

Abstract
Background and Objectives
We evaluated the infusion accuracy and device-related safety of implantable drug infusion pumps in subjects with chronic pain or severe spasticity.
Methods
Nine centers in the United States enrolled patients receiving intrathecal drug delivery systems to manage chronic pain and/or severe spasticity. Infusion accuracy was assessed at 6 and 12 months by comparing syringe-measured delivered volumes to programmer-predicted volumes. Safety was evaluated through analysis of adverse events. Separate laboratory testing conducted by the manufacturer also evaluated infusion accuracy.
Results
Eighty of 82 enrolled subjects were implanted. Sixty-five and 54 subjects, respectively, were analyzable for accuracy at 6 and 12 months. On average at 6 months, the pumps were measured to have delivered 1% more than the programmed delivery volume. Analyzed on a per-refill basis, the pumps delivered, on average, 2.5% more than the programmed delivery volume. Differences between per-refill means versus per-subject cumulative means were due to limitations in clinicians’ ability to precisely visualize single small syringe-volume differences, or possibly incomplete withdrawal of fluid from the pump. Laboratory testing demonstrated a per-refill mean accuracy error of minus 2.4%. Because average observed flow-rate error at 6 and 12 months (1% overinfusion) was derived from pump residual volume measurements by syringe and carried out in a clinical setting, clinical volume ratios were larger than direct volume measurements by weight observed in the laboratory. No deaths, permanent injuries, or unanticipated adverse device effects occurred.
Conclusions
The pump accurately delivered intrathecal medication in the clinical setting of this study. Adverse events were similar in nature and severity to those described in the product labeling and literature.
Implantable drug delivery systems have been used to treat chronic pain and spasticity for 3 decades. 1 Approved drugs for continuous intrathecal administration include preservative-free morphine sulfate 2 and ziconotide for chronic pain, 3 and baclofen for severe spasticity. 4–9 Programmable pumps, such as the SynchroMed (Medtronic, Inc., Minneapolis, Minnesota) devices, permit dosage adjustments by increasing or decreasing the flow rate and/or by altering the mode of administration (continuous mode, scheduled boluses, and/or step-function dosing), as required by an individual patient.
Infusion systems must deliver accurate and steady dosing, or patients will experience adverse effects related to overdosing or underdosing. In some cases, the effects can be fatal or life-threatening. 10 Clinicians evaluate a pump’s drug-dispensing accuracy by comparing the actual (measured) volume, inferred from the volume placed initially in the pump reservoir minus the volume of drug withdrawn from the reservoir, to the predicted volume of drug delivered since the previous refill to the current refill session. A volume discrepancy outside the labeled accuracy specification—too large or too small infused volume—may suggest a pump (or less commonly, catheter) malfunction.
The investigators performed a prospective clinical study to determine the flow-rate accuracy and safety of the SynchroMed II pump over 12 months. Accuracy calculations were based on clinician measurements of drug reservoir residual volumes by syringe—a method that replicated routine medical practice. The manufacturer also conducted separate laboratory testing with 23 SynchroMed II pumps to establish the benchmark device flow-rate accuracy under ideal, controlled conditions. The bench data allowed us to compare true device errors (genuine overinfusion or underinfusion) versus variations in technique or errors in clinician volume measurements.
METHODS
The US Food and Drug Administration and institutional review boards approved the clinical investigational plan before study initiation. All patients or their legal guardians provided written informed consent. The trial was sponsored by the manufacturer and registered as NCT00773019 at clinicaltrials.gov on August 21, 2008. The study was conducted using commercially available pumps and refill kits as a condition of approval required by the US Food and Drug Administration.
Devices under study were the SynchroMed II Model 8637 20- and 40-mL pumps (Medtronic, Inc, Minneapolis, Minnesota). Eligible subjects were at least 18 years old, were willing to return for follow-up visits, met labeled indications with no contraindications, and received their initial or replacement system for infusion of intrathecal medication to treat severe spasticity or chronic pain. Subjects who received a replacement system were allowed to use their existing catheter or have a new one implanted at the time of pump replacement surgery. Subjects were excluded if they had an ongoing infection, insufficient body mass to accept the pum, a condition in which the pump could not be implanted close enough to the skin surface to ensure proper telemetry communication, or a life expectancy shorter than 1 year. Subjects were required to return for refill visits (at minimum) at 1, 6, and 12 months after implantation. If additional refills were needed, data from those visits also were collected. Each site’s principal investigator was a physician responsible for all study activities who ensured that any delegated study-specific activities were performed by individuals qualified by education and training. Pump flow rates and drug dosages were prescribed by the study physicians and allowed to be changed as needed to meet therapeutic needs. Adverse events (AEs) were assessed at all scheduled and unscheduled visits.
Accuracy
Drug residual volumes were obtained using the syringe supplied in the commercially available refill kit. The primary outcome variable was each subject’s cumulative 6-month accuracy ratio, calculated as the ratio of the aggregate clinic-measured volume of drug dispensed divided by the aggregate pump calculated volume dispensed over the consecutive refill sessions through 6 months (Eq. 1). Accuracy ratios were also calculated through 12 months.
Eq. 1. Primary End Point: Per Subject Accuracy Ratio at 6 Months
Formula.

The end point required that more than 90% of subjects have an accuracy ratio between 0.75 and 1.25. Using a 1-sample binomial test with p 0 = 0.1 versus p 1 = 0.025, a 1-sided significance level of 0.05 and at least 80% power, a minimum sample size of 61 subjects was required. An accuracy ratio of 1.00 would indicate that the observed (clinic-measured) volume dispensed matched the pump-calculated volume exactly and would correspond to a 100% measured-to-calculated ratio and a 0% flow-rate error. Using the same 6-month dataset, an additional per-refill analysis was carried out on the clinic data using Eq. 2.
Eq. 2. Additional Analysis: Per-Refill Accuracy Ratio
Formula.
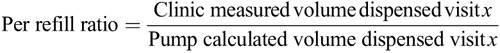
Laboratory testing of 23 separate SynchroMed II pumps for 4 consecutive days compared the actual output compared with the pump calculated output using Eq. 3, at various infusion rates (0.1, 0.3, 0.5, and 0.7 mL/d) and reservoir volumes (full, half-full, and low [1–2 mL residual volume]).A mix of 20- and 40-mL pumps were tested at body temperature (37°C ±1°C) using an analytical balance (Model HM-300, A&D Company, Tokyo, Japan) which measures to 0.0001 g to determine the infused volume of sterile water for injection by weight.
Eq. 3. Laboratory Testing: Accuracy Error
Formula.

Safety
Investigators reported all AEs during the 12-month study, categorizing each as either device system related or non–system related. System-related AEs included those related to the pump, catheter, incisional sites, programming/refill procedures, or surgery/anesthesia. Non–system-related events included those related to the intrathecal drug(s), concomitant medications, and new illnesses or worsening of preexisting conditions. An AE was categorized as serious if it resulted in inpatient hospitalization, prolongation of existing hospitalization, a life-threatening situation (immediate risk of death), persistent or significant disability or incapacity, permanent impairment of body function or permanent damage to a body structure, necessity for medical or surgical intervention to preclude permanent impairment of a body function or permanent damage to a body structure, patient death, or a congenital anomaly/birth defect. Terms were standardized using MedDRA (Medical Dictionary for Regulatory Activities Maintenance, version 8.0, Maintenance and Support Services Organization [MSSO], McLean, VA).
RESULTS
Eighty-two patients were enrolled from December 22, 2004, to November 7, 2007. Two subjects discontinued before device implantation, resulting in 80 SynchroMed II infusion pumps implanted at 9 investigational centers (41 receiving 20-mL pumps and 39 receiving 40-mL pumps). Mean subject age was 51.4 years (range, 22–89 years); 44 were male (55%), and 36 were female (45%). Forty-three subjects were being treated for spasticity, 32 for pain, and 5 were treated with intrathecal drugs for both spasticity and pain. Twelve subjects (15%) discontinued participation in the study prior to the 12-month visit. Figure 1 summarizes study compliance and the reasons for subject discontinuation prior to the 12-month visit.
FIGURE 1.
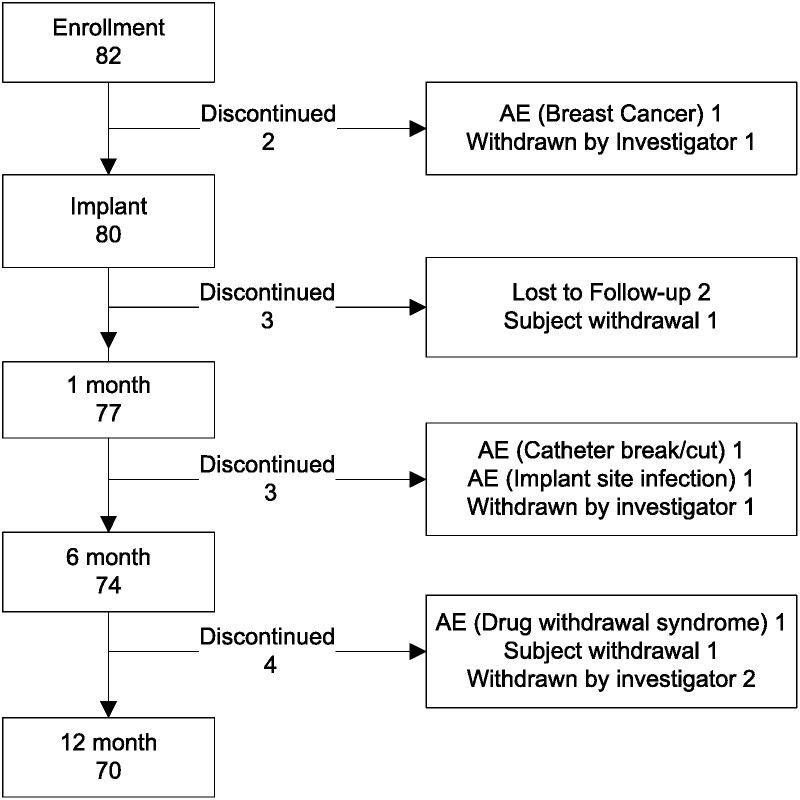
Study compliance.
Flow-Rate Accuracy
Seventy-four subjects completed the 6-month visit, with 65 subjects having sufficient data (193 refills in 65 subjects) for the primary analysis. The mean flow rate for subjects in the primary analysis data set was 0.330 mL/d, with a minimum of 0.048 and maximum of 1.959 (2 of 65 subjects were on flex dosing, and their flow rate was not calculated for this statistic). To be included in the primary analysis, subjects underwent at least 2 refills with no missing refill data elements. Among subjects with incomplete data sets, 2 subjects missed 1 visit each, and 7 who attended all visits were missing a critical data element. A summary of the 6-month per-subject clinical accuracy ratios appears in Figure 2. All ratios were within 0.75 to 1.25 and met the primary success criterion. The mean per-subject accuracy ratio for the 65 subjects was 1.01 (SD, 0.05; 101% of the programmed volume) for a plus 1% error at 6 months. The 12-month results in 54 subjects with complete data were similar.
FIGURE 2.
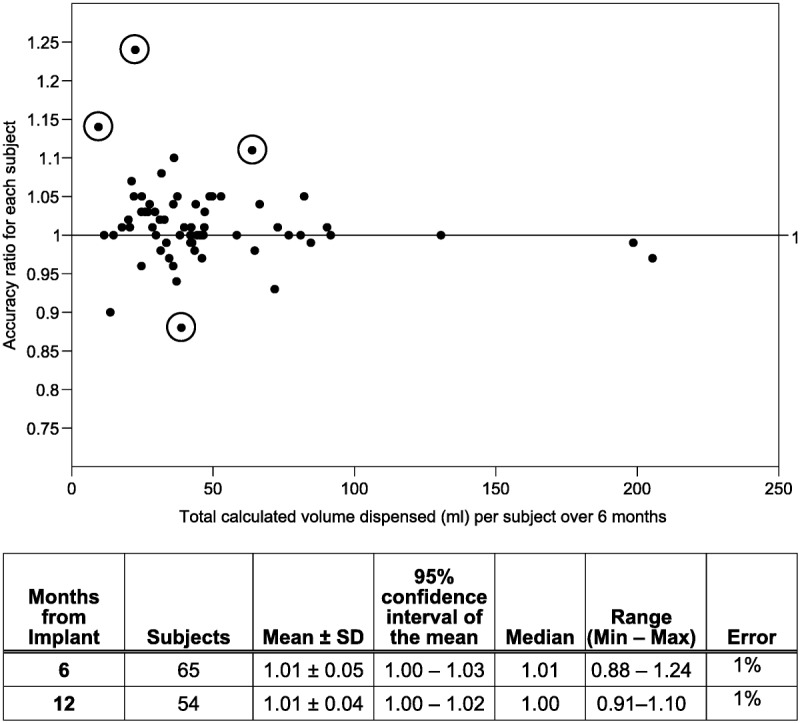
Per-subject 6-month accuracy ratios (n = 65 patients). Details of 4 subjects with 6-month ratios farthest from 1.0 (circled values) are described from the top of graph downward. Ratio = 1.24. Five refills of 20-mL pain pump with per-visit ratios ranging from 1.13 to 2.5, observed delivery volumes of 10.6, 2.4, 1.0, 2.2, and 11.6 mL. During this period, 2 adverse events of drug toxicity/adverse effects were reported (nausea and vomiting, and peripheral edema). Ratio = 1.14: 2 refills of 40-mL spasticity pump per-visit ratios of 0.93 and 1.31, observed delivery volumes of 4.0 and 6.8 mL. Twenty-nine days after implantation, catheter dislodgment was reported. Pump programmed to minimum rate and catheter replacement was carried out prior to 6-month visit. Ratio = 1.11: 2 refills of 40-mL pain pump, per-visit ratios of 1.14 and 1.07, observed delivery volumes of 39.8 and 31 mL. Eleven days after implantation, the patient reported increased pain. Magnetic resonance imaging confirmed no inflammatory mass, and radiographs confirmed catheter tip location at T11-T12. Ratio = 0.88. Two refills of 40-mL spasticity pump during the 6-month period, per-visit ratios of 0.93 and 0.71, observed delivery volumes of 27 and 7.0 mL. Following the 6-month visit, the patient experienced muscle spasms. The intrathecal baclofen dosage was increased at 4 consecutive visits.
Accuracy ratios relatively far from the 1.01 mean (or 1.00 median) tended to be associated with relatively small total delivery volumes—a situation that can magnify otherwise small visual (syringe) measurement errors or variations.
Because the per-subject accuracy ratios combine data from multiple visits, an additional analysis was conducted to examine the refill ratio at each visit. Figure 3 illustrates the complete per-refill data, with the accompanying table showing calculations with and without the 4 outlier values (open boxes). The mean per-refill accuracy ratio without outliers was 1.025 (SD, 0.121; mean error of plus 2.5% overinfusion) in 189 refills at varying intervals and volumes. The mean per-refill accuracy is very similar to the mean per-subject accuracy, but there is a wider range and larger SD associated with these single observations. As illustrated in Figure 3, more discrepant accuracy ratios are seen in cases where smaller volumes were dispensed—a situation where small measurement variations or errors can be greater than the volume of drug that was delivered and magnify otherwise small visual (syringe) measurement errors or variations. Incomplete medication removal from the pump may provide an additional explanation of discrepant ratios, particularly those greater than 1.
FIGURE 3.
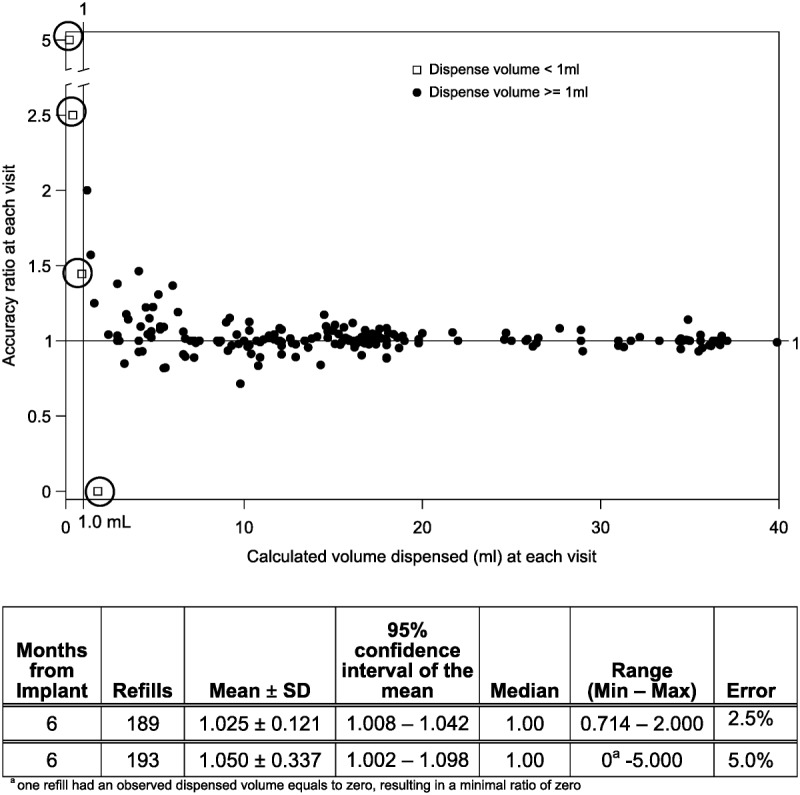
Per-refill accuracy ratios, from implant to 6 months after implantation (n = 193 refills). Each refill visit’s data were used to calculate a ratio of observed volume dispensed to programmer-calculated amount dispensed. The individual ratios are plotted and summarized in 2 ways, including the circled outlier values (193 refills) and excluding them (189 refills).
Laboratory testing of 23 pumps over 92 refill cycles demonstrated an average flow-rate accuracy of 0.976 (SD, 0.019 (97.6% of the programmed volume; minus 2.4% average delivery error).
Safety
Fifty-eight system-related AEs were reported in 38 (48%) of the 80 implanted subjects. These events were attributed to the pump, catheter, implant sites, the implant procedure, and programming. Table 1 summarizes system-related AEs in order of frequency. Most events resolved without hospitalization or surgical intervention. Thirteen subjects experienced system-related events that met serious criteria that were resolved through surgical intervention on the catheter or pump or medical intervention involving hospitalization. No deaths or permanent injuries occurred.
TABLE 1.
System-related AEs

One possibly pump-related event demonstrates the importance of dose titration with a replacement pump. A subject entering the study received her study pump as a replacement for a previous pump. For some time prior to pump replacement, the patient had experienced symptoms consistent with loss of intrathecal baclofen effects. Oral baclofen relieved her symptoms and observed (old) pump residual volumes matched programmer expected volumes. The old pump (6 years old; SynchroMed EL) was replaced. Review of the operative report and programming printouts reveal that intraoperative priming bolus may have caused 0.169 mL of drug (338 μg of Lioresal) to be administered unintentionally. In addition, the new pump was programmed to the same dose as the previous pump. Initially, the subject’s symptoms were thought to be related to anesthesia. However, after the subject returned 4 days postoperatively with ongoing nausea, vomiting, sedation, and lethargy, sequential decreases in the daily intrathecal baclofen dosage led to symptom resolution (1113.1 μg/d to 849.3 μg/d). No further AEs occurred in this subject.
Three events resulted in pump explant. One event (catheter break/cut) was experienced in a subject whose repeated withdrawal symptoms led to system replacement. At the time of reoperation, a leak in the catheter was discovered so the catheter was repaired, and the pump was replaced at the physician’s discretion. The second event (implant site infection) was an infection detected at 167 days after implantation that led to system removal. Both pumps were returned to the manufacturer, and no defect was found. The third event of repeated drug withdrawal symptoms (drug withdrawal syndrome) was experienced in a subject receiving intrathecal morphine for pain. Although investigations revealed no evidence of device system malfunction, the subject underwent complete infusion system explantation and replacement (per-refill volume ratios were 1.0–1.08, indicating no underinfusion). The explanted pump in question was lost in transit upon attempted return to the manufacturer and could not be analyzed.
Sixty-six events reported in 32 subjects (40%) were related to intrathecal or nonintrathecal drugs. None of these events resulted in surgical intervention on the drug infusion system and were most often treated by adjusting intrathecal or nonintrathecal medications.
DISCUSSION
Flow-Rate Accuracy and Patient Safety
Pump accuracy analyses in the clinical setting, as performed in this study, reflect factors that influence the actual flow rate (temperature, atmospheric pressure, fullness of the reservoir) as well as those that influence flow-rate calculations during a single visit (limitations of visual perception of small syringe volume differences, incomplete withdrawal of fluid from the pump, undetected partial pocket fill at previous visit, motor stall, or catheter obstruction). Having a cumulative aggregate volume calculation based on multiple refills of each pump as the primary end point reduced the effects of perceptual variations and/or measurement errors at individual visits. In clinical practice, single-visit volume measurements remain subject to potentially large errors that should make physicians cautious before changing pump programming (or performing a device or component replacement procedure) in the absence of drug underdose or overdose symptoms. These measurements errors are particularly likely to occur in the first few days or weeks after refill because of the small volumes dispensed in that period. Individual measurement variability is illustrated by the approximately 4-fold difference in the ordinate volume-ratio scale of Figure 3 versus Figure 2, as well as the approximately 5-fold difference in the x-axis volume scales. In Figure 3, several single, small (absolute) volume discrepancies apparently well outside the protocol specification (eg, ratio >1.25) were recorded, but turned out to be artifactual.
In terms of patient safety, a single discrepant refill needs to be considered within the context of patient symptoms and other factors that may have influenced the measurements. Physicians and practitioners should pay close attention to pump refill techniques because an inadvertent deposit of medication into the subcutaneous tissue can result in a dangerous, possibly life-threatening, overdose. A recent consensus paper reviews the pump’s safety issues. 11 Clinicians are reminded that flow rate can vary from pump to pump, and the product labeling directs physicians to closely monitor patients following device implantation or replacement (eg, end-of-life pump replacement) and to titrate daily drug dosages gradually upward based on clinical effects, regardless of the individual’s intrathecal drug dosing history with their previous pump. A replacement pump that delivers a higher mean flow rate than the previous pump could compound problems associated with a patient becoming more or less drug-naive if there had been a lapse in therapy delivery related to the previous pump reaching its end of life. This highlights the importance of dosage titration after pump implant or replacement to account for the potentially different (but consistently delivered) inherent flow rate of the new pump.
This clinical study is not the only source for real-world accuracy data. At the time of manuscript submission, the manufacturer was evaluating newly identified SynchroMed II overinfusion events confirmed by returned product analysis. Of these events, some pumps were explanted and returned because of patient complications indicative of overinfusion, and some were returned for other reasons (eg, end of battery life). None of the events noted were associated with the clinical study reported here. Manufacturer recommendations include closely monitoring pump accuracy at each refill, recording actual and expected pump residual volume, and closely evaluating an increase in volume discrepancy over time (volume withdrawn from the pump is less than expected) or volume discrepancies of more than 2 mL. Evaluate for other causes, such as unrecognized partial pocket fill, self-aspiration of reservoir medication, and less than full reservoir at prior refill. If overinfusion is strongly suspected, clinically monitor the patient and consider pump replacement. The decision to replace the pump should take the following factors into consideration: history of pump volumes, magnitude of the volume discrepancy, presence/severity of overdose symptoms, and the individual patient situation. Information specific to this new risk with the SynchroMed II Infusion System can be found with product advisories at http://professional.medtronic.com/pt/neuro/idd/ind/product-advisories.
Other implantable pump studies have described flow-rate accuracy using similar methods and demonstrated similar results to the present clinical study, albeit with a slightly broader range of individual average accuracy ratios. 12–14 An in vivo study in sheep 13 showed larger average flow-rate error and a smaller SD (9.32% [SD, 9.27%]) in individual refill measurements than either the current study or the 2 studies cited above. Investigators in the sheep study were able to weigh the pump volumes input and the residual volumes, which may be more accurate than the syringe-read volumetric measurements used in the present clinical study. Although the sheep study tightly controlled drug delivery measurements, it did not control for other factors, such as temperature or pressure, which may have influenced accuracy calculations. The sheep investigators also performed bench testing with the pumps explanted from the sheep. Their bench laboratory data using the same devices previously implanted in the sheep demonstrated a mean flow-rate error for 64 refills in 16 pumps of minus 1.05% (SD, 2.55%) under controlled temperature and atmospheric pressure conditions. Laboratory investigation of the SynchroMed II pump revealed a comparable flow-rate error (also by direct weight) of minus 2.4% (SD, 2.1%) under controlled temperature only. Direct fluid weight laboratory measures reduced measurement error especially at low flow rates.
The smaller flow-rate variations in laboratory data compared with clinical data in the present studies illustrate the limitations of clinical measurements and indicate that volume discrepancies from a single refill alone should not be relied upon as a decisive factor to assess pump performance or malfunctions.
The system-related AEs reported during the clinical study were similar in nature to those reported in the drug infusion therapy literature with predicate pumps. 15–17 In the current clinical study, complications requiring surgical intervention on the infusion system were experienced by 14% of subjects, whereas the remainder of system-related AEs resolved following medical intervention. Ten percent of subjects experienced catheter complications. The manufacturer has subsequently introduced a new catheter designed to prevent common catheter complications and posts product performance reports annually at http://professional.medtronic.com/ppr/intrathecal-drug-delivery-systems/index.htm.
Clinical measurements of accuracy demonstrate real-world experience. These measurements are susceptible to sources of error unrelated to pump function. Bench data provide the pump’s actual accuracy, without the sources of error such as refill technique and imprecise measurement tools. Although individual pumps vary slightly in their average flow rates, the programmable pump allows the clinician to adjust flow rate (and thus the dose) according to the patient’s symptoms. The current study demonstrated that the SynchroMed II pump accurately and safely delivered intrathecal medications in the clinical setting of this study.
ACKNOWLEDGEMENTS
The following study investigators conducted the study at their respective sites, participated on the publication committee, and reviewed the manuscript: Peter Kosek, MD, Pain Consultants of Oregon, PC, Eugene, Oregon; Cindy Ivanhoe, MD, The Institute for Rehabilitation and Research–Memorial Hermann Hospital, Houston, TX; Steven Charapata, MD, Pain Management Associates, Independence, MO; Scott McLanahan, MD, Carolina Neurosurgery and Spine Associates, Charlotte, NC; Edgar Ross, MD, Brigham and Women’s Hospital, Boston, MA; Michael Turner, MD, Goodman Campbell Brain and Spine, Indianapolis, IN. Thomas Silvestrini, MD, St Mary’s/Duluth Clinic Health conducted the study at his site. The engineer/author of the laboratory bench report was Brian Ball, Medtronic, Inc. The authors also thank Maria Breitenfeldt, PhD, Medtronic, Inc, for data analysis discussions and manuscript contributions.
Footnotes
The work was sponsored by Medtronic, Inc.
Some authors, as indicated, are employees of Medtronic.
The work was conducted at the following institutions: Carolina Neurosurgery and Spine Associates, Charlotte, NC; Brigham and Women’s Hospital, Boston, MA; Goodman Campbell Brain and Spine, Indianapolis, IN; Pain Consultants of Oregon, PC, Eugene, OR; Pain Management Associates, Independence, MO; Rush University Medical Center, Chicago, IL; St Mary’s/Duluth Clinic Health, Duluth, MN; The Institute for Rehabilitation and Research—Memorial Hermann Hospital, Houston, TX; and University of California San Diego, La Jolla, CA.
REFERENCES
- 1.Onofrio BM, Yaksh TL, Arnold PG. Continuous low-dose intrathecal morphine administration in the treatment of chronic pain of malignant origin. Mayo Clinic Proc. 1981;56:516–520 [PubMed] [Google Scholar]
- 2.Turner JA, Sears JM, Loeser JD. Programmable intrathecal opioid delivery systems for chronic noncancer pain: a systematic review of effectiveness and complications. Clin J Pain. 2007;23:180–195 [DOI] [PubMed] [Google Scholar]
- 3.Wallace MS, Charapata SG, Fisher R, et al. Intrathecal ziconotide in the treatment of chronic nonmalignant pain: a randomized, double-blind, placebo-controlled clinical trial. Neuromodulation. 2006;9:75–86 [DOI] [PubMed] [Google Scholar]
- 4.Gilmartin R, Bruce D, Storrs BB, et al. Intrathecal baclofen for management of spastic cerebral palsy: multicenter trial. J Child Neurol. 2000;15:71–77 [DOI] [PubMed] [Google Scholar]
- 5.Meythaler JM, DeVivo MJ, Hadley M. Prospective study on the use of bolus intrathecal baclofen for spastic hypertonia due to acquired brain injury. Arch Phys Med Rehabil. 1996;77:461–466 [DOI] [PubMed] [Google Scholar]
- 6.Nance P, Schryvers O, Schmidt B, et al. Intrathecal baclofen therapy for adults with spinal spasticity: therapeutic efficacy and effect on hospital admissions. Can J Neurol Sci. 1995;22:22–29 [DOI] [PubMed] [Google Scholar]
- 7.Coffey RJ, Cahill D, Steers W, et al. Intrathecal baclofen for intractable spasticity of spinal origin: results of a long-term multicenter study. J Neurosurg. 1993;78:226–232 [DOI] [PubMed] [Google Scholar]
- 8.Penn RD. Intrathecal baclofen for spasticity of spinal origin: seven years of experience. J Neurosurg. 1992;77:236–240 [DOI] [PubMed] [Google Scholar]
- 9.Penn RD, Savoy SM, Corcos D, et al. Intrathecal baclofen for severe spinal spasticity. N Engl J Med. 1989;320:1517–1521 [DOI] [PubMed] [Google Scholar]
- 10.Watve SV, Sivan M, Raza WA, Jamil FF. Management of acute overdose or withdrawal state in intrathecal baclofen therapy. Spinal Cord. 2012;50:107–111 [DOI] [PubMed] [Google Scholar]
- 11.Prager J, Deer T, Levy R, et al. 2014. Best practices for intrathecal drug delivery for pain. Neuromodulation. 2014; E-pub ahead of print. DOI: 10.1111/ner.12146 [DOI] [PubMed] [Google Scholar]
- 12.Rauck R, Deer T, Rosen S, et al. 2012. Long-term follow-up of a novel implantable programmable infusion pump. Neuromodulation. 2013;16:163–167 [DOI] [PubMed] [Google Scholar]
- 13.Venugopalan R, Ginggen A, Bork T, Anderson W, Buffen E. 2011. In vivo study of flow-rate accuracy of the MedStream Programmable Infusion System. Neuromodulation. 2011;14:235–241 [DOI] [PubMed] [Google Scholar]
- 14.Rauck R, Deer T, Rosen S, et al. Accuracy and efficacy of intrathecal administration of morphine sulfate for treatment of intractable pain using the Prometra Programmable Pump. Neuromodulation. 2010;13:102–108 [DOI] [PubMed] [Google Scholar]
- 15.Hayek SM, Deer TR, Pope JE, Panchal SJ, Patel V. Intrathecal therapy for cancer and non-cancer pain. Pain Physician. 2011;14:219–248 [PubMed] [Google Scholar]
- 16.Atli A, Theodore BR, Turk DC, Loeser JD. Intrathecal opioid therapy for chronic nonmalignant pain: a retrospective cohort study with 3-year follow-up. Pain Med. 2010;11:1010–1016 [DOI] [PubMed] [Google Scholar]
- 17.Flückiger B, Knecht H, Grossmann S, Felleiter P. Device-related complications of long-term intrathecal drug therapy via implanted pumps. Spinal Cord. 2008;46:639–643 [DOI] [PubMed] [Google Scholar]