

Abstract
Traumatic brain injury (TBI) is a leading cause of mortality and disability. Acute postinjury insults after TBI, such as hypoxia, contribute to secondary brain injury and worse clinical outcomes. The functional and neuroinflammatory effects of brief episodes of hypoxia experienced following TBI have not been evaluated. Our previous studies have identified interleukin 6 (IL-6) as a potential mediator of mild TBI–induced pathology. In the present study, we sought to determine the effects of brief hypoxia on mild TBI and whether IL-6 played a role in the neuroinflammatory and functional deficits after injury. A murine model of mild TBI was induced by a weight drop (500 g from 1.5 cm). After injury, mice were exposed to immediate hypoxia (Fio2 = 15.1%) or normoxia (Fio2 = 21%) for 30 min. Serum and brain samples were analyzed for inflammatory cytokines 24 h after TBI. Neuron-specific enolase was measured as a serum biomarker of brain injury. Evaluation of motor coordination was performed for 5 days after TBI using a rotarod device. In some animals, anti–IL-6 was administered following TBI and hypoxia to neutralize systemic IL-6. Mice undergoing TBI had significant increases in brain injury. Exposure to brief hypoxia after TBI resulted in a more than 5-fold increase in serum neuron-specific enolase. This increase was associated with increases in serum and brain cytokine expression, suggesting that brief hypoxia exacerbates systemic and brain inflammation. Neutralization of IL-6 suppressed postinjury neuroinflammation and neuronal injury. In addition, TBI and hypoxia induced significant motor coordination deficits that were completely abrogated by IL-6 blockade. Exposure to hypoxia after TBI induces neuroinflammation and brain injury. These changes can be mitigated by neutralization of systemic IL-6. Interleukin 6 blockade also corrected the TBI-induced deficit in motor coordination. These data suggest that systemic IL-6 modulates the degree of neuroinflammation and contributes to reduced motor coordination after mild TBI.
Keywords: Traumatic brain injury, neuroinflammation, motor coordination, hypoxia, trauma, inflammation
INTRODUCTION
Traumatic brain injury (TBI) is a serious public health concern for both civilian and military populations. In the civilian setting, there are approximately 1.7 million cases of TBI each year in the United States (1–3). In the military, TBI is a signature injury of the conflicts in the Middle East (4, 5). In both settings, a majority of the cases reported are mild TBI (mTBI) (6, 7). Furthermore, because of the minimal symptoms of mTBI, many cases are not reported. A variety of complications that often occur after TBI, including hypotension and hypoxia, can contribute to secondary brain injury and are associated with worse functional outcomes (8–10). Many patients experience brief (<30 min) episodes of hypoxia before their hospitalization that may be caused secondary to other injuries or medical evacuation by rotary wing aircraft (8). Complications from these brief hypoxic episodes include increased neuronal damage (11), worsened axonal pathology, exacerbated neuroinflammatory response (12), augmented brain edema, and sensorimotor and cognitive deficits (11, 13).
Our previous work has demonstrated that a component of the inflammatory response to mTBI is increased expression of the cytokine, interleukin 6 (IL-6), systemically and within the brain (12, 14). The expression of IL-6 in our model, as well as in various clinical studies of TBI, is directly associated with the degree of brain injury and outcome (14–16). However, there have been no studies that have examined whether IL-6 is simply a biomarker of injury or a mediator of the pathophysiology. In the current study, we sought to determine the effects of brief hypoxia on the pathophysiology of mTBI and whether IL-6 plays a functional role in the injury response.
METHODS
Animals
Male C57BL/6 mice (Jackson, Me) aged 8 to 10 weeks, weighing 28 to 30 g, were used in the experiments. Animals were acclimated for at least 1 week and were housed in controlled conditions with 12-h light-dark cycle. The mice had free access to water and standard chow. All experiments were approved by the Institutional Animal Care and Use Committee of the University of Cincinnati.
Head injury model
Blunt TBI was induced using a modification of our previously described model (15). Briefly, mice were anesthetized for 2 min with 2% isoflurane in 100% oxygen at 1 L/min. Animals were then placed in a prone position on a platform below the head injury device so that the central region between coronal and lambdoid sutures was centered beneath the weight drop device. Head injury was delivered by dropping a 500-mg weight from a 1.5-cm height. Sham-injury mice were anesthetized and positioned in the same manner as injured mice but were not subjected to head injury.
IL-6 neutralization
Rat monoclonal anti–IL-6 (R&D Systems, Minneapolis, Minn) was administered at a dose of 1 μg by intraperitoneal injection 10 min after head injury and immediately before hypoxia exposure (17). Control animals received 1 μg of nonspecific rat IgG1.
Simulated hypoxia
Simulated hypoxia experiments were performed using a hypoxia chamber. The study protocols were modified using a hypoxia-altitude simulation test (18, 19). Simulation utilizes hypoxic gas mixtures (15.1% oxygen) representing an altitude of 8,000 ft above sea level (18, 19). Fifteen minutes after TBI (n = 6) or sham injury (n = 6), animals were exposed to 30 min of 15.1% or 21% Fio2 then recovered at room air.
Serum/tissue analysis
Animals were killed 24 h after injury. Blood samples were obtained by cardiac puncture. Blood was obtained by cardiac puncture, and the brain was excised by immediate postmortem craniectomy. Serum and left cortical brain samples were analyzed for levels of inflammatory cytokines using customized multiplex enzyme-linked immunosorbent assay (ELISA) technology (Quansys, Logan, Utah). Thirteen cytokines and chemokines were evaluated in the ELISA analysis, including Granulocyte-macrophage colony-stimulating factor, interferon γ, IL-1 α, IL-1β, IL-6, IL-10, keratinocyte-derived chemokine (KC), macrophage inflammatory protein-1alpha (MIP-1α), and tumor necrosis factor α. Cortical samples were processed as previously described and stored at −80°C until analysis (12). Cerebral cytokine levels were normalized to cortical protein content using a BCA Protein Assay Kit (Thermo Scientific, Rockford, Ill). Neuron-specific enolase (NSE) was measured as a serum biomarker of head injury severity using an ELISA kit (Immuno-Biological Laboratories, Inc, Minneapolis, Minn).
Motor evaluation
A rotarod device (IITC Life Science, Woodland Hills, Calif) was utilized to test motor deficits. Groups included in the experiment were sham injury (n = 6), TBI with normoxia (n = 8), TBI with hypoxia (n = 8), and TBI with hypoxia and anti–IL-6 (n = 8). All mice were trained daily for 5 consecutive days. Head injury was induced on day 6, and training resumed on day 7 for an additional 5 days. Mice were evaluated at two different settings: an initial speed of 5 revolutions/min (rpm) and accelerated to 24 rpm over 90 s, or accelerated to 36 rpm over 180 s. Each animal performed the task three times at each setting daily. Each trial on the rod was terminated when the animal fell off the rod, held on to the rod and completed two complete revolutions, or remained on the rod for 600 s. Mean time to trial termination was recorded and expressed as a ratio of daily performance to initial performance (ratio to baseline).
Statistical analysis
Statistical analysis was completed by using SigmaPlot (Systat Software Inc, San Jose, Calif) for two-tailed Student t test and one-way analysis of variance with Tukey analysis. Data are reported as mean ± SEM. In two instances, there were cytokine data points that appeared to be outliers in an otherwise normally distributed data set. For these data points, we applied the Grubbs test (20) to determine if they were outliers and could be excluded. P < 0.05 was considered significant.
RESULTS
Brain injury and neuroinflammation caused by mTBI are exacerbated by brief hypoxia
We first evaluated the effects of brief hypoxia on brain injury induced by mTBI. Hypoxia alone in sham animals did not increase NSE compared with normoxic sham mice (Fig. 1). Under normoxic conditions, mTBI significantly increased serum NSE levels compared with sham animals. However, hypoxia exposure after mTBI resulted in a more than 5-fold increase in NSE compared with normoxic mTBI (Fig. 1).
Fig. 1. Brain injury after TBI with or without brief hypoxia.

Serum NSE was used as biochemical marker of brain injury. Serum samples were collected at 24 h after injury, and samples were analyzed by ELISA. Data are mean ± SEM with n = 5 per group. *P < 0.05 compared with sham. #P < 0.05 compared with all other groups.
To evaluate whether the increased brain injury observed in mice exposed to brief hypoxia was related to increased systemic or local (neuro)inflammation, we measured cytokine levels in the serum and brain tissues. In this model, we have previously shown that the cytokines, IL-6, KC, and MIP-1α are indicative of this response in both the periphery (serum) and brain environments (14, 21). Interestingly, hypoxia alone, without any mTBI, caused significant increases in serum levels of KC and MIP-1α, but had no effect on IL-6 (Fig. 2) or any other cytokine in our multiplex assay (Table 1). Mild TBI under normoxic conditions resulted in a marked increase in serum IL-6 and increased KC to a similar degree as hypoxia. Mice undergoing mTBI with hypoxia had serum IL-6 levels that were the same as normoxic mTBI. However, mice undergoing TBI with hypoxia had greater serum levels of KC and MIP-1α than either hypoxia or TBI alone (Fig. 2).
Fig. 2. Systemic inflammation after TBI with or without brief hypoxia.
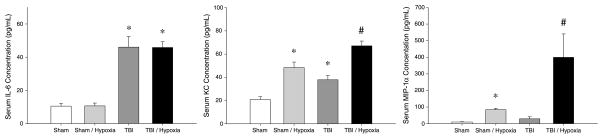
Serum cytokines were evaluated 24 h after injury by ELISA. Data are mean ± SEM with n = 5 per group. *P < 0.05 compared with sham. #P < 0.05 compared with all other groups.
Table 1.
Cytokine levels in serum and brain
| Cytokine | Serum
|
Brain
|
||
|---|---|---|---|---|
| Sham | TBI | Sham | TBI | |
| Granulocyte-macrophage colony-stimulating factor | 3.3 ± 3.7 | 0.5 ± 1.0 | 15.0 ± 3.6 | 20.9 ± 4.1 |
| Interferon γ | 17.7 ± 7.7 | 14.1 ± 6.0 | 37.8 ± 5.2 | 53.7 ± 5.0 |
| IL-1α | 4.4 ± 1.7 | 5.7 ± 4.4 | 21.0 ± 2.7 | 63.1 ± 12.9 |
| IL-1β | 24.0 ± 14.8 | 21.7 ± 4.1 | 46.6 ± 9.3 | 62.9 ± 6.3 |
| IL-10 | 6.7 ± 4.5 | 4.2 ± 4.7 | 15.6 ± 3.3 | 53.1 ± 9.9 |
| Tumor necrosis factor α | 12.6 ± 5.0 | 17.4 ± 13.2 | 17.5 ± 3.1 | 31.0 ± 4.9 |
Data are mean ± SEM with n = 5 per group.
In brain tissue, hypoxia alone did not cause an increase in any of these cytokines (Fig. 3). Mild TBI under normoxic conditions induced increases in brain IL-6 and MIP-1α, but not KC (Fig. 3) or any other cytokine in our multiplex assay (Table 1). Mild TBI with brief hypoxia showed marked increases in all three cytokines, all having significantly greater expression than mTBI without hypoxia (Fig. 3).
Fig. 3. Brain inflammation after TBI with or without brief hypoxia.
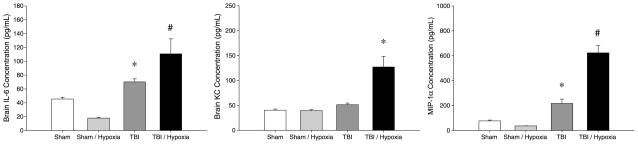
Cerebral cytokines were evaluated 24 h after injury by ELISA. Data are mean ± SEM with n = 5 per group. *P < 0.05 compared with sham. #P < 0.05 compared with all other groups.
Neutralization of IL-6 reduces neuroinflammation, brain injury, and deficits in motor coordination
To determine if IL-6 mediates the hypoxia-induced increases in the inflammatory and injury responses after mTBI, we administered neutralizing antibodies to IL-6 after mTBI, but just before hypoxia. Treatment with anti–IL-6 abrogated increases in serum IL-6 in mice undergoing mTBI with brief hypoxia (Fig. 4). In addition, anti–IL-6 treatment significantly reduced serum levels of KC and MIP-1α induced by mTBI with hypoxia (Fig. 4). Similar results were observed in brain tissue, with anti–IL-6 treatment completely suppressing IL-6 and markedly reducing KC and MIP-1α expression (Fig. 5).
Fig. 4. Effect of IL-6 neutralization on systemic inflammation after TBI and brief hypoxia.
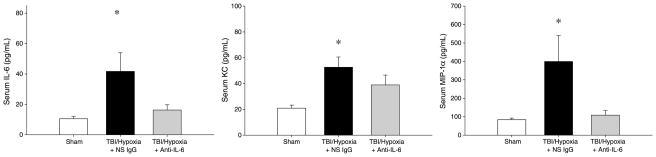
Serum cytokines were evaluated 24 h after injury by ELISA. Data are mean ± SEM with n = 5 per group *P < 0.05 compared with sham. #P < 0.05 compared with all other groups.
Fig. 5. Effect of IL-6 neutralization on brain inflammation after TBI and brief hypoxia.
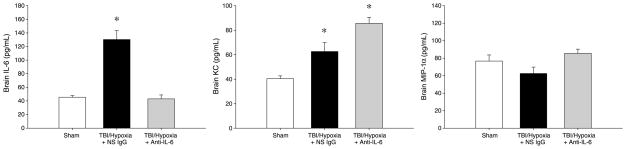
Cerebral cytokines were evaluated 24 h after injury by ELISA. Data are mean ± SEM with n = 5 per group. *P < 0.05 compared with sham. #P < 0.05 compared with all other groups.
We next examined whether the reduced systemic and neuroinflammation observed with anti–IL-6 treatment resulted in decreased brain injury. Analysis of serum NSE levels confirmed that treatment of mice undergoing mTBI and brief hypoxia with anti–IL-6 reduced the amount of brain injury by more than 50% compared with the vehicle control group (Fig. 6). In addition to decreased brain injury, we found that neutralization of IL-6 restored motor coordination after mTBI. As shown in Figure 7, mice undergoing mTBI with and without brief hypoxia had a significant deficit in motor coordination, as determined by time on the rotarod. Notably, treatment with anti–IL-6 restored motor coordination to a level indistinguishable from sham control mice (Fig. 7).
Fig. 6. Effect of IL-6 neutralization on brain injury after TBI and brief hypoxia.

Serum samples were collected at 24 h after injury, and samples were analyzed by ELISA. Data are mean ± SEM with n = 5 per group. *P < 0.05 compared with sham. #P < 0.05 compared with all other groups.
Fig. 7. Effect of IL-6 neutralization on motor coordination deficits induced by mTBI with or without brief hypoxia.

Rotarod testing was used to evaluate motor coordination. All mice were initially pretrained from days 1 to 5 to obtain a baseline skill level on rotarod. Mice received TBI on day 6, and training was resumed 24 h after injury on day 7 and continued for 5 additional days. Data are mean ± SEM with n = 6 per group postinjury. *P < 0.05 compared with sham. #P < 0.05 compared with all other groups.
DISCUSSION
The current study is an extension of our previous work evaluating aeromedical evacuation and the effects of hypobaric hypoxia on systemic and cerebral inflammatory responses (12). We previously demonstrated that simulated flight at 8,800 ft (hypobaric hypoxia) for 5 h results in an augmented brain injury if the exposure to hypobaric hypoxia occurs shortly after injury (12). Those studies modeled the transport of mTBI patients from the combat theater to regional medical centers in Europe, requiring a long period of exposure to hypobaric hypoxia. Our current work models more acute transport, as might occur via rotary wing aircraft in both military and civilian prehospital settings. To do this, we investigated the effects of a brief hypoxic exposure (30 min) with 15.1% oxygen, which is equivalent to oxygen saturations of 90%, at normobaric pressures (18, 19). This represents clinically relevant desaturation. Hypoxia is frequently seen before hospital admission; trauma patients with suspected brain injuries during helicopter transport were shown to have hypoxia episodes 30% of the time (8). Although our use of 30 min of hypoxia could be viewed clinically as a somewhat extended period, a previous study of helicopter transport of TBI patients showed a wide variability in the duration of hypoxia, with the average period of hypoxia being 17 min, but several instances of hypoxic episodes lasting 30 min or more (8). Furthermore, other studies have shown that 15 or 30 min of hypoxia had similar effects on TBI-induced neuronal cell death (22).
We found that a 30-min exposure to hypoxia leads to a robust neuroinflammatory response. Significant increases in brain IL-6, KC, and MIP-1α were observed after mTBI and hypoxia. Neuronal damage and death, evaluated by NSE, showed a significant increase after TBI/hypoxia exposure. The combination of increased neuroinflammatory response and increased brain damage also leads to a greater functional deficit. These results are supported by other studies showing that hypoxic episodes contribute to brain inflammation and injury (22, 23). However, our study is unique in that it is the first to identify IL-6 as a driving factor for the neuroinflammatory response as well as a primary contributor to the motor coordination deficit that occurs after mTBI.
A recent report from our laboratory fully characterized our model of mTBI and demonstrated that serum IL-6 levels are increased proportionally with the degree of head injury (14). However, whether IL-6 was simply a biomarker of injury or an active participant in the resulting pathology was not determined. In this study, we directly assessed the role of IL-6. We found that neutralization of IL-6 mitigated the neuroinflammatory response, significantly reduced brain injury, and completely abrogated loss of motor coordination after mTBI and hypoxia. The precise mechanism by which IL-6 regulates these effects requires further experimentation.
The brain is thought to be immunologically privileged because of the blood-brain barrier (BBB). However, the breakdown of the BBB after TBI may allow leakage of some humoral factors as well as facilitate recruitment of immune cells. The leakage of the BBB followed by TBI and a hypoxic event is increased, and this may allow for more factors to cross the disrupted barrier (24, 25). Increased levels of proinflammatory cytokines, IL-6 and IL-8, are seen in human serum after TBI and have been correlated to poor outcomes (15, 16). Our study clearly demonstrates a markedly increased neuroinflammatory response that is further exacerbated by subsequent hypoxic exposure. Neutralization of systemic IL-6 mitigates this response. While the cellular sources of cytokines expressed in the serum and brain tissue are clearly different, we posit that systemic IL-6 is able to modulate brain cytokine expression. Whether IL-6 leaks into the brain shortly after TBI due to transient breakdown of the BBB, acts on brain endothelial cells or microglial cells, or through some other mechanism to modulate the neuroinflammatory response remains to be determined. However, our data support the concept that systemic IL-6 serves as an amplification signal for the inflammatory response and motor coordination deficits. The fact that systemic neutralization resulted in a complete inhibition of brain IL-6 and complete restoration of normal motor coordination is supportive of this hypothesis. Additional investigation into the nature of IL-6 receptor location and signaling in neuronal cells may provide additional valuable insights.
In summary, the current study demonstrates that brief exposure to hypoxia after mTBI results in exacerbated brain inflammation and injury. Systemic neutralization of IL-6 mitigates these effects and reverses loss of motor coordination after mTBI. The data suggest that IL-6 may be a rational target for therapeutic intervention in cases of mTBI.
Acknowledgments
This work was supported by US Air Force grant FA8650-10-2-6B01 and NIH training grant T32 GM08478.
Footnotes
This article was presented at the 36th Annual Conference on Shock as one of the finalists of the New Investigator Awards Competition, San Diego, California, June 1–4, 2013.
References
- 1.Coronado VG, Faul M, Wald MM, Xu L National Center for Injury Prevention and Control (U.S.). Division of Injury Response. Traumatic Brain Injury in the United States: Emergency Department Visits, Hospitalizations, and Deaths, 2002–2006. Atlanta: Dept. of Health and Human Services, Centers for Disease Control and Prevention, National Center for Injury Prevention and Control; 2010. [Google Scholar]
- 2.Fujimoto ST, Longhi L, Saatman KE, Conte V, Stocchetti N, McIntosh TK. Motor and cognitive function evaluation following experimental traumatic brain injury. Neurosci Biobehav Rev. 2004;28:365–378. doi: 10.1016/j.neubiorev.2004.06.002. [DOI] [PubMed] [Google Scholar]
- 3.Thurman DJ, Alverson C, Dunn KA, Guerrero J, Sniezek JE. Traumatic brain injury in the United States: a public health perspective. J Head Trauma Rehabil. 1999;14:602–615. doi: 10.1097/00001199-199912000-00009. [DOI] [PubMed] [Google Scholar]
- 4.Okie S. Traumatic brain injury in the war zone. N Engl J Med. 2005;352:2043–2047. doi: 10.1056/NEJMp058102. [DOI] [PubMed] [Google Scholar]
- 5.Wojcik BE, Stein CR, Bagg K, Humphrey RJ, Orosco J. Traumatic brain injury hospitalizations of U.S. army soldiers deployed to Afghanistan and Iraq. Am J Prev Med. 2010;38:S108–S116. doi: 10.1016/j.amepre.2009.10.006. [DOI] [PubMed] [Google Scholar]
- 6.Langlois JA, Rutland-Brown W, Wald MM. The epidemiology and impact of traumatic brain injury: a brief overview. J Head Trauma Rehabil. 2006;21:375–378. doi: 10.1097/00001199-200609000-00001. [DOI] [PubMed] [Google Scholar]
- 7.Elder GA, Cristian A. Blast-related mild traumatic brain injury: mechanisms of injury and impact on clinical care. Mt Sinai J Med. 2009;76:111–118. doi: 10.1002/msj.20098. [DOI] [PubMed] [Google Scholar]
- 8.Chi JH, Knudson MM, Vassar MJ, McCarthy MC, Shapiro MB, Mallet S, Holcroft JJ, Moncrief H, Noble J, Wisner D, et al. Prehospital hypoxia affects outcome in patients with traumatic brain injury: a prospective multicenter study. J Trauma. 2006;61:1134–1141. doi: 10.1097/01.ta.0000196644.64653.d8. [DOI] [PubMed] [Google Scholar]
- 9.Davis DP, Meade W, Sise MJ, Kennedy F, Simon F, Tominaga G, Steele J, Coimbra R. Both hypoxemia and extreme hyperoxemia may be detrimental in patients with severe traumatic brain injury. J Neurotrauma. 2009;26:2217–2223. doi: 10.1089/neu.2009.0940. [DOI] [PubMed] [Google Scholar]
- 10.Chesnut RM, Marshall LF, Klauber MR, Blunt BA, Baldwin N, Eisenberg HM, Jane JA, Marmarou A, Foulkes MA. The role of secondary brain injury in determining outcome from severe head injury. J Trauma. 1993;34:216–222. doi: 10.1097/00005373-199302000-00006. [DOI] [PubMed] [Google Scholar]
- 11.Bauman RA, Widholm JJ, Petras JM, McBride K, Long JB. Secondary hypoxemia exacerbates the reduction of visual discrimination accuracy and neuronal cell density in the dorsal lateral geniculate nucleus resulting from fluid percussion injury. J Neurotrauma. 2000;17:679–693. doi: 10.1089/089771500415427. [DOI] [PubMed] [Google Scholar]
- 12.Goodman MD, Makley AT, Huber NL, Clarke CN, Friend LA, Schuster RM, Bailey SR, Barnes SL, Dorlac WC, Johannigman JA, et al. Hypobaric hypoxia exacerbates the neuroinflammatory response to traumatic brain injury. J Surg Res. 2011;165:30–37. doi: 10.1016/j.jss.2010.05.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Clark RS, Kochanek PM, Dixon CE, Chen M, Marion DW, Heineman S, DeKosky ST, Graham SH. Early neuropathologic effects of mild or moderate hypoxemia after controlled cortical impact injury in rats. J Neurotrauma. 1997;14:179–189. doi: 10.1089/neu.1997.14.179. [DOI] [PubMed] [Google Scholar]
- 14.Yang SH, Gustafson J, Gangidine M, Stepien D, Schuster R, Pritts TA, Goodman MD, Remick DG, Lentsch AB. A murine model of mild traumatic brain injury exhibiting cognitive and motor deficits. J Surg Res. 2013;184:981–988. doi: 10.1016/j.jss.2013.03.075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Minambres E, Cemborain A, Sanchez-Velasco P, Gandarillas M, Diaz-Reganon G, Sanchez-Gonzalez U, Leyva-Cobian F. Correlation between transcranial interleukin-6 gradient and outcome in patients with acute brain injury. Crit Care Med. 2003;31:933–938. doi: 10.1097/01.CCM.0000055370.66389.59. [DOI] [PubMed] [Google Scholar]
- 16.Hergenroeder GW, Moore AN, McCoy JP, Jr, Samsel L, Ward NH, 3rd, Clifton GL, Dash PK. Serum IL-6: a candidate biomarker for intracranial pressure elevation following isolated traumatic brain injury. J Neuroinflammation. 2010;7:19. doi: 10.1186/1742-2094-7-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.LaSpina M, Tripathi S, Gatto LA, Bruch D, Maier KG, Kittur DS. An interleukin-6–neutralizing antibody prevents cyclosporine-induced nephrotoxicity in mice. J Surg Res. 2008;148:121–125. doi: 10.1016/j.jss.2007.12.786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dillard TA, Moores LK, Bilello KL, Phillips YY. The preflight evaluation. A comparison of the hypoxia inhalation test with hypobaric exposure. Chest. 1995;107:352–357. doi: 10.1378/chest.107.2.352. [DOI] [PubMed] [Google Scholar]
- 19.Dine CJ, Kreider ME. Hypoxia altitude simulation test. Chest. 2008;133:1002–1005. doi: 10.1378/chest.07-1354. [DOI] [PubMed] [Google Scholar]
- 20.Grubbs F. Procedures for detecting outlying observations in samples. Technometrics. 1969;11:1–21. [Google Scholar]
- 21.Goodman MD, Makley AT, Lentsch AB, Barnes SL, Dorlac GR, Dorlac WC, Johannigman JA, Pritts TA. Traumatic brain injury and aeromedical evacuation: when is the brain fit to fly? J Surg Res. 2010;164:286–293. doi: 10.1016/j.jss.2009.07.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Grissom CK, Weaver LK, Clemmer TP, Morris AH. Theoretical advantage of oxygen treatment for combat casualties during medical evacuation at high altitude. J Trauma. 2006;61:461–467. doi: 10.1097/01.ta.0000221699.71596.9d. [DOI] [PubMed] [Google Scholar]
- 23.Feng JF, Zhao X, Gurkoff GG, Van KC, Shahlaie K, Lyeth BG. Post-traumatic hypoxia exacerbates neuronal cell death in the hippocampus. J Neurotrauma. 2012;29:1167–1179. doi: 10.1089/neu.2011.1867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chodobski A, Zink BJ, Szmydynger-Chodobska J. Blood-brain barrier pathophysiology in traumatic brain injury. Transl Stroke Res. 2011;2:492–516. doi: 10.1007/s12975-011-0125-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ballabh P, Braun A, Nedergaard M. The blood-brain barrier: an overview: structure, regulation, and clinical implications. Neurobiol Dis. 2004;16:1–13. doi: 10.1016/j.nbd.2003.12.016. [DOI] [PubMed] [Google Scholar]