

Abstract
Nucleic acid hybridization is widely used for the specific capture of complementary sequences from complex samples. It is useful for both analytical methodologies, such as array hybridization (e.g. transcriptome analysis, genetic variation analysis), and preparative strategies such as exome sequencing and sequence-specific proteome capture and analysis (PICh, HyCCAPP). It has not generally been possible to selectively elute particular captured subsequences, however, as the conditions employed for disruption of a duplex can lack the specificity needed to discriminate between different sequences. We show here that it is possible to bind and selectively release multiple sets of sequences by using toehold-mediated DNA branch migration. The strategy is illustrated for simple mixtures of oligonucleotides, for the sequence-specific capture and specific release of crosslinked yeast chromatin, and for the specific release of oligonucleotides hybridized to DNA microarrays.
Keywords: DNA, Nucleic acid hybridization, Microarray, Toehold-mediated branch migration, Selective elution
The exquisite binding specificity of a nucleic acid sequence to its complement is fundamental to life. Since the first discovery of the double helix1, a detailed understanding of the energetics of DNA duplex formation has developed, providing a foundation for predictions of nucleic acid hybridization, folding and dynamics2–6. Recently, the process of toehold-mediated DNA branch migration has been described, in which displacement of an incumbent strand hybridized to a substrate strand is thermodynamically driven by enhanced Watson-Crick base pairing of an invader strand7, 8. We show here that this process enables the programmable, selective, and quantitative release of targeted subsets of nucleic acid sequences from solid supports. The approach is demonstrated for simple mixtures of synthetic oligonucleotides, for the selective release of individual target regions in crosslinked yeast chromatin and for oligonucleotides hybridized to DNA microarrays.
The toehold-mediated capture and release strategy employed here for programmable, multiplexed release of multiple captured DNA sequences is depicted in Figure 1. Capture and release oligonucleotides are designed for each target sequence. The capture oligonucleotide contains 30 nt of sequence complementary to the target oligonucleotide as well as an 8 nt toehold that is not complementary. The release oligonucleotide is completely complementary to the capture oligonucleotide and is therefore 38 nt in length. The unique sequence of the toehold allows for multiplexed programmable release of multiple captured sequences: for every target sequence a unique toehold sequence is utilized.
Figure 1.
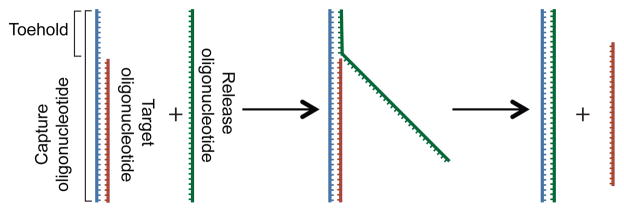
Toehold-mediated capture/release strategy of target DNA sequences. For a given target DNA sequence, a capture oligonucleotide is designed to complement the sequence with an additional 8 nt of unique sequence (toehold). The release oligonucleotide is 100% complementary to the capture oligonucleotide, allowing for release of the target sequence
Toehold-mediated strand displacement is described well by bimolecular reactions9, where the kinetics of release are dependent upon the concentration of both the capture:target duplex and the release oligonucleotide. In order to optimize the release kinetics for the capture strategy outlined below, we empirically tested and determined the concentration of release oligonucleotide necessary to release the majority of target oligonucleotides within less than one hour. The release kinetics were characterized using a biotinylated capture oligonucleotide and fluorescently labeled target oligonucleotide (Figure 2A). The capture and target oligonucleotides were hybridized and the resulting duplex was captured on streptavidin-coated magnetic particles. Release efficiency was determined by measuring the fluorescence in solution after addition of the corresponding release oligonucleotide. As shown in Figure 2B, the release rate increases with molar excess of release oligonucleotide. Addition of low molar excesses of release oligonucleotide (5X and 10X) results in less than 50% of the total target oligonucleotide released after one hour. A molar excess of 50X or greater results in very fast (<2 minutes) release of more than 50% of the target oligonucleotide. In fact, with a 500X molar excess of release oligonucleotide all of the target oligonucleotide is released within the first five minutes of incubation. When no release oligonucleotide is added, the target oligonucleotide does not dissociate from the duplex.
Figure 2.

Capture/release of fluorescently labelled target oligonucleotides. (A) For each target sequence, a biotinylated capture oligonucleotide was designed. (B) The kinetics of release were measured using 8 nt capture/release oligonucleotides and a molar excess of release oligonucleotide ranging from 0-1000X. (C) Capture/release oligonucleotides containing different toehold lengths were used to measure the release kinetics with a molar excess of 1000X release oligonucleotide. Fluctuations in the 8 nt data are likely a result of noise in the assay due to handling of small volumes of magnetic particles. (D) Sequential release of two target oligonucleotides was performed from aliquots of the same sample using both combinations of release oligonucleotide order. (E) The fluorescence signal for both Alexa 488 and Texas Red was measured in all four release samples.
The kinetics of toehold-mediated exchange are strongly dependent upon toehold length7, 10. We sought to measure the effect of toehold length on release kinetics in the above system, in order to ascertain the minimum toehold length required for fast release. Using the same capture/release strategy described above, we determined the release kinetics for capture oligonucleotides with 0 nt, 4 nt, 6 nt and 8 nt toehold lengths, using 1000X molar excess of release oligonucleotide. Capture oligonucleotides with an 8 nt toehold exhibited rapid kinetics, releasing around 80% of the target oligonucleotide within the first 5 minutes of incubation with release oligonucleotide (Figure 2C). The capture oligonucleotide containing a 6 nt toehold had slower release kinetics, with about 50% of the total target oligonucleotide released after an hour. Consistent with previous reports that toeholds shorter than 5 nt have substantially slower release kinetics7, the capture oligonucleotides with a 4 nt toehold did not release any measurable amount of target oligonucleotide over the course of one hour. The capture oligonucleotide with no toehold did not release any target oligonucleotide in a one hour incubation, as expected.
The optimal conditions determined above for the capture/release strategy (1000X molar excess release oligonucleotide and 8 nt toehold length) were employed to test the selectivity and specificity of sequential release of two target sequences. Two fluorescently labeled oligonucleotides were designed with unique sequences and non-interfering fluorophores (Alexa 488 and Texas Red). Both target oligonucleotides were combined with corresponding biotinylated capture oligonucleotides, each containing a unique toehold sequence, and allowed to hybridize. The resulting duplexes were combined with a suspension of streptavidin-coated magnetic particles for capture, and the resultant mixture was aliquoted equally into two tubes (Fig. 2D). The release oligonucleotide for the Alexa 488 labeled target was added to one tube and the release oligonucleotide for the TexasRed target was added to the other. After a 15 minute incubation at room temperature, the beads were magnetically immobilized, the supernatants were removed, and solutions containing the alternate release oligonucleotide were added to each tube, followed by a second 15 minute incubation and removal of the supernatants. Fluorescence was measured in each of the four supernatants for both Alexa 488 (λex = 501 nm, λem =527 nm) and Texas Red (λex = 596 nm, λem =620 nm). As shown in Figure 2E, the designated target oligonucleotide was released preferentially in each case, with a selectivity factor of intended oligonucleotide release fluorescence divided by the fluorescence from the alternative oligonucleotide ranging from 8-fold to 126-fold (Fig. S1). The differences in release efficiencies are probably due to the different toehold sequences employed, as described in ref. 7. This demonstrates the selectivity of release and hence the multiplexing capability of the approach.
The capture/release strategy was used to sequence-specifically capture and selectively release targeted regions of formaldehyde crosslinked chromatin in the yeast genome. This is useful for the mass spectrometric identification of locus-specific DNA-associated proteins11–13, a powerful emerging technology. Current methodologies to sequence-specifically capture crosslinked chromatin regions utilize desthiobiotinylated oligonucleotides and soluble biotin for subsequent release11, 13. This strategy is not amenable to multiplexing, however, as all capture oligonucleotides will be released similarly upon addition of biotin, without discrimination as to target sequence. We first designed experiments to compare the rates of release of 25s rDNA13 chromatin by toehold-mediated exchange and desthiobiotin/biotin exchange, using qPCR to measure release of the target DNA.
Cell lysate containing fragmented chromatin was prepared from 3% formaldehyde crosslinked cells13 (see SI and ref. 13 for sample preparation details). Hybridization using either toehold-mediated exchange or desthiobiotin-modified oligonucleotides was carried out at 37°C, followed by capture using streptavidin-coated magnetic particles and washing steps to remove non-specifically bound material. The release moiety was then added to solution (release oligonucleotide or biotin) and aliquots of the supernatant were periodically removed for analysis by qPCR. As shown in Figure 3A, the target DNA captured by the toehold-mediated strategy was completely released in 15 minutes, whereas the desthiobiotin capture oligonucleotide required two hours for 80% release using standard conditions. In both cases, the capture of the target sequence was specific, as shown by the low level of capture by a scrambled oligonucleotide sequence (92-fold higher capture using the toehold-biotin oligonucleotide and 73-fold higher capture using the desthiobiotin oligonucleotide).
Figure 3.

Multiplexed capture/release of target genomic loci in yeast. (A) The kinetics of release of the 25S rDNA locus was measured by qPCR using both toehold-mediated release and biotin release of desthiobiotin. (B) Six hybridization samples were subjected to all possible combinations of release oligonucleotide order. The release oligonucleotides are indicated by color: blue=X-element, red=5S rDNA and green=25S rDNA. (C) The qPCR signal for each of the three genes was measured in each release sample.
The toehold-mediated capture/release strategy was then utilized to capture three genomic loci in yeast in parallel: 25S rDNA14, 5S rDNA14 and the X-element15. Three capture oligonucleotides, each containing unique toehold sequences, were added to cell lysate containing fragmented, crosslinked chromatin and allowed to hybridize. The capture/target complexes were isolated using streptavidin-coated magnetic particles, the resultant sample was split into six aliquots, and sequential release of each of the target loci was performed in all possible release orders (Figure 3B). The amount of DNA released was measured by qPCR for each of the target loci. As shown in Figure 3C, in each case, only the designated target region was released while the other sequences remained bound to the support particles. The specificity of release was determined by dividing the qPCR signal obtained from samples with the target release oligonucleotide added by the qPCR signals for each of the other off-target genes (see Table S1). The specificity ratios range from 8.5–7800, with an average specificity ratio of 290. Due to the relatively fast release kinetics, the total release time for all three target regions was less than the release time required for release of a single region when using the desthiobiotin/biotin release strategy.
We next used this strategy to specifically release captured oligonucleotides from DNA microarrays, which are commonly used to capture RNA, DNA, or proteins from solution for various types of analyses16, 17. DNA microarrays have also been used in enzymatic reactions enabling diverse technologies such as gene assembly and DNA computation18–20. Thus, the ability to control the specific release of given subsets of captured DNA sequences could provide new possibilities for these and other DNA microarray applications.
A DNA microarray was synthesized on a custom built maskless array synthesizer21 to contain two different 38 nt length sequences, one patterned in the shape of a fern and the second comprising the background. Both sequences contained an identical 30 nt region and differed only by an 8 nt toehold region at their 3′-termini. Thus, upon hybridization with a FAM-labeled probe complementary to the 30 nt region, the array displayed a uniform level of fluorescence (Figure 4B). After addition of a Cy3-labeled release oligonucleotide complementary to the full 38 nt sequence corresponding to the fern shape, only a specific subset of the original FAM-labeled oligonucleotides were released to reveal the fern pattern shown in Figures 4E and 4F. This is an example of the ability of the toehold mediated branch migration strategy to reveal hidden information encrypted in a DNA microarray.
Figure 4.
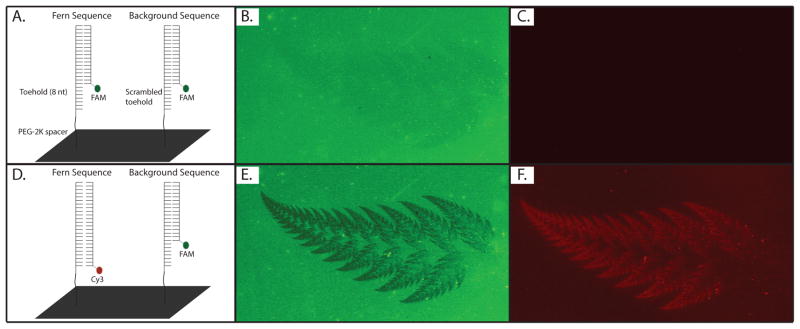
Selective release of a DNA oligomer hybridized to a microarray. (A) schematic of the DNA array after hybridization with a FAM-labelledoligonucleotide; (B) FAM and (C) Cy3 images of the surface; (D) schematic of the array after addition of a Cy3-labeled release oligonucleotide containing a toehold complementary to the fern sequence; (E) FAM and (F) Cy3 images taken after this exchange illustrating that the replacement of the FAM sequence with the Cy3 release oligonucleotide occurs only at features containing the designated toehold. The fern in panel E (green, negative image) shows the absence of FAM-labelled target oligonucleotide, while the fern in panel F (red, positive image) shows the presence of the displacing Cy3-labeled release oligonucleotide. The fern depicted in (E) and (F) is 9 mm in length and is comprised of individual DNA features that are 14 × 14 μm in size. We have observed no differences in elution efficiency from identical array features. Images (B), (C), (E) and (F) were enhanced for visibility (contrast+40% and brightness+40%).
We show here that toehold-mediated branch migration permits the selective release of target sequences from solid supports, making it possible to multiplex strategies that involve the sequence-specific capture of nucleic acids on solid supports such as PICh11, GENECAPP12, HyCCAPP13 and DNA enrichment for sequencing22. The strategy is characterized and feasibility is demonstrated on a model oligonucleotide system, the locus-specific capture of crosslinked yeast chromatin and on DNA microarrays. The approach shows high release efficiency and rapid release kinetics.
Experimental Section
Please see Supporting Information for experimental details.
Supplementary Material
Acknowledgments
This work was supported by the Wisconsin Center of Excellence in Genomics Science, USA, through NIH/NHGRI grant 1P50HG004952 and by NIGMS grant 5T32GM08349
References
- 1.Watson JD, Crick FH. Molecular structure of nucleic acids; a structure for deoxyribose nucleic acid. Nature. 1953;171:737–738. doi: 10.1038/171737a0. [DOI] [PubMed] [Google Scholar]
- 2.Santa Lucia J, Allawi HT, Seneviratne A. Improved nearest-neighbor parameters for predicting DNA duplex stability. Biochemistry. 1996;35:3555–3562. doi: 10.1021/bi951907q. [DOI] [PubMed] [Google Scholar]
- 3.Bonnet G, Tyagi S, Libchaber A, Kramer FR. Thermodynamic basis of the enhanced specificity of structured DNA probes. Proceedings of the National Academy of Sciences of the United States of America. 1999;96:6171–6176. doi: 10.1073/pnas.96.11.6171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Owczarzy R, Vallone PM, Gallo FJ, Paner TM, Lane MJ, Benight AS. Predicting sequence dependent melting stability of short duplex DNA oligomers. Biopolymers. 1997;44:217–239. doi: 10.1002/(SICI)1097-0282(1997)44:3<217::AID-BIP3>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- 5.Antao VP, Lai SY, Tinoco I. A thermodynamic study of unusually stable RNA and DNA hairpins. Nucleic Acids Research. 1991;19:5901–5905. doi: 10.1093/nar/19.21.5901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Antao JM, Mason JM, Dejardin J, Kingston RE. Protein landscape at Drosophila melanogaster telomere-associated sequence repeats. Molecular and Cellular Biology. 2012;32:2170–2182. doi: 10.1128/MCB.00010-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhang DY, Winfree E. Control of DNA Strand Displacement Kinetics Using Toehold Exchange. Journal of the American Chemical Society. 2009;131:17303–17314. doi: 10.1021/ja906987s. [DOI] [PubMed] [Google Scholar]
- 8.Yurke B, Turberfield AJ, Mills AP, Simmel FC, Neumann JL. A DNA-fuelled molecular machine made of DNA. Nature. 2000;406:605–608. doi: 10.1038/35020524. [DOI] [PubMed] [Google Scholar]
- 9.Zhang DY, Turberfield AJ, Yurke B, Winfree E. Engineering entropy-driven reactions and networks catalyzed by DNA. Science. 2007;318:1121–1125. doi: 10.1126/science.1148532. [DOI] [PubMed] [Google Scholar]
- 10.Srinivas N, Ouldridge TE, Sulc P, Schaeffer JM, Yurke B, Louis AA, Doye JPK, Winfree E. On the biophysics and kinetics of toehold-mediated DNA strand displacement. Nucleic Acids Research. 2013;41:10641–10658. doi: 10.1093/nar/gkt801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dejardin J, Kingston RE. Purification of proteins associated with specific genomic loci. Cell. 2009;136:175–186. doi: 10.1016/j.cell.2008.11.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wu CH, Chen SY, Shortreed MR, Kreitinger GM, Yuan Y, Frey BL, Zhang Y, Mirza S, Cirillo LA, Olivier M, Smith LM. Sequence-specific capture of protein-DNA complexes for mass spectrometric protein identification. Plos One. 2011;6:14. doi: 10.1371/journal.pone.0026217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kennedy-Darling J, Guillen-Ahlers H, Shortreed MR, Scalf M, Frey BF, Kendziorski C, Olivier M, Gasch AP, Smith LM. Discovery of chromatin-associated proteins via sequence-specific capture and mass spectrometric protein identification in Saccharomyces cerevisiae. Journal of Proteome Research. 2014 doi: 10.1021/pr5004938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Udem SA, Warner JR. Ribosomal-RNA synthesis in Saccharomyces-cerevisiae. Journal of Molecular Biology. 1972;65:227–242. doi: 10.1016/0022-2836(72)90279-3. [DOI] [PubMed] [Google Scholar]
- 15.Louis EJ. The chromosome ends of Saccharomyces cerevisiae. Yeast. 1995;11:1553–1573. doi: 10.1002/yea.320111604. [DOI] [PubMed] [Google Scholar]
- 16.Warren CL, Kratochvil NCS, Hauschild KE, Foister S, Brezinski ML, Dervan PB, Phillips GN, Ansari AZ. Defining the sequence-recognition profile of DNA-binding molecules. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:867–872. doi: 10.1073/pnas.0509843102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Booth S, Drebot M, Tipples G, Ng L. Application of DNA array technology for diagnostic microbiology. Canadian Journal of Infectious Diseases & Medical Microbiology. 2000;11:291–294. doi: 10.1155/2000/127160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wu C-H, Lockett MR, Smith LM. RNA-Mediated Gene Assembly from DNA Arrays. Angewandte Chemie International Edition. 2012;51:4628–4632. doi: 10.1002/anie.201109058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Quan J, Saaem I, Tang N, Ma S, Negre N, Gong H, White KP, Tian J. Parallel on-chip gene synthesis and application to optimization of protein expression. Nature Biotechnology. 2011;29:449–452. doi: 10.1038/nbt.1847. [DOI] [PubMed] [Google Scholar]
- 20.Liu Q, Wang L, Frutos AG, Condon AE, Corn RM, Smith LM. DNA computing on surfaces. Nature. 2000;403:175–179. doi: 10.1038/35003155. [DOI] [PubMed] [Google Scholar]
- 21.Singh-Gasson S, Green RD, Yue Y, Nelson C, Blattner F, Sussman MR, Cerrina F. Maskless fabrication of light-directed oligonucleotide microarrays using a digital micromirror array. Nature Biotechnology. 1999;17:974–978. doi: 10.1038/13664. [DOI] [PubMed] [Google Scholar]
- 22.Gnirke A, Melnikov A, Maguire J, Rogov P, LeProust EM, Brockman W, Fennell T, Giannoukos G, Fisher S, Russ C, Gabriel S, Jaffe DB, Lander ES, Nusbaum C. Solution hybrid selection with ultra-long oligonucleotides for massively parallel targeted sequencing. Nature Biotechnology. 2009;27:182–189. doi: 10.1038/nbt.1523. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.