

Summary
The zebrafish (Danio rerio) has long been used as a model for developmental biology, making it an excellent model to use also in developmental toxicology. The many advantages of zebrafish include their small size, prolific spawning, rapid development, and transparent embryos. They can be easily manipulated genetically through the use of transgenic technology and gene knock-down via morpholino-modified antisense oligonucleotides (MOs). Knocking down specific genes to assess their role in the response to toxicant exposure provides a way to further our knowledge of how developmental toxicants work on a molecular and mechanistic level, while establishing a relationship between these molecular events and morphological, behavioral, and/or physiological effects (i.e. phenotypic anchoring).
In this chapter we address important considerations for using MOs to study developmental toxicology in zebrafish embryos and provide a protocol for their use.
Keywords: (5-10) reverse genetics, morpholino, developmental toxicology, knockdown, antisense, mechanisms of toxicity, oxidative stress, aryl hydrocarbon receptor
1. Introduction
Zebrafish share many cellular and physiological characteristics with other vertebrates and have been widely used as a model in vertebrate developmental biology and genetics since the 1960s. The zebrafish model has since been embraced by the fields of toxicology, pharmacology, neurobiology and behavior, and also is used to investigate mechanisms underlying human diseases (for reviews, see 1-4). As such, it is ideally positioned as a model for the interdisciplinary field of developmental toxicology.
Compared to mammalian models, zebrafish are relatively easy to manipulate genetically, and there are numerous tools available to do so (e.g. generation of mutant and transgenic fish, injection of RNA for overexpression, gene targeting by zinc finger nuclease technology). One of the most rapid and economical techniques for performing reverse genetic analysis is the powerful approach employing morpholino-modified anti-sense oligonucleotides (MOs), which provide transient gene knockdown by binding to RNA and inhibiting protein synthesis (5). MOs are antisense oligonucleotides of typically 25 bases, synthesized with a non-ionic, phosphorodiamidate backbone with morpholine rings substituted for ribose; an excellent description of the chemistry underlying MOs is available elsewhere (6). MOs bind to targeted RNA and function either to block protein translation of the mRNA at the translation start site or to inhibit the splicing of the primary transcript to the mature mRNA (see Figure 1). Both types of MOs will inhibit protein synthesis from zygotic transcripts; start site MOs will also knock down maternally loaded mRNAs, whereas splice-blocking MOs are ineffective on these already mature transcripts. MOs also can be designed to bind to microRNAs and prevent their function (7). Because MOs targeting mRNAs are highly effective through four days post-fertilization (8, 9), they can be used to study gene function during the entire period of zebrafish embryonic development.
Figure 1.

Illustration of how start site (A) and splice site (B) morpholinos work. Under normal conditions, following transcription of DNA to pre-mRNA, introns are spliced out and mature RNA is exported to the cytoplasm for protein translation. A start site morpholino functions by binding to a sequence on or close to the ATG start site of the mRNA transcript, sterically blocking the translation machinery and thus preventing protein synthesis. Splice site morpholinos act upon pre-mRNA by binding to intron/exon junctions and preventing proper splicing. Depending on the sequence, this can result in either retention of an intron as illustrated in pathway B, loss of an exon, or the use of a cryptic splice site causing either truncation of an exon or retention of a partial intron. Upon export to the cytoplasm, these altered transcripts would either be degraded by nonsense-mediated decay, or in some cases could result in synthesis of a truncated protein.
MOs have been widely used in studies of zebrafish development, in part because of the lack (until recently) of methods for generating targeted null mutants as well as the accessibility of the MO technique to most laboratories. In addition, for some applications gene knockdown offers distinct advantages as compared to gene knockout approaches. Gene knockout confers complete inactivation of the gene and loss of functional gene product, whereas gene knockdown is characterized by reduced expression of the gene product without its complete elimination. In cases where a gene is required for development, the MO concentration can be titrated down to a level that is not embryolethal (e.g. 10). Another way to bypass embryolethal gene knockdowns is through the use of “caged” MOs that can be photo-activated to act in specific locations at specific times, thus creating conditional knockdowns in developing zebrafish (11, 12).
In the past few years, the use of MOs to study the mechanistic roles of specific genes in embryo toxicity has grown dramatically (see Table 1). For example, MOs can be used to determine the role of specific transcription factors in mediating toxicity; knock-down of such a transcription factor would provide protection against toxicity. Alternatively, gene knock-downs may sensitize the embryo to effects of a chemical, suggesting that the gene provides a protective effect, for example through its regulation of protective responses or its participation in detoxication reactions. MO knock-down may be used to confirm the identity of a proposed target protein, in which case the phenotype of the morphant will mimic the toxic response caused by chemical exposure. MOs are also excellent tools with which to confirm mechanistic hypotheses generated by exploratory methods such as gene expression profiling by microarrays (10, 13).
Table 1.
Examples of how MOs in zebrafish have been used to understand the role of a particular gene in developmental toxicology of various chemicals. hpf = hours post fertilization; dpf = days post fertilization. TCDD = 2,3,7,8-tetrachlorodibenzo-p-dioxin; PAH= polycyclic aromatic hydrocarbons; BNF= β-naphthoflavone; ANF= α=naphthoflavone; tBOOH= tert butyl hydroperoxide; tBHQ = tert butyl hydroquinone; PCB = polychlorobiphenyl; MPTP = 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine; FICZ = 6-formylindolo[3,2-b]carbazole; PFOS = perfluorooctane sulfonate.
| Gene targeted | Toxicant |
Time of
Exposure |
Toxicant Effect | MO effect on toxicity | Reference |
|---|---|---|---|---|---|
| Ahr2 | TCDD | 3-4 hpf | Pericardial edema at 96 hpf | Protected | 24 |
| PCB-126 | 8-32 hpf | Pericardial edema at 72 hpf | Protected | 33 | |
| FICZ | 48-56 hpf | Non-toxic, induction of Cyp1 genes |
Decreased gene induction |
34 | |
| Tri-cyclic PAHs, crude oil |
4-48 hpf | Cardiac dysfunction | No effect | 35 | |
| PAHs (pyrene) |
4-48 hpf | Cardiac dysfunction | Protected | ||
| PAH (benz[a]ant hracene) |
6-48 hpf | Pericardial edema | Protected | 36 | |
| PAHs (BNF, ANF) |
24-96 hpf | Pericardial edema and truncation of Meckel’s cartilage at 72 and 96 hpf |
Protected | 21 | |
| Ahr1a | PAH (pyrene) |
6-96 hpf | Pericardial edema and hepatomegaly at 96 hpf |
Protected | 35 |
| Cyp1a | PAHs (ANF, BNF) |
24-96 hpf | Pericardial edema and truncation of Meckel’s cartilage at 96 hpf |
Exacerbated | 21 |
| TCDD | 4-5 hpf | Pericardial edema and truncation of Meckel’s cartilage at 72 and 96 hpf |
No effect | 22 | |
| Cyp1b1 | PAHs (ANF, BNF) |
24-96 hpf | Pericardial edema and truncation of Meckel’s cartilage at 96 hpf |
No effect | 37 |
| Arnt1 | TCDD | 3-4 hpf | Pericardial edema at 96 hpf | Protected | 38 |
| Arnt2 | TCDD | 3-4 hpf | Pericardial edema at 96 hpf | No effect | 29 |
| Ahrra | TCDD | 6 hpf | Pericardial edema at 72 hpf | Exacerbated | 17 |
| - | - | - | Mimics TCDD deformities at 72 hpf |
17 | |
| Ahrrb | TCDD | 6 hpf | Pericardial edema at 72 hpf | Exacerbated | 17 |
| p53 | PCB 126 | 5-24 hpf | Cardiac dysmorphogenesis at 72 hpf |
No effect | 39 |
| Sox9b | - | - | - | Mimics TCDD jaw deformities at 72 hpf |
13 |
| R-Spondinl | TCDD | 2 dpf | Inhibition of fin regeneration at 2-3 days post amputation (4-5 dpf) |
Protected | 10 |
| Nrf2 | tBOOH | 24-96 hpf | Mortality at 96 hpf | Exacerbated | 8 |
| tBHQ | 4 dpf | Induction of gstp mRNA | Protected | 30 | |
| PFOS | 4-96 hpf | Increased detection of reactive oxygen species (ROS) and expression of antioxidant genes |
Increased ROS detection with DCF- DA probe; reduced induction of HO-1 gene expression |
40 | |
| cardiac troponin T | PCB 126 | 5-24 hpf | Cardiac dysmorphogenesis at 72 hpf |
Protected | 39 |
| Dopamine transporter (DAT) |
MPTP | 24-96 hpf | Neurodegeneration at 4 dpf | Protected | 41 |
| Cox2 | TCDD | 24-50 hpf | Mesencephalic blood flow reduced at 50 hpf |
Protected | 42 |
| thromboxane synthase |
TCDD | 24-50 hpf | Mesencephalic blood flow reduced at 50 hpf |
Protected | 42 |
| cereblon (CRBN) | - | - | - | Mimics thalidomide toxicity (Exposure 2- 72 hpf results in reduction in size of otic vesicles at 30 hpf, loss or truncation of pectoral fins at 75 hpf,) |
43 |
| Cul4a | |||||
| PTEN (phosphatase/tensi n homolog)- induced putative kinase (PINKl) |
MPTP | 1-4 dpf | Reduced swimming activity, loss of TH immunoreactivity in cells in the parvocellular pretectal nucleus |
Exacerbated | 28 |
There are several important considerations in the design of a MO knockdown experiment when performed in the context of developmental toxicology in zebrafish. First is careful selection of the gene to be targeted. Zebrafish often have duplicated genes (paralogs) that are co-orthologous to a single mammalian gene, a result of a whole genome duplication that occurred in the teleost lineage after its divergence from the lineage leading to tetrapods (14). In some cases, the duplicated fish genes have partitioned the multiple functions of their single mammalian ortholog (14). For example, fish, including zebrafish, have duplicated copies of genes in the aryl hydrocarbon receptor (AHR)-dependent signaling pathway that is involved in the response to dioxin-like chemicals (15). While mammals and birds have a single gene for the aryl hydrocarbon receptor repressor (AHRR), zebrafish have two AHRR genes, AHRRa and AHRRb (16). Studies employing paralog-specific knock-down of AHRRa and AHRRb have revealed that these two proteins have distinct functions in regulating the AHR signaling pathway in the unexposed embryo and in the response to dioxin exposure (17). The zebrafish genome has been fully sequenced, but its assembly and annotation are still underway; it is therefore important to carefully screen for gene paralogs when designing a new MO.
The design of a MO knockdown experiment when performed in the context of developmental toxicology in zebrafish requires careful consideration of timing: timing of MO persistence in relation to the developmental expression of the target gene, timing of chemical exposure relative to the duration of MO action and the windows of toxicant sensitivity during development, and timing of assessment of the effect (see Figure 2). One can often examine in situ hybridization data in the database at www.zfin.org to help determine when and where a particular target gene is expressed.
Figure 2.

Illustration of timing of MO effectiveness and toxicant exposure during development. While period of MO effectiveness will vary depending on the synthesis/degradation kinetics of its target gene product, in general, MO persistence will decrease by dilution as development progresses (solid line). How MO effectiveness overlaps with sensitivity to the toxicant being tested (dashed line), when the exposure occurs, as well as when the effect is observed, should all be considered when designing a MO experiment.
Third, it is important to verify the efficacy of each MO, i.e. how well it is knocking down the desired protein target (9, 18-20). Ideally, this should be done by measuring the amount of the target protein expressed in the MO-injected embryos. This can be accomplished with an existing antibody in conjunction with western blotting or immunohistochemistry, or indirectly, by using protein function assays (21, 22). For splice-site MOs, the effect on the amount of mature mRNA can be measured by quantitative RT-PCR (23). Often, however, suitable antibodies are not available, or a gene has only one exon, precluding the use of splice-blocking MOs. In such cases, an alternative approach is to assess the efficacy of translational inhibition in vitro, by using in vitro transcription and translation to assess the ability of the MO to inhibit protein synthesis directed by a cloned cDNA encoding the target protein (17, 24).
Finally, the specificity of the MO effect must be confirmed. This is generally done by confirming the effect using a second MO, ideally one that does not overlap the sequence of the first one (9). In some cases it is possible to synthesize capped mRNA, co-inject this with your MO, and observe rescue of the MO effect (e.g. 13).
This chapter provides a protocol for the use of MO technology in the context of studies to examine mechanisms of developmental toxicity of chemicals. We provide methods for microinjection of MO oligonucleotides into developing zebrafish embryos, verification of MO function via inhibition of in vitro protein translation, and exposure of embryos to developmental toxicants.
2. Materials
2.1 Zebrafish
Adult zebrafish (Danio rerio) of prime breeding age (3-15 months), from a defined strain such as AB, TL, or Tubingen (see Note 1).
2.2 Microinjection instrumentation and other equipment
Needle puller (Narishige PC-10).
Microinjection apparatus (for example, Narishige IM-300 microinjector) with foot pedal (see Figure 3).
Nitrogen gas tank and regulator.
Micromanipulator (for example, Narishige).
Dissecting microscope with eyepiece micrometer (e.g. Zeiss Stemi 2000C).
Fluorescence microscope with GFP fluorescence filter.
Gel dryer (for example, Bio Rad Model 583).
Figure 3.

Microinjection equipment: dissecting scope and light source, microinjector (on left side of shelf), micromanipulator (next to scope stand), and needle puller (in the back right).
2.3 Small equipment and supplies
Injection egg holder tray made from agarose (see methods 3.2.1). This can be stored in water with methylene blue at 4°C for several months.
Borosilicate glass capillary tubes with microfilament (World Precision Instruments 1B100F-4) pulled into injection needles (see methods 3.2.2). These can be stored in a clean petri dish mounted on a raised platform such as a microscope slide with double-sided sticky tape or modeling clay.
Needle backloader, such as made from a 1 ml syringe (see methods 3.2.3).
2 ml plastic transfer pipettes.
Parafilm.
0.5 ml eppendorf tubes.
Glass plate (2“×3”) to hold eggholder.
Rimmed microscope slide that fits on fluorescent scope.
Ribbon cable to make the eggholder.
2.4 Reagents and solutions
Gene-specific MO and control MO (Gene Tools, LLC, Philomath, OR). Stock solutions can be stored at −20°C, and working solutions at room temperature. If fluorescein tagged, protect from light.
Sterile water for dissolving MOs and making solutions.
Methylene blue.
Danieau’s water (also called “Embryo medium,” recipe found in the Zebrafish Book online at http://zfin.org/zf_info/zfbook/chapt10.html#wptohtml16)
Mineral oil for calibrating needles.
Toxicant of choice and solvent control as necessary.
Agarose.
2.5 Validation Tools
TNT kit and reagents (Promega).
Cloned gene in a T7, SP6, or T3 polymerase-controlled plasmid.
[35S]methionine.
SDS-PAGE gels and buffers.
X-ray film.
Gel fixing solution (10% acetic acid, 7.5% methanol, 5% glycerol).
Amplify fluorography solution (GE Healthcare).
3. Methods
3.1 Target identification and MO design
Identify gene target and sequence (see Note 2). Decide whether a start-site or splice-site MO is more appropriate (see Note 3). You can also choose to knock-down more than one target at a time, for example by using paralog-specific MOs to knock down both copies of a duplicated gene.
Identify control MO to be used. You can use the standard control MO from Gene Tools (targeted to a rare mutation in human beta globin) or a mismatch to your specific target (see Note 4). For this example, the standard control MO was used.
Decide whether you want your MO with a tag. In this example, the carboxyfluorescein tag was used (Note 5).
Order MO(s); it can take a week or two to synthesize. Upon arrival, dissolve in sterile water to make a 1 mM stock solution; aliquot and store at −20°C in sterile eppendorf tubes wrapped in parafilm and protected from light until use. When defrosting an aliquot, heat to 65°C for 5 minutes to ensure that the MO is solubilized before diluting to a working concentration with sterile Danieau’s water. In this example, we used a working concentration of 0.18 mM (see Note 6).
3.2 Preparation for Injection
3.2.1 Construction of Eggholder Tray
While many variations of this exist (see Note 7), we use double-sided sticky tape to secure ridged ribbon cable to bottom of a 60 mm plastic petri dish.
Prepare a 3% agarose solution with Danieau’s water and pour on top of the ribbon cable, taking care that the cable remains flat.
Allow agarose to set and then cut agarose lengthwise down the center of the ribbon cable to make two molds; cut off agarose to make rectangular shaped molds (see Figure 4).
Store in water with methylene blue at 4°C.
Prior to use, warm to room temperature and place on glass plate for egg mounting and injection.
Figure 4.

(A) Egg holder on glass plate. The ribbon tape mold leaves an imprint of ridges surrounded by three agarose walls. Embryos are held in place by the walls and ridges during injection as shown in (B).
3.2.2 Making Needles
Insert borosilicate glass capillary tube into needle-puller instrument and secure.
Use machine to pull the needle using optimized settings (see Note 8); we use a temperature setting of 60°C with one weight removed.
Break tip by holding a razor blade at a 45° angle to the needle and cutting off the end of the tip to a 1 μm diameter.
3.2.3 Filling needles
Make a back-filler. While there are several different methods for filling a needle (see Note 9), we use a modified 1 ml sterile plastic syringe. With the plunger removed, hold the syringe in a horizontal position and roll the syringe over a flame, targeting the flame tip to the 0.3 to 0.4 measurement indicators on the syringe until the plastic just begins to melt. Be careful not to let the plastic burn. Then turn the syringe to a vertical position and gently pull the unflamed bottom opening of the syringe such that it drops to the floor and stretches the plastic in between into a string. Continue to pull the string slowly until the plastic hardens. Use scissors to cut the plastic string about 4” from the syringe. Test to make sure that air will flow through your backfiller by inserting the plunger into the syringe. You will also need to test whether your backfiller is thin enough to fit into the back of your needle (see Figure 5).
Dispense 2 μl of your MO onto a square of parafilm and draw it up into your backfiller by slowly pulling the plunger up.
Holding the needle horizontally, insert the backloader into the end pore of the needle until it reaches the point at which the internal needle tip tapers. Slowly depress plunger down and withdraw backloader as the solution fills the needle.
The microfilament inside the capillary tube should wick the MO solution into the tip of the needle (see Note 10).
Check needle under microscope to be sure there are no air bubbles in the tip (see Note 11).
Figure 5.

Example of backloader (below) made from a 1 ml plastic syringe (above).
3.2.4 Calibrating needles
Set microscope magnification to the same setting that you will use to microinject; we generally use a 3.2× objective lens.
Load needle into micromanipulator.
Turn on microinjector, open nitrogen gas regulator. Adjust psi settings on microinjector; we use 5-7 psi with an injection time of 10-30 msec.
Place a drop of mineral oil onto a piece of parafilm.
Inject a bolus of your MO into a drop of mineral oil.
Measure the diameter of the sphere with an eyepiece micrometer; volume of the bolus V = 4/3 πr3
Be sure to note the size of the bolus with respect to the micrometer, and use this to continually monitor the bolus size to be sure that it does not change during the course of injection (see Note 12). For example, a bolus with a 160 μM diameter is equivalent to 2.1 nl (17)
3.2.5 Embryo collection and mounting for injection
Collect embryos from matings in 20 minute intervals; zebrafish generally breed best at morning’s first light.
Rinse eggs well with DI water.
Maintain eggs in Danieau’s water and screen for cytoplasmic streaming or cell division as indications of fertilization.
Place eggholder onto a glass plate.
Array 0-4 cell stage zygotes onto eggholder; our eggholder holds 50 (see Figure 4).
Remove any water that has transferred onto the eggholder with the eggs; this is important, as the surface tension between the chorion and the agarose wall is required to prevent the egg from moving while withdrawing the needle after injection.
3.3 Injection Process
Position needle next to chorion, approximately halfway between the top and bottom of the chorion, at a 35-45° angle to the egg holder floor.
Use micromanipulator to insert needle through chorion into the yolk.
Aim to place bolus into the center of the yolk underneath the cell(s) up to the 4-cell stage (see Note 13).
Press foot pedal to inject MO.
Withdraw needle gently, avoiding damage to the yolk, cells, and membranes.
To determine how many embryos to inject, you will need to first consider the number needed for your experimental design. Then take into account the background fertilization and deformity rate of the fish strain (see Note 14). Add an extra 10% to allow for variation in MO incorporation into the embryo tissue. When first using the microinjection technique, you will likely need to inject double the number of embryos you would like to use; these numbers will likely decrease as proficiency with the technique increases. With practice, it is possible for one person to inject 1200 embryos over the course of 3 hours.
Retain non-injected clutch control embryos.
3.4 Screening
Remove any dead or undeveloped embryos daily.
Screen for fluorescence incorporation. This can be done 3-4 hours post injection or on the following day around 24 hpf, depending on the timing of toxicant exposure. We prefer to use the 24 hpf timepoint for screening when possible as it allows us to better distinguish weak vs. strong fluorescence incorporation (see Note 15).
Place embryos in individual droplets on a rimmed slide.
Assess fluorescence on each embryo (see Figure 6).
Remove any embryos that have weak fluorescence or only partially distributed fluorescence.
Figure 6.

Examples of a successful (above) vs. failed (below) incorporation of a fluorescently tagged morpholino at 24 hpf (50×), following injection with ~2 nl of 0.18 mM of Ahr2-MO at the 1-4 cell stage.
3.5 Validation
3.5.1 Efficacy validation with TnT
While there are several ways to confirm that your MO has indeed knocked down your target (see Note 16), we describe here the use of in vitro transcription and translation as a convenient approach for validating the efficacy of start-site MOs. This is a procedure in which the DNA template is transcribed and translated in the presence of [35S]methionine (see Note 17). The resulting radiolabeled protein can be visualized by SDS-PAGE and autoradiography. The efficacy of a MO can be determined by its ability to attenuate protein synthesis. The template for the reaction is a T7, SP6 or T3 polymerase-controlled expression vector that contains the cDNA for the target gene, including the 5′ untranslated region complementary to the MO. Since the template does not contain any introns, this method is not suitable for testing splice-site MOs.
Combine 20 μl TnT-quick lysate (Promega), 1 μg template plasmid, 1 μl [35S]methionine (10μCi/μl), 0.5 μl gene-specific or control MO (25μM) or water, and water up to 25 μl. The final concentration of MO is 500 nM.
Incubate at 30 °C for 90 minutes.
Resolve 5 μl aliquots of each reaction on an SDS-PAGE gel.
Soak gel in fixing solution 2 hours or overnight, shaking gently.
Soak in fluorography (Amplify (GE Healthcare) solution for 30 minutes, shaking gently.
Vacuum-dry gel at 80 °C for 2 hours and expose to X-ray film.
Measure intensity of the protein bands by densitometry, or by scintillation counting of the excised bands (see Figure 7).
Figure 7.

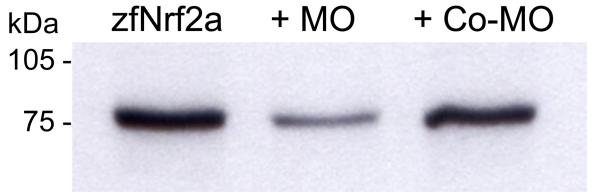
Assessment of MO efficacy by in vitro transcription and translation. An expression construct encoding zebrafish Nrf2a (30) was transcribed and translated in vitro in the absence of any MO (lane 1) or in the presence of Nrf2-MO (500 nM; lane 2) or a control MO (500 nM; lane 3). Translated Nrf2 protein was labeled with [35S]methionine.
3.5.2 Demonstration of MO Specificity
It is important to confirm that any effect observed is due to a specific interaction between the MO and the target sequence, and not due to non-specific effects (see Note 18). To do this, a second MO, ideally of non-overlapping sequence, should be tested. If the same effect is observed, it is highly likely that this is an effect specific to knockdown of your target gene (see Note 19). It is also advisable to use a mismatch control, that is one that is specific to your target sequence with exception of a few basepairs.
3.6 Exposure of embryos and assessment of effects
The details regarding embryo exposure and phenotypic assessment will vary depending on the toxicant of interest, solvent control, and effects of concern. The latter may include gene expression (assessed by quantitative, real-time RT-PCR, for example), morphological (gross) and histological (microscopic) changes, and/or behavioral changes.
We describe here, as an example, a protocol for exposing Ahr2 MO-injected embryos to PCB-126, with an endpoint altered morphology (deformities) assessed at 72 hpf (See Note 21). In this experiment, 150 embryos were injected with ~2 μL of 0.18 mM Ahr2-MO (3.6 ng), and 150 embryos were injected with the same amount of control-MO. Embryos were screened by fluorescence microscopy for MO incorporation and for the background incidence of deformities, as described above.
Conduct preliminary experiments with uninjected embryos to identify a dose of chemical (PCB-126) that causes deformities (in this case, pericardial edema and truncation of Meckel’s cartilage), the window of susceptibility, and the timing of onset of effects.
Expose triplicate pools of 50 embryos to either PCB-126 or solvent (DMSO). Prepare a 15-cm glass Petri dish containing 50 embryos in 100 mL of 0.3× Danieau’s water. Add 10 μL of a 50-μM solution of PCB-126 in DMSO; stir with pipette to mix well. Expose for 24 hr.
At the end of the exposure, sample the embryos. If sampling for gene expression, immediately after exposure, transfer embryos to a 1.5 ml eppendorf tube, remove any dosing solution, and snap freeze in liquid nitrogen. For later sampling or observation, wash the embryos three times with clean 0.3× Danieau’s water and transfer to a clean dish containing fresh 0.3× Danieau’s water and maintain under standard light and temperature conditions (see Note 20).
Assess embryos with microscopy for MO-associated changes in deformity severity or incidence (see Figure 8).
Figure 8.

Rescue of PCB-126-induced pericardial edema by Ahr-MO. Embryos were microinjected with 2 nl of 0.18 mM Ahr2-MO at the 1-4 cell stage, screened for incorporation of the fluorescently tagged MO at 24 hpf, and exposed in triplicate groups of 50 embryos to either 5 nM of PCB-126 or a DMSO solvent control from 24-48 hpf. At 72 hpf, embryos were mounted in 3% methylcellulose and imaged. Embryos exposed to DMSO show no deformities (left column). Non-injected and control-MO injected embryos exposed to PCB-126 suffer from pericardial edema, while Ahr2 morphant embryos are protected from this deformity (right column).
Acknowledgements
We would like to thank Gale Clark and Brandy Joyce for fish care, and Elwood Linney and Nicole Roy for microinjection training, and Bruce Woodin for imaging assistance.
All experiments were conducted using protocols approved by the WHOI IACUC.
This work was supported in part by National Institutes of Health grants F32ES017585 (AT-L) and R01ES016366 (MEH) and by Walter A. and Hope Noyes Smith.
Footnotes
Although any strain of zebrafish can be used with this technique, it is important to use a defined strain, such as those described by the Zebrafish International Resource Center (ZIRC; http://zebrafish.org/zirc/fish/lineAll.php), and to sequence the ATG start site or splice site region of the gene that you are targeting, as some sequences differ among strains. For this work, the TL strain was used.
Gene duplication is common in fish as a result of the whole genome duplication that occurred in the teleost lineage following its divergence from the tetrapod lineage (14), so it is important to consider the possible existence of a closely related paralog of your target. To screen GenBank and Ensembl for unidentified, potential gene paralogs, you can do a tBlastn search with your gene’s protein sequence. In addition, unless the published sequence was from the same strain of fish you are using, it is a good idea to sequence the proposed MO target sites in the gene you are attempting to knock down, because there can be variations in sequence among strains.
Start site MOs will target both maternal and zygotic transcripts, whereas splice site MOs will only target zygotic transcripts and so are not effective if the embryo has maternally supplied transcripts for the targeted gene. To assess the in vivo effectiveness of start site MOs, an antibody or functional assay for the target protein is needed; the in vivo effectiveness of splice site MOs is easier to assess because one can use PCR with primers that distinguish between mature and defectively spliced transcripts. To design a start site MO, you will need to know the sequence covering 25 base pairs on either side of the start codon. Design of a splice site MO requires careful consideration of the exon to be targeted. Targeting early exons is preferred because it is more likely to result in a nonfunctional protein through translational frameshift and premature stop codon caused by the deleted exon or a retained intron. Detailed discussions on MO design are available elsewhere (9, 18, 25; http://www.gene-tools.com/node/18).
To demonstrate the specificity of your MO for its target, it is preferable to use a mismatch control MO that contains 4 or 5 mismatches (18, 26).
Alternatively, phenol red can be added to your MO solution to monitor injections; however, this will not tell you whether the MO has been incorporated and evenly distributed throughout the embryo tissue.
Injection of 2 nl of 0.18 mM solution yields 3.6 ng MO per embryo. When using a new MO, it is advisable to test several concentrations of the MO and then use the lowest concentration that produces a consistent, specific effect. For most genes, injected amounts of ≤5 ng should be sufficient; larger amounts (>6 ng) may increase the chances of mistargeting leading to nonspecific effects (20,26)
Several egg-holder options have been described, such as one found in The Zebrafish Book at http://www.zfin.org/zf_info/zfbook/zfbk.html.
A good needle is key to successful injection. If it is too long, it will tend to bend or break rather than penetrate the chorion; if it is too short, the steep taper of the tip can damage the embryo; see p. 233 of Methods in Molecular Biology v.546 (27).
Alternatively, backloaders can be purchased, e.g. the Femtotip microloaders from Eppendorf.
In practice, the fluid is not always wicked into the needle tip as it should be. If this occurs, hold the needle in a horizontal position and roll it between your fingers until fluid flows to the tip. You can also gently flick the back end of the needle to move the solution.
It is extremely important to avoid getting an air bubble in the tip of your needle because a bubble can prevent the solution from being dispensed by the microinjector. You can try gently flicking the back end of the needle to dislodge the bubble, or load the needle into the micromanipulator and press the “clear” button on the microinjector. If the air bubble is very close to the tip with some liquid already at the tip, it is sometimes possible to gently brush a kimwipe against the tip of the loaded needle and wick the liquid to the tip and remove air bubbles that way, but it may be necessary to load a new needle.
Needles can become partially or fully clogged with yolk during injections, and this can change the size of the bolus. The needle will either have to be cleared by pressing the “clear” button on the microinjector, or adjustments to the injection pressure or time can be made to maintain the bolus size.
Cytoplasmic bridges will distribute the MO throughout the cells up through the 8-cell stage; however, we do not recommend injections beyond the 4-cell stage. When practicing this technique, it is possible to increase your injection time window by slowing down embryo development; this can be done by placing your petri dish with the embryos on top of ice and cooling the embryos down. However, this will introduce cold shock as a factor in your experiment, and this can cause changes in gene expression.
We have observed that different strains of zebrafish have different background rates of deformity. Thus, depending on which strain of zebrafish is used, you may need to inject 10-30% more embryos than you will need for your experimental design. For example, the AB strain may only need 10% extra, while the TL strain may need 30% extra.
Alternatively, you can screen using a lower fluorescence power setting to distinguish those embryos that have less successful MO incorporation. Occasionally you may also see embryos in which the MO bolus has remained in the yolk and has not been distributed into the tissues at all (see Figure 6).
There are several ways to validate MO efficacy. You can use an antibody to your target protein if one is available (22, 28, 29). In some cases you can examine alterations in protein function as an indicator of MO efficacy, as has been done with Cyp1a using in vivo ethoxyresorufin-o-deethylase (EROD) reaction (21, 22). You can also measure the expression of a known downstream target gene (8, 30). Splice-site MOs can be verified with RT-PCR with primers that allow one to distinguish between mature transcripts and those resulting from altered splicing (23, 25).
If non-radioactive approaches are preferred, two other possibilities are the Transcend non-radioactive translation detection system (Promega) or the Fluorotect Green in vitro translation labeling system (Promega). These alternatives do not require the use of the gel dryer or X-ray films, but do require the appropriate imaging system.
Some common, non-specific effects of morpholinos include cell death (often in the brain) and developmental delay (20). Many non-specific effects are mediated by p53, and can be rescued by co-injection with a p53-MO (31). This approach may be helpful in confirming whether your MO alone is producing these effects; however the inhibition of the p53-dependent cell death pathway in combination with a chemical exposure should only be done after careful consideration of the chemical, dosing, and target effects.
An alternative approach is to use injection of capped mRNA together with the MO to rescue the effect. However, the rescue construct must be designed so that it does not contain the MO target sequence. In some cases, rescue may only be partial (i.e. occur only in a subset of embryos; (13) or the expressed protein may not last long enough to rescue the morphant phenotype (17).
For highly hydrophobic compounds such as PCBs, glass dishes are preferred over plastic ones, because the compounds will adsorb more to plastic, reducing the bioavailability of the chemical. Chemicals are first dissolved in an organic solvent such as DMSO, acetone, ethanol, or isooctane. Solvent controls (embryos exposed to the same concentration of solvent, but without the toxicant of interest) must always be run concurrenty. Some chemicals may be so hydrophobic that, even in the presence of a carrier solvent such as DMSO, it is not possible to achieve the desired exposure concentrations in aqueous solution. In this case, chemicals can be injected directly into the embryo yolk, using the same microinjection apparatus described for MO injection (e.g. 32).
References
- 1.Grunwald DJ, Eisen JS. Headwaters of the zebrafish- emergence of a new model vertebrate. Nature Reviews Genetics. 2002;3:717–724. doi: 10.1038/nrg892. [DOI] [PubMed] [Google Scholar]
- 2.Linney E, Upchurch L, Donerly S. Zebrafish as a neurotoxicological model. Neurotoxicology and teratology. 2004;26:709–718. doi: 10.1016/j.ntt.2004.06.015. [DOI] [PubMed] [Google Scholar]
- 3.Zon LI. Zebrafish: a new model for human disease. Genome Res. 1999;9:99–100. [PubMed] [Google Scholar]
- 4.Zon LI, Peterson RT. In vivo drug discovery in the zebrafish. Nature Reviews Drug Discovery. 2005;4:35–44. doi: 10.1038/nrd1606. [DOI] [PubMed] [Google Scholar]
- 5.Nasevicius A, Ekker SC. Effective targeted gene ‘knockdown’ in zebrafish. Nat Genet. 2000;26:216–220. doi: 10.1038/79951. [DOI] [PubMed] [Google Scholar]
- 6.Moulton JD, Yan YL. Using Morpholinos to control gene expression. In: Ausubel Frederick M., et al., editors. Current protocols in molecular biology. 2008. p. 28. Chapter 26, Unit 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Flynt AS, Li N, Thatcher EJ, Solnica-Krezel L, Patton JG. Zebrafish miR-214 modulates Hedgehog signaling to specify muscle cell fate. Nat Genet. 2007;39:259–263. doi: 10.1038/ng1953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Timme-Laragy AR, Van Tiem LA, Linney EA, Di Giulio RT. Antioxidant responses and NRF2 in synergistic developmental toxicity of PAHs in zebrafish. Toxicol Sci. 2009;109:217–227. doi: 10.1093/toxsci/kfp038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bill BR, Petzold AM, Clark KJ, Schimmenti LA, Ekker SC. A primer for morpholino use in zebrafish. Zebrafish. 2009;6:69–77. doi: 10.1089/zeb.2008.0555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mathew LK, Sengupta SS, Ladu J, Andreasen EA, Tanguay RL. Crosstalk between AHR and Wnt signaling through R-Spondin1 impairs tissue regeneration in zebrafish. Faseb J. 2008;22:3087–3096. doi: 10.1096/fj.08-109009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shestopalov IA, Sinha S, Chen JK. Light-controlled gene silencing in zebrafish embryos. Nature chemical biology. 2007;3:650–651. doi: 10.1038/nchembio.2007.30. [DOI] [PubMed] [Google Scholar]
- 12.Ouyang X, Shestopalov IA, Sinha S, Zheng G, Pitt CL, Li WH, Olson AJ, Chen JK. Versatile synthesis and rational design of caged morpholinos. Journal of the American Chemical Society. 2009;131:13255–13269. doi: 10.1021/ja809933h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Xiong KM, Peterson RE, Heideman W. Aryl hydrocarbon receptor-mediated down-regulation of sox9b causes jaw malformation in zebrafish embryos. Molecular pharmacology. 2008;74:1544–1553. doi: 10.1124/mol.108.050435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Postlethwait J, Amores A, Cresko W, Singer A, Yan YL. Subfunction partitioning, the teleost radiation and the annotation of the human genome. Trends Genet. 2004;20:481–490. doi: 10.1016/j.tig.2004.08.001. [DOI] [PubMed] [Google Scholar]
- 15.Hahn ME, Karchner SI, Evans BR, Franks DG, Merson RR, Lapseritis JM. Unexpected diversity of aryl hydrocarbon receptors in non-mammalian vertebrates: insights from comparative genomics. Journal of experimental zoology. 2006;305:693–706. doi: 10.1002/jez.a.323. [DOI] [PubMed] [Google Scholar]
- 16.Evans BR, Karchner SI, Franks DG, Hahn ME. Duplicate aryl hydrocarbon receptor repressor genes (ahrr1 and ahrr2) in the zebrafish Danio rerio: structure, function, evolution, and AHR-dependent regulation in vivo. Arch Biochem Biophys. 2005;441:151–167. doi: 10.1016/j.abb.2005.07.008. [DOI] [PubMed] [Google Scholar]
- 17.Jenny MJ, Karchner SI, Franks DG, Woodin BR, Stegeman JJ, Hahn ME. Distinct roles of two zebrafish AHR repressors (AHRRa and AHRRb) in embryonic development and regulating the response to 2,3,7,8-tetrachlorodibenzo-p-dioxin. Toxicol Sci. 2009;110:426–441. doi: 10.1093/toxsci/kfp116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Eisen JS, Smith JC. Controlling morpholino experiments: don’t stop making antisense. Development (Cambridge, England) 2008;135:1735–1743. doi: 10.1242/dev.001115. [DOI] [PubMed] [Google Scholar]
- 19.Sumanas S, Larson JD. Morpholino phosphorodiamidate oligonucleotides in zebrafish: a recipe for functional genomics? Briefings in functional genomics & proteomics. 2002;1:239–256. doi: 10.1093/bfgp/1.3.239. [DOI] [PubMed] [Google Scholar]
- 20.Ekker SC, Larson JD. Morphant technology in model developmental systems. Genesis. 2001;30:89–93. doi: 10.1002/gene.1038. [DOI] [PubMed] [Google Scholar]
- 21.Billiard SM, Timme-Laragy AR, Wassenberg DM, Cockman C, Di Giulio RT. The role of the aryl hydrocarbon receptor pathway in mediating synergistic developmental toxicity of polycyclic aromatic hydrocarbons to zebrafish. Toxicol Sci. 2006;92:526–536. doi: 10.1093/toxsci/kfl011. [DOI] [PubMed] [Google Scholar]
- 22.Carney SA, Peterson RE, Heideman W. 2,3,7,8-Tetrachlorodibenzo-p-dioxin activation of the aryl hydrocarbon receptor/aryl hydrocarbon receptor nuclear translocator pathway causes developmental toxicity through a CYP1A-independent mechanism in zebrafish. Molecular pharmacology. 2004;66:512–521. doi: 10.1124/mol.66.3.. [DOI] [PubMed] [Google Scholar]
- 23.Draper BW, Morcos PA, Kimmel CB. Inhibition of zebrafish fgf8 pre-mRNA splicing with morpholino oligos: a quantifiable method for gene knockdown. Genesis. 2001;30:154–156. doi: 10.1002/gene.1053. [DOI] [PubMed] [Google Scholar]
- 24.Prasch AL, Teraoka H, Carney SA, Dong W, Hiraga T, Stegeman JJ, Heideman W, Peterson RE. Aryl hydrocarbon receptor 2 mediates 2,3,7,8-tetrachlorodibenzo-p-dioxin developmental toxicity in zebrafish. Toxicol Sci. 2003;76:138–150. doi: 10.1093/toxsci/kfg202. [DOI] [PubMed] [Google Scholar]
- 25.Morcos PA. Achieving targeted and quantifiable alteration of mRNA splicing with Morpholino oligos. Biochemical and biophysical research communications. 2007;358:521–527. doi: 10.1016/j.bbrc.2007.04.172. [DOI] [PubMed] [Google Scholar]
- 26.Ekker SC. Nonconventional antisense in zebrafish for functional genomics applications. Methods in cell biology. 2004;77:121–136. doi: 10.1016/s0091-679x(04)77007-7. [DOI] [PubMed] [Google Scholar]
- 27.Pase L, L GJ. Validating microRNA Target Transcripts Using Zebrafish Assays. In: Lieschke GJ, Oates AC, Kawakami K, editors. Zebrafish, Methods in Molecular Biology. Humana Press; New York: 2009. [DOI] [PubMed] [Google Scholar]
- 28.Sallinen V, Kolehmainen J, Priyadarshini M, Toleikyte G, Chen YC, Panula P. Dopaminergic cell damage and vulnerability to MPTP in Pink1 knockdown zebrafish. Neurobiology of disease. 2010;40:93–101. doi: 10.1016/j.nbd.2010.06.001. [DOI] [PubMed] [Google Scholar]
- 29.Prasch AL, Heideman W, Peterson RE. ARNT2 is not required for TCDD developmental toxicity in zebrafish. Toxicol Sci. 2004;82:250–258. doi: 10.1093/toxsci/kfh235. [DOI] [PubMed] [Google Scholar]
- 30.Kobayashi M, Itoh K, Suzuki T, Osanai H, Nishikawa K, Katoh Y, Takagi Y, Yamamoto M. Identification of the interactive interface and phylogenic conservation of the Nrf2-Keap1 system. Genes Cells. 2002;7:807–820. doi: 10.1046/j.1365-2443.2002.00561.x. [DOI] [PubMed] [Google Scholar]
- 31.Robu ME, Larson JD, Nasevicius A, Beiraghi S, Brenner C, Farber SA, Ekker SC. p53 activation by knockdown technologies. PLoS genetics. 2007;3:e78. doi: 10.1371/journal.pgen.0030078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Colman JR, Ramsdell JS. The type B brevetoxin (PbTx-3) adversely affects development, cardiovascular function, and survival in Medaka (Oryzias latipes) embryos. Environmental health perspectives. 2003;111:1920–1925. doi: 10.1289/ehp.6386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jonsson ME, Jenny MJ, Woodin BR, Hahn ME, Stegeman JJ. Role of AHR2 in the expression of novel cytochrome P450 1 family genes, cell cycle genes, and morphological defects in developing zebra fish exposed to 3,3′,4,4′,5-pentachlorobiphenyl or 2,3,7,8-tetrachlorodibenzo-p-dioxin. Toxicol Sci. 2007;100:180–193. doi: 10.1093/toxsci/kfm207. [DOI] [PubMed] [Google Scholar]
- 34.Jonsson ME, Franks DG, Woodin BR, Jenny MJ, Garrick RA, Behrendt L, Hahn ME, Stegeman JJ. The tryptophan photoproduct 6-formylindolo[3,2-b]carbazole (FICZ) binds multiple AHRs and induces multiple CYP1 genes via AHR2 in zebrafish. Chemico-biological interactions. 2009;181:447–454. doi: 10.1016/j.cbi.2009.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Incardona JP, Carls MG, Teraoka H, Sloan CA, Collier TK, Scholz NL. Aryl hydrocarbon receptor-independent toxicity of weathered crude oil during fish development. Environmental health perspectives. 2005;113:1755–1762. doi: 10.1289/ehp.8230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Incardona JP, Day HL, Collier TK, Scholz NL. Developmental toxicity of 4-ring polycyclic aromatic hydrocarbons in zebrafish is differentially dependent on AH receptor isoforms and hepatic cytochrome P4501A metabolism. Toxicol Appl Pharmacol. 2006;217:308–321. doi: 10.1016/j.taap.2006.09.018. [DOI] [PubMed] [Google Scholar]
- 37.Timme-Laragy AR, Noyes PD, Buhler DR, Di Giulio RT. CYP1B1 knockdown does not alter synergistic developmental toxicity of polycyclic aromatic hydrocarbons in zebrafish (Danio rerio) Marine environmental research. 2008;66:85–87. doi: 10.1016/j.marenvres.2008.02.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Prasch AL, Tanguay RL, Mehta V, Heideman W, Peterson RE. Identification of zebrafish ARNT1 homologs: 2,3,7,8-tetrachlorodibenzo-p-dioxin toxicity in the developing zebrafish requires ARNT1. Molecular pharmacology. 2006;69:776–787. doi: 10.1124/mol.105.016873. [DOI] [PubMed] [Google Scholar]
- 39.Grimes AC, Erwin KN, Stadt HA, Hunter GL, Gefroh HA, Tsai H-J, Kirby ML. PCB126 Exposure Disrupts ZebraFish Ventricular and Branchial but Not Early Neural Crest Development. Toxicol. Sci. 2008;106:193–205. doi: 10.1093/toxsci/kfn154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shi X, Zhou B. The role of Nrf2 and MAPK pathways in PFOS-induced oxidative stress in zebrafish embryos. Toxicol Sci. 2010;115:391–400. doi: 10.1093/toxsci/kfq066. [DOI] [PubMed] [Google Scholar]
- 41.McKinley ET, Baranowski TC, Blavo DO, Cato C, Doan TN, Rubinstein AL. Neuroprotection of MPTP-induced toxicity in zebrafish dopaminergic neurons. Molecular Brain Research. 2005;141:128–137. doi: 10.1016/j.molbrainres.2005.08.014. [DOI] [PubMed] [Google Scholar]
- 42.Teraoka H, Kubota A, Dong W, Kawai Y, Yamazaki K, Mori C, Harada Y, Peterson RE, Hiraga T. Role of the cyclooxygenase 2-thromboxane pathway in 2,3,7,8-tetrachlorodibenzo-p-dioxin-induced decrease in mesencephalic vein blood flow in the zebrafish embryo. Toxicology and Applied Pharmacology. 2009;234:33–40. doi: 10.1016/j.taap.2008.09.021. [DOI] [PubMed] [Google Scholar]
- 43.Ito T, Ando H, Suzuki T, Ogura T, Hotta K, Imamura Y, Yamaguchi Y, Handa H. Identification of a primary target of thalidomide teratogenicity. Science (New York, N.Y. 2010;327:1345–1350. doi: 10.1126/science.1177319. [DOI] [PubMed] [Google Scholar]