

Abstract
Neuro-glucostasis is required for normal expression of the steroid positive-feedback - induced preovulatory pituitary luteinizing hormone (LH) surge, a critical element of female reproduction. Glucoprivic signals from the caudal hindbrain restrain this surge, but the cellular source of this stimulus is unclear. Norepinephrine (NE) exerts well-defined stimulatory effects on the reproductive neuroendocrine axis. Our studies show that medullary A2 noradrenergic neurons are both estrogen- and glucoprivic-sensitive. Here, we investigated the premise that the LH surge is inhibited by A2 cell reactivity to hindbrain glucopenia and diminished preoptic NE neurotransmission. Estradiol- and progesterone-primed ovariectomized (OVX) female rats were injected into the caudal fourth ventricle (CV4) with the glucose anti-metabolite, 5-thioglucose (5TG) or saline (SAL) prior to onset of the LH surge. Pretreatment by intra-CV4 delivery of the selective catecholamine neurotoxin, 6-OHDA, attenuated LH output, but prevented inhibition by 5TG. 5TG modified patterns of steroid feedback-associated Fos staining of A2, but not other medullary catecholamine cell groups. Intra-preoptic administration of the alpha1-adrenergic receptor agonist, methoxamine, elicited site-specific reversal of hindbrain glucoprivic suppression of gonadotropin-releasing hormone (GnRH) neuron Fos labeling and LH release. Western blotting of laser-microdissected A2 neurons revealed glucoprivic stimulation of Fos, but inhibition of the catecholamine synthetic enzyme, dopamine-β-hydroxylase; 5TG also diminished A2 estrogen receptor (ER)-α and progesterone receptor profiles, but augmented ER-β protein. Intriguingly, A2 AMPK activity was decreased in 5TG-treated rats, despite down-regulation of GLUT3 and no change in MCT2 protein expression. Rostral preoptic GnRH neurons also exhibited decreased AMPK activation simultaneous with apparent reduction of neuropeptide signaling to the pituitary. The present studies demonstrate that hindbrain glucoprivation inhibits the LH surge, in part, by reducing preoptic noradrenergic input, and furthermore implicate A2 neurons as a source of this altered signal. Results also suggest that AMPK sensor deactivation does not supersede the impact of pharmacological inhibition of glucose catabolism on A2 cell function nor afferent signaling of hindbrain glucopenia on GnRH neurons. Further studies are needed to determine if decreased AMPK activation in these cell populations reflect compensatory gain in positive energy balance and/or direct effects of estrogen on AMPK.
Keywords: luteinizing hormone, gonadotropin-releasing hormone, 5-thioglucose, dorsal vagal complex, Western blot, AMPK, A2 noradrenergic neurons, estrogen receptor-alpha, estrogen receptor-beta
Introduction
Clinical and experimental studies confirm the tightly-coupled regulation of reproduction and energy balance. Inadequate energy to support bodily metabolic needs results in diminished fecundity in women, food animals, and laboratory species [Wade and Schneider, 1992]. Oxidation of substrate fuels, particularly glucose, is a critical cue for reproduction. Metabolic fuel control of reproduction hormone secretion is rapid, as these hormone profiles very quickly adjust to acute reductions in glucose oxidation [Wade and Jones, 2004]. The preovulatory LH surge is a crucial event for female reproduction. This transient elevation in hormone release is initiated by steroid positive-feedback and triggers resumption of primary oocyte meiosis, ovulation, and transformation of Graafian follicles into corpora lutea. Energy shortage is a key factor in decreased frequency or cessation of ovulation in women [Crosignani, 2006].
The hindbrain, rather than the hypothalamus, is the primary source of glucoprivic stimuli that inhibit pituitary luteinizing hormone (LH) secretion [Ohkura et al., 2000]. Nerve cell metabolic status is monitored in the hindbrain dorsal vagal complex (DVC) [Oomura and Yoshimatsu, 1984]. Caudal fourth ventricular administration of the glucose anti-metabolite, 5-thioglucose (5TG), reverses steroidal activation of preoptic gonadotropin-releasing hormone (GnRH) neurons (the final common pathway for neural control of reproductive hormone secretion), thereby diminishing the LH surge [Singh and Briski, 2004]. DVC A2 neurons are a logical conduit of metabolic input to the GnRH-pituitary LH neuroendocrine axis as norepinephrine (NE) has a well-defined role in its regulation and these cells express molecular biomarkers for metabolo-sensory function, e.g. glucokinase, KATP, and the ultra-sensitive energy sensor, adenosine 5’-monophosphate-activated protein kinase (AMPK) [Briski et al., 2009; Cherian and Briski, 2011; Ibrahim et al., 2013]. It is also likely that A2 cells are direct substrates for steroid regulatory actions as they express estrogen receptor-alpha (ERα) and -beta (ERβ) mRNAs and proteins [Tamrakar et al., 2012; Ibrahim et al., 2013]. Our evidence that A2 neurons are both estrogen-and glucoprivic-sensitive supports the possibility that these cells may adjust estrogen-mediated stimulatory noradrenergic input to the reproductive neuroendocrine axis according to prevailing energy status. The present studies utilized pharmacological tools to address the hypothesis that hindbrain glucoprivic inhibition of the LH surge requires DVC catecholamine nerve cell function. High-sensitivity Western blot techniques were applied to laser catapult microdissected A2 neurons [Koshy Cherian and Briski, 2011; 2012] to evaluate effects of hindbrain glucoprivation on A2 AMPK activity and indicators of A2 functional status, e.g. Fos, a reliable marker for transcriptional activation, and the glucoprivic-sensitive catecholamine biosynthetic enzyme, dopamine-β-hydroxylase (DβH). NE acts within the medial preoptic area to mediate steroid feedback regulation of LH; indeed, divergent estrogen negative- and positive-feedback actions on this hormone are achieved, in part, by corresponding patterns of basal versus elevated NE release into that area [Demling et al., 1985; Mohankumar et al., 1994; Szawka et al., 2013]. The current studies examined the premise that hindbrain glucoprivic suppression of the LH surge involves diminished preoptic NE release.
ERα and ERβ exert divergent, e.g. stimulatory versus inhibitory effects, respectively, on catecholamine enzyme transcription in vitro [Maharjan et al., 2005]. Reports that increases in plasma estradiol coincide with augmented A2 ERα-ir [Haywood et al., 1999] imply that estrogenic control of these cells may involve, in part, distinct steroid concentration-dependent receptor signaling mechanisms for stimulatory versus inhibitory hormone actions on noradrenergic neurotransmission. We investigated here whether hindbrain glucopenia suppresses steroid positive-feedback stimulation of ERα and/or increases ERβ protein profiles in A2 cells.
Connectivity of preoptic GnRH neurons with extra-preoptic metabolic sensors is affirmed by the capacity of hindbrain glucopenia to reverse steroid positive-feedback activation of those cells and suppress the LH surge [Briski and Sylvester, 1998]. Recent work suggests that GnRH neurons may also engage in energy self-monitoring and undergo functional adjustments accordingly. In vitro studies utilizing preoptic area slice preparations report that media glucose decrements inhibit GnRH nerve cell firing [Zhang et al., 2007], a response that is abolished by AMPK inhibition [Roland and Moenter, 2011]. Moreover, immortalized mouse hypothalamic GT1-7 cells express AMPK, and exhibit diminished GnRH release when treated with AMPK activators [Coyral-Castel et al., 2088; Wen et al., 2008; Cheng et al., 2011]. It is well documented that in addition to nutrient status, hypothalamic AMPK is also regulated by endocrine signals of peripheral energy status [Ronnett et al., 2009]. Our recent studies suggest that AMPK functionality in separate metabolo-sensory cell groups may be functionally linked, as we found that manipulation of hindbrain metabolic fuel availability altered hypothalamic AMPK activity independently of blood glucose profiles [Gujar et al., 2012]. The present studies utilized the combinatory laser-microdissection/Western blot approach outline above to investigate the hypothesis that GnRH neurons express AMPK in vivo, and that glucoprivic input from the caudal hindbrain augments activity of this sensor.
Methods and Materials
Animal Model for Steroid Positive-Feedback Induction of the LH Surge [Singh and Briski, 2004]
Adult female Sprague Dawley rats were from the ULM College of Pharmacy in-house breeding colony and treated in accordance with the Guide for Use and Care of Animals. Breeders were purchased from Harlan Laboratories, Inc. Animals were maintained under a 14hr light:10 hr dark schedule, and allowed free access to standard laboratory rat chow and water. Rats were bilaterally OVX on day 1 under ketamine:xylazine anesthesia (0.1 mL/100g bw ip; 100 mg ketamine:10 mg xylazine/mL), and implanted with a hand-held PE-20 cannula into the caudal fourth ventricle (CV4). On day 5, accuracy of cannula placement was verified by assessment of blood glucose levels following CV4 administration of the glycolytic enzyme inhibitor, 5-thioglucose (5TG; 200 ug). Rats were injected sc with estradiol benzoate (E; 2 μg/0.1 mL safflower oil) at 10.00 hr on days 14 and 17, and progesterone (P; 2.0 mg/0.2 mL safflower oil) at 11.00 hr on day 18. At 14.00 hr (time zero; to) on day 18, OVX + EP rats were injected into the CV4 with 200 ug 5TG or the vehicle, saline (SAL).
Experimental Design
Experiment 1: Effects of DVC catecholamine nerve cell destruction on hindbrain glucoprivic inhibition of the steroid positive-feedback induction of the LH surge
In addition to treatments administered according to the schedule described above, sets of rats were injected into the CV4 on days 7 and 9 with either 6-OHDA (50 μg/uL/day [van den Buuse et al., 1984]; n=10) or vehicle containing 0.2% ascorbic acid (n=10). On day 14, animals were implanted with intracardiac venous silastic catheters [Briski and Sylvester, 1988]. On day 18, serial blood samples were collected at hourly intervals between 13.00 and 20.00 hr from OVX + EP rats injected into the CV4 at to with SAL (group 1; n=5 vehicle-pretreated; group 2; n=5 6-OHDA-pretreated) or 5TG (group 3; n=5 vehicle-pretreated; group 4; n=5 6-OHDA-pretreated). Plasma LH concentrations were measured by radioimmunoassay, as described [Briski and Sylvester, 1988]. After the final sample collection, animals were sacrificed by transcardial perfusion with 2.0% sodium nitrite in 0.9% saline, followed by 4.0% paraformaldehyde and 0.2% picric acid in 0.1 M potassium phosphate buffer, pH 7.6, under deep sodium pentobarbital anesthesia. At the conclusion of perfusion, the brain was postfixed overnight, sunk in 30% sucrose, and cut into 20 μm serial sections. Accuracy of cannula placement was verified histologically; animals with misplaced cannulas were replaced. For each animal, two sections were processed, per level, from the rostral (-13.28 to -13.60 mm), commissural (-13.76 to -14.16 mm), and caudal (-14.36 to -14.86 mm) DVC by avidin-biotin peroxidase immunochemistry for tyrosine hydroxylase (TH) immunoreactivity (-ir) [Briski, 1997] to confirm catecholamine nerve cell lesioning in this structure by 6-OHDA. Tissues were processed with mouse monoclonal anti-TH primary antibodies (prod. no. 22941, 1:10K; ImmunoStar, Inc., Hudson, WI) and Vector Laboratories (Burlingame, CA) Vectastain Elite ABC mouse IgG (prod. no. PK-6102) and DAB (3,3’-diaminobenzidene) peroxidase substrate (SK-4100) kit reagents. Images were captured with a Zeiss AxioScope outfitted with an AxioCam HR camera, and processed with AxioVision software (Carl Zeiss Microimaging, Inc., Thornwood, NY). For each animal, bilateral counts of TH-ir neurons were taken from each DVC region.
Experiment 2: Effects of CV4 5TG Administration on Steroid Positive-Feedback - Associated Fos Immunoexpression by DVC (A2, C2), Dorsomedial Medullary C3, and Ventrolateral Medullary A1 Catecholamine Neurons
On day 18, groups of OVX + EP rats were injected with 5TG (group 1; n=4) or SAL (group 2; n=4) at 14.00 hr and sacrificed by transcardial perfusion 2 hr at 16.00 hr for brain tissue sectioning. For each animal, three sections through the C2 [collected between -13.28 to -13.60 mm] and A2 [collected between -14.36 to -14.86 mm] cell groups were processed by dual-label avidin-biotin-peroxidase immunocytochemistry for Fos- and TH-ir [Briski et al., 2001]. In the first staining series, nuclear Fos-ir was identified by a rabbit polyclonal anti-Fos antiserum (prod. no. PC38, 1:100K; EMD Millipore, Billerica, MA) and visualized by black-brown nickel-intensified diaminobenzidine reaction product. In the second series, mouse anti-TH antibodies and diaminobenzidine were used, as described for Experiment 1 above, to label cytoplasmic TH antigenicity as gold-brown. Sections containing C3 neurons [collected between -11.80 to -12.80 mm] were processed to determine effects of intra-CV4 5TG delivery on extra-DVC dorsomedial hindbrain catecholamine cells. A1 cells [collected between -14.00 to -14.60 mm] were evaluated as they function as an alternative source of noradrenergic input to the reproductive neuroendocrine axis [Wright and Jennes, 1993], and are innervated by A2 neurons [Pacák and Palkovits; 2001]. For each animal, bilateral counts were made for total number of TH-ir nerve cells and number of TH-/Fos-ir cells in individual catecholamine cell groups.
Experiment 3: Effects of Intra-Preoptic Administration of the Alpha1 Receptor Agonist Methoxamine on Hindbrain Glucoprivic Inhibition of the LH surge and Fos Expression by GnRH neurons
On day 1, after bilateral OVX and CV4 cannula placement, rats were implanted with a midline 26-gauge stainless-steel guide cannula (prod. no. C315G/SP; Plastic One, Inc., Roanoke, VA) directed dorsal to (group 1) the rostral preoptic area (coordinates: 0.00 mm posterior to bregma; 0.00 mm lateral to midline; 6.8 mm ventral to skull surface) or (group 2) median preoptic nucleus (MEPO; group 2; 0.12 mm posterior to bregma; 0.00 mm lateral to midline; 6.4 mm ventral to skull surface) or (group 3) with double-guide cannulas (prod. no. C235G-0.8 S7]P; Plastics One, Inc.) aimed above the anteroventral periventricular nucleus (AVPV; group 3; 0.26 mm posterior to bregma; 0.4 mm lateral to midline; 7.1 mm ventral to skull surface), as described [Briski and Vogel, 1995]. On day 14, rats were implanted with intracardiac silastic catheters. On day 18, OVX + EP animals were pretreated at 13.50 hr with the alpha1 receptor agonist, methoxamine (ME; 0.1 ug/0.2 uL [Vetrivelan et al., 2005; 2006]) or vehicle (artificial cerebrospinal fluid; aCSF) into the rPO, MEPO, or AVPV. At 14.00 hr, aCSF-pretreated rats received intra-CV4 injections of SAL or 5TG; ME-pretreated animals were injected with 5TG; thus, groups 1-3 consisted of 3 subsets each (n=5 animal/subset). Animals were sacrificed by transcardial perfusion at 16.00 hr for brain tissue collection. Accuracy of cannula placement was verified histologically; animals with misplaced cannulas were replaced. Immediately before perfusion, trunk blood was obtained by cardiac puncture for LH radioimmunoassay. Tissue sections of 20 μm thickness were collected at 160 μm intervals through the rostral preoptic area at the level of the organum vasculosum of the lamina terminalis for dual-label immunocytochemical labeling of GnRH- and Fos-ir [Singh and Briski, 2004]. This subpopulation of GnRH cells was targeted for analysis as it is characterized by transcriptional reactivity to glucoprivation [Briski and Sylvester, 1998; Singh and Briski, 2004]. The sequential staining procedure outlined for Experiment 2 above was carried out here using a rabbit polyclonal antiserum against GnRH (1:25K; kindly provided by Dr. R. Benoit, Quebec, Canada) in the second staining series.
Experiment 4: Effects of Hindbrain Glucopenia on Genomic Activation, AMPK Activity, and Catecholamine Biosynthetic Enzyme/Neurotransmitter Protein Expression in Caudal DVC A2 and Rostral Preoptic Area GnRH Neurons
On day 18, groups of OVX + EP rats were injected with 5TG (group 1; n=8) or SAL (group 2; n=8) at 14.00 hr, and sacrificed by decapitation at 16.00 hr for brain and trunk blood collection. Blood glucose levels were measured using an AccuCheck Advantage glucometer (Roche Diagnostics, Indianapolis, IN), as described [Kale et al., 2006]. Dissected brains were snap-frozen in liquid nitrogen-cooled isopentane. Fresh-frozen 10 μm serial sections were cut from the caudal DVC between -14.36 to -14.86 mm, mounted on PEN membrane-coated slides, and labeled for TH-ir by avidin-biotin peroxidase immunochemistry, as described [Briski et al., 2009]. The forebrain preoptic area was cut into 10 μm serial sections between +0.40 to -0.40 mm. Rostral preoptic area GnRH neurons were immunolabeled using rabbit polyclonal antibodies against GnRH I precursor (sc-20941, 1:300; Santa Cruz Biotechnol.). Caudal DVC TH- and rostral preoptic area GnRH-ir neurons were harvested individually with a Zeiss P.A.L.M. UV microlaser. For each protein of interest, n=50 neurons (n=12-13 cells per rat) were harvested per treatment group. Pooled nerve cell lysates were separated on 4–12% gradient Tris-glycine gels and transferred to 0.45 um PVDF-Plus membranes [Ibrahim et al., 2013]. Membranes were treated with Quentix Western blot signal enhancer (product No. 21050; Pierce, Rockford, IL) and blocked with 0.1% Tween-20 and 2% bovine serum albumin prior to sequential incubation with primary antisera, peroxidase-conjugated goat anti-mouse, goat anti-rabbit, or rabbit anti-goat secondary antibodies, and Supersignal West Femto Maximum Sensitivity substrate. A2 cell lysates were probed for Fos, phosphoAMPKα1/2 (pAMPK), ERα, ERβ, PR (progesterone receptor), monocarboxylate transporter-2 (MCT2), glucose transporter-3 (GLUT3), glucose transporter-4 (GLUT4), and α-tubulin protein expression [Ibrahim et al., 2013]. AMPKα1/2 and DβH proteins were also evaluated, using rabbit polyclonal antisera (sc-25792, 1:500; sc-15318, 1:500, respectively) from Santa Cruz Biotechnol., Inc. (Santa Cruz, CA). GnRH neurons were analyzed for Fos, AMPK, pAMPKα1/2, GnRH, and α-tubulin protein expression. GnRH I precursor protein blots were probed with the aforementioned primary antiserum at a dilution of 1:500. Chemiluminescent signals were visualized in a Syngene G:box Chemi. Protein band optical densities (O.D.) were normalized to α-tubulin. Protein molecular weight markers were included in each Western blot analysis. Each protein analysis was performed in triplicate.
Statistical Analyses
Mean plasma LH concentrations in Experiment 1 were evaluated by two-way ANOVA for repeated measures and Duncan’s multiple range test. LH levels and numbers of rostral preoptic GnRH-ir and GnRH-/Fos-ir neurons in Experiment 3 were evaluated by one-way ANOVA and Student-Newman-Keuls test. Protein O.D. values, blood glucose levels, and numbers of TH-ir and TH-/Fos-ir neurons analyzed by Student’s t test. The criterion for significance was set to p = 0.05 in all comparisons.
Results
Figure 1 depicts effects of 6-OHDA pretreatment on patterns of LH secretion following intra-CV4 administration of 5TG or vehicle at 14.00 hr to steroid-primed OVX female rats. A significant group effect on LH release was observed (F8,152 = 5.02; p<0.0001). Main effects were due to time (F = 4.68; p<0.0001) and drug treatment (F = 3.19; p<0.0001); interaction between these factors was significant (F=4.94; p<0.0001. In vehicle-pretreated, SAL-injected controls [filled circles], mean plasma LH levels were significantly elevated over a five hr interval, e.g. from 15.00 to 19.00 hr, compared to values measured before (13.00 hr) and at to (14.00 hr). Treatment with the glucose anti-metabolite, 5TG [open circles], attenuated this elevation in hormone release, as this group had significantly lower LH output between 15.00 and 19.00 hr compared to the vehicle + SAL group. The LH surge was diminished by 6-OHDA pretreatment, but hormone secretion was equivalent over the duration of the experiment in 6-OHDA + 5TG [open triangles] versus 6-OHDA + SAL groups [filled triangles]. The data in Table 1 indicate that 6-OHDA significantly reduced mean TH-ir-positive nerve cell counts within the rostral (F3,16=14.77; p<0.0001), commissural (F=16.52; p<0.0001), and caudal DVC (F=13.94; p<0.0001). Included below the table are photomicrographs showing typical patterns of TH immunolabeling in the caudal DVC of a vehicle- [bottom left] versus 6-OHDA [bottom right]-treated animal, which depict a decline in TH-ir cells after neurotoxin pretreatment.
Figure 1. Effects of Caudal Fourth Ventricular (CV4) Pretreatment with the Catecholamine Neurotoxin, 6-Hydroxydopamine (6-OHDA) on Hindbrain Glucoprivic Inhibition of Luteinizing Hormone (LH) Secretion in Estrogen (E) Plus Progesterone (P)-Primed Ovariectomized (OVX) Female Rats.
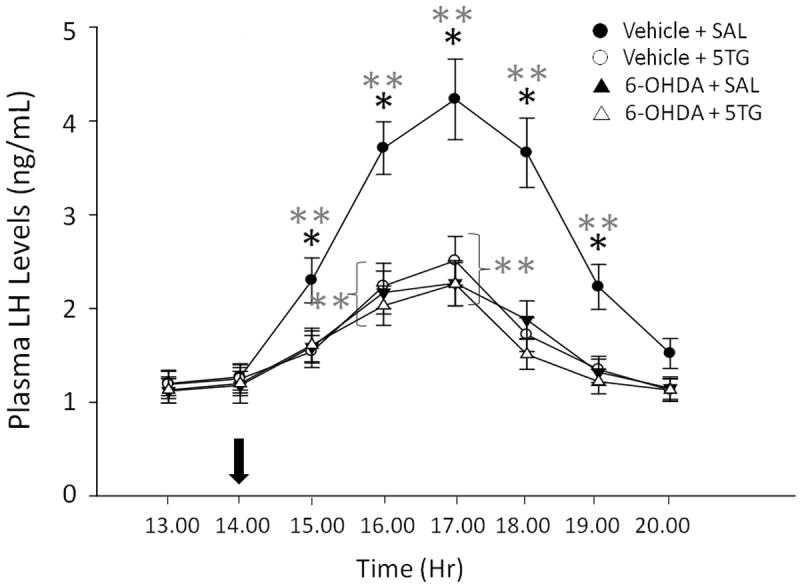
Groups of 6-OHDA- and vehicle-pretreated OVX+EP rats were injected with the glucose antimetabolite, 5-thioglucose (5TG; 200 ug), or saline (SAL) into the CV4 at 14.00 hr (arrow at abscissa). Blood samples were obtained hourly between 13.00 and 20.00 hr. Mean plasma LH concentrations ± S.E.M. are depicted for the following treatment groups (n=5 rats/group): vehicle + SAL [
 ]; vehicle + 5TG [
]; vehicle + 5TG [
 ]; 6-OHDA + SAL [
]; 6-OHDA + SAL [
 ]; and 6-OHDA + 5TG [
]; and 6-OHDA + 5TG [
 ]. Data show mean values for n=5 rats/group. *p<0.05, compared to other treatment groups at the same time point. **p<0.05, compared to 13.00 and 14.00 hr. Results show that 6-OHDA – pretreated animals exhibited reduced LH output during the steroid feedback-induced surge, but that these hormone profiles were resistant to inhibition by 5TG.
]. Data show mean values for n=5 rats/group. *p<0.05, compared to other treatment groups at the same time point. **p<0.05, compared to 13.00 and 14.00 hr. Results show that 6-OHDA – pretreated animals exhibited reduced LH output during the steroid feedback-induced surge, but that these hormone profiles were resistant to inhibition by 5TG.
Table 1. Effects of 6-Hydroxydopamine (6-OHDA) on Dorsal Vagal Complex (DVC) Tyrosine Hydroxylase-Ir Nerve Cell Counts.
| Treatment Groups | Cell Counts (x ± S.E.M.)a
|
||
|---|---|---|---|
| Rostral DVC | Commissural DVC | Caudal DVC | |
| Vehicle + Saline | 33 ± 4 | 72 ± 8 | 41 ± 5 |
| Vehicle + 5-thioglucose (5TG) | 37 ± 4 | 78 ± 9 | 38 ± 5 |
| 6-OHDA + Saline | 8 ± 2b | 20 ± 3b | 12 ± 2b |
| 6-OHDA + 5TG | 11 ± 2b | 18 ± 3b | 13 ± 2b |
n=5 rats per treatment group
p<0.05; versus vehicle controls

The data in Figure 2 portray effects of CV4 administration of 5TG [grey bars] versus SAL [white bars] to steroid-primed OVX female rats at 14.00 hr on patterns of Fos immunolabeling of catecholamine nerve cell populations located internal (C2, A2) and external (C3, A1) to the DVC at 16.00 hr. Dual-label avidin-biotin peroxidase immunocytochemical processing for TH- and Fos-immunoreactivity (-ir) showed that glucose anti-metabolite treatment did not alter total numbers of TH-ir neurons [solid bars] in any cell group, but significantly increased co-expression of Fos protein [cross-hatched bars] in A2 [t(6)=5.52]; p<0.005), but not C2 [t(6)=1.46], C3 [t(6)=1.38], or A1 [t(6)=1.21] neurons. Total numbers of Fos-ir nerve cell nuclei in the A2 area were elevated in 5TG- (114 ± 12) versus SAL- (72 ± 8) injected rats [t(6)=4.66; p<005]; drug treatment did not alter Fos-ir neuron counts in other examined sites. Mean blood glucose levels were 102.6 ± 3.0 versus 133.5 ± 8.4 mg/dL in SAL- and 5TG-treated groups, respectively, indicating significant 5TG-induced augmentation of circulating glucose levels [t(12)=3.85; p<0.005]. Characteristic A2 neurons exhibiting gold-brown diaminobenzidine staining of the cytoplasm are shown in Figure 3 to illustrate the presence [Panel 3A] versus absence [Panel 3B] of co-labeling for Fos-ir, e.g. localization of black-brown nickel-intensified reaction product to the nuclear compartment.
Figure 2. Effects of CV4 5TG Administration on Fos Immunolabeling of Hindbrain Medullary Catecholamine Cell Groups in Steroid-Primed OVX Female Rats.
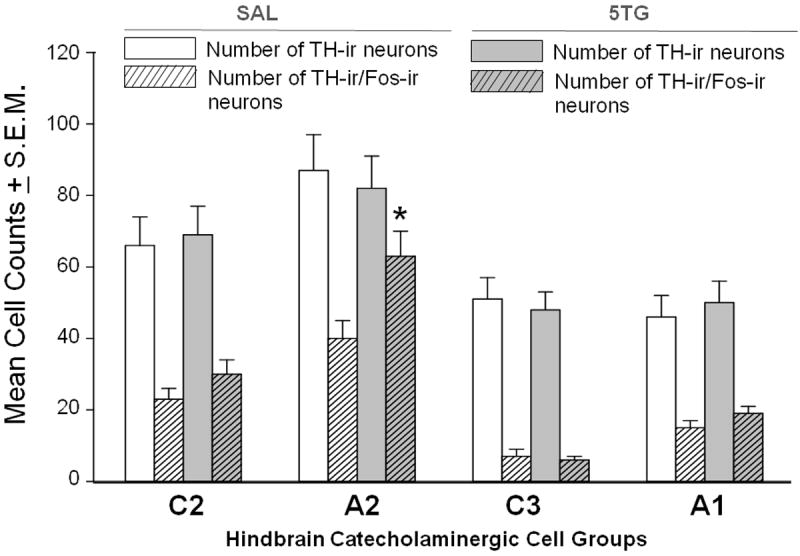
Groups of OVX+EP rats were sacrificed 2 hr after CV4 delivery of SAL (n=4) or 5TG (n=4), and sections through A2, C2, C3, and A1 cell groups were processed by dual-label avidin-biotin peroxidase immunocytochemistry for tyrosine hydroxylase- (TH)- and Fos-immunoreactivity (-ir). For each cell population, bars depict mean bilateral counts of TH-ir neurons [SAL: white bar; 5TG: gray bar] and TH-/Fos-ir cells [SAL: white diagonal-striped bar; 5TG: gray diagonal-striped bar]. *p <0.05, compared to SAL. Data show that 5TG significantly increased Fos labeling of A2, but not other hindbrain catecholamine cell groups evaluated.
Figure 3. Dual Immunoperoxidase Labeling of Tyrosine Hydroxylase (TH)- and Fos-ir in Caudal Hindbrain A2 Neurons.
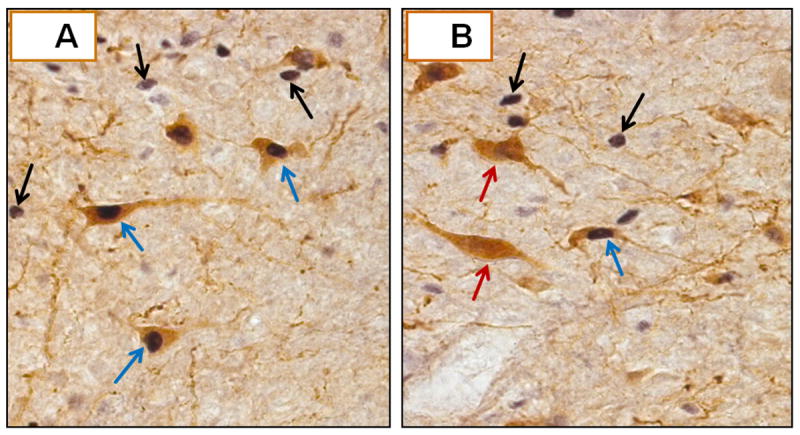
A depicts several TH-ir-positive A2 neurons, identifiable by gold-brown cytoplasmic staining, that also exhibit black-brown nuclear labeling for Fos (blue arrows). Fos-positive nuclei of nearby TH-ir-negative nerve cells are indicated by black arrows. B shows two TH-ir-positive A2 cells that lack Fos staining (red arrows), as indicated by the absence of nuclear label.
Figure 4 depicts effects of targeted delivery of the alpha1-adrenergic receptor agonist, ME, into the rPO [Figure 4.A], AVPA [Figure 4.B], or MEPO [Figure 4.C] on hindbrain glucoprivic inhibition of LH secretion. Results show that inhibitory effects of 5TG [grey bars] on circulating LH, relative to SAL-injected controls [white bars], were reversed by prior administration of ME into the AVPA (F2,12=8.74; p<0.005), but not the rPO (F2,12=1.93) or MEPO (F2,12=2.28) [diagonal-striped grey bars]. The data in Table 2 indicate that glucose anti-metabolite treatment had no effect on mean numbers of rostral preoptic GnRH-ir neurons, but did significantly reduce mean numbers of GnRH neurons that were co-labeled for Fos-ir. ME administration into the AVPV (F2,12=9.59; p<0.005), but not the rPO (F2,12=1.47) or MEPO (F2,12=2.61) reversed hindbrain glucoprivic repression of numbers of GnRH- plus Fos-ir neurons. Representative Fos-ir-positive versus -negative GnRH neurons are depicted in Figure 5.A and 5.B, respectively.
Figure 4. Effects of Intra-Preoptic Administration of the Alpha1-Adrenergic Receptor Agonist, Methoxamine (ME), on Hindbrain Glucoprivic Inhibition of the Steroid Feedback – Induced LH Surge.

Groups of OVX+EP rats were pretreated 10 minutes prior to CV4 injection of 5TG or SAL at 14.00 hr by delivery of ME (0.1 ug) or vehicle, artificial cerebrospinal fluid (aCSF), into either the rostral preoptic area (rPO; Panel 3.A), anteroventral periventricular nucleus (AVPV; Panel 3.B), or median preoptic nucleus (MEPO; Panel 3.C). Trunk blood was collected at 16.00 hr for LH radioimmunoassay. For each pre-treatment site, mean plasma LH concentrations ± S.E.M. are depicted for aCSF + SAL [white bar]; aCSF + 5TG [gray bar]; and ME + 5TG [gray diagonal-striped bar] groups (n=5/group). Data show that ME delivery to the AVPA, but not the rPO or MEPO reversed inhibitory effects of intra-CV4 5TG delivery on LH release.
Table 2. Effects of Site-Specific Intra-Preoptic Administration of the Alpha1 Receptor Agonist, Methoxamine on Hindbrain Glucoprivic Inhibition of Rostral Preoptic GnRH Nerve Cell Fos Expression in Steroid-Primed Ovariectomized Female Rats.
| Treatment Group | Cell Countsa
|
|
|---|---|---|
| GnRH-ir Neurons | GnRH-/Fos-ir Neurons | |
| A. Rostral Preoptic Area (Pre-Treatment of aCSF vs. ME) | ||
| aCSF + SALCV4 | 81 ± 9 | 53 ± 6 |
| aCSF + 5TG CV4 | 68 ± 7 | 29 ± 3b |
| ME + 5TGCV4 | 75 ± 9 | 26 ± 3b |
| B. Anteroventral Periventricular Nucleus (Pre-Treatment of aCSF vs. ME) | ||
| aCSF + SALCV4 | 93 ± 10 | 64 ± 7 |
| aCSF + 5TGCV4 | 85 ± 9 | 25 ± 3b |
| ME + 5TGCV4 | 78 ± 9 | 50 ± 6 |
| C. Median Preoptic Nucleus (Pre-Treatment of aCSF vs. ME) | ||
| aCSF + SALCV4 | 69 ± 8 | 40 ± 5 |
| aCSF + 5TGCV4 | 80 ± 9 | 24 ± 3b |
| ME + 5TGCV4 | 73 ± 9 | 21 ± 3b |
Mean number ± S.E.M. for n=5 rats/group
p<0.05 compared to aCSF + SAL group
Figure 5. Immuno-Co-Localization of Fos Protein to Rostral Preoptic Gonadotropin-Releasing Hormone (GnRH) Neurons.
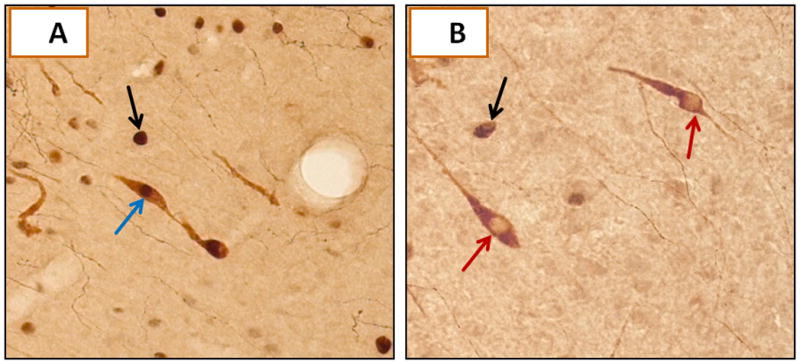
A illustrates a characteristic GnRH- plus Fos-ir-positive neuron (blue arrow), indicated by gold-brown and black-brown staining of cytoplasmic GnRH and nuclear Fos proteins. The black arrow indicates a Fos-positive neuron that lacks GnRH-ir (black arrow). B depicts two GnRH-ir-positive neurons that lack labeling for Fos (red arrows).
A2 neurons identified by TH-immunolabeling were harvested from the caudal DVC 2 hrs after intra-CV4 administration of 5TG or SAL to steroid-primed OVX rats. Figure 6 depicts representative immunoblots of Fos [Row A] and DβH [Row B] proteins with corresponding levels of the housekeeping protein, α-tubulin [Row K] in SAL- [left-hand column] versus 5TG- [right-hand column] treated rats. Mean normalized O.D. measures presented in Figure 7 show that glucose anti-metabolite treatment significantly increased A2 Fos [Panel A; t(4)=7.24; p<0.001], but decreased DβH [Panel B; t(4)=4.326; p<0.01] expression. Western blots [Figure 6; Rows C and D, respectively] and mean normalized O.D. measures [Figure 7; Panels C and D] of A2 AMPK and pAMPK protein profiles show that 5TG had no impact on AMPK levels [t(4)=0.72], but caused a decline in A2 pAMPK content [t(4)=5.03; p<0.005]. Rows E, F, and G of Figure 6 depict characteristic immunoblots of ERα, ERβ, and PR, respectively. In Figure 7, Panels E, F, and G show that 5TG-treated animals exhibited a decline in mean O.D. measures for ERα [t(4)=2.38; p<0.05] and PR proteins [t(4)=5.15; p<0.01], but that ERβ expression [t(4)=11.58; p<0.001]. was augmented relative to SAL controls. Immunoblots of A2 MCT2, GLUT4, and GLUT3 proteins are shown in corresponding Rows H, I, and J. Panels H, I, and J of Figure 7 reveal that MCT2 [t(4)=1.71] and GLUT4 protein [t(4)=1.87] profiles were unaffected, whereas GLUT3 expression was reduced by glucose anti-metabolite treatment [t(4)=7.37; p<0.001]. Figure 8 illustrates microlaser catapult-dissection of caudal DVC A2 neurons. Panel 8.B shows individual A2 cells situ immunolabeled in situ prior to two-step dissection involving circumnavigation [Panel 8.C; red circles denote precision Zeiss P.A.L.M. microlaser alignment external to individual TH-ir A2 neurons] and ejection [Panel 8.D; empty spaces denote prior cell locations] by focused laser beam.
Figure 6. Effects of CV4 5TG on A2 Noradrenergic Nerve Cell Fos, Dopamine-β-Hydroxylase (DβH), Steroid Hormone Receptor, and Substrate Fuel Transporter Protein Expression and AMPK Activity in OVX+EP Female Rats.
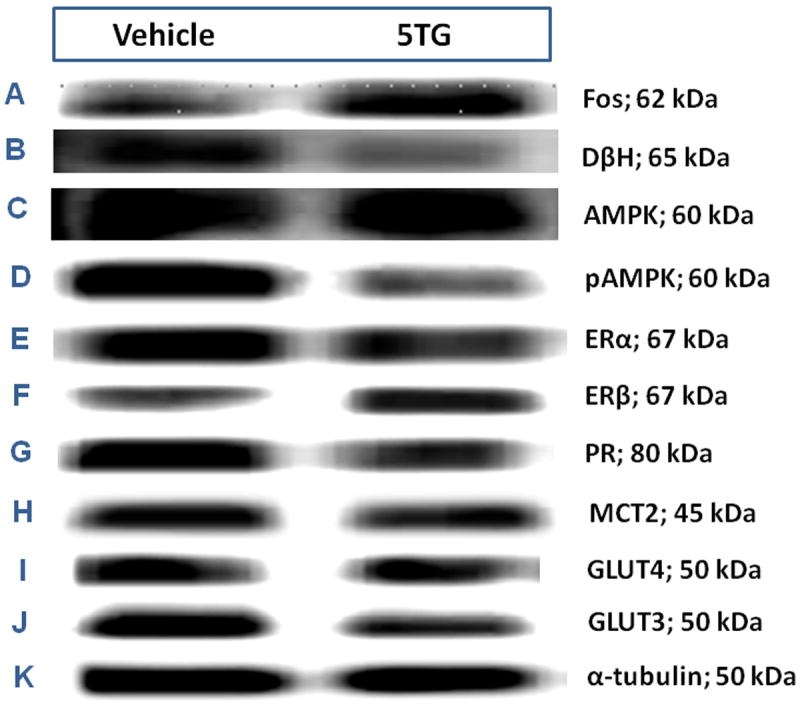
Individual TH-ir A2 neurons were laser-catapult microdissected from 10 μm-thick sections of the caudal dorsal vagal complex of OVX+EP rats 2 hr after intra-CV4 administration of 5TG (200 ug; n=8) or SAL (n=8). For each treatment group, separate lysates of pooled n=50 A2 cells (n=12-13/rat) were analyzed by Western blot for Fos (Row A), DβH (Row B), phosphoAMPK (Row C); AMPK (Row D), estrogen receptor-alpha (ERα; Row E), ER-beta (ERβ; Row F), progesterone receptor (Row G), monocarboxylate transporter-2 (MCT2; Row H), glucose transporter-4 (GLUT4; Row I), glucose transporter-3 (GLUT3; Panel Row J), or α-tubulin (Row K). For each protein of interest, immunoblot analyses were performed in triplicate.
Figure 7. Effects of CV4 5TG Treatment on Normalized A2 Nerve Cell Protein Levels in OVX+EP Female Rats.
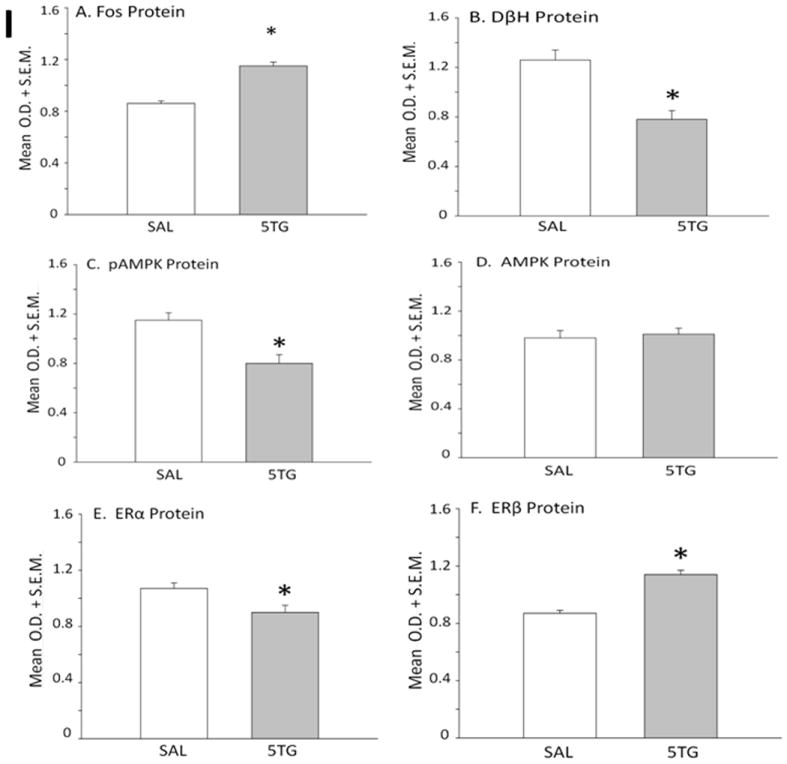
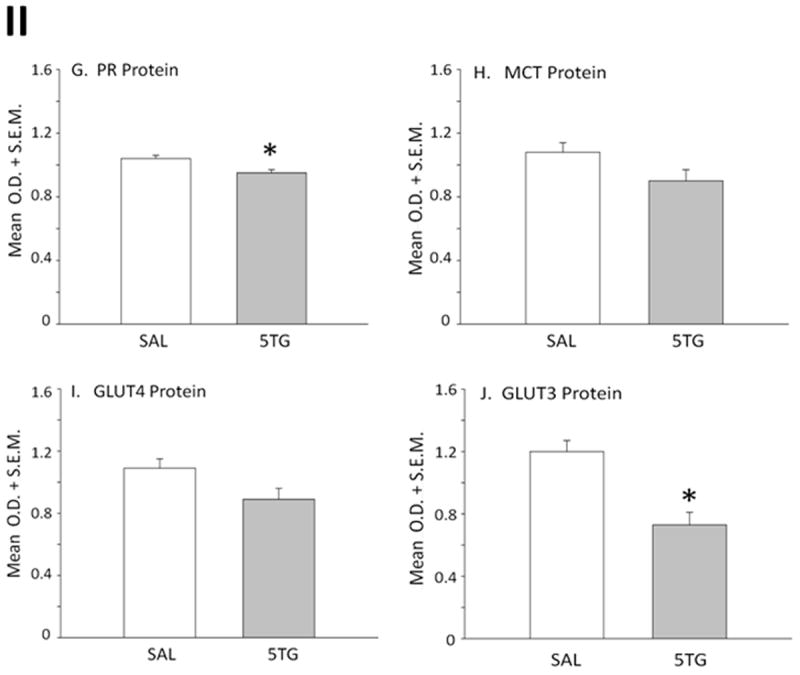
Protein band optical densities (O.D.) of immunoblots of n=50 neurons/treatment group from SAL- (white bars) or 5TG- (gray bars) treated rats were quantified with Alpha-Imager HP V 5.0.1 software, and expressed relative to the housekeeping protein, α-tubulin. Panels depict mean normalized O.D. values ± S.E.M. for A2 nerve cell Fos (Panel A), DβH (Panel B), pAMPK (Panel C), AMPK (Panel D), ERα (Panel E), ERβ (Panel F), PR (Panel G), MCT2 (Panel H), GLUT4 (Panel I), and GLUT3 (Panel J) protein content 2 hr after treatments. *p <0.05, compared to SAL.
Figure 8. Laser-Catapult Microdissection (LCM) of A2 Neurons from the Caudal DVC of Steroid-Primed OVX Female Rats Following CV4 Administration of 5TG or SAL.
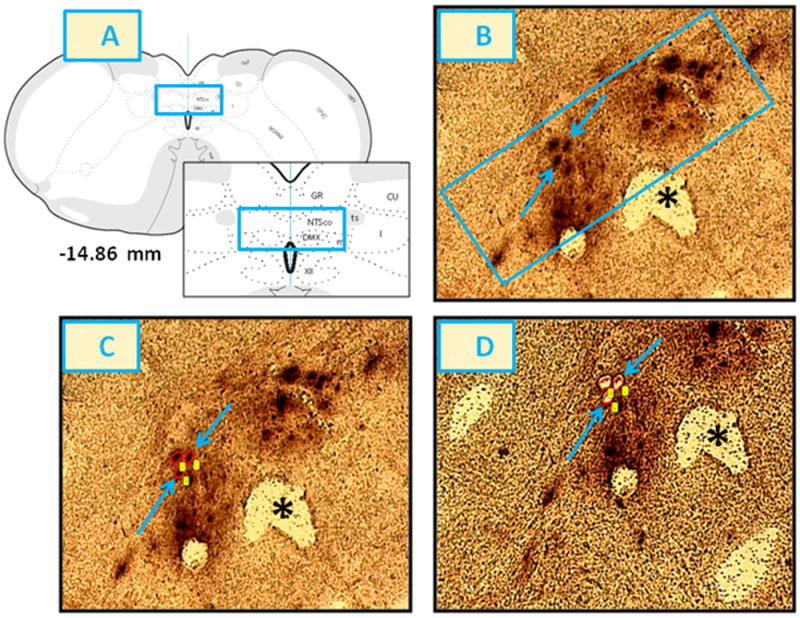
The blue rectangles on the brain map (-14.86 mm) and within the inset of Panel 8.A illustrate the location of harvested A2 cells. Blue arrows in Panel 8.B indicate two representative TH-ir neurons in that site. The rectangle-enclosed area in Panel 8.B was re-photographed after positioning of circular laser cuts, depicted in red, around individual nerve cell bodies [Panel 8.C] and ejection of circumdissected cells by laser-pulse, leaving vacant spaces correspondingly to single A2 neurons [Panel 8.D]. Note that this microdissection technique causes negligible destruction of surrounding tissue and minimal inclusion of adjacent non-nerve cell tissue. The asterisk indicates the central canal. Abbreviations in Panel 8.A: NTSco, m, l: commissural, medial and lateral parts of the nucleus of the solitary tract; ts: solitary tract; C: central canal; GR: gracile nucleus; CU: cuneate nucleus.
Rostral preoptic GnRH neurons were laser-microdissected 2 hrs after intra-CV4 treatment of steroid-primed OVX rats with 5TG or SAL. Figure 9 shows representative Western blots of GnRH nerve cell Fos [Row A], GnRH [Row B], AMPK [Row C], pAMPK [Row D], and α-tubulin proteins [Row E] in SAL- [left-hand column] versus 5TG- [right-hand column] treated rats. Normalized O.D. measures presented in Figure 10 indicate that glucose anti-metabolite delivery to the CV4 caused a decline in Fos [t(4)=3.43; p<0.01] and pAMPK [t(4)=5.46; p<0.005] protein expression in GnRH neurons, while augmenting GnRH I precursor [t(4)=3.37; p<0.02] and AMPK [t(4)=18.19; p<0.0001] protein profiles in these cells. Figure 11 depicts laser-catapult microdissection of GnRH neurons. Panel 8.B shows in situ immunolabeling of GnRH cells prior to isolation [Panel 8.C] and ejection [Panel 8.D] of single neurons by focused laser beam.
Figure 9. Effects of Hindbrain Glucoprivation on Steroid Positive-Feedback Patterns of Fos, GnRH, AMPK, and pAMPK Protein Expression in Rostral Preoptic Gonadotropin-Releasing Hormone (GnRH) Neurons.
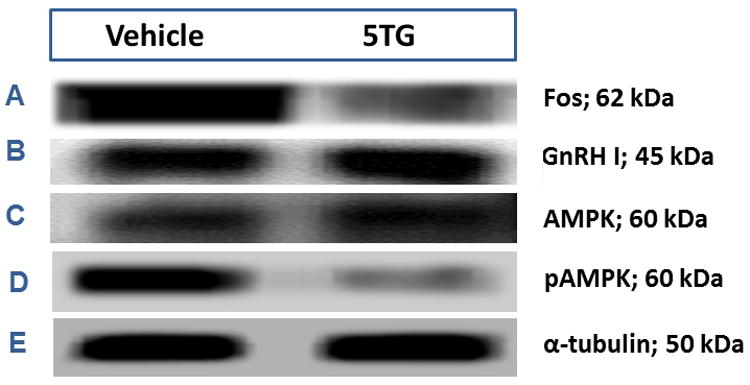
GnRH I precursor-ir neurons were laser-microdissected from 10 μm sections of the rostral preoptic area of OVX+EP rats injected 2 hr earlier with 5TG (200 ug; n=8) or SAL (n=8). For each treatment group, separate lysates of n=50 GnRH cells (n=12-13/rat) were analyzed by Western blotting for Fos (Row A), GnRH (Row B), AMPK (Row C), pAMPK (Row D), or α-tubulin (Row E). For each protein of interest, immunoblot analyses were performed in triplicate.
Figure 10. Effects of CV4 5TG on Normalized GnRH Neuron Protein Levels in OVX+EP Female Rats.
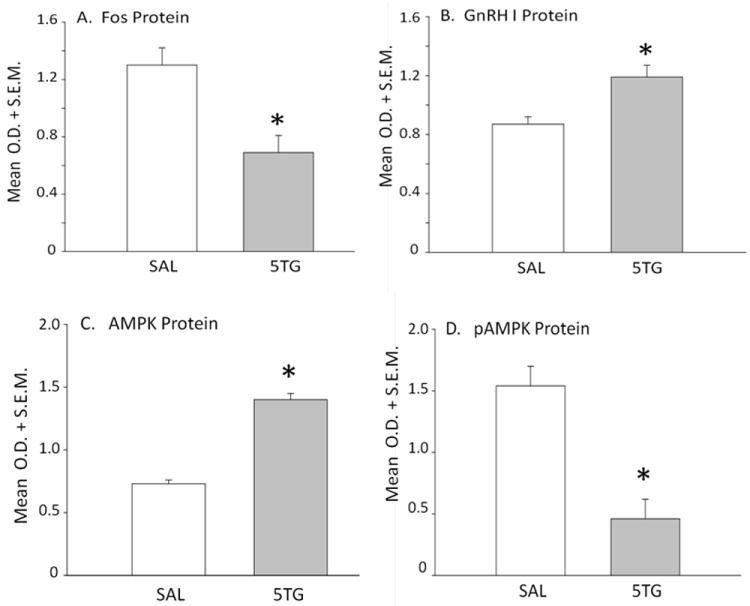
Mean normalized O.D. values of immunoblots (n=50 neurons/treatment group) of Fos (Panel A), GnRH I precursor (Panel B), AMPK (Panel C), and pAMPK (Panel D) from SAL- (white bars) or 5TG- (gray bars) treated rats are shown. *p<0.05, compared to SAL. Data show that 5TG significantly decreased Fos and pAMPK, while increasing GnRH I precursor and AMPK expression in GnRH neurons.
Figure 11. LCM of Rostral Preoptic GnRH Neurons after CV4 5TG or SAL Administration to Steroid-Primed OVX Female Rats.
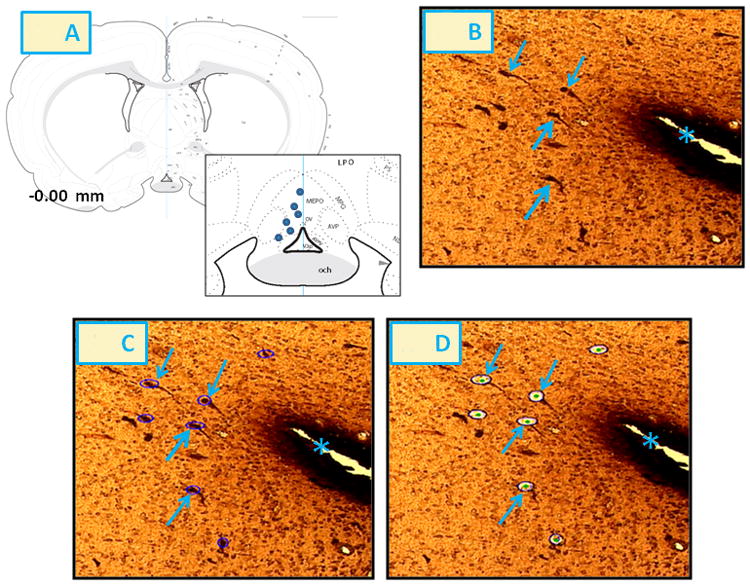
The inset to Panel 11.A depicts the location of harvested GnRH I precursor-ir neurons (blue circles) at -0.00 mm relative to bregma. Blue arrows in Panel 11.B indicate representative rostral preoptic GnRH-ir neurons. The area shown in Panel 11.B was re-photographed after positioning of circular laser cuts (depicted in purple) for circumdissection of individual nerve cell bodies [Panel 11.C] and single-cell ejection by laser-pulse [Panel 11.D]. The asterisk in Panels 11.B-11.D indicates the third ventricle. Abbreviations in Panel 11.A: OV: vascular organ of the lamina terminalis; V3p: third ventricle, preoptic recess; och: optic chiasm; AVPV: anteroventral periventricular nucleus hypothalamus; AVP: anteroventral preoptic nucleus; MEPO: median preoptic nucleus; MPO: medial preoptic area.
Discussion
Female reproductive function is closely controlled by cues of substrate fuel availability, derived chiefly from the caudal hindbrain [Ohkura et al., 2000]. The present studies sought to identify the cellular source of hindbrain signals that restrain the GnRH-pituitary LH axis during pharmacological disruption of glucose catabolism in this area of the brain. Considerable experimental evidence supports hindbrain NE as an obligatory stimulatory signal to the forebrain medial preoptic area for steroid positive-feedback activation of the preovulatory LH surge [Barraclough and Wise, 1982; Herbison, 1997]. The current work extends those findings with new evidence that hindbrain glucoprivation attenuates the LH surge, in part, by suppressing NE input to the anteroventral periventricular nucleus. Our studies show that DVC A2 noradrenergic neurons express estrogen receptors as well as biomarkers for metabolic sensory function [Tamrakar et al., 2012; Ibrahim et al., 2013]. Here, Fos immunocytocytochemical mapping data reveal that A2, but not other medullary catecholamine cell groups respond to intra-CV4 glucose antimetabolite administration, thus implicating the former as a source of glucoprivic-driven patterns of NE neurotransmission. New findings that 5TG decreased A2 DβH enzyme protein levels while increasing Fos expression imply that catecholamine neurotransmitter production may be down-regulated despite net amplification of transcription activity. Hindbrain glucopenia correspondingly reduced and augmented ERα- and ERβ-mediated estrogen input to A2 cells, and diminished A2 AMPK activity despite down-regulated GLUT3 and no change in MCT2 protein profiles. This research also provides unique evidence that preoptic GnRH neurons express AMPK, and that hindbrain glucopenia causes deactivation of this sensor coincident with diminished neuroendocrine stimulation of pituitary LH secretion. Collectively, the present work suggests that hindbrain glucoprivic suppression of the LH surge is mediated, in part, by repressed A2 signaling. Evidence that decreased glucose catabolism in A2 neurons and communication of that metabolic disturbance to GnRH cells correlate with suppressed signaling in each population, despite decreased AMPK activity, suggests that these inhibitory stimuli may override AMPK sensor status in regulating neurotransmission by these cells.
The current studies show that neurotoxin-mediated destruction of DVC catecholamine neurons abolishes the capacity of 5TG to inhibit LH release in steroid-primed OVX female rats, results that demonstrate a critical function for these cells in caudal hindbrain metabolic sensor communication with the reproductive neuroendocrine axis. DVC adrenergic C2 neurons are unlikely to be involved in hindbrain glucoprivic suppression of the LH surge, as transcriptional activity of these cells was unaltered by glucose anti-metabolite treatment, which implies a lack of response to local glucopenia. Brainstem noradrenergic innervation of the preoptic area is derived from A1 and A2 cell groups [Fernandez-Galaz et al., 1994]. Disruption of A2 input to the preoptic area is likely to impair the LH surge as preoptic-projecting A2, but not A1 neurons undergo transcriptional activation in response to steroid positive-feedback [Szawka et al., 2013]. The present work documents changes in genomic activity in A2, but not A1 cells after CV4 glucose anti-metabolite administration to steroid-primed OVX female rats, suggesting that hindbrain glucoprivic modifications in NE input to preoptic and other projection sites, after onset of the LH surge, involve adjustments in A2 signaling. These results are consistent with earlier evidence of A2 nerve cell reactivity to glucopenia [Briski and Marshall, 2000; Briski et al., 2009; Koshy Cherian and Briski, 2011]. The corollary notion that glucopenia inhibits A2 neurotransmission is bolstered by our Western blot evidence for down-regulated levels of the catecholamine biosynthetic enzyme, DβH, in these cells post-5TG treatment. A plausible outcome of this reduced protein profile is that synaptic vesicular norepinephrine content is decreased, which corresponds, in turn, to diminished neurotransmitter release. However, the possibility that neurotransmitter storage, but not release is decreased due to a decline in catecholamine production cannot be discounted. Our data differ from reports of elevated A2 DβH gene expression following systemic glucose antimetabolite treatment [Li et al., 2006]. This discrepancy could reflect, in part, 1) sex or more specifically, estrogen-dependent differences in A2 cell reactivity to glucopenia; 2) dissimilar, e.g. intraventricular versus subcutaneous routes of anti-metabolite administration, which would elicit localized versus body-wide glucopenia; and/or 3) divergent effects of impaired A2 cell glucose catabolism on DβH protein versus mRNA expression. Our Western blot analyses extend immunocytochemical evidence for expanded co-labeling of A2 neurons for Fos in 5TG-treated rats with new evidence that this treatment enhances Fos protein expression in pooled A2 cells, results that reflect transcriptional augmentation. Further studies are needed to identify gene products, in addition to DβH, that are up- or down-regulated as a result of this heightened genomic activity and to determine consequential effects on A2 cell function.
NE release into the medial preoptic area is causally related to expression of the LH surge [Honma and Wuttke, 1980; Demling et al., 1985; Mohankumar et al., 1994; Szawka et al., 2013]. Here, an alpha1-adrenergic receptor agonist pretreatment strategy was used to determine if diminished receptor activation in this site is germane to hindbrain glucoprivic repression of the GnRH-pituitary LH axis. Our studies show that ME delivery into the AVPV attenuated 5TG-induced inhibition of both GnRH neuron genomic activation and LH secretion in steroid-primed OVX rats, suggesting that disrupted NE action on AVPV substrates is a key element of hindbrain metabolic sensor regulation of the LH surge. Further studies are needed to determine how the AVPV relays hindbrain glucoprivic signals to the reproductive neuroendocrine axis. AVPV kisspeptinergic cells plausibly fulfill this function as GnRH neurons express receptors for this neurotransmitter [Messager et al., 2005], which mediates steroid positive-feedback induction of the LH surge [Smith et al., 2006; Dungan et al., 2007] and is subject to metabolic control [Kalamatianos et al., 2008; Castellano et al., 2009]. A2 neurons project to the vicinity of rostral preoptic GnRH neurons in mice [Campbell and Herbison, 2007], but our data imply that during hindbrain glucoprivation, adrenergic receptor stimulation in that site influences neither GnRH activation state nor LH output.
A2 cells are likely direct substrates for steroid regulatory actions as they express estrogen receptor-alpha (ERα) and -beta (ERβ) mRNAs and proteins [Tamrakar et al., 2012; Ibrahim et al., 2013]. A2 tyrosine hydroxylase gene expression and Fos immunolabeling [Jennes et al., 1992; Curran-Rauhut and Petersen, 2004] are up-regulated in response to estrogen positive-feedback. ERα is implicated in positive-feedback induction of the LH surge, as ERα knockdown prevents this episodic burst of hormone release [Wintermantel et al., 2006; Christian et al., 2008]. The present data demonstrate that hindbrain glucoprivation alters A2 ERα, ERβ, and PR protein expression, exerting divergent effects on ERα and ERβ protein profiles in these cells. Recent work also implicates progesterone in estrogen positive-feedback action on the GnRH-pituitary LH axis [Micevych and Sinchak, 2011]. Ongoing studies seek to determine if glucoprivic inhibition of A2 nerve cell signaling involves reductions in ERα- and PR- and/or augmented ERβ-mediated hormonal signaling to A2 neurons. Observed differences in ERα versus ERβ N-terminal domains support the possibility that these variants exert dissimilar effects on A2 cell functions. As ERα and ERβ enhance or inhibit, respectively, catecholamine enzyme gene expression in vitro [Maharjan et al., 2005], it is not inconceivable that these variants elicit divergent responses from A2 neurons in vivo. We speculate that glucoprivic up-regulation of ERβ, alone or relative to ERα, may allow rapid superimposition of inhibitory effects of that metabolic disturbance on steroid positive-feedback activation of A2 cells. Estradiol exerts neuro-protective actions that prevent injury to vulnerable CNS structures from bio-energetic insults, including stimulation of energy metabolism pathways [Nilsen and Brinton, 2004]. In the brain, this hormone stimulates key glycolytic, tricarboxylic acid cycle, and respiratory chain enzyme activity [Kostanyan and Nazaryan, 1992; Nilsen et al., 2007; Chen et al., 2009] and oxidative respiration [Irwin et al., 2008]. Our studies show that estradiol up-regulates substrate fuel catabolic pathways [Tamrakar et al., 2012] and preserves energy stability, e.g. prevents AMPK activation in A2 cells during hypoglycemia [Koshy Cherian and Briski, 2012]. Further research is needed to characterize the molecular mechanisms that mediate glucoprivic adjustments in ER isoform protein profiles. It would also be informative to learn how changes in ERα- versus ERβ-mediated hormone input control adaptive cellular responses to energetic imbalance due to diminished glucose catabolism, and if protective actions of estrogen on A2 cells are guided by direction, magnitude, and/or temporal alterations in ERβ protein expression alone or relative to changes in ERα.
A2 AMPK sensor activity decreased after 5TG treatment of steroid-primed OVX rats, data that align with evidence for diminished pAMPK expression in these cells in hypoglycemic estrogen-treated OVX animals [Koshy Cherian and Briski, 2012]. One explanation for the present outcome is that over the interval between drug treatment and cell sampling, cellular compensation for pharmacologic impairment of glycolysis, including adjustments in energy yield and expenditure, may elicit a more positive energy state. Verification of this assumption awaits development of analytical methods of requisite sensitivity for measurement of AMP/ATP levels in single neurons in vivo; such techniques would provide critical insight on energy production capabilities of A2 cells during metabolic stress. Alternatively, 5TG may deactivate A2 AMPK despite ATP shortage by increasing cell sensitivity to one or more neural or endocrine signals that inhibit enzyme activity. Our evidence that the AMP mimic, 5-aminoimidazole-4-carboxamide-riboside (AICAR), increases A2 pAMPK expression in the presence of estrogen [Ibrahim et al., 2013] supports the view that the present decline in AMPK activity may indeed reflect a reduction in AMP/ATP ratio. A2 neurons from 5TG-treated rats exhibited a significant decrease in GLUT3 transporter protein levels, indicative of decreased glucose catabolism by glycolysis; however, neither MCT2 nor GLUT4 profiles were changed, indicating that this treatment did not likely alter either lactate internalization or this transporter-specific means of glucose uptake. Ongoing studies seek to determine how utilization of these substrates and alternative fuels, e.g. fatty acids, may be affected by experimental manipulation of glycolysis, as performed here, and if changes are estrogen-dependent. Interestingly, A2 DβH and pAMPK protein profiles were both decreased after glucose anti-metabolite treatment. This concurrence may reflect abatement of neurotransmitter production, despite positive energy balance, due to non-AMPK – generated signals of metabolic imbalance such as decline in glycolytic pathway product yield owing to pharmacological suppression of glucose catabolism, or to prioritized energy partitioning that favors alternative uses of available energy. If catecholamine biosynthesis is indeed sensitive to multiple metabolic indicators, neurotransmitter output may thus serve as a more accurate reflection of net shifts in A2 energy supply versus demand equilibrium than AMPK activity alone. In light of evidence that A2 neurons are responsive to steroid positive-feedback [Jennes et al., 1992; Curran-Rauhut and Petersen, 2004] and engage in metabolic sensing [Brisk et al., 2009; Koshy Cherian and Briski, 2011; 2012], our findings that hindbrain glucopenia regulates markers of A2 nerve cell function support the view that these hindbrain neurons may function as a common pathway for opposing steroidal stimulatory versus metabolic inhibitory input to the GnRH-pituitary LH axis. The present research is thus significant for advancing the notion that A2 cells are able to control the LH surge according to prevailing energy status, namely to inhibit that critical event in response to acute reductions in substrate fuel-derived energy. This work reveals divergent effects of 5TG on A2 DβH and Fos protein expression. Augmented A2 transcriptional activity, denoted by Fos protein up-regulation, alongside diminished catecholamine biosynthesis may reflect metabolic functional adaptations designed to counteract pharmacologic suppression of glycolysis and/or to divert energy to activities unassociated with synaptic firing.
Western blot data demonstrating hindbrain glucoprivic inhibition of Fos protein expression by GnRH neurons from steroid-primed OVX rats confirms prior reports of reduced co-labeling of these cells for For-ir [Briski and Sylvester, 1998; Singh and Briski, 2004]. The current work provides novel evidence for AMPK and pAMPK expression in vivo by rostral preoptic GnRH neurons, revealing that these proteins are up- and downregulated, respectively, by hindbrain glucoprivation. Previously, extracellular recordings demonstrated a decline in GnRH nerve cell firing rates upon brain slice incubation with AICAR, data that demonstrated a negative correlation between AMPK activation and GnRH neurotransmission in vitro [Roland and Moenter, 2011]. Here, GnRH nerve cell sensor deactivation occurred paradoxically at a time point coinciding with reduced pituitary LH secretion [an indirect indicator of suppressed GnRH neuropeptide signaling to the pituitary] in 5TG-treated rats. This reduction in GnRH pAMPK expression may reflect, in part, augmented cellular glucose uptake and metabolism, as pharmacological induction of hindbrain glucopenia elevates blood glucose levels and those processes are likely unimpeded by 5TG as GnRH cells reside far from the caudal hindbrain drug delivery site. Anti-metabolite – induced hyperglycemia may indirectly suppress activity of this sensor by enhancing input to GnRH neurons by central or peripheral stimuli that convey a status of ample energy. GnRH I precursor protein levels in GnRH neurons were elevated in 5TG-treated rats, results likely reflect a reduction in post-translational processing, e.g. cleavage, and/or axonal transport of this parent molecule, leading to decreased neurotransmitter release. This scenario is consistent with the measured decline in LH release. As we did not investigate rates of amino acid incorporation rate into GnRH-I in the presence versus absence of 5TG, we cannot speculate on whether synthesis of this precursor is regulated by hindbrain glucoprivation. Taken together, the present studies support the novel concept that negative glucoprivic signaling from hindbrain metabolic sensors to GnRH neurons can suppress neuropeptide outflow to the pituitary, despite concurrent intracellular sensor detection of a more positive internal metabolic state. Thus, it is plausible that signals of metabolic deficiency, of either internal or external origin, may supersede or negate simultaneous signals of metabolic abundance in regulating GnRH neuroendocrine signaling. The current study highlights a potential critical caveat of glucose anti-metabolite treatment for experimental disruption of local or systemic metabolic stasis, namely effects of secondary hyperglycemia on body-wide metabolic sensors.
In summary, the current studies demonstrate that hindbrain glucoprivation suppresses the LH surge in steroid-primed OVX female rats, in part, by reducing noradrenergic input to the preoptic AVPV, and this work implicates DVC A2 neurons as a source of this metabolic inhibitory signal [Figure 12]. Ongoing efforts to identify AVPV substrates that convey glucoprivic input to GnRH neurons are needed. Present results also suggest that AMPK deactivation, indicative of an intracellular state of increased positive metabolic balance, does not override inhibitory effects of pharmacological suppression of caudal hindbrain glucose catabolism or its physiological sequelae on A2 cell function or of afferent hindbrain glucoprivic signals on GnRH neurons. Further studies are needed to determine if decreased AMPK activation in these cell populations reflects compensatory gain in positive energy balance and/or direct effects of estrogen or other indicators of metabolic status on enzyme activity.
Figure 12. Medullary-Preoptic Circuitry Mediating Glucoprivic Regulatory Input to the Reproductive Neuroendocrine Axis.
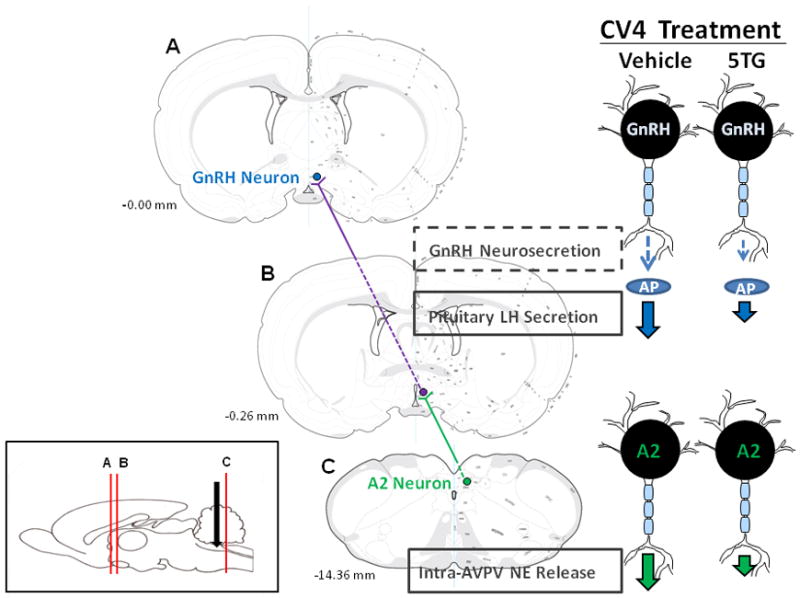
Maps of coronal brain sections A (-0.00 mm), B (-0.26 mm), and C (-14.36 mm) depict key elements in the ascending pathway that links hindbrain metabolo-sensory and preoptic neuroendocrine motor neurons, the sole conduit of neural outflow to pituitary gonadotropes. The sagittal rat brain image in the lower left corner illustrates rostrocaudal levels of sections A-C (vertical red lines) and caudal fourth ventricular cannula implantation site (black arrow) used for localized induction of neuroglucopenia by glucose antimetabolite administration. A2 neurons are estrogen- (they express classical estrogen receptors, e.g. -α and −β, and are a source of steroid feedback-driven norepinephrine signaling to the reproductive neuroendocrine axis) as well as glucoprivic-sensitive. Among hindbrain catecholamine cell groups, dorsal vagal complex A2 cells (green circle; section C) are singularly sensitive to hindbrain neuroglucopenia in steroid-primed OVX female rats, resulting in diminished AVPV alpha1 adrenergic receptor activation (section B) and pituitary LH output volume (right of section C). Current research supports the notion of A2 neurons are a common substrate for opposing steroid activational and glucoprivic effects on LH. The current studies did not assess hypothalamic GnRH release during hindbrain glucoprivation, but evidence for 5TG-induced truncation of the LH surge provides plausible indirect evidence for inhibition of this hypophysiotropic signal to pituitary gonadotropes. Comprehensive understanding of the metabolic regulatory circuitry revealed here will require insight on connectivity between glucoprivic-sensitive AVPV substrates and rostral preoptic GnRH neurons (blue circle; section C).
Highlights.
Glucoprivic cues from the caudal hindbrain restrain the preovulatory LH surge
Hindbrain glucoprivation altered Fos expression in A2, but not other catecholamine cell groups
Intra-preoptic methoxamine reversed glucoprivic effects on GnRH neuron Fos labeling and LH release
5TG stimulated Fos, but inhibited DBH protein profiles in A2 cells
Hindbrain glucoprivation modified GnRH neuron AMPK and pAMPK protein expression.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Balfour RH, Hansen AMK, Trapp S. Neuronal responses to transient hypoglycaemia in the dorsal vagal complex of the rat brainstem. J Physiol. 2006;570:469–484. doi: 10.1113/jphysiol.2005.098822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barraclough CA, Wise PM. The role of catecholamines in the regulation of pituitary luteinizing hormone and follicle stimulating hormone secretion. Endocrine Reviews. 1982;3:91–119. doi: 10.1210/edrv-3-1-91. [DOI] [PubMed] [Google Scholar]
- Briski KP, Sylvester PW. Effects of specific acute stressors on LH release in ovariectomized and ovariectomized estrogen-treated female rats. Neuroendocrinology. 1988;47:194–202. doi: 10.1159/000124913. [DOI] [PubMed] [Google Scholar]
- Briski KP, Vogel KL. Role of endogenous opioid peptides in central glucocorticoid receptor (GR) - induced decreases in circulating LH. Neuropeptides. 1995;28:175–181. doi: 10.1016/0143-4179(95)90113-2. [DOI] [PubMed] [Google Scholar]
- Briski KP, Sylvester PW. Effects of the glucose antimetabolite, 2-deoxy-D-glucose (2-DG), on the LH surge and Fos expression by preoptic GnRH neurons in ovariectomized, steroid-primed rats. J Neuroendocrinol. 1998;10:769–776. doi: 10.1046/j.1365-2826.1998.00262.x. [DOI] [PubMed] [Google Scholar]
- Briski KP, Marshall ES. Caudal brainstem Fos expression is restricted to periventricular catecholaminergic loci following intraventricular administration of 2-deoxy-D-glucose. Exper Brain Res. 2000;133:547–551. doi: 10.1007/s002210000448. [DOI] [PubMed] [Google Scholar]
- Briski KP, Marshall ES, Sylvester PW. Effects of estradiol on glucoprivic transactivation of catecholaminergic neurons in the female rat caudal brainstem. Neuroendocrinology. 2001;73(6):369–377. doi: 10.1159/000054655. [DOI] [PubMed] [Google Scholar]
- Briski KP, Kale AY, Vavaiya KV. Impact of recurring insulin-induced hypo-glycemia on corticotropin-releasing hormone, oxytocin, vasopressin, and glucokinase gene expression in hypothalamic paraventricular nucleus: role of type II glucocorticoid receptors. Exper Brain Res. 2009;195:499–507. doi: 10.1007/s00221-009-1787-4. [DOI] [PubMed] [Google Scholar]
- Briski KP. Induction of cFos expression in hypothalamic dopaminergic neurons by the glucose antimetabolite, 2-deoxy-D-glucose: site-specific modulation by estradiol. NeuroReport. 1997;9:289–295. [Google Scholar]
- Briski KP, Koshy Cherian A, Genabai NK, Vavaiya KV. In situ coexpression of glucose and monocarboxylate transporter mRNAs in metabolic-sensitive dorsal vagal complex catecholaminergic neurons: transcriptional reactivity to insulin-induced hypoglycemia (IIH) and caudal hindbrain glucose or lactate repletion during IIH. Neuroscience. 2009;164:1152–1160. doi: 10.1016/j.neuroscience.2009.08.074. [DOI] [PubMed] [Google Scholar]
- Campbell RE, Herbison AE. Definition of brainstem afferents to gonadotropin-releasing hormone neurons in the mouse using conditional viral tract tracing. Endocrinology. 2007;148:5884–5890. doi: 10.1210/en.2007-0854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castellano JM, Roa J, Luque RM, Diequez C, Aguilar E, Pinilla A, Tena-Sempere M. KiSS- 1/kisspeptins and the metabolic control of reproduction: physiologic roles and putative physiopathological implications. Peptides. 2009;30:139–145. doi: 10.1016/j.peptides.2008.06.007. [DOI] [PubMed] [Google Scholar]
- Chen JQ, Brown TR, Russo J. Regulation of energy metabolism pathways by estrogens and estrogenic chemicals and potential implications in obesity associated with increased exposure to endocrine disruptors. Biochim Biophys Acta. 2009;1793:1128–1143. doi: 10.1016/j.bbamcr.2009.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen MD, O’Byrne KT, Chiappini SE, Hotchkiss J, Knobil E. Hypoglycemia ‘stress’ and gonadotropin-releasing hormone pulse generator activity in the rhesus monkey: role of the ovary. Neuroendocrinology. 1992;56:666–673. doi: 10.1159/000126291. [DOI] [PubMed] [Google Scholar]
- Christian CA, Glidewell-Kenney C, Jameson JL, Moenter SM. Classical estrogen receptor alpha signaling mediates negative and positive feedback on gonadotropin-releasing hormone neuron firing. Endocrinology. 2008;149:5328–5334. doi: 10.1210/en.2008-0520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crosignani PG. Nutrition and reproduction in women. The ESHRE Capri Workshop. Human Reproduction Update. 2006;12:193–207. doi: 10.1093/humupd/dmk003. [DOI] [PubMed] [Google Scholar]
- Coyral-Castel S, Tosca L, Ferreira G, Jeanpierre E, Rame C, Lomet D, Caraty A, Monget P, Chabrolle C, Dupont J. the effect of AMP-activated kinase activation on gonadotrophin-releasing hormone secretion in GT1-7 cells and its potential role in hypothalamic regulation of the oestrous cyclicity in rats. J Neuroendocrinol. 2008;20:335–346. doi: 10.1111/j.1365-2826.2007.01643.x. [DOI] [PubMed] [Google Scholar]
- Curran-Rauhut MA, Petersen SL. Oestradiol-dependent and -independent modulation of tyrosine hydroxylase mRNA levels in subpopulations of A1 and A2 neurones with oestrogen receptor (ER)α and ERβ gene expression. J Neuroendocrinol. 2003;15:296–303. doi: 10.1046/j.1365-2826.2003.01011.x. [DOI] [PubMed] [Google Scholar]
- Demling J, Fuchs E, Baumert M, Wuttke W. Preoptic catecholamine, GABA, and glutamate release in ovariectomised and ovariectomised estogen-primed rats utilising a push-pull cannula technique. Neuroendocrinology. 1985;41:212–218. doi: 10.1159/000124180. [DOI] [PubMed] [Google Scholar]
- Dungan HM, Gottsch ML, Zeng H, Gragerov A, Bergmann JE, Vassilatis DK, Clifton DK, Steiner RA. The role of kisspeptin-GPR54 signaling in the tonic regulation and surge release of gonadotropin-releasing hormone/luteinizing hormone. J Neurosci. 2007;27:12088–12095. doi: 10.1523/JNEUROSCI.2748-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez-Galaz C, Dyer RG, Herbison AE. Analysis of brainstem A1 and A2 noradrenergic inputs to the preoptic area using microdialysis in the rat. Brain Res. 1994;636:227–232. doi: 10.1016/0006-8993(94)91021-9. [DOI] [PubMed] [Google Scholar]
- Gujar AD, Ibrahim BA, Tamrakar P, Briski KP. Soc Neurosci. New Orleans, LA: Oct, 2012. Hindbrain nutrient status regulates hypothalamic AMPK activity and metabolic neurotransmitter expression; pp. 13–17. Abst. 284.10/UU12. [Google Scholar]
- Hardie DG. Minireview: The AMP-activated protein kinase cascade: the key sensor of cellular energy status. Endocrinology. 2003;144:5179–5183. doi: 10.1210/en.2003-0982. [DOI] [PubMed] [Google Scholar]
- Haywood SA, Simonian SX, van der Beek EM, Bicknell RJ, Herbison AE. Fluctuating estrogen and progesterone receptor expression in brainstem norepinephrine neurons through the rat estrous cycle. Endocrinology. 1999;1407:3255–3263. doi: 10.1210/endo.140.7.6869. [DOI] [PubMed] [Google Scholar]
- Herbison AE. Noradrenergic regulation of cyclic GnRH secretion. Rev Reprod. 1997;2:1–6. doi: 10.1530/ror.0.0020001. [DOI] [PubMed] [Google Scholar]
- Honma K, Wuttke W. Norepinephrine and dopamine turnover rates in the medial preoptic area and the mediobasal hypothalamus of the rat brain after various endocrinological manipulations. Endocrinology. 1980;106:1848–1853. doi: 10.1210/endo-106-6-1848. [DOI] [PubMed] [Google Scholar]
- Ibrahim BA, Tamrakar P, Gujar AD, Koshy Cherian A, Briski KP. Caudal fourth ventricular AICAR regulates glucose and counterregulatory hormone profiles, dorsal vagal complex metabolo-sensory neuron function, and hypothalamic Fos expression. J Neurosci Res. 2013;91:1226–1238. doi: 10.1002/jnr.23230. [DOI] [PubMed] [Google Scholar]
- Irwin RW, Yao J, Hamilton RT, Cadenas E, Brinton RD, Nilsen J. Progesterone and estrogen regulate oxidative metabolism in brain mitochondria. Endocrinology. 2008;149:3167–3175. doi: 10.1210/en.2007-1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jennes L, Jennes ME, Purvis C, Nees M. c-fos expression in noradrenergic A2 neurons of the rat during the estrous cycle and after steroid hormone treatments. Brain Res. 1992;586:171–175. doi: 10.1016/0006-8993(92)91391-q. [DOI] [PubMed] [Google Scholar]
- Kalamatianos T, Grimshaw SE, Poorun R, Hahn JD, Coen CW. Fasting reduces KiSS-1 expression in the anteroventral periventricular nucleus (AVPV): effects of fasting on the expression of KiSS-1 and neuropeptide Y in the AVPV or arcuate nucleus of female rats. J Neuroendo. 2008;20:1089–1097. doi: 10.1111/j.1365-2826.2008.01757.x. [DOI] [PubMed] [Google Scholar]
- Kale AY, Paranjape SA, Briski KP. Intracerebroventricular administration of the nonsteroidal glucocorticoid receptor antagonist, CP472555, prevents exacerbation of hypo-glycemia during repeated insulin administration. Neuroscience. 2006;140:555–565. doi: 10.1016/j.neuroscience.2006.02.041. [DOI] [PubMed] [Google Scholar]
- Koshy Cherian A, Briski KP. Quantitative real-time RT PCR and high-sensitivity immunoblot analyses of laser-microdissected A2 noradrenergic neurons reveal adaptation of phosphoAMPK, glucokinase, substrate fuel transporter, and dopamine-beta-hydroxylase responses to intermediate insulin-induced hypoglycemia. J Neurosci Res. 2011;89:1114–1124. doi: 10.1002/jnr.22632. [DOI] [PubMed] [Google Scholar]
- Koshy Cherian A, Briski KP. A2 noradrenergic nerve cell metabolic transducer and nutrient transporter adaptation to hypoglycemia: Impact of estrogen. J Neurosci Res. 2012;90:1347–1358. doi: 10.1002/jnr.23032. [DOI] [PubMed] [Google Scholar]
- Kostanyan A, Nazaryan K. Rat brain glycolysis regulation by estradiol-17 beta. Biochim Biophys Acta. 1992;1133:301–3-6. doi: 10.1016/0167-4889(92)90051-c. [DOI] [PubMed] [Google Scholar]
- Li AJ, Wang Q, Ritter S. Differential responsiveness of dopamine-beta-hydroxylase gene expression to glucoprivation in different catecholamine cell groups. Endocrinology. 2006;147:3428–3434. doi: 10.1210/en.2006-0235. [DOI] [PubMed] [Google Scholar]
- Maharjan S, Serova L, Sabban EL. Transcriptional regulation of tyrosine hydroxylase by estrogen: opposite effects with estrogen receptors alpha and beta and interactions with cyclic AMP. J Neurochem. 2005;93:1502–14. doi: 10.1111/j.1471-4159.2005.03142.x. [DOI] [PubMed] [Google Scholar]
- Messager S, Chatzidaki EE, Ma D, Hendrick AG, Zahn D, Dixon J, Thresher RR, Malinge I, Lomet D, Carlton MB, Colledge WH, Caraty A, Aparicio SA. Kisspeptin directly stimulates gonadotropin-releasing hormone release via G protein-coupled receptor 54. Proc Natl Acad Sci. 2005;102:1761–1776. doi: 10.1073/pnas.0409330102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Micevych P, Sinchak K. The Neurosteroid Progesterone Underlies Estrogen Positive Feedback of the LH Surge. Front Endocrinol. 2011 doi: 10.3389/fendo.2011.00090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizuno Y, Oomura Y. Glucose responding neurons in the nucleus tractus solitarius of the rat: in vitro study. Brain Res. 1984;307:109–116. doi: 10.1016/0006-8993(84)90466-9. [DOI] [PubMed] [Google Scholar]
- Mohankumar PS, Thyagarajan S, Quadri SK. Correlations of catecholamine release in the medial preoptic area with proestrous surge. Endocrinology. 1994;135:119–126. doi: 10.1210/endo.135.1.8013343. [DOI] [PubMed] [Google Scholar]
- Nagatani S, Bucholtz DC, Murahashi K, Estacio MAC, Tsukamura H, Foster DL, Maeda MI. Reduction of glucose availability suppresses pulsatile luteinizing hormone release in female and male rats. Endocrinology. 1996;137:1166–1170. doi: 10.1210/endo.137.4.8625885. [DOI] [PubMed] [Google Scholar]
- Nilsen J, Brinton RD. Mitochondria as therapeutic targets of estrogen action in the central nervous system. Curr Drug Targets – CNS & Neurol Disorders. 2004;3:297–313. doi: 10.2174/1568007043337193. [DOI] [PubMed] [Google Scholar]
- Nilsen J, Irwin RW, Gallaher TK, Brinton RD. Estadiol in vivo regulation of brain mitochondrial proteome. J Neurosci. 2007;27:14069–14077. doi: 10.1523/JNEUROSCI.4391-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohkura S, Tanaka T, Nagatani S, Bucholtz DC, Tsukamura H, Maeda K, Foster DL. Central, but not peripheral, glucose-sensing mechanisms mediate glucoprivic suppression of pulsatile luteinizing hormone secretion in the sheep. Endocrinology. 2000;141:4472–4480. doi: 10.1210/endo.141.12.7853. [DOI] [PubMed] [Google Scholar]
- Oomura Y, Yoshimatsu H. Neural network of glucose monitoring system. J Auton Nerv Syst. 1984;10:359–372. doi: 10.1016/0165-1838(84)90033-x. [DOI] [PubMed] [Google Scholar]
- Pacák K, Palkovits M. Stressor specificity of central neuroendocrine responses: implications for stress-related disorders. Endocr Rev. 2001;22:502–548. doi: 10.1210/edrv.22.4.0436. [DOI] [PubMed] [Google Scholar]
- Roland AV, Moenter SM. Regulation of gonadotropin-releasing hormone neurons by glucose. Trends Endocrinol Metab. 2011;22:443–449. doi: 10.1016/j.tem.2011.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ronnett GV, Ramamurthy S, Kleman AM, Landree LE, Aja S. AMPK in the brain: its roles in energy balance and neuroprotection. J Neurochem. 2009;109:17–23. doi: 10.1111/j.1471-4159.2009.05916.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silver IA, Erecinska M. Glucose-induced intracellular ion changes in sugar-sensitive hypothalamic neurons. J Neurophysiol. 1998;79:1733–1745. doi: 10.1152/jn.1998.79.4.1733. [DOI] [PubMed] [Google Scholar]
- Singh SR, Briski KP. Septopreoptic mu opioid receptor mediation of hindbrain gluco-privic inhibition of reproductive neuroendocrine function in the female rat. Endocrinology. 2004;145:5322–5331. doi: 10.1210/en.2004-0130. [DOI] [PubMed] [Google Scholar]
- Smith JT, Clifton DK, Steiner RA. Regulation of the neuroendocrine reproductive axis by kisspeptin- GPR54 signaling. Reproduction. 2006;131:623–630. doi: 10.1530/rep.1.00368. [DOI] [PubMed] [Google Scholar]
- Szawka RE, Poletini MO, Leite CM, Bernuci MP, Kalil BK, Mendonca LBD, Carolino ROG, Helena CVV, Bertram R, Franci CR, Anselmo-Franci JA. Release of norepinephrine in the preoptic area activates anteroventral periventricular nucleus neurons and stimulates the surge of luteinizing hormone. Endocrinology. 2013;154:363–374. doi: 10.1210/en.2012-1302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamrakar P, Ibrahim BA, Briski KP. Soc Neurosci. New Orleans, LA: Oct, 2012. Estradiol is neuroprotective of hindbrain A2 noradrenergic metabolic sensory neurons during insulin-induced hypoglycemia; pp. 13–17. Abst. 903.05/XX6. [Google Scholar]
- Vetrivelan R, Mallick HN, Kumar VM. Unmasking of alpha1 adrenoreceptor induced hypnogenic response from medial preoptic area. Physiol Behav. 2005;84:641–650. doi: 10.1016/j.physbeh.2005.02.013. [DOI] [PubMed] [Google Scholar]
- Vetrivelan R, Mallick HN, Kumar VM. Sleep induction and temperature lowering by medial preoptic alpha(1) adrenergic receptors. Physiol Behav. 2006;87:707–713. doi: 10.1016/j.physbeh.2006.01.015. [DOI] [PubMed] [Google Scholar]
- Wade GN, Jones JE. Neuroendocrinology of nutritional infertility. Amer J Physiol. 2004;287:R1227–1296. doi: 10.1152/ajpregu.00475.2004. [DOI] [PubMed] [Google Scholar]
- Wade GN, Schneider JE. Metabolic fuels and reproduction in female mammals. Neurosci Biobehav Rev. 1992;16:235–272. doi: 10.1016/s0149-7634(05)80183-6. [DOI] [PubMed] [Google Scholar]
- van den Buuse M, Versteeg DH, de Jong W. Role of dopamine in the development of spontaneous hypertension. Hypertension. 1984;6:899–905. doi: 10.1161/01.hyp.6.6.899. [DOI] [PubMed] [Google Scholar]
- Wen JP, Lu WS, Yang J, Nie AF, Cheng XB, Yang Y, Ge Y, Li XY, Ning G. Globular adiponectin inhibits GnRH secretion from GT1-7 hypothalamic GnRH neurons by induction of hyperpolarization of membrane potential. Biochem Biophys Res Commun. 2008;371:756–761. doi: 10.1016/j.bbrc.2008.04.146. [DOI] [PubMed] [Google Scholar]
- Wintermantel TM, Campbell RE, Porteous R, Bock D, Gröne HJ, Todman MG, Korach KS, Greiner E, Pérez CA, Schülz G, Herbison AE. Definition of estrogen receptor pathway critical for estrogen positive feedback to gonadotropin-releasing hormone neurons and fertility. Neuron. 2006;52:271–280. doi: 10.1016/j.neuron.2006.07.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright DE, Jennes L. Origin of noradrenergic projections to GnRH perikarya-containing areas in the medial septum-diagonal band and preoptic area. Brain Res. 1993;621:27u–278. doi: 10.1016/0006-8993(93)90116-5. [DOI] [PubMed] [Google Scholar]
- Zhang C, Bosch M, Levine JE, Ronnekleiv O, Kelly M. GnRH neurons express KATP channels that are regulated by estrogen and responsive to glucose and metabolic inhibition. J Neurosci. 2007;27:10153–10164. doi: 10.1523/JNEUROSCI.1657-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]