

Abstract
Glioblastoma is one of the most common malignant tumors in the nervous system in both adult and pediatric patients. Studies suggest that abnormal activation of receptor tyrosine kinases contributes to pathological development of glioblastoma. However, current therapies targeting tyrosine kinase receptors have poor therapeutic outcomes. Here, we examined anticancer effects of ponatinib, a multi-targeted tyrosine kinase inhibitor, on glioblastoma cells both in the U87MG cell line and in the mouse xenograft model. We showed that ponatinib treatment reduced cell viability and induced cell apoptosis in a dose-dependent manner in U87MG cells. In addition, ponatinib suppressed migration and invasion of U87MG cells effectively. Furthermore, ponatinib-treated tumors showed an obvious reduction of tumor volume and an increase of apoptosis as compared with vehicle-treated tumors in the mouse xenograft model. These findings support a potential application of ponatinib as a chemotherapeutic option against glioblastoma cells.
Keywords: cancer therapy, glioma, glioblastoma multiforme
Introduction
Gliomas are the most common solid tumors of the nervous system.1–3 Gliomas can be divided into astrocytomas, oligodendrogliomas, ependymomas, and oligoastrocytomas according to different cell types.1 Gliomas are further categorized into four grades, which depend on the aggressiveness of the tumors.2 Grade I and grade II gliomas are low-grade gliomas with slow growth and low rate of recurrence.3 Grade III (malignant glioma) and grade IV (glioblastoma multiforme [GBM]) gliomas are more aggressive tumors, which invade other parts of the brain.3 According to molecular signatures, there are four different subtypes of GBMs.4
Various signaling pathways are involved in pathological development of GBM, including growth factor/tyrosine kinase receptor pathway, Raf-MAPKK-ERK, p53-MDM2-p14ARF pathway, RB1-p16INK4a pathway, and PTEN/Akt-1 pathway.5 Among those pathways, tyrosine kinase receptor signaling pathways play a central role in oncogenesis of GBM.6,7 Deregulations of tyrosine kinase receptor signaling in GBM cause robust alterations of proliferation, invasion, and tumor-induced neovascularization.6,8 Epidermal growth factor receptor (EGFR), a typical receptor tyrosine kinase (RTK), is strongly associated with pathological development of GBM.9–11 Particularly, a mutated form of EGFR, which results in a constitutively autophosphorylated receptor, enhances growth, proliferation, migration, and tumor neovascularization.12,13 Studies in vitro further confirm that EGFR plays important roles in gliomagenesis.14 Considering the central role of EGFR in gliomagenesis, it has been an attractive candidate for chemotherapy targeting.
Although RTKs are promising candidates for targeted therapies of GBM, drugs such as gefitinib (targeted to EGFR), have not shown a significant improvement of survival rate compared with standard therapy. Several reasons contribute to the poor efficiency of current RTK inhibitors. First, only a few GBM patients with overexpression of RTK could have been targeted because of molecular heterogeneity between individuals.15 Second, many GBM tumors are not sensitive to RTK inhibitors in RTK-mutated tumors.15 Third, inhibitors targeting to single RTK are ineffective, since other RTKs still drive tumor growth.16 Therefore, a current challenge is to develop new drugs to overcome the drug resistance during GBM treatment. As a first step, it is important to find other potential chemical components against glioblastoma cells. Ponatinib (AP24534), a potent multi-targeted tyrosine kinase inhibitor,17 has shown robust inhibitory activity against tyrosine kinases including BCR-ABL, PDGFR, KIT, and FGFR in chronic myeloid leukemia and Philadelphia chromosome-positive acute lymphoblastic leukemia.18,19 However, the inhibitory efficiency of ponatinib in glioblastoma cells is not examined.
To elucidate the roles of ponatinib in glioblastoma cells, we examined the efficiency of ponatinib against U87MG cells in vitro and in vivo. We found that ponatinib treatment inhibited cell viability and induced cell apoptosis in U87MG human cell line. We also demonstrated that ponatinib prevented U87MG cell migration and invasion in vitro. Moreover, we confirmed that ponatinib could restrict tumor growth and promote cell apoptosis in mouse xenograft model. Taken together, our findings demonstrate that ponatinib is a promising candidate against glioblastoma U87MG cells.
Materials and methods
Materials and reagents
U87MG, a commonly studied grade IV glioma cell line, was purchased from American Type Culture Collection (ATCC), Manassas, VA, USA. Ponatinib was bought from Thermo Fisher Scientific (Waltham, MA, USA; Catalog No: NC0565468).
Cell culture
U87MG cells were grown in Dulbecco’s Modified Eagle’s Medium (DMEM)/F12 medium supplemented with 10% heat-inactivated fetal bovine serum and 100 U/mL penicillin-streptomycin. Cells were cultured in a humidified incubator at 37°C in 5% CO2.
Cell viability assay
Cells were seeded in 96-well plates. Cells were treated with different doses of ponatinib (0.78–200 nM) for 72 hours, or with 20 nM ponatinib for 24, 48, or 72 hours. Then, cell viability of U87MG cells was analyzed using CellTiter-Glo Assay kit (Promega Corporation, Fitchburg, WI, USA) according to the manufacturer’s instructions. Experiments were repeated in triplicate.
Flow cytometry analysis
Cells were treated with ponatinib at different concentrations for 48 hours. Cells were harvested, followed by staining with propidium iodide using CycleTEST plus DNA reagent kit (Becton Dickinson, Franklin Lakes, NJ, USA). The apoptotic cells with DNA content in sub-G1 were detected with a FACSCalibur flow cytometry. The results were analyzed using CellQuest Pro software (Becton Dickinson).
Morphological analysis of apoptotic cells
U87MG cells were treated with ponatinib at different concentrations for 48 hours. Cells with condensed or fragmented nuclei were identified by Hoechst staining.
Cell invasion assay
One-million cells were plated into the upper chambers of the 24-transwell Boyden chamber wells (Costar, Bedford, MA, USA). After treatment with ponatinib at different concentrations for 8 hours, the cells were fixed and stained with 0.1% crystal violet. After images were taken, the migrated cells were lysed with 10% acetic acid. The absorbance of lysate was measured at 595 nm.
Efficacy of ponatinib in mouse xenograft model in vivo
U87MG cells (2×106 cells/mouse) were injected into the axillary regions of mice. The mice were randomly separated into control group and ponatinib-treatment groups (n=6 per group) after the tumor grew to 70 mm3 in volume. For ponatinib-treatment groups, low-dose (5 mg/kg) or high-dose (10 mg/kg) ponatinib was injected into the mice every day. The tumor size and body weight of the mice were measured every day. The tumor volume was calculated according to the following formula: volume (mm3) = (width × width × length)/2.
TUNEL staining
After treatment, tumor samples were harvested from the mice. The tumors were trimmed into 5 mm3 pieces and immediately fixed in 4% paraformaldehyde. Then, samples were transferred to 70% ethanol, embedded into paraffin blocks, and sectioned (5 mm thickness). The apoptosis of cells was identified by the terminal deoxynucleotidyl transferase-mediated dUTP-digoxigenin nick end-labeling (TUNEL) method. In brief, cells were incubated with TdT enzyme solution for 60 minutes, washing with phosphate-buffered saline twice, followed by 30 minutes incubation with DAPI (4′,6-diamidino-2-phenylindole). Images of the staining were taken with a fluorescent microscope.
Data analysis and presentation
All experiments were repeated at least three times independently. Graphs were drawn using the SPSS® statistical software, version 17.0 (SPSS Inc., Chicago, IL, USA). Data are expressed as means ± standard deviation. P<0.05 was considered as significance.
Results
Ponatinib suppresses cell viability of U87MG cells
To measure whether ponatinib impacts growth of U87MG cells, cell viability assay was performed after cells were treated with different concentrations of ponatinib for 72 hours. We found that the cell viability was reduced gradually as the concentration of ponatinib was increased (Figure 1A). Notably, the cell viability was inhibited completely after treatment with 200 nM ponatinib for 72 hours (Figure 1A). To confirm the inhibitory efficiency of ponatinib on U87MG cells, we treated cells with 20 nM ponatinib for 24, 48, and 72 hours. The time-course study showed that the inhibitory efficiency of ponatinib against U87MG cell viability was significantly increased following ponatinib treatment for longer time (Figure 1B). Taken together, our data demonstrate that ponatinib impacts U87MG cell viability in vitro.
Figure 1.
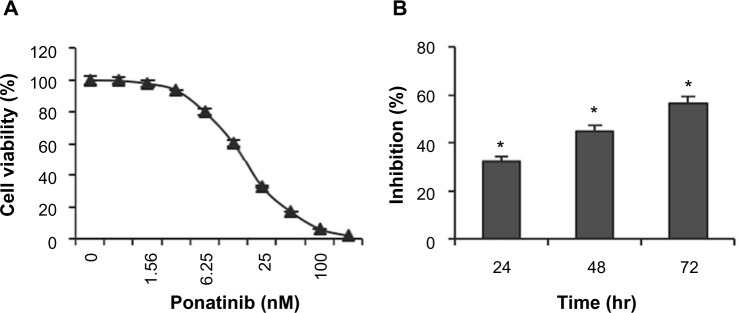
Ponatinib suppresses U87MG cell viability.
Notes: (A) U87MG cells were treated with different doses of ponatinib as indicated for 72 hours, followed by measurement with cell viability assay. Viability of cells without ponatinib treatment was set to 100%. (B) U87MG cells were treated with ponatinib (20 nM) for 24, 48, or 72 hours, followed by measurement with cell viability assay. Inhibition of control cells was measured as decrease in viability (ATP content in cells). *P<0.05 (one-way ANOVA).
Abbreviations: ANOVA, analysis of variance; ATP, adenosine triphosphate.
Ponatinib induces cell apoptosis of U87MG cells
To explore whether ponatinib impacts cell viability via regulating cell apoptosis, we first examined the fraction of apoptotic cells with sub-G1 DNA content after cells were treated with ponatinib. We found that the percentage of cells with sub-G1 DNA content was elevated in a dose-dependent manner when cells were incubated with different doses of ponatinib (Figure 2A). This result suggests that ponatinib causes cell death or cell apoptosis in U87MG cells. Consistently, ponatinib induced condensed or fragmented nuclei, a typical phenotype in apoptotic cells, in U87MG cells (Figure 2B). The statistical analysis showed that the number of condensed or fragmented nuclei was increased dramatically in U87MG cells after ponatinib treatment (Figure 3C). Meanwhile, this increase was gradually induced by addition of ponatinib concentration (Figure 3C). Collectively, these results demonstrate that ponatinib induces cell apoptosis of U87MG cells.
Figure 2.
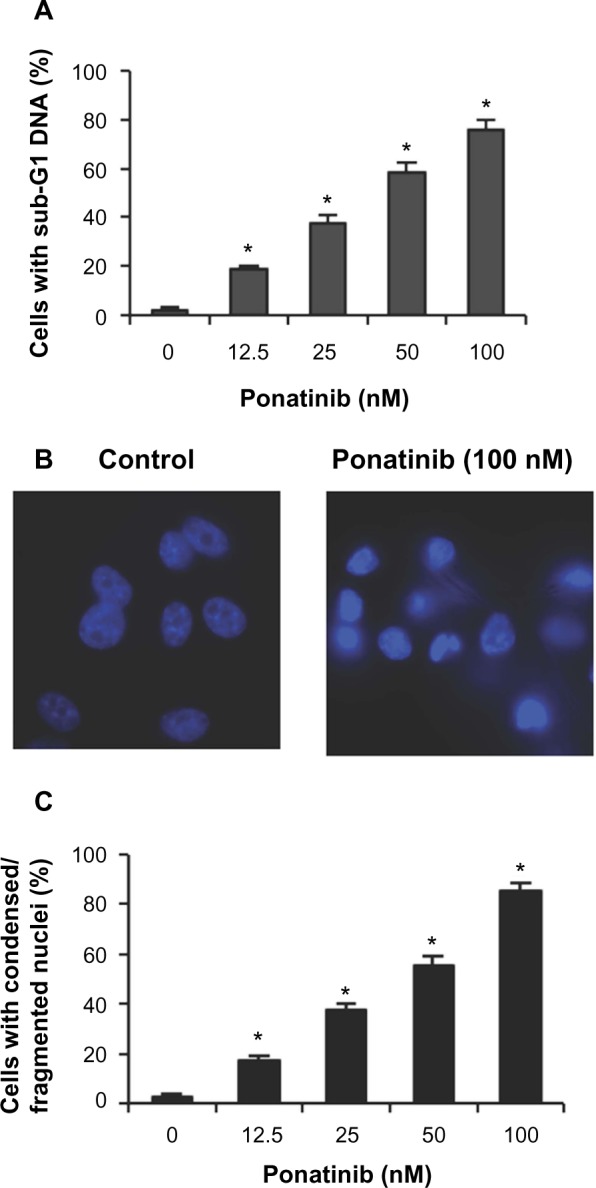
Ponatinib induces apoptosis of U87MG cells.
Notes: (A) U87MG cells were treated with ponatinib at the indicated concentrations for 48 hours, followed by propidium iodide staining and flow cytometry analysis. (B) U87MG cells were incubated with ponatinib for 48 hours. The nuclei were stained with Hoechst, and analyzed using a fluorescent microscope. The representative images are shown. (C) The percentage of cells with condensed/fragmented nuclei was calculated by counting in seven random fields. *P<0.05 (one-way ANOVA) for (A and C).
Abbreviation: ANOVA, analysis of variance.
Figure 3.
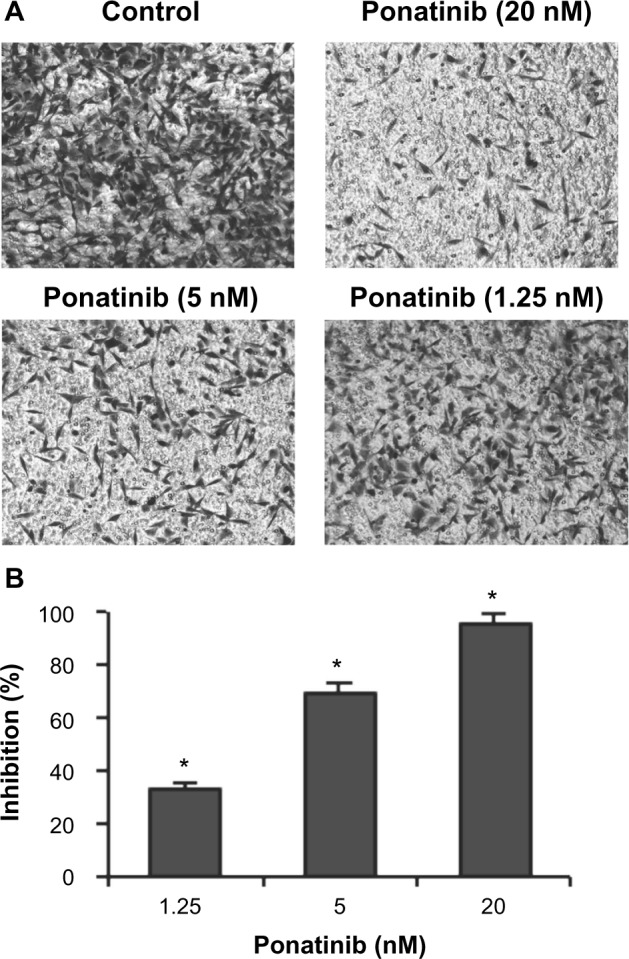
Ponatinib inhibits migration of U87MG cells.
Notes: (A) U87MG cells were treated with different concentrations of ponatinib (1.25, 5, or 20 nM) for 8 hours. The non-migrated cells on the upper surface of the filter were removed, and the migrated cells on the lower side were stained and photographed. The representative images are shown, then cells were lysed, and colorimetric determination was made at 595 nm. (B) Quantitation of the inhibition from Transwell assay. The inhibition efficiency (%) was compared with control cells without ponatinib treatment. *P<0.05 (one-way ANOVA).
Abbreviation: ANOVA, analysis of variance.
Ponatinib inhibits cell invasion in vitro
Next, we wondered whether ponatinib affects invasion ability of U87MG cells. Therefore, Matrigel invasion assays were performed using 8.0 μm pore-size transwells, which allow cell migration across the filter. The data showed that the number of U87MG cells migrated across both the Matrigel and the insert was obviously decreased as concentration of ponatinib was increased from 1.25 to 20 nM (Figure 3A). Statistically, ponatinib significantly enhanced inhibitory efficacy against migration of U87MG cells in a dose-dependent manner (Figure 3B). Therefore, the results indicate that ponatinib prevents invasion and migration of U87MG cells in vitro.
Ponatinib antagonizes tumor growth and induces cell apoptosis in vivo
Considering the obvious role of ponatinib against cancer cells in vitro, it is worth addressing whether ponatinib has similar effects in vivo. We studied the antitumor activity of ponatinib in xenograft models using U87MG cells. After treatment with ponatinib, mice tumor growth was clearly repressed in a dose-dependent manner (Figure 4A). Notably, the growth rate of tumors was significantly decreased after continuing injections of ponatinib for 13 days (Figure 4A). Consistent with reduction of tumor volume, the cell apoptosis in tumor tissues identified by TUNEL staining was increased after injection of ponatinib (Figure 4B). We also noticed that the body weight of mice was not obviously changed after ponatinib treatment (Figure 4C), suggesting ponatinib may not have other side effects to these mice. Herein, our data implicate that ponatinib antagonizes tumor growth by inducing cell apoptosis in vivo.
Figure 4.

Ponatinib suppresses glioblastoma multiforme tumor growth in vivo.
Notes: (A) After inoculation of U87MG cells, ponatinib (5 or 10 mg/kg) was injected into the mice every day. The tumors were measured every other day, and tumor volumes are shown as indicated. *P<0.05 (two-way ANOVA). (B) Ponatinib (5 or 10 mg/kg)-induced cell apoptosis in tumor tissues in vivo was measured by TUNEL assay. The representative images are shown with TUNEL signals (green) and the nuclei staining (blue). (C) The body weights of the mice during the treatments with ponatinib (5 or 10 mg/kg).
Abbreviations: ANOVA, analysis of variance; TUNEL, terminal deoxynucleotidyl transferase-mediated dUTP-digoxigenin nick end-labeling.
Discussion
GBM is one of the most common and aggressive brain tumors.1–3 Current therapies for glioblastoma consist of surgical resection, radiotherapy, and chemotherapy.20,21 For elderly patients, chemotherapy is usually employed.22,23 The most commonly used drug for elderly patients is temozolomide, which enhances MGMT promoter methylation in GBM patients.23 In addition to temozolomide, the RTK, EGFR has become a potential target for treatment because of its frequent mutations in GBM.24–26 EGFR-targeting tyrosine kinase inhibitors, including gefitinib and erlotinib, have shown some promising results in preclinical studies in GBM.27,28 However, the rate of GBM responses to gefitinib and erlotinib in patients is pretty low, which may be due to small populations with EGFR overexpression or alteration of pathways in EGFR-mutated GBMs.27,28 Therefore, other inhibitors have to be developed. Here, we reported ponatinib, a multi-targeted tyrosine kinase inhibitor, against glioblastoma cells in vitro and in vivo. We demonstrated that ponatinib efficiently decreased cell viability and promoted cell apoptosis in U87MG cells (Figures 1 and 2). Meanwhile, ponatinib inhibited cell migration in U87MG cells (Figure 3). In U87MG cell-derived mouse xenograft model, ponatinib obviously reduced tumor growth and induced tumor cell apoptosis (Figure 4). These findings revealed a role of ponatinib against glioblastoma cells both in vitro and in vivo. It has been reported that there were four different subtypes of GBMs, which had specific molecular signatures.4 Though the efficiency of ponatinib is promising in the U87MG glioblastoma cell line, its effects in specific subtypes of GBMs should be examined in future. In addition, we demonstrated the role of ponatinib in mouse xenograft model in axillary regions of mice, but did not examine its role on in situ brain tumor. Although the non-orthotopic model is widely used in glioblastoma studies,29–32 the effects of ponatinib in an orthotopic model should be further examined. Reports have also suggested that the current anti-RTK drugs such as gefitinib and erlotinib usually need high dose (1–10 μM) to induce dramatic decrease of cell viability and increase of cell apoptosis in U87MG cells;33,34 while in this study we noticed that even a very low dose of ponatinib (12.5 nM) could obviously decrease cell viability and induce cell apoptosis (Figures 1–3). These findings may implicate the different efficiency of ponatinib and other RTK inhibitors against glioblastoma cells. Thus, it is interesting to compare the efficiency of ponatinib with current anti-RTK drugs in preclinical experiments.
In chronic myelogenous leukemia (CML) and Philadelphia-positive acute lymphoblastic leukemia (Ph+ ALL), the primary target of ponatinib is BCR-ABL, an abnormal tyrosine kinase.17 BCR-ABL protein inhibits apoptosis35 and accelerates cell division in CML.36 Here, we found that ponatinib induced cell apoptosis and prevented cell growth in GBM (Figures 1–4). This behavior of ponatinib is similar to its role in CML and Ph+ ALL, suggesting that ponatinib may antagonize glioblastoma cells through a similar mechanism. There are several advantages for ponatinib as a potential drug in clinical treatment of leukemia. First, the efficiency of ponatinib against resistant or intolerant CML and Ph+ ALL has been reported.37 Second, ponatinib can be used in CML and Ph+ ALL with a variety of known mutations and no mutations.38 Considering the advantages and the similar effects of ponatinib in CML/Ph+ ALL and GBM, ponatinib may be potentially applied in resistant GBM and heterogeneous GBM with different mutations. On the other side, there are also potential disadvantages of ponatinib because of its risk in threatening blood clots and narrowing of blood vessels as reported by the US Food and Drug Administration. Thus, the application of ponatinib as a clinical drug against tumors should be carefully evaluated in future.
In conclusion, our study assessed the effects of ponatinib in reduction of cell viability and induction of cell apoptosis in a human glioblastoma cell line and in a glioblastoma cell-derived mouse xenograft model. These results support ponatinib as a chemotherapeutic option against glioblastoma cells.
Acknowledgments
We thank all other members in Dr Xiaoqun Gao’s laboratory for their technical support and helpful discussions.
Footnotes
Disclosure
The authors declare that they have no conflicts of interest in this work.
References
- 1.Fuller GN. The WHO Classification of Tumours of the Central Nervous System. 4th ed. Arch Pathol Lab Med. 2008;132(6):906. doi: 10.5858/2008-132-906-TWCOTO. [DOI] [PubMed] [Google Scholar]
- 2.Louis DN, Holland EC, Cairncross JG. Glioma classification: a molecular reappraisal. Am J Pathol. 2001;159(3):779–786. doi: 10.1016/S0002-9440(10)61750-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Okada H, Kohanbash G, Zhu X, et al. Immunotherapeutic approaches for glioma. Crit Rev Immunol. 2009;29(1):1–42. doi: 10.1615/critrevimmunol.v29.i1.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Verhaak RG, Hoadley KA, Purdom E, et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell. 2010;17(1):98–110. doi: 10.1016/j.ccr.2009.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kanu OO, Hughes B, Di C, et al. Glioblastoma multiforme oncogenomics and signaling pathways. Clin Med Oncol. 2009;3:39–52. doi: 10.4137/cmo.s1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Carrasco-Garcia E, Saceda M, Martinez-Lacaci I. Role of receptor tyrosine kinases and their ligands in glioblastoma. Cells. 2014;3(2):199–235. doi: 10.3390/cells3020199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Vlahovic G, Crawford J. Activation of tyrosine kinases in cancer. Oncologist. 2003;8(6):531–538. doi: 10.1634/theoncologist.8-6-531. [DOI] [PubMed] [Google Scholar]
- 8.Zhu H, Acquaviva J, Ramachandran P, et al. Oncogenic EGFR signaling cooperates with loss of tumor suppressor gene functions in gliomagenesis. Proc Natl Acad Sci U S A. 2009;106(8):2712–2716. doi: 10.1073/pnas.0813314106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ohgaki H, Dessen P, Jourde B, et al. Genetic pathways to glioblastoma: a population-based study. Cancer Res. 2004;64(19):6892–6899. doi: 10.1158/0008-5472.CAN-04-1337. [DOI] [PubMed] [Google Scholar]
- 10.Wong AJ, Bigner SH, Bigner DD, Kinzler KW, Hamilton SR, Vogelstein B. Increased expression of the epidermal growth factor receptor gene in malignant gliomas is invariably associated with gene amplification. Proc Natl Acad Sci U S A. 1987;84(19):6899–6903. doi: 10.1073/pnas.84.19.6899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ekstrand AJ, Sugawa N, James CD, Collins VP. Amplified and rearranged epidermal growth factor receptor genes in human glioblastomas reveal deletions of sequences encoding portions of the N- and/or C-terminal tails. Proc Natl Acad Sci U S A. 1992;89(10):4309–4313. doi: 10.1073/pnas.89.10.4309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sugawa N, Ekstrand AJ, James CD, Collins VP. Identical splicing of aberrant epidermal growth factor receptor transcripts from amplified rearranged genes in human glioblastomas. Proc Natl Acad Sci U S A. 1990;87(21):8602–8606. doi: 10.1073/pnas.87.21.8602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nagane M, Levitzki A, Gazit A, Cavenee WK, Huang HJ. Drug resistance of human glioblastoma cells conferred by a tumor-specific mutant epidermal growth factor receptor through modulation of Bcl-XL and caspase-3-like proteases. Proc Natl Acad Sci U S A. 1998;95(10):5724–5729. doi: 10.1073/pnas.95.10.5724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cichowski K, Jacks T. NF1 tumor suppressor gene function: narrowing the GAP. Cell. 2001;104(4):593–604. doi: 10.1016/s0092-8674(01)00245-8. [DOI] [PubMed] [Google Scholar]
- 15.Szerlip NJ, Pedraza A, Chakravarty D, et al. Intratumoral heterogeneity of receptor tyrosine kinases EGFR and PDGFRA amplification in glioblastoma defines subpopulations with distinct growth factor response. Proc Natl Acad Sci U S A. 2012;109(8):3041–3046. doi: 10.1073/pnas.1114033109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bianco R, Garofalo S, Rosa R, et al. Inhibition of mTOR pathway by everolimus cooperates with EGFR inhibitors in human tumours sensitive and resistant to anti-EGFR drugs. Br J Cancer. 2008;98(5):923–930. doi: 10.1038/sj.bjc.6604269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.O’Hare T, Shakespeare WC, Zhu X, et al. AP24534, a pan-BCR-ABL inhibitor for chronic myeloid leukemia, potently inhibits the T315I mutant and overcomes mutation-based resistance. Cancer Cell. 2009;16(5):401–412. doi: 10.1016/j.ccr.2009.09.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gozgit JM, Wong MJ, Wardwell S, et al. Potent activity of ponatinib (AP24534) in models of FLT3-driven acute myeloid leukemia and other hematologic malignancies. Mol Cancer Ther. 2011;10(6):1028–1035. doi: 10.1158/1535-7163.MCT-10-1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lierman E, Smits S, Cools J, Dewaele B, Debiec-Rychter M, Vandenberghe P. Ponatinib is active against imatinib-resistant mutants of FIP1L1-PDGFRA and KIT, and against FGFR1-derived fusion kinases. Leukemia. 2012;26(7):1693–1695. doi: 10.1038/leu.2012.8. [DOI] [PubMed] [Google Scholar]
- 20.Sathornsumetee S, Reardon DA, Desjardins A, Quinn JA, Vredenburgh JJ, Rich JN. Molecularly targeted therapy for malignant glioma. Cancer. 2007;110(1):13–24. doi: 10.1002/cncr.22741. [DOI] [PubMed] [Google Scholar]
- 21.Furnari FB, Fenton T, Bachoo RM, et al. Malignant astrocytic glioma: genetics, biology, and paths to treatment. Genes Dev. 2007;21(21):2683–2710. doi: 10.1101/gad.1596707. [DOI] [PubMed] [Google Scholar]
- 22.Glantz M, Chamberlain M, Liu Q, Litofsky NS, Recht LD. Temozolomide as an alternative to irradiation for elderly patients with newly diagnosed malignant gliomas. Cancer. 2003;97(9):2262–2266. doi: 10.1002/cncr.11323. [DOI] [PubMed] [Google Scholar]
- 23.Malmstrom A, Gronberg BH, Marosi C, et al. Temozolomide versus standard 6-week radiotherapy versus hypofractionated radiotherapy in patients older than 60 years with glioblastoma: the Nordic randomised, phase 3 trial. Lancet Oncol. 2012;13(9):916–926. doi: 10.1016/S1470-2045(12)70265-6. [DOI] [PubMed] [Google Scholar]
- 24.Karpel-Massler G, Schmidt U, Unterberg A, Halatsch ME. Therapeutic inhibition of the epidermal growth factor receptor in high-grade gliomas: where do we stand? Mol Cancer Res. 2009;7(7):1000–1012. doi: 10.1158/1541-7786.MCR-08-0479. [DOI] [PubMed] [Google Scholar]
- 25.Berezowska S, Schlegel J. Targeting ErbB receptors in high-grade glioma. Curr Pharm Des. 2011;17(23):2468–2487. doi: 10.2174/138161211797249233. [DOI] [PubMed] [Google Scholar]
- 26.Hegi ME, Rajakannu P, Weller M. Epidermal growth factor receptor: a re-emerging target in glioblastoma. Curr Opin Neurol. 2012;25(6):774–779. doi: 10.1097/WCO.0b013e328359b0bc. [DOI] [PubMed] [Google Scholar]
- 27.Taylor TE, Furnari FB, Cavenee WK. Targeting EGFR for treatment of glioblastoma: molecular basis to overcome resistance. Curr Cancer Drug Targets. 2012;12(3):197–209. doi: 10.2174/156800912799277557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Vivanco I, Robins HI, Rohle D, et al. Differential sensitivity of glioma-versus lung cancer-specific EGFR mutations to EGFR kinase inhibitors. Cancer Discov. 2012;2(5):458–471. doi: 10.1158/2159-8290.CD-11-0284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yang G, Han D, Chen X, et al. MiR-196a exerts its oncogenic effect in glioblastoma multiforme by inhibition of IkappaBalpha both in vitro and in vivo. Neurooncology. 2014;16(5):652–661. doi: 10.1093/neuonc/not307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bonavia R, Inda MM, Vandenberg S, et al. EGFRvIII promotes glioma angiogenesis and growth through the NF-kappaB, interleukin-8 pathway. Oncogene. 2012;31(36):4054–4066. doi: 10.1038/onc.2011.563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Milanovic D, Braun F, Weber W, Grosu AL, Behe M, Niedermann G. The influence of the combined treatment with Vadimezan (ASA404) and taxol on the growth of U251 glioblastoma xenografts. BMC Cancer. 2012;12:242. doi: 10.1186/1471-2407-12-242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Karmakar S, Banik NL, Ray SK. Combination of all-trans retinoic acid and paclitaxel-induced differentiation and apoptosis in human glioblastoma U87MG xenografts in nude mice. Cancer. 2008;112(3):596–607. doi: 10.1002/cncr.23223. [DOI] [PubMed] [Google Scholar]
- 33.Ramis G, Thomas-Moya E, Fernandez de Mattos S, Rodriguez J, Villalonga P. EGFR inhibition in glioma cells modulates Rho signaling to inhibit cell motility and invasion and cooperates with temozolomide to reduce cell growth. PloS One. 2012;7(6):e38770. doi: 10.1371/journal.pone.0038770. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 34.Learn CA, Hartzell TL, Wikstrand CJ, et al. Resistance to tyrosine kinase inhibition by mutant epidermal growth factor receptor variant III contributes to the neoplastic phenotype of glioblastoma multiforme. Clin Cancer Res. 2004;10(9):3216–3224. doi: 10.1158/1078-0432.ccr-03-0521. [DOI] [PubMed] [Google Scholar]
- 35.Bedi A, Zehnbauer BA, Barber JP, Sharkis SJ, Jones RJ. Inhibition of apoptosis by BCR-ABL in chronic myeloid leukemia. Blood. 1994;83(8):2038–2044. [PubMed] [Google Scholar]
- 36.Jonuleit T, Peschel C, Schwab R, et al. Bcr-Abl kinase promotes cell cycle entry of primary myeloid CML cells in the absence of growth factors. Br J Haematol. 1998;100(2):295–303. doi: 10.1046/j.1365-2141.1998.00564.x. [DOI] [PubMed] [Google Scholar]
- 37.Shamroe CL, Comeau JM. Ponatinib: a new tyrosine kinase inhibitor for the treatment of chronic myeloid leukemia and Philadelphia chromosome-positive acute lymphoblastic leukemia. Ann Pharmacother. 2013;47(11):1540–1546. doi: 10.1177/1060028013501144. [DOI] [PubMed] [Google Scholar]
- 38.Breccia M, Alimena G. Second-generation tyrosine kinase inhibitors (Tki) as salvage therapy for resistant or intolerant patients to prior TKIs. Mediterr J Hematol Infect Dis. 2014;6(1):e2014003. doi: 10.4084/MJHID.2014.003. [DOI] [PMC free article] [PubMed] [Google Scholar]