

Abstract
The use of biotherapeutics, such as monoclonal antibodies, has markedly increased in recent years. It is thus essential that biotherapeutic production pipelines are as efficient as possible. For the production process, one of the major concerns is the propensity of a biotherapeutic antibody to aggregate. In addition to reducing bioactive material recovery, protein aggregation can have major effects on drug potency and cause highly undesirable immunological effects. It is thus essential to identify processing conditions which maximize recovery while avoiding aggregation. Heat resistance is a proxy for long-term aggregation propensity. Thermal stability assays are routinely performed using various spectroscopic and scattering detection methods. Here, we evaluated the potential of macro attenuated total reflection Fourier transform infrared (ATR-FT-IR) spectroscopic imaging as a novel method for the high-throughput thermal stability assay of a monoclonal antibody. This chemically specific visualization method has the distinct advantage of being able to discriminate between monomeric and aggregated protein. Attenuated total reflection is particularly suitable for selectively probing the bottom of vessels, where precipitated aggregates accumulate. With focal plane array detection, we tested 12 different buffer conditions simultaneously to assess the effect of pH and ionic strength on protein thermal stability. Applying the Finke model to our imaging kinetics allowed us to determine the rate constants of nucleation and autocatalytic growth. This analysis demonstrated the greater stability of our immunoglobulin at higher pH and moderate ionic strength, revealing the key role of electrostatic interactions. The high-throughput approach presented here has significant potential for analyzing the stability of biotherapeutics as well as any other biological molecules prone to aggregation.
In 2011, sales of 40 therapeutic monoclonal antibodies (mAbs) accounted for $45 billion, around 5% of the global drug market.1,2 Currently, there are more than 350 mAbs in clinical trials and 60% of new patent applications for drugs are biotherapeutics.3 However, there are a number of challenges to the production of these drugs in a suitable state for delivery to patients. One key issue is the propensity of the proteins to nonspecifically aggregate during expression, isolation, final formulation, and storage. Protein aggregation reduces bioactive material yields and can have major effects on drug potency. Protein aggregates can also provoke highly undesirable immunological effects such as anaphylactic shock.4−8
Protein aggregation can occur in one of two ways: through chemical aggregation by formation of new covalent bonds or by physical association. Chemical aggregation is commonly a result of nonspecific disulfide bond formation,9,10 although other chemical modifications such as deamination can also increase aggregation propensity.11 Physical aggregation is considered to be caused by the formation of partially unfolded intermediates. The partial unfolding exposes hydrophobic regions, usually buried in the core of the protein, which are much more prone to nucleate aggregation. Many factors can affect the aggregation state of proteins including protein structure and environmental factors such as temperature, pH, and concentration.12 Temperatures of 40 to 70 °C typically result in loss of protein activity due to protein unfolding and aggregation.13 Since the probability of protein denaturation increases with temperature,14 heat resistance is indicative of long-term stability at storage temperatures.15 Hence, heat is routinely used as a source of stress for assessing the thermal stability of protein using a range of detection methods.
There are a number of methods available for identifying protein aggregation including size exclusion chromatography (SEC),16,17 negative stain electron microscopy,18 multiangle light scattering,19 and sedimentation velocity analytical ultracentrifugation (SV-AUC) and flow field flow fractionation20 as well as the ultracentrifugation sedimentation dispersity assay combining ultracentrifugation and SDS-PAGE analysis of high concentration (10 mg/mL) protein samples.21 However, none of these techniques are able to monitor the aggregation process in situ.
Using only visible light, dynamic light scattering (DLS) is a nondamaging laboratory technique which can detect protein aggregation over a wide range of length scales,22,23 but measurements are restricted to a narrow concentration range and are also time-consuming.24 In dynamic scanning fluorescence (DSF), the fluorescence of specially designed dyes can be used to specifically probe protein domains during protein unfolding and subsequent aggregation.25 Although DSF is very sensitive and high-throughput,26 measurements are indirect and quantification of aggregates is therefore challenging.
Highly sensitive to protein secondary structure, circular dichroism (CD) can detect subtle conformation changes occurring during protein unfolding and subsequent aggregation.27 Despite being compatible with a wide pH range, the buffer used must be transparent in the far-UV and data collection is also slow.
Vibrational spectroscopy techniques such as Raman and infrared spectroscopy provide information on protein secondary structure based on protein molecular vibrations absorbing at specific frequencies. Although Raman spectroscopy informs on the protein secondary structure in solution and can be performed through glass vessels, fluorescence often interferes with the signal and quantification can be challenging.28 The measurement times in Raman spectroscopy are usually much longer compared to infrared spectroscopy, important for studying dynamic systems. Infrared spectroscopy does not suffer from fluorescence baseline distortions and has the further advantage that the strong absorption of infrared light by proteins results in high signal-to-noise spectra collected in seconds for fast kinetic measurements.22,29 Folded, unfolded, and aggregated protein samples give characteristic FT-IR spectra; thus, this method is particularly well suited for the detection of heat induced aggregates.22,27,30−32
The main limitation of FT-IR spectroscopy for probing aqueous solution is the strong absorption of water which overlaps with the protein amide bands causing aqueous samples thicker than several micrometers to saturate the absorption in the mid infrared range.33,34 Using a high refractive index infrared transparent internal reflection element (IRE) can help to solve this issue. If directed at an angle of incidence larger than the critical angle as defined by Snell’s law, the infrared beam will be internally reflected, generating an emerging evanescent wave.35 When a sample is placed in close contact with the IRE, only a thin layer is probed by the evanescent wave through the attenuated total reflection (ATR) without suffering from H2O absorption saturation.36,37 Aggregates smaller than 100 nm are typically soluble; once they grow larger, however, they start to precipitate.38 ATR has thus the additional advantage of probing the bottom surface layer of aqueous solutions where protein adsorbs and aggregates concentrate due to precipitation.39,40
Although external modules allow the automation of spectra collection,41 conventional infrared spectroscopy is low-throughput as only one sample can be measured at a time. However, spectral output can be dramatically increased by using focal plane array (FPA) detectors which allow spectroscopic imaging by simultaneously collecting a thousand spectra in one measurement. The use of ATR-FT-IR spectroscopic imaging for high-throughput analysis of many samples was first introduced for studies of polymer/drug formulations.42,43 Applications of ATR-FTIR spectroscopic imaging to biological systems has been proposed for studying multiple protein solutions44 and have been recently reviewed.36,37 Using ATR-FT-IR spectroscopic imaging, protein crystallization has been monitored under various solvent conditions simultaneously45 while protein adsorption and crystallization was studied under a gradient of surface properties.46 The use of a microscope allowed the detection of protein crystal seeds as small as 6 μm.47
For these combined reasons, we developed a high-throughput thermal stability assay based on ATR-FT-IR spectroscopic imaging. Because immunoglobulin G (IgG) is a widely used biotherapeutic and is prone to aggregation,28 we chose IgG gamma 4 as a model system to demonstrate the applicability of ATR-FT-IR spectroscopic imaging for the study of the heat induced aggregation kinetic. Since the size of aggregates are much smaller than the diffraction limit of infrared light, a macro ATR-FT-IR imaging system with a large field of view45 was employed. The thermal stability of 12 samples with different buffer conditions could thus be assessed in a single experiment. This allowed the screening of the harsh conditions typically experienced by therapeutic antibodies during the isolation process. Conveniently, the 1.4 mm wide wells designed for this study could be loaded by common laboratory pipetting.
The results obtained by this high-throughput imaging approach showed that the aggregation kinetic follows a sigmoidal function. The screening of different pH and ionic strength conditions revealed that IgG4 is less stable around the protein’s isoelectric point, as reported by previous studies.28,48 The kinetic also revealed that insoluble aggregates are formed faster at moderate salt concentration than at low and high ionic strength. These results stressed the impact of electrostatic interactions on the stability of the IgG in solution. Hence, our novel screening method can investigate the effect of these interactions by mainly probing the formation of insoluble protein aggregates under a range of buffer conditions simultaneously.
Materials and Methods
Immunoglobulin G Sample Preparation
Glutamine Synthetase Chinese Hamster Ovary (GS-CHO) cell lines that express the chimeric B72.3 immunoglobulin G gamma 4 (cB72.3 IgG4) were provided by Lonza (Lonza Biologics, Basel, Switzerland). Cultures were maintained in CD-CHO medium (Invitrogen, UK) with 25 M MSX (Sigma, UK) at 36.5 °C with 8% CO2 humidified air while shaking on an orbital shaker at 140 rpm. Cells were routinely subcultured every 3 or 4 days with a seeding cell density of 2 × 105 cells/mL, with batch cultures conducted in 1 L Erlenmeyer flasks with a working volume of 300 mL. The cB72.3 IgG4 is secreted into the culture media, and thus, the spent media presented a source of protein for further studies. The IgG concentration in the different media samples was quantified by ELISA (Montgomery, TX, US) and UV–vis spectroscopy using a Nanodrop Lite system (Thermo, Wilmington, DE, USA) prior to storage at −80 °C until further use.
For isolation of the IgG, the media samples were defrosted, prior to filtration through a 0.45 μm filter disk to remove aggregates and other large particles. The media containing the protein was then desalted using a HiPrep 26/10 desalting column (GE Healthcare Life Sciences) equilibrated with Buffer A (20 mM Tris-HCl, pH 8.5). Subsequently, the protein solution was loaded onto a HiTrap Q FF prepacked with Q Sepharose (GE Healthcare Life Sciences) equilibrated with Buffer A and the bound proteins eluted using a buffer elution gradient from 0 to 1000 mM of NaCl. Thereafter, the protein was loaded onto a Superdex 200 HR 10/30 size exclusion chromatography (SEC) column (GE Healthcare Life Sciences) with the Buffer A. The SEC chromatogram presented in the Supporting Information is strongly indicative that the purified IgG 4 is monodisperse. As a final step, the protein was further desalted using a HiPrep 26/10 desalting column (GE Healthcare Life Sciences) with type I demineralized water (resistivity at 25 °C > 10 MΩ·cm). The purified protein was then concentrated by centrifugal filtration using a molecular weight cut off of 100 kDa to 2 mg/mL and divided into aliquots which were stored at −80 °C.
Experimental Setup
Infrared Spectroscopy Experimental Setup
Infrared spectra were measured using a Varian 670 spectrometer (Agilent Technologies, Wokingham, UK) coupled to a Varian external large sample compartment. The spectra presented in below were collected using an uncooled single element DTGS detector with a diamond ATR accessory Golden Gate (Specac, Orpington, UK). The macro ATR-FT-IR spectroscopic images of the high-throughput grid were measured using a cooled MCT focal plane array (FPA) displaying 128 by 128 elements.37 16 384 infrared spectra were thus collected simultaneously. To reduce memory requirements, images were aggregated to 64 by 64 by averaging groups of 4 pixels. Spectra were acquired in continuous scan mode by coadding 32 scans at a 4 cm–1 spectral resolution, taking 135 s. Spectra were then truncated to cover only the 4000 to 800 cm–1 range. The experimental setup located in the large sample compartment of the spectrometer is shown in Figure 1. It comprises a 20 mm circular zinc selenide (ZnSe) single bounce internal reflection element prism (PIKE Technologies, Madison, WI, USA). In this configuration, the attenuated total reflection (ATR) accessory allowed a 7 by 9.8 mm field of view for imaging studies46 and a depth of penetration (dp) of 1.2 μm at 1600 cm–1 and at a 45° angle of incidence.49 The temperature of the ZnSe element was set using a nichrome wire based heating controller. The ZnSe top surface was covered by a Sylgard 184 poly(dimethylsiloxane) (PDMS) elastomeric substrate (Dow Corning Corporation, Midland, MI, USA) cast on top of a 12 well PMMA 3D printed master (Shapeways, New York, USA). The PDMS wells were sealed with a 2 mm PDMS film mounted onto a laser cut 13 mm thick PMMA plate which was bolted onto the steel bottom plate. Each well is a 1.4 by 1.4 by 1.5 mm deep rounded box holding up to 2.6 μL of solution.
Figure 1.

Cut through schematic of the experimental setup comprised the ZnSe internal reflection element, the PDMS well grid, the PDMS lid, and the PMMA top plate.
Experimental Procedure
Because the infrared beam is spread over thousands of detecting elements in an FPA detector when performing ATR-FT-IR spectroscopic imaging, each pixel receives less infrared light, resulting in a lower signal-to-noise ratio than can be obtained using a single element detector. Although a germanium crystals can be used over a wider pH range, ZnSe was preferred because it offered a ∼5 times greater absorbance of the spectra bands. The pH was thus kept between pH 5 and 10; measurements were restricted to this range to avoid prism degradation. For each sample, 1.5 μL of a 2 mg/mL IgG 4 stock solution was mixed with 0.75 μL of 40 mM phosphate buffer pH 5, 6, 7, 8, 9, or 10 and 0.75 μL of sodium chloride at 125, 250, 500, 1000, 2000, or 4000 mM. When ignoring contributions from protein charges, the resulting solutions had ionic strengths of between 0.041 and 1.01 M. The sample solution constituents were subsequently mixed by brief centrifugation for a few seconds before loading a 2.2 μL aliquot in each well. Once all samples were loaded, a background image was collected at room temperature (25 °C) to calculate the absorbance. To prevent evaporation, wells were then hermetically sealed and the temperature was rapidly increased to 60 °C before the next 90 or so images were collected over approximately 2.5 h.
Data Analysis
Raw interferograms were collected using Resolution Pro 5.2 (Agilent Technologies Ltd., Santa Clara, CA, USA), and all subsequent data analysis operations were performed by MATLAB (MatWorks, Natick, MA, USA). Fourier transformed single beam data were offset to zero thereafter using the average value between 700 and 600 cm–1. The single beam intensity between 1900 and 1850 cm–1 was also set to a value of 1 since no absorption should have been observed in this region. An offset using the 1900 to 1850 cm–1 region was then performed before correcting for the penetration depth variation depending on the wavelength.35 Since no absorbance variation is expected between the PDMS wells, these regions were selected to offset the spectra of the images over time. The offset absorbance values for the pixels in the gaps were interpolated linearly to reduce the signal fluctuation between each spectroscopic image captured. Afterward, spectra with pixel positions corresponding to the bottom of each of the 12 wells were averaged. To reduce the spectra to a single variable proportional to the local protein concentration at the bottom layer of the wells, the amide II band was integrated between 1590 and 1495 cm–1.
Results and Discussion
ATR-FT-IR Spectroscopy
To assess the potential of ATR-FT-IR spectroscopy for thermal stability assays, the behavior of our IgG was monitored with single element detection (DTGS) offering high spectrum reproducibility. Since previous studies have reported distinct infrared absorption for native protein and aggregates,22,27,30−32 we first tested the ability of ATR-FT-IR spectroscopy to discriminate between native IgG4 and protein aggregates. To obtain a high absorbance spectrum and assign bands without the contribution of water, IgG solutions without buffer salt were simply dried under vacuum to cast a protein film onto the measuring surface of the diamond IRE.
Represented by the blue curve in Figure 2a, the IgG sample has an amide I band peaking at 1637 cm–1, representative of proteins with a secondary structure comprising mainly β-sheets,50 which is the predominant secondary structure as determined by X-ray crystallography.48 At lower frequencies, the amide II and III bands peaked at 1533 and 1220 cm–1, respectively. In between, the infrared spectra of the pure IgG also gave rise to four maxima at 1434, 1388, 1332, and 1301 cm–1 assigned to protein side chain vibration modes.50−52 Additional bands observed at 1066 cm–1 were assigned to polysaccharide because of the known glycosylated state of the IgG.53 These IgG band assignments are listed in Table 1.
Figure 2.
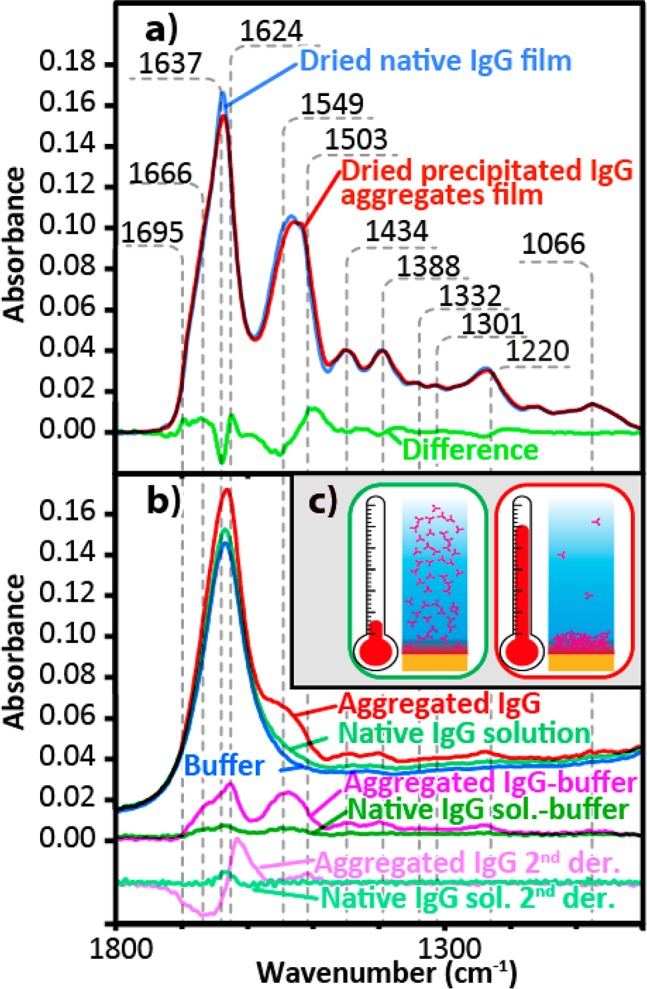
(a) ATR-FT-IR spectra of cast native IgG dried film from a demineralized solution (blue), cast film from thermally induced aggregated IgG solution (red), and the difference between them (green). The dried native film was normalized for comparison using the average absorbance between 1300 and 1350 cm–1. (b) ATR-FT-IR spectra of the buffer (blue), 1 mg/mL native IgG solution (light green), precipitated aggregates induced by heating a 1 mg/mL IgG solution (red), difference between the native IgG solution and the buffer (dark green) and its second derivative (teal), and the difference between the aggregated IgG and the buffer (fuchsia) and its second derivative (pink). (c) Schematics illustrating the native IgG (green box) and the increased local concentration in the volume probed by ATR-FT-IR spectroscopy caused by heat induced protein aggregates precipitation (red box).
Table 1. Assignment of the Main Protein Bands Present between 1700 and 1315 cm–1 a.
| cm–1 | assignment |
|---|---|
| 1695 | amide I, β-sheets50 |
| 1666 | amide I, turns54 |
| 1636 | δ(O–H), water33,34 |
| 1637 | amide I, β-sheets50 |
| 1624 | amide I, β-sheets, aggregates29,30,55−57 |
| 1549 | amide II, unordered58,59 |
| 1503 | amide II, β-sheets, aggregates59−61 |
| 1434 | δas(CH3), β-sheets50,54 |
| 1388 | δS(CH3), Ala, Val50,51 |
| 1332 | δ(CH)51,52 |
| 1301 | w(CH2), Gly51 |
| 1217 | amide III, β-sheets50,62 |
| 1066 | υ(C–O–C), polysaccharides53 |
δ is for bending; υ is for stretching; w is for wagging.
Because the hydrogen bonding strength affects the wavenumber of the amide C=O groups, the wavenumber position of the amide I band is sensitive to the hydrogen bonding network difference between β-sheets in native protein and aggregates.59−61 The red trace in Figure 2a shows the ATR-FT-IR spectrum of a dried IgG film obtained from heat induced precipitated protein aggregates. Spectra were normalized using the 1350 to 1300 cm–1 region which is independent of the protein conformation in order to compensate for any concentration difference between measurements. The difference spectrum (light green) shows a sharp negative peak at 1637 cm–1 as well as a positive peak at 1624 cm–1 indicating the loss of native β-sheets and gain of intermolecular β-sheet aggregates.30,31,55−57 In addition, the positive peaks at 1666 and 1695 cm–1 suggest that aggregated proteins have a higher proportion of turns and antiparallel β-sheets.50 In the amide II region, the negative peak at 1549 cm–1 and the positive component at 1503 cm–1 indicate disordered structure and aggregated β-sheet, respectively.59−61 These differences corroborate conformation discriminating power of the technique.
At 1 mg/mL, the ATR-FT-IR spectrum of solution (dark green curve) is barely distinguishable from the spectrum of the phosphate buffer (blue curve), Figure 2b. As shown, protein bands are only clearly identifiable once the buffer is subtracted. The low signal-to-noise ratio (SNR) suggests that this concentration is close to the detection limit. Depending on their solvent, proteins will undergo a denaturation transition upon heating, resulting in greater backbone mobility.14 The protein thus unfolds and aggregates through the formation of nonspecific hydrophobic interactions or anomalous disulfide bridges.10 As the aggregates grow larger than 100 nm, they typically become insoluble and precipitate.38 The SNR of ATR-FT-IR spectroscopy can therefore be improved by the heat induced precipitation of protein aggregates concentrating at the probed bottom layer as illustrated in Figure 2c.
After heating a sealed 1 mg/mL IgG solution to 60 °C, the red curve of Figure 2b shows a ∼5-fold increase in the local protein concentration. As a result of the higher concentration, the IgG main vibration bands become clearly identifiable in the difference spectrum presented in fuchsia (Figure 2b). Other spectroscopic techniques probing sample bulk such as CD or UV–vis would lose signal upon protein precipitation. This feature makes ATR-FT-IR spectroscopy particularly well suited for protein thermal stability assays.27 Since the concentration of the protein increases in the bottom of the wells where it has been probed, it was impossible to deconvolute the amount of soluble native protein in bulk solution undergoing denaturation and denatured protein precipitating into the probed volume. The later phenomenon should be predominant, since we observed a many fold increase in protein absorbance signal. The single reflection ATR approach required for imaging results in lower sensitivity compared to multireflection ATR. Hence, we only determined the total protein content in the probed volume instead of attempting to quantify the protein conformation using the second derivative or peak deconvolution.
Noncovalent interactions such as hydrogen-bonding, van der Waals, hydrophobic, and electrostatic interactions govern the stability of proteins in solution.63 Since these interactions appear to be highly context dependent,64 predicting their effect is challenging. The protein net charge can be affected by the pH of the buffer as the net charge varies with gained or lost protons (H+). Most proteins are less stable and tend to form aggregates around the pH at which the protein carries no net electrical charge (pI).65 Since ions can shield side chain charges,64,66 controlling the buffer ionic strength is equality important. Because the charge distribution also depends on protein conformation, the pI can only be determined accurately by experimental titration. To find the optimum buffer conditions for protein purification, storage, or handling, high-throughput screening assays are therefore highly favorable.67
Unlike dynamic scanning fluorimetry (DSF) performed in qPCR instruments which can probe many samples in a single heating ramp,25 conventional ATR-FT-IR spectroscopy can only probe one sample at a time. In contrast, mercury cadmium telluride (MCT) focal plane array (FPA) detectors43,68,69 deliver high-throughput by allowing multiple samples to be measured simultaneously. Using such FPA with its 128 by 128 pixels and a field of view of 7.0 mm by 9.8 mm, it was thus possible to probe 12 sample wells simultaneously in this study.
ATR-FT-IR Spectroscopic Imaging
Because of the interfering water OH bending mode at 1636 cm–1, the amide II bands offers the largest absorbance difference, providing a better signal-to-noise ratio than the amide I band for ATR-FT-IR spectroscopic imaging. Shortly after increasing the temperature suddenly to 60 °C, the absorbance across the image does not vary much as spectra are close to zero absorbance. However, after 2000 s (see Figure 3), the relative concentration at the bottom of the well becomes much higher. It is also possible to observe that the relative concentration is much higher for wells containing pH 5 than those with pH 10, suggesting that more aggregates precipitated at low pH.
Figure 3.
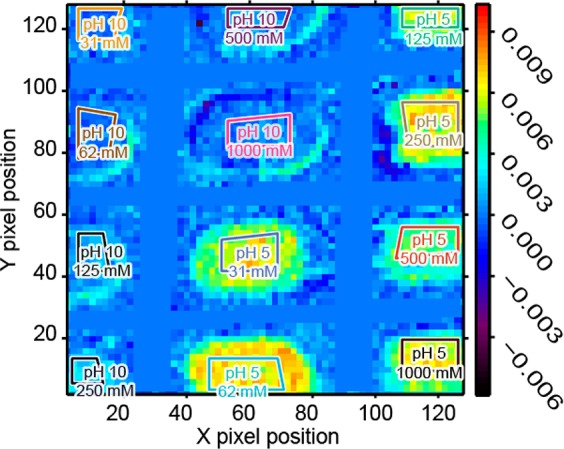
ATR-FT-IR spectroscopic image generated using the amide II band area after 2000 s. All the images collected during the thermal stability assay are presented as a spectroscopic movie in the Supporting Information. The unit of the scale bar is integrated absorbance in cm–1. The polygons represent the integrated region of interest of each well. The field of view covered approximately 9.8 mm along the Y axis and 7.0 mm long the X axis.
Precipitation Kinetics
The pixels contained in the 12 color coded polygons shown in Figure 3 were averaged together to obtain the one spectrum per well region for every image collected at a 150 s interval. The absorbance difference of the amide II band could thus be plotted as a function of time to easily visualize the IgG aggregate precipitation kinetic. Following the abrupt temperature rise to 60 °C, the absorbance of the amide II band increased due to higher concentration at the bottom of the wells caused by protein precipitation. Figure 4a shows two examples of these aggregate precipitation kinetics, at pH 5 (light blue) and pH 10 (dark orange). It is clear that protein precipitated faster and resulted in a greater final amide II absorbance after 12 000 s at pH 5 than at pH 10. Because the cumulative distribution functions for many common probability distributions are sigmoidal, these curves are typically the result of a probabilistic mechanism.14 Such a profile is typical for heat induced protein denaturation and aggregation.14,70 Fitting such kinetic data using a sigmoidal function can thus reduce the data set to a few informative parameters to quickly assess the effect of different buffer conditions on the protein thermal stability.
Figure 4.
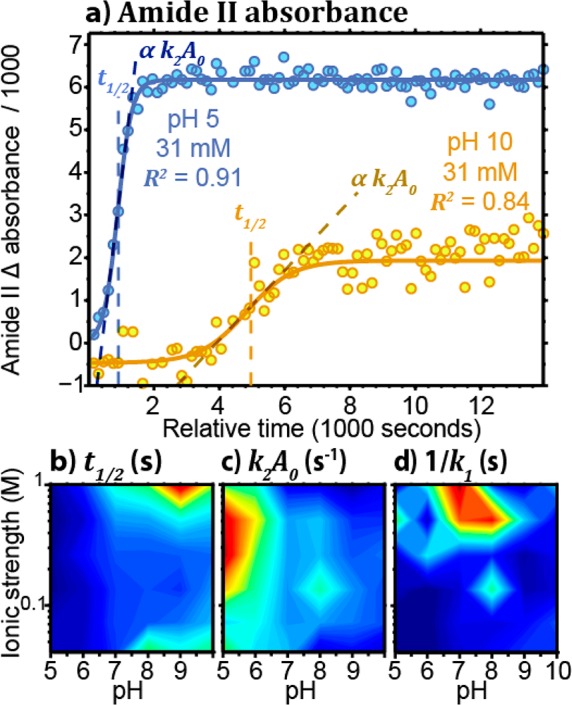
(a) Integrated absorbance difference of the amide II band (1590–1495 cm–1) as a function of time for pH 5 (light blue) and 10 (dark orange) with 31 mM NaCl. The solid lines represent the result of the Finke-Watzky model fitting. (b) t1/2 as a function of the pH and ionic strength. (c) k2[A]0 as a function of the pH and ionic strength. (d) 1/k1 as a function of the pH and ionic strength.
Several kinetic models are sigmoidal, but it is preferable to choose one which provides physically relevant quantities. This is why we selected the Finke-Watzky model, which was previously used to describe protein aggregation kinetics.70−73 Also known as the Minimal/“Ockham’s Razor” model, it assumes a two-step mechanism whereas the native protein slowly nucleates Amonomer → Baggregate at the same time as the aggregates grow autocatalytically Amonomer + Baggregate → 2Baggregate. The rate of native protein depletion −d[A]/dt is dependent on the rate constant of the nucleation (k1) and autocatalytic growth (k2). By integrating over t, the concentration of the protein aggregates [B]t is given by72
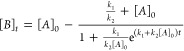 |
1 |
By fitting our kinetic data using eq 1, we were able to calculate the initial concentration [A]0 as well as the rate constants k1 and k2 for the buffer conditions tested. For all kinetic fitting, we calculated a median coefficient of determination R2 of 0.94, indicating that this sigmoidal model can accurately describe the aggregate precipitation mechanism. In Figure 4a, fitted curves are presented with solid lines. Such high coefficients of determination suggest that ATR-FT-IR spectroscopic imaging can be as informative as other techniques for assessing the thermal stability of multiple protein solutions simultaneously. Since 1/k1 is proportional to the induction period tind or lag before the growth, this metric can inform on the nucleation period.72 Other simple sigmoidal models often use the X and Y positions of the inflection point at the half process as fitting parameters. To calculate the position of the inflection point from k1, k2 and [A]0, the time of the midpoint t1/2 was calculated using eq 2.
| 2 |
The t1/2 calculated for the aggregation kinetics presented in Figure 4a is shown as dashed lines. Given in seconds, t1/2 has the advantage of being independent from the absolute absorbance value and thus the initial protein concentration in the wells. Another concentration independent parameter is the normalized slope of the curve at the inflection point, which is then given by eq 3.
| 3 |
k2 × [A]0 is thus given in reciprocal time units such as s–1.
Effect of Buffer Conditions
Using our experimental setup for the high-throughput thermal stability screening assay, we investigated the stability effect of the buffer on the IgG solution for every combination of pH 5, 6, 7, 8, 9, and 10 and ionic strength 0.041, 0.072, 0.135, 0.26, 0.51, and 1.01 M. Unfortunately, pH lower than 5 and higher than 10 could not be studied due to the stability of the ZnSe IRE. For each of the 36 conditions tested, the data from the kinetic was fitted with eq 2 to calculate t1/2 and k2A0.
Figure 4b shows the aggregation half time t1/2 for the 1 mg/mL IgG solution under a range of pH and ionic strength buffer conditions. Around the isoelectric point (pI), charges are neutralized, making intermolecular interaction more favorable, typically resulting in lower stability and solubility.65 Since the pI of the IgG is between 5.5 and 6.0,74 it is known to be less stable at low pH.28 The smaller t1/2 values measured at low pH by our thermal stability assay were thus expected.
The effect of charge shielding on the thermal stability also needed to be determined experimentally as it is highly system dependent and can also depend on the salt bridge content.64,66 To test this effect of the thermal stability of our protein, we varied the ionic strength by adding sodium chloride to the buffer. At low pH, the ionic strength did not appear to affect the t1/2. At higher pH, the IgG carries a net negative charge making the protein more susceptible to electrostatic interactions and charge shielding. At pH 8 to 10, moderate salt concentrations (0.135 and 0.26 M) appear to be less stabilizing than low (0.041 and 0.072 M) or high salt concentration (0.51 and 1.01 M). Although the effect of salt is less marked than the pH, the result is interesting as it indicates that the ionic strength does not have a monotonic effect on the thermal stability of the protein. With smaller t1/2, the lower stability could be the result of a net attractive potential when charges are optimally shielded at moderate ionic strength.75 Such a net attractive potential could result from the remaining van der Waals interactions. At extreme salt concentration, the remaining electrostatic interactions could thus still overcome the attractive interaction and stabilize the protein in solution.
k2[A]0 was also calculated to evaluate the relative rate at which the rapid growth of the aggregates occurs. Figure 4c shows that the relative aggregation rate is roughly the inverse of t1/2. It is the most rapid at low pH and 0.26 to 0.51 M ionic strength while the slope of the kinetic is the smallest at either low or high salt concentration solution or high pH. This result was expected since the t1/2 and k2[A]0 are both dependent on the autocatalytic growth rate k2.
In contrast, tind = 1/k1 shown in Figure 4d does not tell the same story. The lower value of 1/k1 indicates that nucleation occurs much faster at low pH and ionic strength. At high pH, however, the nucleation rate is comparable across the ionic strength range studied. This observation implies that the charge shielding effect observed at high pH has a greater effect on aggregate growth than the nuclei formation rate. By allowing both measurement of protein precipitation in multiple samples in situ and calculation of rate constants, this ATR-FT-IR spectroscopic imaging assay could provide valuable new insights into nonspecific interactions causing undesirable precipitation of protein aggregates.
Conclusions
By taking advantage of FPA detection, this study represents the first example of a high-throughput thermal stability assay based on ATR-FT-IR spectroscopic imaging. Tested using a common immunoglobulin G, this experimental setup allowed the measurement of 12 different buffer conditions simultaneously, allowing studies that are impossible by conventional infrared spectroscopy. In addition, our approach could be extended to standard size 96 well plates with the appropriate magnification.43 With its high-throughput and direct distinct signal measurement, ATR-FT-IR spectroscopic imaging offers many advantages over other techniques such as DSF, DSC, and CD to assess the thermal stability of protein in solution. The precipitation of insoluble aggregates had the benefit of increasing by several fold the local concentration and thus markedly improving the SNR when using ATR. Since the infrared absorption signature of protein aggregates is distinct from the native IgG,29 these experiments allowed us to selectively monitor the formation of insoluble IgG aggregates. Another advantage of using FPA was the opportunity of using the constant regions between the wells as internal reference for the offset of the image absorbance values. With this approach, ATR-FT-IR spectroscopic imaging provided high quality kinetic data of protein aggregate precipitation. By fitting this data to a two-step kinetic model,73 the obtained data from these precipitation experiments agreed with a well-established model for protein aggregation, which includes a nucleation and an autocatalytic growth phase. By testing a wide range of pH and ionic strength conditions, we found that IgG 4 is more stable at high pH but also at either low or high salt concentrations. A high-throughput ATR-FT-IR spectroscopic imaging study of thermal stability assays could thus be considered as a powerful addition to the techniques employed for the direct investigation of protein aggregation in situ. This tool could help address one of the main challenges associated with the development of new therapeutics monoclonal antibody and also any other class of aggregation prone proteins.
Acknowledgments
Funding was provided by the Bioprocessing Research Industry Club (BRIC) (BB/K0111030/1) and the Research Council under the European Community’s Seventh Framework Programme (FP7/2007-2013)/ERC Advanced Grant Agreement [No. 227950]. The authors wish to thank Dr. Cleo Kontoravdi for providing the IgG samples used in this study.
Supporting Information Available
Figure 1S. Size exclusion chromatography (SEC) monodispersity of the purified IgG 4 analyzed by high-throughput thermal stability assay based on ATR-FTIR spectroscopic imaging. Chemical movie of the Amide II band intensity at the bottom of the wells as a function of time during a typical high-throughput thermal stability assay at pH 5 and 10. This material is available free of charge via the Internet at http://pubs.acs.org
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Evans J. B.; Syed B. A. Nat. Rev. Drug Discovery 2014, 13, 413. [DOI] [PubMed] [Google Scholar]
- Cavallo P.; Pagano S.; Boccia G.; De Caro F.; De Santis M.; Capunzo M. Pharmacoepidemiol. Drug Saf. 2013, 22, 130. [DOI] [PubMed] [Google Scholar]
- Philippidis A. Genet. Eng. Biotechnol. News 2012, 32, 10. [Google Scholar]
- Pisal D. S.; Kosloski M. P.; Balu-Iyer S. V. J. Pharm. Sci. 2010, 99, 2557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kayed R.; Head E.; Thompson J. L.; McIntire T. M.; Milton S. C.; Cotman C. W.; Glabe C. G. Science 2003, 300, 486. [DOI] [PubMed] [Google Scholar]
- Hermeling S.; Crommelin D. J. A.; Schellekens H.; Jiskoot W. Pharm. Res. 2004, 21, 897. [DOI] [PubMed] [Google Scholar]
- Sauerborn M.; Brinks V.; Jiskoot W.; Schellekens H. Trends Pharmacol. Sci. 2010, 31, 53. [DOI] [PubMed] [Google Scholar]
- Wang W.; Singh S. K.; Li N.; Toler M. R.; King K. R.; Nema S. Int. J. Pharm. 2012, 431, 1. [DOI] [PubMed] [Google Scholar]
- van den Berg B.; Ellis R. J.; Dobson C. M. EMBO J. 1999, 18, 6927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldberg M. E.; Rudolph R.; Jaenicke R. Biochemistry 1991, 30, 2790. [DOI] [PubMed] [Google Scholar]
- Nilsson M. R.; Driscoll M.; Raleigh D. P. Protein Sci. 2002, 11, 342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szabo Z.; Klement E.; Jost K.; Zarandi M.; Soos K.; Penke B. Biochem. Biophys. Res. Commun. 1999, 265, 297. [DOI] [PubMed] [Google Scholar]
- Nagano T.; Hirotsuka M.; Mori H.; Kohyama K.; Nishinari K. J. Agric. Food Chem. 1992, 40, 941. [Google Scholar]
- Porter D.; Vollrath F. Soft Matter 2008, 4, 328. [DOI] [PubMed] [Google Scholar]
- Roberts C. J. J. Phys. Chem. B 2003, 107, 1194. [Google Scholar]
- Lemieux M. J.; Reithmeier R. A. F.; Wang D. N. J. Struct. Biol. 2002, 137, 322. [DOI] [PubMed] [Google Scholar]
- Kawate T.; Gouaux E. Structure 2006, 14, 673. [DOI] [PubMed] [Google Scholar]
- Horsefield R.; Yankovskaya V.; Tornroth S.; Luna-Chavez C.; Stambouli E.; Barber J.; Byrne B.; Cecchini G.; Iwata S. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2003, 59, 600. [DOI] [PubMed] [Google Scholar]
- Li Y.; Weiss W. F.; Roberts C. J. J. Pharm. Sci. 2009, 98, 3997. [DOI] [PubMed] [Google Scholar]
- Liu J.; Andya J. D.; Shire S. J. AAPS J. 2006, 8, E580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutmann D. A. P.; Mizohata E.; Newstead S.; Ferrandon S.; Henderson P. J. F.; Van Veen H. W.; Byrne B. Protein Sci. 2007, 16, 1422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Militello V.; Casarino C.; Emanuele A.; Giostra A.; Pullara F.; Leone M. Biophys. Chem. 2004, 107, 175. [DOI] [PubMed] [Google Scholar]
- Gimel J. C.; Durand D.; Nicolai T. Macromolecules 1994, 27, 583. [Google Scholar]
- Berne B. J. P. R.Dynamic light scattering: With applications to chemistry, biology, and physics; Wiley: New York, 1976. [Google Scholar]
- Vollrath F.; Hawkins N.; Porter D.; Holland C.; Boulet-Audet M. Sci. Rep. 2014, 4, 5625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garner H. R.; Armstrong B.; Lininger D. M. Biotechniques 1993, 14, 112. [PubMed] [Google Scholar]
- Kask L.; Villoutreix B. O.; Steen M.; Ramesh B.; Dahlback B.; Blom A. M. Protein Sci. 2004, 13, 1356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li C. H.; Narhi L. O.; Wen J.; Dimitrova M.; Wen Z. Q.; Li J.; Pollastrini J.; Nguyen X.; Tsuruda T.; Jiang Y. J. Biochemistry 2012, 51, 10056. [DOI] [PubMed] [Google Scholar]
- Middleton C. T.; Marek P.; Cao P.; Chiu C. C.; Singh S.; Woys A. M.; de Pablo J. J.; Raleigh D. P.; Zanni M. T. Nat. Chem. 2012, 4, 355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shivu B.; Seshadri S.; Li J.; Oberg K. A.; Uversky V. N.; Fink A. L. Biochemistry 2013, 52, 5176. [DOI] [PubMed] [Google Scholar]
- Schwartz D.; Sofia S.; Friess W. Eur. J. Pharm. Biopharm. 2006, 63, 241. [DOI] [PubMed] [Google Scholar]
- Bondos S. E. Curr. Anal. Chem. 2006, 2, 157. [Google Scholar]
- Max J. J.; Chapados C. J. Chem. Phys. 2009, 131, 184505. [DOI] [PubMed] [Google Scholar]
- Bertie J. E.; Lan Z. D. Appl. Spectrosc. 1996, 50, 1047. [Google Scholar]
- Harrick N. J.Internal reflection spectroscopy; John Wiley & Sons: New York, 1967. [Google Scholar]
- Glassford S. E.; Byrne B.; Kazarian S. G. Biochim. Biophys. Acta, Proteins Proteomics 2013, 1834, 2849. [DOI] [PubMed] [Google Scholar]
- Kazarian S. G.; Chan K. L. A. Analyst 2013, 138, 1940. [DOI] [PubMed] [Google Scholar]
- Narhi L. O.; Schmit J.; Bechtold-Peters K.; Sharma D. J. Pharm. Sci. 2012, 101, 493. [DOI] [PubMed] [Google Scholar]
- Krebs M. R. H.; Devlin G. L.; Donald A. M. Biophys. J. 2007, 92, 1336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauer H. H.; Muller M.; Goette J.; Merkle H. P.; Fringeli U. P. Biochemistry 1994, 33, 12276. [DOI] [PubMed] [Google Scholar]
- Sellick C. A.; Hansen R.; Jarvis R. M.; Maqsood A. R.; Stephens G. M.; Dickson A. J.; Goodacre R. Biotechnol. Bioeng. 2010, 106, 432. [DOI] [PubMed] [Google Scholar]
- Chan K. L. A.; Kazarian S. G. J. Comb. Chem. 2005, 7, 185. [DOI] [PubMed] [Google Scholar]
- Chan K. L. A.; Kazarian S. G. Lab Chip 2006, 6, 864. [DOI] [PubMed] [Google Scholar]
- Kazarian S. G. Anal. Bioanal. Chem. 2007, 388, 529. [DOI] [PubMed] [Google Scholar]
- Chan K. L. A.; Govada L.; Bill R. M.; Chayen N. E.; Kazarian S. G. Anal. Chem. 2009, 81, 3769. [DOI] [PubMed] [Google Scholar]
- Glassford S.; Chan K. L. A.; Byrne B.; Kazarian S. G. Langmuir 2012, 28, 3174. [DOI] [PubMed] [Google Scholar]
- Glassford S. E.; Govada L.; Chayen N. E.; Byrne B.; Kazarian S. G. Vib. Spectrosc. 2012, 63, 492. [Google Scholar]
- Davies A. M.; Rispens T.; Ooijevaar-de Heer P.; Gould H. J.; Jefferis R.; Aalberse R. C.; Sutton B. J. J. Mol. Biol. 2014, 426, 630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrick N. J. J. Opt. Soc. Am. 1965, 55, 851. [Google Scholar]
- Moore W. H.; Krimm S. Biopolymers 1976, 15, 2465. [DOI] [PubMed] [Google Scholar]
- Barth A. Prog. Biophys. Mol. Biol. 2000, 74, 141. [DOI] [PubMed] [Google Scholar]
- Colthup N. B.Introduction to infrared and raman spectroscopy; Academic Press: New York, 1964. [Google Scholar]
- Barsberg S. J. Phys. Chem. B 2010, 114, 11703. [DOI] [PubMed] [Google Scholar]
- Barth A. Biochim. Biophys. Acta, Bioenerg. 2007, 1767, 1073. [DOI] [PubMed] [Google Scholar]
- Jackson M.; Haris P. I.; Chapman D. Biochemistry 1991, 30, 9681. [DOI] [PubMed] [Google Scholar]
- Boulet-Audet M.; Buffeteau T.; Boudreault S.; Daugey N.; Pézolet M. J. Phys. Chem. B 2010, 114, 8255. [DOI] [PubMed] [Google Scholar]
- Arrondo J. L. R.; Goni F. M. Prog. Biophys. Mol. Biol. 1999, 72, 367. [DOI] [PubMed] [Google Scholar]
- de Jongh H. H. J.; Goormaghtigh E.; Ruysschaert J. M. Anal. Biochem. 1996, 242, 95. [DOI] [PubMed] [Google Scholar]
- Paquet-Mercier F.; Lefevre T.; Auger M.; Pezolet M. Soft Matter 2013, 9, 208. [Google Scholar]
- Boulet-Audet M.; Terry A. E.; Vollrath F.; Holland C. Acta Biomater. 2013, 10, 776. [DOI] [PubMed] [Google Scholar]
- Sonoyama M.; Nakano T. Appl. Spectrosc. 2000, 54, 968. [Google Scholar]
- Cai S. W.; Singh B. R. Biochemistry 2004, 43, 2541. [DOI] [PubMed] [Google Scholar]
- Dobson C. M. Philos. Trans. R. Soc., B 2001, 356, 133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makhatadze G. I.; Loladze V. V.; Ermolenko D. N.; Chen X. F.; Thomas S. T. J. Mol. Biol. 2003, 327, 1135. [DOI] [PubMed] [Google Scholar]
- Shaw K. L.; Grimsley G. R.; Yakovlev G. I.; Makarov A. A.; Pace C. N. Protein Sci. 2001, 10, 1206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wunderlich M.; Martin A.; Schmid F. X. J. Mol. Biol. 2005, 347, 1063. [DOI] [PubMed] [Google Scholar]
- Zheng B.; Roach L. S.; Ismagilov R. F. J. Am. Chem. Soc. 2003, 125, 11170. [DOI] [PubMed] [Google Scholar]
- Chan K. L. A.; Kazarian S. G. J. Comb. Chem. 2006, 8, 26. [DOI] [PubMed] [Google Scholar]
- Chan K. L. A.; Kazarian S. G.; Vassou D.; Gionis V.; Chryssikos G. D. Vib. Spectrosc. 2007, 43, 221. [Google Scholar]
- Morris A. M.; Watzky M. A.; Finke R. G. Biochim. Biophys. Acta, Proteins Proteomics 2009, 1794, 375. [DOI] [PubMed] [Google Scholar]
- Watzky M. A.; Morris A. M.; Ross E. D.; Finke R. G. Biochemistry 2008, 47, 10790. [DOI] [PubMed] [Google Scholar]
- Morris A. M.; Watzky M. A.; Agar J. N.; Finke R. G. Biochemistry 2008, 47, 2413. [DOI] [PubMed] [Google Scholar]
- Watzky M. A.; Finke R. G. J. Am. Chem. Soc. 1997, 119, 10382. [Google Scholar]
- Melvin T.; Kim Y.; Michael A. F. Am. J. Pathol. 1984, 115, 443. [PMC free article] [PubMed] [Google Scholar]
- Liu Y.; Fratini E.; Baglioni P.; Chen W. R.; Chen S. H. Phys. Rev. Lett. 2005, 95, 118102. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.