiation
The IR-based regulation of Jagged and NICD (both key components of Notch signaling in our model) appears to be very different in the candidate networks. For example, negative IR regulation of Jagged occurs in 100% of the networks. In contrast, negative IR regulation of NICD occurs in only 5% of the networks, while positive IR regulation of NICD occurs in more than 77% of the networks (Table 1). This strongly suggests that signaling downstream of IR stimulation has antagonistic effects on different elements of the Notch pathway. Finally, honokiol activated Jagged, the Notch ligand, in 25 (0.08%) of the networks but inhibited Jagged in almost all the remaining networks (99.00%). Two possible mechanisms of honokiol regulation of Jagged are presented in Figure 4. It is noteworthy that in many of the candidate networks we interrogated, IR inhibition was usually part of a AND gate when honokiol had an activating effect on Jagged (Fig. 4B) but was sometimes part of an OR gate when honokiol had an inhibitory effect on Jagged. When an inhibitor is part of an OR gate, the regulation is much weaker then when it is part of AND gate because, when an activator and inhibitor are joined by the AND operator, the inhibitor will always dominate when both are present. In contrast, when they are joined by an OR operator, the activator will dominate.
Figure 4.
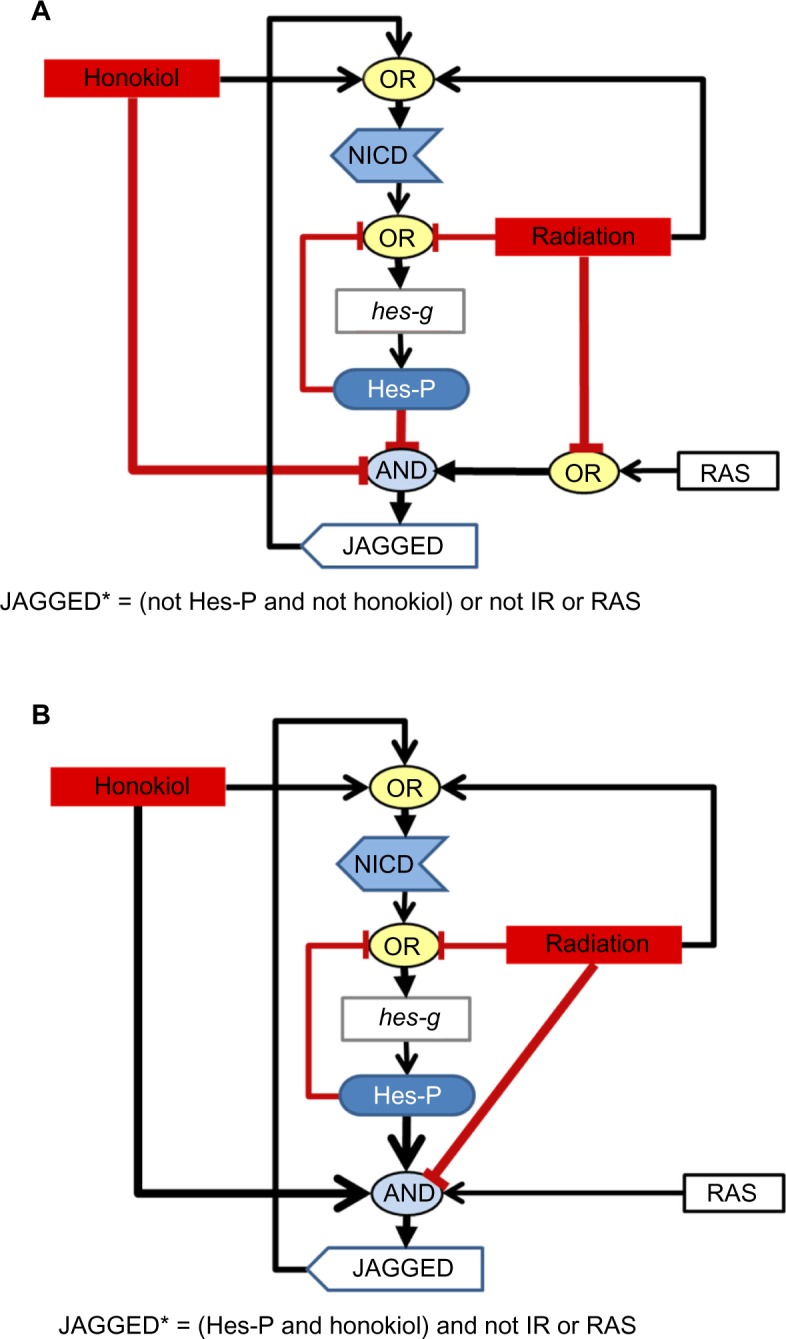
Two potential mechanisms of honokiol regulation of Notch ligands. In the set of 30,347 candidate networks that fit the SW480 data, only 25 (0.08%) networks included honokiol activating Jagged, the node representing Notch ligands in our network. In contrast, 30,044 (99.00%) networks included honokiol inhibiting Jagged. In all networks, IR inhibited Jagged. (A) A representative Notch circuit from the set of candidate networks where honokiol is inhibiting Jagged and (B) a representative circuit from the set of candidate networks where honokiol is activating Jagged are presented. Logic regulations that differ between (A) and (B) are bolded. Black edges with arrows indicate activating regulations and red edges with blunt ends indicate inhibiting regulations.
Discussion
It is increasingly accepted that the inability of many targeted cancer therapies to keep the disease in check or eliminate all cancer cells, despite promising pre-clinical investigations, is due in large part to the complexity of the non-linear intracellular molecular circuitry inside a tumor cell that can evolve through its plasticity as a tangible mechanism for rapid resistance. This evolution can also occur by further genomic mutations that typically occur at longer timescales, in the order of many months to years. Moreover, short-term plasticity can occur as a result of more rapidly occurring epigenetic changes that affect a network’s connectivity over days, weeks, or months.
The ability to rapidly identify the regulatory structure of dysregulated networks would enable the rational development of more precise therapeutic avenues. The objectives of reverse engineering methods are often very different and may include the prediction of elementary reaction mechanisms in a biochemical pathway,47,48 the prediction of gene regulatory networks,49 or the prediction of protein-based signal transduction networks.9 The theoretical approaches employed by these methods also vary widely and may include mass action-based kinetics, discrete Boolean logic, or stochastic probabilistic approaches.50–53 Regardless of the theoretical underpinnings of a reverse engineering methodology, the development of predictive reverse engineering methodologies that also include protein-based signaling networks as well as information about whether a regulatory molecule is activating or inhibiting its target is critically needed.
Our logic-based network approach allowed us to make predictions about the “promiscuity” of honokiol in SW480 cells in the absence of an exact mechanism. Additional experiments (including time course data) are needed in order to predict the underlying logical circuitry with a higher degree of confidence. If such a network is inferred via reverse engineering methods or based on an experimentally data driven hypothesis, a logic network may also be used to make predictions about the drug’s expected potency in specific interactions. If direct or indirect interactions are predicted that contain an OR NOT logic gate, this implies that the drug is effective but relatively weak compared to the activating molecule. In contrast, an AND NOT logic gate would suggest the opposite: a very potent inhibitor that will silence the target under most conditions (see.31,54 for a more detailed discussion).
The data used in this work were based on published WBs performed with or without IR and/or honokiol in SW480 cells by Ponnurangam et al.5 These authors also performed the same experiments on HCT116 cells. While both SW480 and HCT116 cells are human derived colorectal cancer cell lines with K-RAS mutations, there are important oncogenic differences between the two cell lines. For example, a mutation in adenomatous polyposis coli (APC) is found in SW480 but APC is wild type in HCT116.55 APC is a tumor suppressor that acts on the WNT pathway56 and may play an important role in regulating the Notch pathway.57 It is clear from Supplementary Figure S1 (as well as the data from5) that CDK1, MYC, BAX, and HES respond very differently to IR and honokiol in the two cell lines.
In addition to the SW480 simulations described, we also ran network searches to attempt to infer the network in HCT116 cells (Fig. S1B). The best fitting networks found with the HCT116 data scored 0.18. In contrast, we found thousands of candidate networks that scored a perfect 0.00 with the SW480 data. As a consequence, we chose to focus on the SW480 cell line in this work. We conclude that additional signaling pathways not presently included in our model are likely activated or deactivated in HCT116, which account for the very different responses to IR and honokiol in the two cell lines (possibly because of regulatory differences related to the tumor suppressor APC). Access to more detailed molecular data collected from both cell lines would facilitate additional predictions for both cell lines. In an ideal experimental setup, data from several perturbation conditions collected at more than one time point (eg, 1, 24, and 48 hours) will be available as well as data from phospho-WBs to provide more accurate readouts of protein activation.
From a general reporting perspective, the development and wide adoption of a set of minimal reporting standards for publication of WB data58 will greatly improve the experimental repeatability of published WB data as well as make it easier to use these data for simulation purposes (and to propose networks that reflect the data). In the Ponnurangam et al study,5 the authors were interested in studying the effect of a specific chemical agent (honokiol). In cases where a pharmacological inhibitor is used with the implicit or explicit intent to perturb a node of interest (eg, inhibition of MEK by UO126 or PD0352901) and observe the downstream effects of silencing the node, the use of highly specific inhibitors will almost always be preferred to less specific inhibitors, to limit non-specific interactions. In addition, the use of clustered regularly interspaced short palindromic repeats (CRISPER) to generate true molecular knockouts will likely be preferred in the near future as the method of choice over short hairpin RNA knockdowns for the collection of experimental data to use as part of reverse engineering computational methods.
It must be emphasized that all modeling is an approxi mation of some type. Criticisms of the Boolean approach are frequently related to the reality that logic models provide only qualitative approximations of molecular regulation. Although this is a limitation, the majority of experimental data available on molecular regulations are also qualitative. Logic models have arbitrary time units, which need to be standardized to biological time. The standardization is non-trivial and is dependent on the model size and structure.31 Building more detailed kinetic models of signaling pathways requires knowing a priori the mechanism of interaction between proteins and guessing unknown kinetic constants or estimating parameters of the interactions. Thus far, such information is available only for a limited number of well-characterized proteins. Once this information is available for most of the protein and enzymes in signaling pathways, it can be built into more realistic models of cancer.59 In the meantime logic models represent a parameter free compromise between mass action kinetics based models and correlative statistical models.31
In many cases, cells use digital yes-or-no decisions, such as transmission of electric impulses by neurons, which are either ON or OFF depending on the signals a neuron received.60 The activation of cellular stress is also triggered in an on-or-off fashion.61 In other cases, cellular signals are analog, with levels of gradation.62 These continuous changes in protein concentrations enable fine control of metabolism or gene expression. The reality is that cells use a hybrid approach to information processing.60 Although, we do not yet fully understand how the hybrid information processing system works inside cells, it would not be surprising if cells turn all signals into a digital form. Analog signals can be turned into digital form by sampling the signal at some interval. The more frequent the sampling is, the better digital signals approximate their analog counterparts. The evolutionary advantage of digital signal processing is that responses will always be more robust to noise and repeatable. For the same reasons that manufactured electronic circuits evolved from analog circuits to almost exclusively digital ones (which are now ubiquitous in our everyday lives), it is conceivable that biological evolution has also favored digital processing.
Ideally, in the not too distant future, we will routinely be able to sample cells from a variety of locations within a patient’s tumor, collect key molecular readouts, and run algorithms that accurately predict how each area of the tumor is likely to respond to a given therapy. It may be that most of a tumor will respond well to standard therapy but a specific sub-clonal population may possess network adaptions that will render it resistant to the therapy. Through the course of treatment, only these cells would survive, leading ultimately to recurrence of a highly resistant tumor. Thus, continuing to develop computational methods that integrate experimental readouts of molecular state to infer intracellular circuitry is of vital importance for the realization of rationally driven personalized medicine.
Methods
The general methodology followed is summarized in Figure 5.
Figure 5.
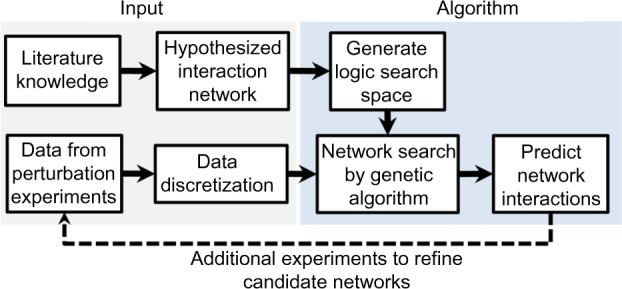
General workflow of methodology used. Data from Ponnurangam et al.5 were discretized into binary ON or OFF values and served as input to the network search algorithm. An initial interaction network was hypothesized from literature knowledge and also used as input to the network search algorithm. The algorithm consists of a constraint-based search over all possible logic network configurations that can be made from the nodes in the interaction network. A set of networks is predicted that match the experimental data. The dotted line indicates the need for additional experiments to distinguish between the set of candidate networks predicted.
Logic network modeling
Logic network simulations used an asynchronous random order update,28,31 which samples from many timescales and is appropriate for simulating the variation in a population of cells.31,63–65 Additional details on the asynchronous modeling approach used are provided in the Supplementary file. The output of an asynchronous simulation with n replicate networks is a probability a node will be activated (ON) in the steady state. Nodes that are oscillating as part of a limit cycle will produce non-zero probabilities that are less than 1.0. The data used for fitting in this work only include a single time point. Without additional time points, it is not possible to make predictions about oscillatory behavior from the published data.
Discretization of data
The predicted model of Notch signaling relied on the published WB data collected from SW480 cells in Ponnurangam et al.5 Before proceeding with network fitting (via the GA search described below), it was necessary to discretize the experimental input data. WB is a qualitative assay, and efforts to exactly quantitate protein levels by WB can be misleading for a variety of reasons.58,66 Because logic models are qualitative models based on digital signals, they are well suited for qualitative data. A two-state logic model assumes that for each node in a network, there is a threshold above which the node is active (ON) and below which it is mostly inactive (OFF). In general, faint or absent blots can be easily assigned OFF values and prominent blots can be assigned ON values. These assignments cannot always be easily made via visual inspection, however. The approach used to infer the state of each protein in this work relied on ImageJ67 and was based on a semi-quantitative densitometric method (which is described in detail in Supplementary file). From these data, an adjusted relative density for each protein under each condition was calculated. In order for a protein to be considered ON in an experimental condition, the following two conditions were required to be true of the protein blot: (1) the total signal for the protein in a given condition was at least 25% of the average signal measured for the protein across all four experimental conditions and (2) the adjusted relative density was at least 20%. The threshold approach used is similar to discretization approaches used elsewhere.68,69
Supplementary Tables S1 and S2 summarize the densitometric data used to infer whether a protein was ON or OFF in the published SW480 and HCT116 WB data,5 respectively. From examining the WB data in Ponnurangam et al.5 as well as the discrete form of these data (Supplementary Fig. S1), it is clear that the SW480 and HCT116 cells respond very differently to the four experimental conditions.
Comparing network output to observed data
A scaled penalty was assessed based on how well simulated model output matched experimentally observed data:
where n is the node, t is the test condition, N is the total number of nodes evaluated, T is the total number of test conditions, Sim(n, t) is the simulated value of the n-th node in the t-th test condition, Obs(n, t) is the experimentally observed value of the n-th node in the t-th test condition. The scaled penalty is a value that ranges from 0.0 to 1.0. Values of 0.0 indicate a perfect fit with observed data, while values of 1.0 indicate that the fit was wrong for all nodes in all test conditions. For the network and test conditions evaluated in this paper, N = 8 and T = 4. Therefore, the penalty was always scaled by 32.
Network searches
Because the search space of all possible logic networks for a set of hypothesized interactions is extremely large, a constraint-based search was used to identify candidate networks that fit the experimental data. The search was implemented as a GA45 where a fitness function assessed the penalty of each simulated network (using the asynchronous logic simulation described above and in Supplementary file). GAs were run for at least 100 generations and repeated at least three times to attempt to identify as many candidate networks (with a penalty of 0.0) as possible. Logic search space generation was based on the number of possible regulators each node in the network could potentially have (Supplementary Fig. S1). For each node in the network, all logic regulation rules that could be formed between potential regulators of the node (as wells as all subsets of these regulators) by the AND, OR, and NOT logic operators were generated. The total network search space is defined by the number of ways the set of potential regulations for all nodes in the network can be combined to make a unique network. As the number of potential regulators increase in a network, the network search space grows exponentially large. Additional details are provided in the Supplementary file.
Computing environment
A Python-based library was written to support the network inference search and implementation of the GA. The library supports distributed computing using message passing interface (MPI) for Python. All distributed simulations were run on the University of Michigan’s high performance computing cluster using Intel Nehalem/i7 Cores.
Supplementary File
Supplementary File 1. This document, which includes Supplementary Tables S1–S2 and Supplementary Figures S1–S3, describes asynchronous logic network simulations, data discretization, and genetic algorithm and network fitting.
Footnotes
Author Contributions
Conceived and designed the experiments: MLW, NC. Analyzed the data: MLW. Wrote the first draft of the manuscript: MLW. Contributed to the writing of the manuscript: MLW, NC, SS. Agree with manuscript results and conclusions: MLW, NC, SDM, SS. Jointly developed the structure and arguments for the paper: MLW, SDM, SS. Made critical revisions and approved final version: MLW, NC, SDM, SS. All authors reviewed and approved of the final manuscript.
ACADEMIC EDITOR: JT Efird, Editor in Chief
COMPETING INTERESTS: Authors disclose no potential conflicts of interest.
FUNDING: This work is supported in part by the James McDonnell Foundation (MLW, SS), the Avon Foundation (SDM), the Breast Cancer Research Foundation (SDM), the METAvivor Foundation (SDM), NIH R25 DK088752 (NC and SS), and the University of Michigan Protein Folding Diseases Initiative (SS). The authors confirm that the funder had no influence over the study design, content of the article, or selection of this journal.
Paper subject to independent expert blind peer review by minimum of two reviewers. All editorial decisions made by independent academic editor. Prior to publication all authors have given signed confirmation of agreement to article publication and compliance with all applicable ethical and legal requirements, including the accuracy of author and contributor information, disclosure of competing interests and funding sources, compliance with ethical requirements relating to human and animal study participants, and compliance with any copyright requirements of third parties.
REFERENCES
- 1.McCormick F. Mutant onco-proteins as drug targets: successes, failures, and future prospects. Curr Opin Genet Dev. 2011;21(1):29–33. doi: 10.1016/j.gde.2010.12.002. [DOI] [PubMed] [Google Scholar]
- 2.Wynn ML, Ventura AC, Sepulchre JA, García HJ, Merajver SD. Kinase inhibitors can produce off-target effects and activate linked pathways by retroactivity. BMC Syst Biol. 2011;5:156. doi: 10.1186/1752-0509-5-156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Leong KG, Karsan A. Recent insights into the role of Notch signaling in tumorigenesis. Blood. 2006;107(6):2223–33. doi: 10.1182/blood-2005-08-3329. [DOI] [PubMed] [Google Scholar]
- 4.Al-Hussaini H, Subramanyam D, Reedijk M, Sridhar SS. Notch signaling pathway as a therapeutic target in breast cancer. Mol Cancer Ther. 2011;10(1):9–15. doi: 10.1158/1535-7163.MCT-10-0677. [DOI] [PubMed] [Google Scholar]
- 5.Ponnurangam S, Mammen JM, Ramalingam S, et al. Honokiol in combination with radiation targets notch signaling to inhibit colon cancer stem cells. Mol Cancer Ther. 2012;11(4):963–72. doi: 10.1158/1535-7163.MCT-11-0999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sundaram MV. The love-hate relationship between Ras and Notch. Genes Dev. 2005;19(16):1825–39. doi: 10.1101/gad.1330605. [DOI] [PubMed] [Google Scholar]
- 7.Zhang GL, DeLuca DS, Brusic V. Database resources for proteomics-based analysis of cancer. Methods Mol Biol. 2011;723:349–64. doi: 10.1007/978-1-61779-043-0_22. [DOI] [PubMed] [Google Scholar]
- 8.Kholodenko BN, Kiyatkin A, Bruggeman FJ, Sontag E, Westerhoff HV, Hoek JB. Untangling the wires: a strategy to trace functional interactions in signaling and gene networks. Proc Natl Acad Sci U S A. 2002;99(20):12841–6. doi: 10.1073/pnas.192442699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Stelniec-Klotz I, Legewie S, Tchernitsa O, et al. Reverse engineering a hierarchical regulatory network downstream of oncogenic KRAS. Mol Syst Biol. 2012;8:601. doi: 10.1038/msb.2012.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Voit E, Neves AR, Santos H. The intricate side of systems biology. Proc Natl Acad Sci U S A. 2006;103(25):9452–7. doi: 10.1073/pnas.0603337103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang YK, Hurley DG, Schnell S, Print CG, Crampin EJ. Integration of steady-state and temporal gene expression data for the inference of gene regulatory networks. PLoS One. 2013;8(8):e72103. doi: 10.1371/journal.pone.0072103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Stolovitzky G, Monroe D, Califano A. Dialogue on reverse-engineering assessment and methods – the DREAM of high-throughput pathway inference. Reverse Eng Biol Networks. 2007;1115:1–22. doi: 10.1196/annals.1407.021. [DOI] [PubMed] [Google Scholar]
- 13.Ferrell JE., Jr Tripping the switch fantastic: how a protein kinase cascade can convert graded inputs into switch-like outputs. Trends Biochem Sci. 1996;21(12):460–6. doi: 10.1016/s0968-0004(96)20026-x. [DOI] [PubMed] [Google Scholar]
- 14.Koshland DE, Jr, Goldbeter A, Stock JB. Amplification and adaptation in regulatory and sensory systems. Science. 1982;217(4556):220–5. doi: 10.1126/science.7089556. [DOI] [PubMed] [Google Scholar]
- 15.Ferrell JE, Jr, Machleder EM. The biochemical basis of an all-or-none cell fate switch in Xenopus oocytes. Science. 1998;280(5365):895–8. doi: 10.1126/science.280.5365.895. [DOI] [PubMed] [Google Scholar]
- 16.Huang J, Brameshuber M, Zeng X, et al. A single peptide-major histocompatibility complex ligand triggers digital cytokine secretion in CD4(+) T cells. Immunity. 2013;39(5):846–57. doi: 10.1016/j.immuni.2013.08.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wertek F, Xu C. Digital response in T cells: to be or not to be. Cell Res. 2014;24(3):265–6. doi: 10.1038/cr.2014.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gordon WR, Arnett KL, Blacklow SC. The molecular logic of Notch signaling--a structural and biochemical perspective. J Cell Sci. 2008;121(pt 19):3109–19. doi: 10.1242/jcs.035683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Grandbarbe L, Bouissac J, Rand M, Hrabé deAngelis M, Artavanis-Tsakonas S, Mohier E. Delta-Notch signaling controls the generation of neurons/glia from neural stem cells in a stepwise process. Development. 2003;130(7):1391–402. doi: 10.1242/dev.00374. [DOI] [PubMed] [Google Scholar]
- 20.Ron D, Walter P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat Rev Mol Cell Biol. 2007;8(7):519–29. doi: 10.1038/nrm2199. [DOI] [PubMed] [Google Scholar]
- 21.Schnell S. A model of the unfolded protein response: pancreatic beta-cell as a case study. Cell Physiol Biochem. 2009;23(4–6):233–44. doi: 10.1159/000218170. [DOI] [PubMed] [Google Scholar]
- 22.Davey RE, Onishi K, Mahdavi A, Zandstra PW. LIF-mediated control of embryonic stem cell self-renewal emerges due to an autoregulatory loop. FASEB J. 2007;21(9):2020–32. doi: 10.1096/fj.06-7852com. [DOI] [PubMed] [Google Scholar]
- 23.Lahav G, Rosenfeld N, Sigal A, et al. Dynamics of the p53-Mdm2 feedback loop in individual cells. Nat Genet. 2004;36(2):147–50. doi: 10.1038/ng1293. [DOI] [PubMed] [Google Scholar]
- 24.Tamsir A, Tabor JJ, Voigt CA. Robust multicellular computing using genetically encoded NOR gates and chemical ‘wires’. Nature. 2011;469(7329):212–5. doi: 10.1038/nature09565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ribba B, Colin T, Schnell S. A multiscale mathematical model of cancer, and its use in analyzing irradiation therapies. Theor Biol Med Model. 2006;3:7. doi: 10.1186/1742-4682-3-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Saez-Rodriguez J, Alexopoulos LG, Zhang M, Morris MK, Lauffenburger DA, Sorger PK. Comparing signaling networks between normal and transformed hepatocytes using discrete logical models. Cancer Res. 2011;71(16):5400–11. doi: 10.1158/0008-5472.CAN-10-4453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhang R, Shah MV, Yang J, et al. Network model of survival signaling in large granular lymphocyte leukemia. Proc Natl Acad Sci U S A. 2008;105(42):16308–13. doi: 10.1073/pnas.0806447105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Albert I, Thakar J, Li S, Zhang R, Albert R. Boolean network simulations for life scientists. Source Code Biol Med. 2008;3:16. doi: 10.1186/1751-0473-3-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Glass L, Kauffman SA. The logical analysis of continuous, non-linear biochemical control networks. J Theor Biol. 1973;39(1):103–29. doi: 10.1016/0022-5193(73)90208-7. [DOI] [PubMed] [Google Scholar]
- 30.Thomas R. Circular causality. Syst Biol. 2006;153(4):140–53. doi: 10.1049/ip-syb:20050101. [DOI] [PubMed] [Google Scholar]
- 31.Wynn ML, Consul N, Merajver SD, Schnell S. Logic-based models in systems biology: a predictive and parameter-free network analysis method. Integr Biol. 2012;4(11):1323–37. doi: 10.1039/c2ib20193c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yao CJ, Lai GM, Yeh CT, et al. Honokiol eliminates human oral cancer stem-like cells accompanied with suppression of Wnt/beta-catenin signaling and apoptosis induction. Evidence-based Complement Alternat Med. 2013;2013:146136. doi: 10.1155/2013/146136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Baldin V, Lukas J, Marcote MJ, Pagano M, Draetta G. Cyclin D1 is a nuclear protein required for cell cycle progression in G1. Genes Dev. 1993;7(5):812–21. doi: 10.1101/gad.7.5.812. [DOI] [PubMed] [Google Scholar]
- 34.Coqueret O. New roles for p21 and p27 cell-cycle inhibitors: a function for each cell compartment? Trends Cell Biol. 2003;13(2):65–70. doi: 10.1016/s0962-8924(02)00043-0. [DOI] [PubMed] [Google Scholar]
- 35.Yang W, Shen J, Wu M, et al. Repression of transcription of the p27(Kip1) cyclin-dependent kinase inhibitor gene by c-Myc. Oncogene. 2001;20(14):1688–702. doi: 10.1038/sj.onc.1204245. [DOI] [PubMed] [Google Scholar]
- 36.Basu A, Haldar S. The relationship between BcI2, Bax and p53: consequences for cell cycle progression and cell death. Mol Hum Reprod. 1998;4(12):1099–109. doi: 10.1093/molehr/4.12.1099. [DOI] [PubMed] [Google Scholar]
- 37.Abkhezr M, Keramati AR, Ostad SN, Davoodi J, Ghahremani MH. The time course of Akt and ERK activation on XIAP expression in HEK 293 cell line. Mol Biol Rep. 2010;37(4):2037–42. doi: 10.1007/s11033-009-9658-4. [DOI] [PubMed] [Google Scholar]
- 38.Tari AM, Lopez-Berestein G. Serum predominantly activates MAPK and Akt kinases in EGFR- and ErbB2-over-expressing cells, respectively. Int J Cancer. 2000;86(2):295–7. doi: 10.1002/(sici)1097-0215(20000415)86:2<295::aid-ijc22>3.0.co;2-k. [DOI] [PubMed] [Google Scholar]
- 39.Park KS, Jeon SH, Kim SE, et al. APC inhibits ERK pathway activation and cellular proliferation induced by RAS. J Cell Sci. 2006;119(pt 5):819–27. doi: 10.1242/jcs.02779. [DOI] [PubMed] [Google Scholar]
- 40.Fried LE, Arbiser JL. Honokiol, a multifunctional antiangiogenic and antitumor agent. Antioxid Redox Signal. 2009;11(5):1139–48. doi: 10.1089/ars.2009.2440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kim BH, Cho JY. Anti-inflammatory effect of honokiol is mediated by PI3K/Akt pathway suppression. Acta Pharmacol Sin. 2008;29(1):113–22. doi: 10.1111/j.1745-7254.2008.00725.x. [DOI] [PubMed] [Google Scholar]
- 42.Garcia A, Zheng Y, Zhao C, et al. Honokiol suppresses survival signals mediated by Rasdependent phospholipase D activity in human cancer cells. Clin Cancer Res. 2008;14(13):4267–74. doi: 10.1158/1078-0432.CCR-08-0102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Giudicelli F, Ozbudak EM, Wright GJ, Lewis J. Setting the tempo in development: an investigation of the zebrafish somite clock mechanism. PLoS Biol. 2007;5(6):e150. doi: 10.1371/journal.pbio.0050150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pourquie O. Vertebrate segmentation: from cyclic gene networks to scoliosis. Cell. 2011;145(5):650–63. doi: 10.1016/j.cell.2011.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mitchell M. An introduction to genetic algorithms. Complex adaptive systems. viii. Cambridge, MA: MIT Press; 1996. p. 205. [Google Scholar]
- 46.Dotto GP. Crosstalk of Notch with p53 and p63 in cancer growth control. Nat Rev Cancer. 2009;9(8):587–95. doi: 10.1038/nrc2675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Srividhya J, Crampin EJ, McSharry PE, Schnell S. Reconstructing biochemical pathways from time course data. Proteomics. 2007;7(6):828–38. doi: 10.1002/pmic.200600428. [DOI] [PubMed] [Google Scholar]
- 48.Srividhya J, Mourão MA, Crampin EJ, Schnell S. Enzyme catalyzed reactions: from experiment to computational mechanism reconstruction. Comput Biol Chem. 2010;34(1):11–8. doi: 10.1016/j.compbiolchem.2009.10.007. [DOI] [PubMed] [Google Scholar]
- 49.Wagner A. Circuit topology and the evolution of robustness in two-gene circadian oscillators. Proc Natl Acad Sci U S A. 2005;102(33):11775–780. doi: 10.1073/pnas.0501094102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Crampin EJ, Schnell S, McSharry PE. Mathematical and computational techniques to deduce complex biochemical reaction mechanisms. Prog Biophys Mol Biol. 2004;86(1):77–112. doi: 10.1016/j.pbiomolbio.2004.04.002. [DOI] [PubMed] [Google Scholar]
- 51.Liang S, Fuhrman S, Somogyi R. Reveal, a general reverse engineering algorithm for inference of genetic network architectures. Pac Symp Biocomput. 1998;3:18–29. [PubMed] [Google Scholar]
- 52.Repsilber D, Liljenstrom H, Andersson SG. Reverse engineering of regulatory networks: simulation studies on a genetic algorithm approach for ranking hypotheses. Biosystems. 2002;66(1–2):31–41. doi: 10.1016/s0303-2647(02)00019-9. [DOI] [PubMed] [Google Scholar]
- 53.Saeed M, Ijaz M, Javed K, Babri HA. Reverse engineering Boolean networks: from Bernoulli mixture models to rule based systems. PLoS One. 2012;7(12):e51006. doi: 10.1371/journal.pone.0051006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Arkin A, Ross J. Computational functions in biochemical reaction networks. Biophys J. 1994;67(2):560–78. doi: 10.1016/S0006-3495(94)80516-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kaur M, Velmurugan B, Tyagi A, Agarwal C, Singh RP, Agarwal R. Silibinin suppresses growth of human colorectal carcinoma SW480 cells in culture and xenograft through down-regulation of beta-catenin-dependent signaling. Neoplasia. 2010;12(5):415–24. doi: 10.1593/neo.10188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Minde DP, Radli M, Forneris F, Maurice MM, Rüdiger SG. Large extent of disorder in adenomatous polyposis coli offers a strategy to guard Wnt signalling against point mutations. PLoS One. 2013;8(10):e77257. doi: 10.1371/journal.pone.0077257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Rodilla V, Villanueva A, Obrador-Hevia A, et al. Jagged1 is the pathological link between Wnt and Notch pathways in colorectal cancer. Proc Natl Acad Sci U S A. 2009;106(15):6315–20. doi: 10.1073/pnas.0813221106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ghosh R, Gilda JE, Gomes AV. The necessity of and strategies for improving confidence in the accuracy of western blots. Expert Rev Proteom. 2014;11(5):549–60. doi: 10.1586/14789450.2014.939635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wynn ML, Merajver SD, Schnell S. Unraveling the complex regulatory relationships between metabolism and signal transduction in cancer. Adv Exp Med Biol. 2012;736:179–89. doi: 10.1007/978-1-4419-7210-1_9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sarpeshkar R. Analog versus digital: extrapolating from electronics to neurobiology. Neural Comput. 1998;10(7):1601–38. doi: 10.1162/089976698300017052. [DOI] [PubMed] [Google Scholar]
- 61.Sandefur CI, Schnell S. A model of threshold behavior reveals rescue mechanisms of bystander proteins in conformational diseases. Biophys J. 2011;100(8):1864–73. doi: 10.1016/j.bpj.2011.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Daniel R, Rubens JR, Sarpeshkar R, Lu TK. Synthetic analog computation in living cells. Nature. 2013;497(7451):619–23. doi: 10.1038/nature12148. [DOI] [PubMed] [Google Scholar]
- 63.Chaves M, Albert R, Sontag ED. Robustness and fragility of Boolean models for genetic regulatory networks. J Theor Biol. 2005;235(3):431–49. doi: 10.1016/j.jtbi.2005.01.023. [DOI] [PubMed] [Google Scholar]
- 64.Garg A, Di Cara A, Xenarios I, Mendoza L, De Micheli G. Synchronous versus asynchronous modeling of gene regulatory networks. Bioinformatics. 2008;24(17):1917–25. doi: 10.1093/bioinformatics/btn336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Thomas R. Boolean formalization of genetic control circuits. J Theor Biol. 1973;42(3):563–85. doi: 10.1016/0022-5193(73)90247-6. [DOI] [PubMed] [Google Scholar]
- 66.Taylor SC, Berkelman T, Yadav G, Hammond M. A defined methodology for reliable quantification of western blot data. Mol Biotechnol. 2013;55(3):217–26. doi: 10.1007/s12033-013-9672-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nat Methods. 2012;9(7):671–75. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Di Camillo B, Sanchez-Cabo F, Toffolo G, Nair SK, Trajanoski Z, Cobelli C. A quantization method based on threshold optimization for microarray short time series. BMC Bioinformatics. 2005;6(suppl 4):S11. doi: 10.1186/1471-2105-6-S4-S11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Samaga R, Saez-Rodriguez J, Alexopoulos LG, Sorger PK, Klamt S. The logic of EGFR/ErbB signaling: theoretical properties and analysis of high-throughput data. PLoS Comput Biol. 2009;5(8):e1000438. doi: 10.1371/journal.pcbi.1000438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Thomas R, D’Ari R. Biological feedback. Boca Raton, Fla: CRC Press; 1990. p. 316. [Google Scholar]
- 71.Deb K. An introduction to genetic algorithms. Sadhana. 1999;24(4–5):293–15. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary File 1. This document, which includes Supplementary Tables S1–S2 and Supplementary Figures S1–S3, describes asynchronous logic network simulations, data discretization, and genetic algorithm and network fitting.