

Abstract
We have developed a stereospecific, nickel-catalyzed cross coupling of benzylic pivalates with aryl boroxines. The success of this reaction relies on the use of Ni(cod)2 as catalyst and NaOMe as a uniquely effective base. This reaction has broad scope with respect to the aryl boroxine and benzylic pivalate, enabling the synthesis of a variety of diarylalkanes and triarylmethanes in good to excellent yields and ee's.
Diarylalkanes and triarylmethanes are important molecules in organic synthesis and pharmaceutical development.1 A highly efficient and direct route to these valuable chiral compounds is the metal-catalyzed cross coupling of a benzylic electrophile with an organometallic reagent.2 Such couplings have been accomplished in both enantioselective and enantiospecific fashion.3,4,5 Of particular note, the Jarvo group has demonstrated that nickel-catalyzed cross couplings of enantioenriched benzylic ethers with Grignard reagents occur with excellent levels of chirality transfer.6 Jarvo's work highlights the convenience of using readily available, enantioenriched benzylic alcohol derivatives as starting materials. However, a limitation to the state-of-the-art in this field is the requirement for highly nucleophilic coupling partners (Grignards or organozincs), which restricts the range of functional groups that are compatible with these reactions. To our knowledge, no enantioselective couplings of benzylic electrophiles with the more mild organoboranes are yet known, and stereospecific couplings with boronic reagents to deliver these products are rare.7,8,9,10,11 The ability to employ organoboranes as coupling partners would lead to greatly expanded scope and functional group tolerance within this important class of reactions.
Recognizing the wide accessibility of enantioenriched benzylic alcohol derivatives and the advantage of using mild and functional group-tolerant organoborane coupling partners, we sought to develop a stereospecific cross coupling of these reagents. Within our research program focused around the development of cross coupling reactions of non-traditional electrophiles, we have been drawn to the use of pivalate substrates for aryl C–O bond activation.12,13 We en-visioned that the use of benzylic pivalates would enable the desired nickel-catalyzed coupling of enantioenriched benzylic alcohol derivatives with organoborane coupling partners (Scheme 1). Herein, we report the stereospecific and high-yielding coupling of benzylic pivalates with aryl boroxines in the presence of a simple nickel(0) catalyst. This cross coupling leads to a wide variety of diarylalkanes and triarylmethanes.
Scheme 1.
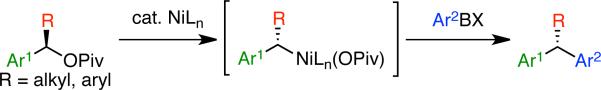
Stereospecific Coupling of Benzylic Pivalates with Aryl Boroxines
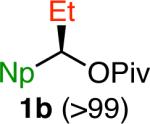
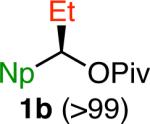
We selected the cross coupling of pivalate 1a, which is readily prepared in >99% ee,14 as our model reaction for optimization. Given the precedent for activation of benzylic C–O bonds5e, f, 6 and our prior experience in Suzuki cross couplings of benzylic electrophiles,8a we anticipated that a Ni(0) catalyst supported by an electron-rich phosphine ligand would enable this transformation. However, with Ni(cod)2/PCy3 as the catalyst system and phenyl boronic acid as the coupling partner, we observed low yields of the desired product using a variety of bases commonly employed in Suzuki cross couplings (Table 1, entries 1 and 2). β-Hydride elimination was also observed under these reaction conditions. However, when NaOMe was employed as base, product 2 was formed in 78% yield (entry 3). Further optimization showed that the use of PCy2Ph as ligand resulted in 93% yield (entry 4), but product 2 was formed in only 54% ee under these conditions. In contrast, much higher chirality transfer was obtained without detrimental effect on yield when Ni(cod)2 alone was used as catalyst (93% ee, entry 5). Notably, the absolute configuration of the major enantiomer was opposite when phosphine ligand was not employed. Using higher equivalents of boronic acid and at lower reaction temperature (70 °C), higher extent of chirality transfer was observed without a significant drop in yield (entry 6). The nature of the counter-ion of the methoxide base had a significant impact on the reaction outcome. With KOMe, the product was formed in lower ee, whereas no reaction occurred with LiOMe (entries 7 and 8). These results suggest that the solubility of the base is important; with more base in solution (as occurs with KOMe), lower product ee is observed, but LiOMe may not be soluble enough in PhMe to promote the reaction. This hypothesis is consistent with our solvent studies; with more polar solvents, lower ee's of product are observed.14 By increasing the concentration of the reaction mixture, we obtained 87% yield of product 2 with excellent chirality transfer using PhB(OH)2 and only 5 mol % Ni(cod)2 (entry 9). Nearly quantitative yield was observed when phenyl boroxine was employed as the coupling partner under these conditions (entry 10). Although the difference in yield was not particularly significant for this model reaction, as we examined other aryl boronic reagents, we generally observed better yields and higher levels of chirality transfer with boroxines, indicating that water likely has a detrimental effect on this reaction. Indeed, the addition of H2O (1.0 equiv) to the reaction resulted in diminished yield and chirality transfer (entry 11).14 Finally, we performed control reactions in the absence of Ni(cod)2 and found that no cross coupling was observed with either phenyl boronic acid or phenyl boroxine (entries 12 and 13). In these reactions, pivalate 1a was recovered quantitatively in >99% ee.
Table 1.
Optimization of Cross Couplinga
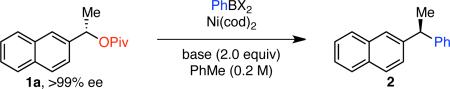
| ||||||
|---|---|---|---|---|---|---|
| entry | mol % Ni | PhBX2 (equiv) | base | temp (°C) | yield (%)b | ee (%)c |
| 1d | 10 | PhB(OH)2 (3.0) | CsF | 100 | 14 | n.d.e |
| 2d | 10 | PhB(OH)2 (3.0) | K3PO4 | 100 | 14 | n.d.e |
| 3d | 10 | PhB(OH)2 (3.0) | NaOMe | 100 | 78 | n.d.e |
| 4f | 10 | PhB(OH)2 (2.0) | NaOMe | 100 | 93 | 54 (R) |
| 5 | 10 | PhB(OH)2 (2.0) | NaOMe | 100 | 99 | 93 (S) |
| 6 | 10 | PhB(OH)2 (3.0) | NaOMe | 70 | 83 | 99 (S) |
| 7 | 10 | PhB(OH)2 (3.0) | KOMe | 70 | 74 | 69 (S) |
| 8 | 10 | PhB(OH)2 (3.0) | LiOMe | 70 | 0 | n.d.e |
| 9g | 5 | PhB(OH)2 (2.5) | NaOMe | 70 | 87 | 98 (S) |
| 10g | 5 | (PhBO)3 (0.83) | NaOMe | 70 | 98 | 97 (S) |
| 11g,h | 5 | (PhBO)3 (0.83) | NaOMe | 70 | 59 | 94 (S) |
| 12g | 0 | PhB(OH)2 (2.5) | NaOMe | 70 | 0 | n.d. |
| 13g | 0 | (PhBO)3 (0.83) | NaOMe | 70 | 0 | n.d. |
Conditions: pivalate 1a (0.10 mmol, 1.0 equiv), PhBX2, Ni(cod)2, base (2.0 equiv), PhMe (0.2 M), unless otherwise noted.
Determined by 1H NMR analysis using 1,3,5-trimethoxybenzene as internal standard.
Determined by chiral HPLC. Absolute configuration of the major enantiomer in parentheses.
Performed with racemic 1a. PCy3 (24 mol %) added.
n.d. = not determined.
PCy2Ph (22 mol %) added.
0.4 M.
1.0 equiv H2O added.
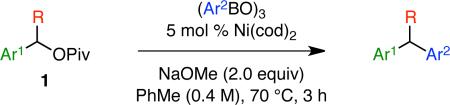
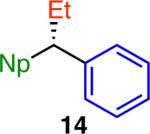
Under our optimized conditions (Table 1, entry 10), we observed broad scope with respect to the aryl boroxine (Scheme 2).15 High yields and excellent chirality transfer were observed with both electron-rich and electron-poor aryl groups. Further, a wide range of functional groups was tolerated, including amino (3), ether (4), fluoro (7, 12), chloro (8), trifluoromethyl (9), and acetal (10) functionalities. The formation of chloride 8 is particularly impressive and highlights the advantage of using a boronic coupling partner and mildness of these reaction conditions. Increased steric hindrance on the aryl fragment was well tolerated, as demonstrated by the formation of product 6 in 94% yield and 96% ee. By comparison of product 5 to an authentic sample recently prepared in our laboratory,8a we confirmed the absolute configuration of this product, which demonstrates that this reaction occurs with overall inversion of configuration at the benzylic stereocenter. The absolute configuration of the other products was assigned by analogy.
Scheme 2.

Scope of Boroxinea
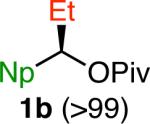
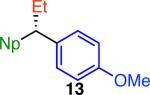
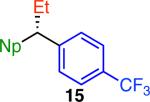
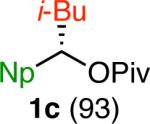
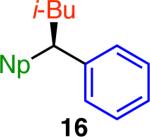
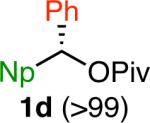
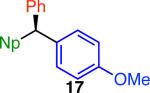
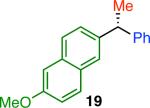
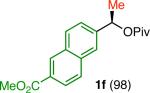
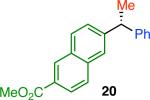
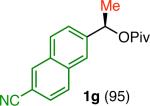
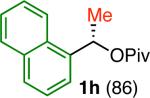
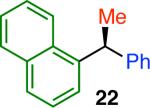
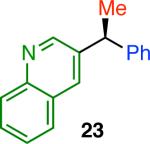
Variation of the pivalate partner led to a variety of diarylal-kane and triarylmethane products in good to excellent yields and enantiospecificities (Table 2). In the formation of diarylalkanes, increased steric bulk of the alkyl substituent (R) was well tolerated, giving excellent levels of chirality transfer in the cross coupling (entries 1–4). Notably, even pivalate 1c with a bulky i-butyl substituent underwent the reaction (entry 4). The coupling of diaryl-substituted pivalate 1d also proceeded in excellent yields to deliver triarylmethanes 17 and 18 with good levels of chirality transfer (entries 5 and 6). A variety of functional groups were well tolerated on the naphthyl fragment. The coupling of 6-methoxynapthyl-substituted pivalate 1e gave diarylethane 19 in 87% yield and 93% ee (entry 7). In particular, the successful couplings of ester- and nitrile-substituted pivalates 1f and 1g highlight the increased functional group tolerance enabled by the use of an aryl boroxine coupling partner (entries 8 and 9). The couplings of benzylic pivalates with aryl groups other than 2- naphthyl were also successful. The reaction of 1-naphthyl-substituted pivalate 1h proceeded to give diarylethane in 73% yield and 85% es (entry 10). We hypothesize that the slightly lower level of chirality transfer in this case may be due to the increased steric hindrance of having ortho substitution on the aromatic group. Both electron-rich and electron-poor heteroaromatics underwent the desired coupling (entries 11 and 12). Finally, we were pleased to observe that the cross coupling of biphenyl-substituted pivalate proceeded in 90% es (entry 13). The diminished yield in this case clearly demonstrates the importance of the aryl group in the oxidative addition of nickel to the pivalate.
Table 2.
Scope of Pivalatea
| |||||
|---|---|---|---|---|---|
| entry | 1 (% ee)b | Product | yield (%)c | ee (%)d | es (%)e |
| 1f,g |
|
|
71 | 94 | ≥94 |
| 2g |
|
|
93 | 97 | ≥97 |
| 3g |
|
|
94 | 96 | ≥96 |
| 4h |
|
|
72 | 91 | 98 |
| 5f,i |
|
|
85 | 86 | ≥86 |
| 6f,i,j |
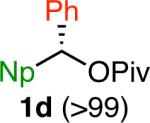
|
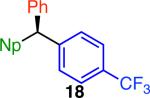
|
96 | 80 | ≥80 |
| 7k |
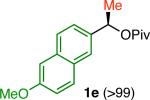
|
|
87 | 93 | ≥93 |
| 8 |
|
|
84 | 94 | 96 |
| 9 |
|
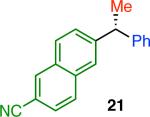
|
45 | 89 | 94 |
| 10 |
|
|
73 | 73 | 85 |
| 11 |

|
|
94 | 58 | 71 |
| 12l,m |
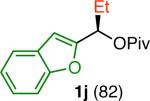
|
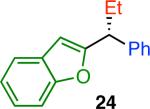
|
49 | 72 | 88 |
| 13m,n |
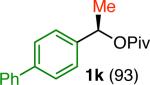
|
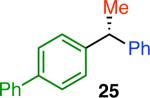
|
33 | 84 | 90 |
Conditions: pivalate (0.20 mmol, 1.0 equiv), boroxine (0.83–1.3 equiv), Ni(cod)2 (5 mol %), NaOMe (2.0 equiv), PhMe (0.4 M), 70 °C, 3 h, unless otherwise noted.
Determined by chiral HPLC.
Average isolated yield of duplicate experiments (±0–2%), unless otherwise noted.
Determined by chiral HPLC. Average ee of duplicate experiments reported (±0–1%).
% es = enantiospecificity = (prod ee)/(SM ee)*100.
10 mol % Ni(cod)2.
80 °C.
12 h.
90 °C.
Result of a single experiment.
50 °C.
32 °C.
24 h.
100 °C.
We hypothesize that this reaction proceeds via oxidative addition of the nickel catalyst into the benzylic C–O bond with subsequent transmetallation and reductive elimination delivering the C–C bond of the arylated product. In an effort to understand the stereochemical outcome of the oxidative addition, we have attempted a series of β-hydride elimination experiments using deuterated pivalate 1l (Scheme 3).16 If the oxidative addition occurs with retention of configuration at the benzylic carbon, then products 26 and/or 27 will result. However, if inversion of configuration occurs upon oxidative addition, then products 28 and/or 29 will be observed. We first attempted to induce β-hydride elimination using stoichiometric Ni(cod)2. However, we observed no reaction, even at reaction temperatures up to 130 °C. In contrast, when a phosphine ligand is added, facile β-hydride elimination is observed. With PCy3, 80% yield of styrene 26 and 5% yield of styrene 28 are observed after only 15 min at 70 °C (Scheme 3). This result is consistent with the predominant pathway for oxidative addition proceeding with retention of configuration at the benzylic carbon when a phosphine-supported nickel catalyst is used. Although it seems somewhat surprising that the C–D bond is selectively cleaved to form 26, this result is likely due to the significant steric hindrance between the naphthyl and methyl groups in the transition state leading to product 27. As shown in Table 1, entry 4 and in the Supporting Information, the cross coupling occurs with overall retention of configuration with phosphine-supported nickel catalysts, suggesting that the transmetallation and reductive elimination also occur with retention of configuration. Such retention of configuration during transmetallation and reductive elimination is well precedented with similar organometallic intermediates.16-17 However, retention of configuration upon oxidative addition of a benzylic electrophile is rare and suggests that the oxidative addition may be more complicated with this catalyst system. In contrast, using only Ni(cod)2 without any phosphine ligand, we observe overall inversion of configuration at the benzylic carbon, suggesting that the oxidative addition step likely occurs with inversion of configuration via an SN2 pathway under these conditions.
Scheme 3.
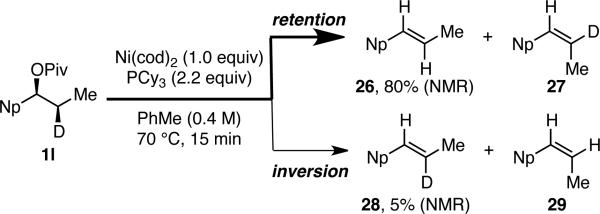
β-Hydride Elimination Experiment
In summary, we have developed a stereospecific, nickel-catalyzed cross coupling of enantioenriched benzylic pivalates with aryl boroxines. The successful development of this reaction depended on the identification of Ni(cod)2 as the optimal catalyst and NaOMe as a uniquely effective base. This reaction has broad scope with respect to the aryl boroxine and benzylic pivalate, enabling the synthesis of a variety of diarylalkanes and triarylmethanes in good to excellent levels of chirality transfer. Our current efforts are directed towards expanding the versatility and scope of this cross coupling, as well as deepening our mechanistic understanding to enable rationale improvements to the catalyst system.
Supplementary Material
ACKNOWLEDGMENT
We gratefully acknowledge Prof. E. R. Jarvo for informing us of her work in this area prior to publication. We also thank D. M. Shacklady-McAtee for insightful discussions. Research reported in this publication was supported by NSF (CHE 1151364) and an Institutional Development Award (IDeA) from the National Institute of General Medical Sciences of the NIH under grant number P20GM103541. NMR and other data were acquired at UD on instruments obtained with the assistance of NSF and NIH funding (NSF MIR 0421224, NIH P20 RR017716).
Footnotes
Funding Sources
No competing financial interests have been declared.
Supporting Information. Experimental procedures, characterization data and spectra of new compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
REFERENCES
- 1.a Zhang J, Bellomo A, Creamer AD, Dreher SD, Walsh PJ. J. Am. Chem. Soc. 2012;134:13765. doi: 10.1021/ja3047816. [DOI] [PubMed] [Google Scholar]; b Cheltsov AV, Aoyagi M, Aleshin A, Yu EC-W, Gilliland T, Zhai D, Bobkov AA, Reed JC, Liddington RC, Abagyan R. J. Med. Chem. 2010;53:3899. doi: 10.1021/jm901446n. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Huang Z, Ducharme Y, Macdonald D, Robichaud A. Curr. Opin. Chem. Biol.;2001;5:432. doi: 10.1016/s1367-5931(00)00224-6. [DOI] [PubMed] [Google Scholar]; d Alami M, Messauoudi S, Hamze A, Provot O, Brion J-D, Liu J-M, Bignon J, Bakala J. Dihydro-Iso-Ca-4 and Analogues: Potent Cytotoxics, Inhibitors of Tubulin Polymerization. 2009 WO2009/147217.; e Messaoudi S, Hamze A, Provot O, Tréguier B, Rodrigo De Losada J, Bignon J, Liu J-M, Wdzieczak-Bakala J, Thoret S, Dubois J, Brion J-D, Alami M. ChemMedChem. 2011;6:488. doi: 10.1002/cmdc.201000456. [DOI] [PubMed] [Google Scholar]; f Moree WJ, Li B-F, Jovic F, Coon T, Yu J, Gross RS, Tucci F, Marinkovic D, Zamani-Kord S, Malany S, Bradbury MJ, Hernandez LM, O'Brien Z, Wen J, Wang H, Hoare SRJ, Petroski RE, Sacaan A, Madan A, Crowe PD, Beaton G. J. Med. Chem. 2009;52:5307. doi: 10.1021/jm900933k. [DOI] [PubMed] [Google Scholar]; g Beaton G, Moree WJ, Jovic F, Coon T, Yu J. Sleep Inducing Compound and Methods Relating Thereto. 2006 US2006/14797.
- 2.For other methods, see Luan Y, Schaus SE. J. Am. Chem. Soc. 2012;134:19965. doi: 10.1021/ja309076g. Fessard TC, Andrews SP, Motoyoshi H, Carreira EM. Angew. Chem., Int. Ed. 2007;46:9331. doi: 10.1002/anie.200702995. Lewis CA, Chiu A, Kubryk M, Balsells J, Pollard D, Esser CK, Murry J, Reamer RA, Hansen KB, Miller SJ. J. Am. Chem. Soc. 2006;128:16454. doi: 10.1021/ja067840j. Pathak TP, Gligorich KM, Welm BE, Sigman MS. J. Am. Chem. Soc. 2010;132:7870. doi: 10.1021/ja103472a. Nave S, Sonawane RP, Elford TG, Aggarwal VK. J. Am. Chem. Soc. 2010;132:17096. doi: 10.1021/ja1084207. Roesner S, Casatejada JM, Elford TG, Sonawane RP, Aggarwal VK. Org. Lett. 2011;13:5740. doi: 10.1021/ol202251p. Bolshan Y, Chen C.-y., Chilenski JR, Gosselin F, Mathre DJ, O'shea PD, Roy A, Tillyer RD. Org. Lett. 2004;6:111. doi: 10.1021/ol0361655.
- 3.For an enantioselective coupling of benzylic bromides with organozinc reagents, see Arp FO, Fu GC. J. Am. Chem. Soc. 2005;127:10482. doi: 10.1021/ja053751f. Binder JT, Cordier CJ, Fu GC. J. Am. Chem. Soc. 2012;134:17003. doi: 10.1021/ja308460z.
- 4.For an enantiospecific coupling of benzylic bromides with Grignard reagents, see López-Peŕez A, Adrio J, Carretero JC. Org. Lett. 2009;11:5514. doi: 10.1021/ol902335c.
- 5.For examples of benzylic cross couplings to give achiral or racemic products, see: Molander GA, Elia MD. J. Org. Chem. 2006;71:9198. doi: 10.1021/jo061699f. Kuwano R. J. Synth. Org. Chem., Jpn. 2011;69:1263. Kofink CC, Knochel P. Org. Lett. 2006;8:4121. doi: 10.1021/ol0616790. McLaughlin M. Org. Lett. 2005;7:4875. doi: 10.1021/ol0517271. Guan B-T, Xiang S-K, Wang B-Q, Sun Z-P, Wang Y, Zhao K-Q, Shi Z-J. J. Am. Chem. Soc. 2008;130:3268. doi: 10.1021/ja710944j. Yu D-G, Wang X, Zhu R-Y, Luo S, Zhang X-B, Wang B-Q, Wang L, Shi Z-J. J. Am. Chem. Soc. 2012;134:14639. doi: 10.1021/ja307045r.
- 6.a Greene MAM, Yonova IMI, Williams FJF, Jarvo ERE. Org. Lett. 2012;14:4293. doi: 10.1021/ol300891k. [DOI] [PubMed] [Google Scholar]; b Taylor BLH, Harris MR, Jarvo ER. Angew. Chem., Int. Ed. 2012;51:7790. doi: 10.1002/anie.201202527. [DOI] [PubMed] [Google Scholar]; c Taylor BLH, Swift EC, Waetzig JD, Jarvo ER. J. Am. Chem. Soc. 2011;133:389. doi: 10.1021/ja108547u. [DOI] [PubMed] [Google Scholar]
- 7.For a stereospecfic coupling of an alpha-cyanohydrin mesylate with a boronic acid, see: He A, Falck JR. J. Am. Chem. Soc. 2010;132:2524. doi: 10.1021/ja910582n.
- 8.For our recently developed stereospecific cross coupling of benzylic ammonium triflates with boronic acids, see: Maity P, Shacklady-McAtee DM, Yap GPA, Sirianni ER, Watson MP. J. Am. Chem. Soc. 2012 doi: 10.1021/ja3089422. accepted Tian has reported the copper-catalyzed coupling of benzylic sulfonimides with Grignard reagents. See: Li M-B, Tang X-L, Tian S-K. Adv. Synth. Catal. 2011;353:1980.
- 9.For examples of a distinct approach, the stereospecific coupling of a benzylic boronic reagent with an aryl halide, see: Imao D, Glasspoole BW, Laberge VS, Crudden CM. J. Am. Chem. Soc. 2009;131:5024. doi: 10.1021/ja8094075. Ohmura T, Awano T, Suginome M. J. Am. Chem. Soc. 2010;132:13191. doi: 10.1021/ja106632j. Awano T, Ohmura T, Suginome M. J. Am. Chem. Soc. 2011;133:20738. doi: 10.1021/ja210025q.
- 10.For examples of stereospecific cross couplings of alkyl boronic reagents with aryl halides to form benzylic stereocenters, see: Sandrock DL, Jean-Geŕard L, Chen C.-y., Dreher SD, Molander GA. J. Am. Chem. Soc. 2010;132:17108. doi: 10.1021/ja108949w. Lee JCH, McDonald R, Hall DG. Nat. Chem. 2011;3:894. doi: 10.1038/nchem.1150.
- 11.Cross couplings of benzylic esters and α-pivaloxyl ketones with boronic reagents to from achiral products has been reported. See: Kuwano R, Yokogi M. Chem. Commun. 2005:5899. doi: 10.1039/b513372f. Yu J-YJ, Kuwano RR. Org. Lett. 2008;10:973. doi: 10.1021/ol800078j. Huang K, Li G, Huang W-P, Yu D-G, Shi Z-J. Chem. Commun. 2011;47:7224. doi: 10.1039/c1cc11193k.
- 12.Ehle AR, Zhou Q, Watson MP. Org. Lett. 2012;14:1202. doi: 10.1021/ol203322v. [DOI] [PubMed] [Google Scholar]
- 13.For other examples of cross couplings of aryl carboxylates, see: Quasdorf KW, Tian X, Garg NK. J. Am. Chem. Soc. 2008;130:14422. doi: 10.1021/ja806244b. Li B-J, Xu L, Wu Z-H, Guan B-T, Sun C-L, Wang B-Q, Shi Z-J. J. Am. Chem. Soc. 2009;131:14656. doi: 10.1021/ja907281f. Guan B-T, Wang Y, Li B-J, Yu D-G, Shi Z-J. J. Am. Chem. Soc. 2008;130:14468. doi: 10.1021/ja8056503.
- 14.See Supporting Information for details.
- 15.Aryl boroxines were easily prepared according to a literature procedure. See: Xiao Q, Tian L, Tan R, Xia Y, Qiu D, Zhang Y, Wang J. Org. Lett. 2012;14:4230. doi: 10.1021/ol301912a.
- 16.Similar β-hydride elimination experiments have been used to probe the oxidative addition step of Suzuki cross-couplings of alkyl tosylates. See: Netherton MR, Fu GC. Angew. Chem., Int. Ed. 2002;41:3910. doi: 10.1002/1521-3773(20021018)41:20<3910::AID-ANIE3910>3.0.CO;2-W.
- 17.Retention of configuration is well precedented in reductive elimination. See: Stille JK. Oxidative Addition and Reductive Elimination. In: Hartley FR, Patai S, editors. The Chemistry of the Metal–Carbon Bond. Vol. 2. John Wiley & Sons, Ltd.; New York: 1985. p. 625.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.